

Abstract
We report a series of molecular dynamics (MD) simulations of up to a microsecond combined simulation time designed to probe epigenetically modified DNA sequences. More specifically, by monitoring the effects of methylation and hydroxymethylation of cytosine in different DNA sequences, we show, for the first time, that DNA epigenetic modifications change the molecule's dynamical landscape, increasing the propensity of DNA toward different values of twist and/or roll/tilt angles (in relation to the unmodified DNA) at the modification sites. Moreover, both the extent and position of different modifications have significant effects on the amount of structural variation observed. We propose that these conformational differences, which are dependent on the sequence environment, can provide specificity for protein binding.
Keywords: DNA methylation, epigenetics, indirect readout, molecular dynamics, recognition
Abbreviations
- AFM
Atomic Force Microscopy
- DDD
Dickerson-Drew Dodecamer
- DFT
Density Functional Theory
- DNA
Deoxyribonucleic Acid
- DNMT
DNA Methyltransferase
- LINEs
Long Interspred Transposable Elements
- MD
Molecular Dynamics
- MeCP
Methylated CpG-binding proteins
- MM
Molecular Mechanics
- PBC
Periodic Boundary Conditions
- QM
Quantum Mechanics
- RDF
Radial Distribution Functions
- RESP
Restrained Electrostatic Potentials Model
- SINEs
Short Interspred Transposable Elements
- SPME
Smooth Particle-Mesh Ewald
- TET
Translocation Proteins
- WT
Wild Type
Introduction
Epigenetic modifications of DNA bases, such as cytosine methylation, hydroxymethylation, formylation, and carboxylation, have challenged the traditional view of the genetic system1,2 (see Fig. 1). These chemical modifications do not affect the base-pairing properties of the nucleotides, i.e., they leave the genetic code of the DNA sequence unchanged.3 Yet, gene expression is altered in different tissues, diseases, and developmental stages, and the modifications are passed on to offspring even through the male germ line.4
Figure 1.
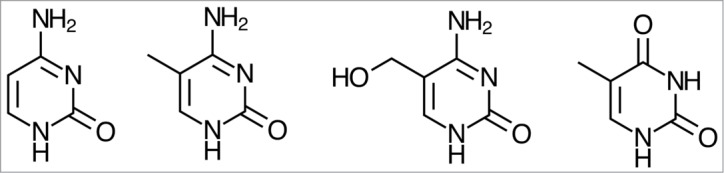
From left to right: cytosine, 5-methylcytosine (5-mC), 5-hydroxymethylcytosine (5-hmC), thymine.
The predominant (and thus far most studied) DNA modification is cytosine methylation at position C5. In mammals, such methylation occurs almost exclusively at CpG dinucleotides,1,5 and 3–6% of all cytosines in the human genome are methylated.6 CpG dinucleotides are mainly found on CpG islands, which are genomic regions with a high G-C content. These islands are associated with gene promoters and tend to be unmethylated, although several cases of tissue-specific and developmental stage-specific methylation of CpG islands have been reported.7 Methylated cytosines mainly prevail in intergenic regions, genomic repetitive elements, and highly copious transposable elements, such as long interspersed transposable elements (LINEs), short interspersed transposable elements (SINEs) and in endogenous retroviruses.5 DNA methylation is preserved after DNA replication or cell division by DNA methyltransferase 1 (DNMT1), which recognizes hemimethylated DNA and turns it into fully methylated DNA, but it can also be formed de novo by members of the DNMT3 family of enzymes.3
Another interesting cytosine modification is hydroxymethylation, which was recently discovered to also be present in significant amounts in the genome. While its functions are still unclear,8 it appears to be involved in gene regulation, and interference with hydroxymethylated cytosines can result in a variety of cancers, including myeloid cancers.9 Even though no protein has been found to interact with hydroxymethylation sites on DNA, 10-11 translocation proteins (TET) have been identified as converters of methylcytosine to hydroxymethylcytosine.10
Functionally, DNA methylation is crucial for both the developmental and adult stages of an organism. Its presence in highly repetitive sequences makes it a powerful mechanism for gene silencing, in order to maintain genome stability and integrity. In the absence of methylation in these regions, illegitimate recombination can occur, with severe mutagenic consequences for the genome, such as duplications, deletions, insertions, and translocations.1 Additionally, inactivation of transposable elements is of extreme importance, because these elements are also highly mutagenic and can self-replicate and be inserted in other regions of the genome, most likely disrupting essential genes for normal cellular function.5
It is believed that DNA methylation leads to gene silencing by 2 different mechanisms: first, methylated CpG-binding proteins (MeCP1 and 2) recognize and bind methylated CpG dinucleotides and themselves alter transcription due to their transcription repression domain. They also recruit other factors involved in chromatin condensation.5 Additionally, methylated CpG nucleotides may directly inhibit the binding of transcription factors and alter gene expression.11 Nevertheless, the precise mechanisms by which epigenetic modifications exert their effects are unclear. For example, adding a methyl group to cytosine alters the hydrophobicity of the major groove and also provides steric hindrance for certain DNA-binding proteins. Crystallographic data suggests C5-methylation has small effects on the overall double-helix structure,12 although IR spectroscopy has demonstrated that C5-methylation shifts the equilibrium between different backbone sugar pucker conformations of DNA in solution.13 Therefore, the main aim of this work is to understand the effects of cytosine modification on the fundamental structural properties of DNA before binding to proteins or other ligands. This is a problem of major biological importance, as the reshaping of DNA methylation patterns throughout the genome is connected to a variety of human diseases, including (but not limited to) imprinting syndromes, autoimmune diseases, and cancer.5 For instance, hypomethylation of transposable elements and hypermethylation of CpG islands flanking gene promoters involved in cell cycle regulation are examples of deregulation of methylation in cancer.6
By now, it is well known that DNA is not a static molecule, but rather a polymorphic one, with access to many energetically similar conformations, even at the base-pair level (although this structural diversity is sequence-dependent).14 That is, the DNA double helix is highly dynamic, a property that is needed for many important cellular events, such as DNA replication, transcription and packaging.15 Consequently, a meaningful conformational analysis of both native and modified DNA requires sampling of the conformational space available to this molecule. Moreover, the binding of proteins to DNA is entirely dependent on its deformability, in that conformational changes and the sequence-dependent twist are essential for the sequence-specific recognition and binding of DNA by several proteins.16
A common challenge when trying to model such conformational diversity is the lack of force field parameters for modified nucleotides that are compatible with generally used biomolecular force fields. For this reason, the number of molecular dynamics (MD) studies with modified DNA is limited. Nevertheless, important insight has come from studies where protein/DNA complexes were analyzed. These studies have shown that cytosine methylation destabilizes nucleosome stability by altering DNA stiffness,17 and another study found that methylating CpG at the major groove could enhance K50-class homeodomain PITX2 binding.18 However, while such studies of modified DNA in complex with proteins are clearly crucial, it is equally important to obtain more detailed insight into the effect of epigenetic modifications on the structure of the free (uncomplexed) DNA. Such studies can show if there are a priori important differences between the normal and the modified DNA that could lead to a different recognition and hence regulatory patterns.
In the present work, we have conducted an extensive study of methylated and hydroxymethylated DNA, performing molecular dynamics (MD) simulations on DNA dodecamers and on a 20 bp long sequence both with and without modifications. The use of MD simulations to sample the conformational space of the DNA made it clear that the methylated (and hydroxymethylated) cytosines have a marked effect on the DNA structural parameters. This effect was most pronounced for symmetrically methylated CpG dinucleotides, which show different values for the twist, roll and tilt parameters near the methylation sites in relation to the unmodified DNA structure. We propose that increased flexibility and accessibility at the modification regions can have important consequences for the recognition of modified cytosines by MeCPs, transcription factors, and other elements involved in gene silencing.
Results
Considerations about the methods employed
A growing interest in the use of theoretical models to obtain a better understanding of DNA structure and its dynamics in general and, in particular, the influence of epigenetic modifications exists.17,19 However, the ability of available methods to represent experimental data with structural accuracy is still unclear. There have been many elegant recent studies of DNA base pairing using fully quantum chemical (DFT)20,21 and hybrid quantum classical (QM/MM) approaches,22 as well as classical force field descriptions of DNA (MD simulations).23,24 In the present work, we are particularly interested in exploring how the dynamical behavior of DNA is potentially affected by epigenetic modifications. Therefore, in order to have an ensemble of structures that most correctly describes such a flexible molecule as DNA, all subsequent work was done with purely classical MD simulations. This approach allows for proper conformational sampling of the DNA strands and for concerted motions of the DNA to be studied.25 Moreover, when performing MD simulations, it is necessary to take into account that the unrestrained conditions that are applied during the simulation lead to a greater elasticity of the terminal part of the structure (the so called “end-effect”26). For these reasons, small oligomers, for example trinucleotides, are inadequate models to predict the reliability of the different methods or whether epigenetic modifications affect the structure of DNA.
DNA flexibility and the differences between WT and modified DNA
DNA is a polymorphic molecule; thus, conformational sampling is required to correctly describe its conformational flexibility. For this reason, we performed most of our analysis on a system large enough to exhibit conformational flexibility without end-effects biasing the simulation outcome.23 Specifically, we started with the well-studied DDD (Dickerson-Drew Dodecamer) structure,27 containing modifications at different cytosines, as illustrated in Figure 2. For these molecules, the initial coordinates were obtained from the reported crystal structures, except for the 3,9-DNA structure that was modeled according to the Methodology section (see also Fig. 2). We also modeled an additional sequence that was previously subjected to AFM experiments to uncover the effect of methylation on strand separation28 (5’-CCGAGATATCCGCACCAACG-3′). The MD trajectories were then analyzed using the 3DNA software, as outlined in the Methods section. A summary of the simulations performed in this work can be found in Table S1.
Figure 2.
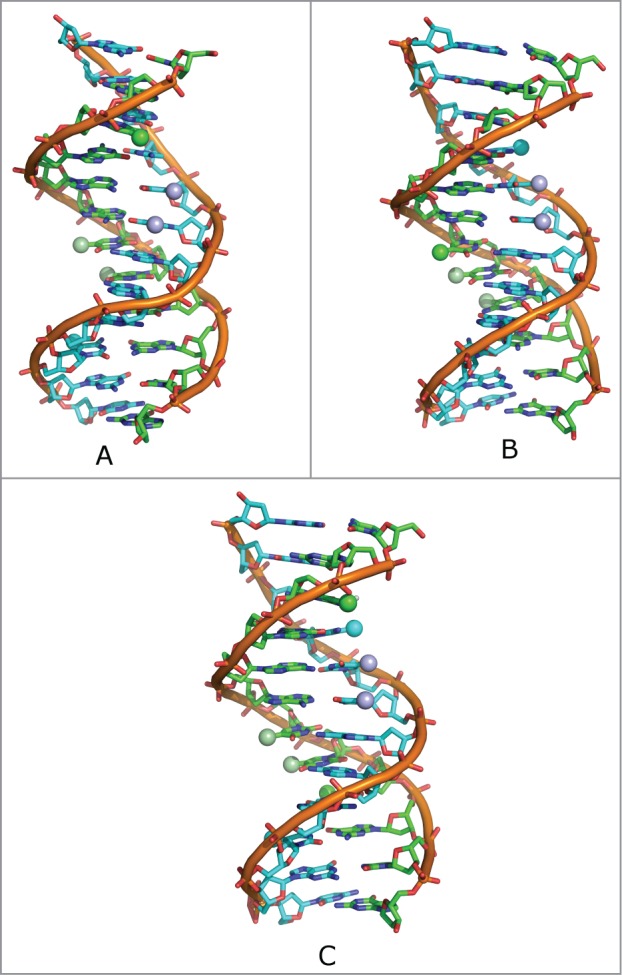
Top: Schematic representation of DNA dodecamer, depicting the positions of the methylated cytosines on both DNA strands (blue and green spheres). Shown here are: (A) 3-DNA, (B) 9-DNA, and (C) 3,9-DNA. The thymines are also depicted in violet and pale green.
Figure 3 shows the frequency of the obtained values for the base step parameters for the unmethylated DD structure and the structure methylated at the 3rd and 9th position (3,9-DNA). Note that Zgarbóva and coworkers have recently explored the impact of base-pair fraying in molecular dynamics simulations of DNA and RNA,29 highlighting the challenges involved in obtaining an accurate physical description of terminal residues using contemporary force fields. Although we did not observe any compromising end-fraying effects on the timescale of our simulations, except for the standard faster base-opening, we have for safety removed the 2 base pairs (bps) at the ends (1 and 12) from the analysis. Similarly, although we show them in the graphs, we did not consider the differences in the bps 2 and 11, due to their proximity to the ends.
Figure 3.
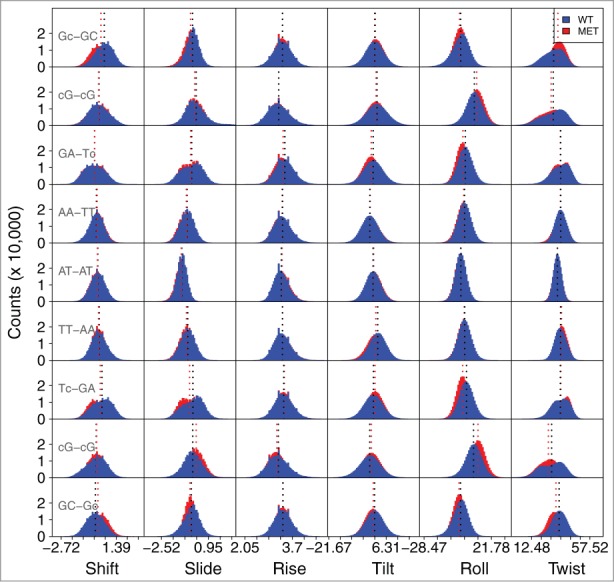
Frequency of values for the base step parameters for the unmethylated DD structure, and for the structure methylated at the 3rd and 9th position (3-DNA). The base step values for the 5′and 3′termini are not shown. Due to space constraints on the figure, the 5-mC modification is represented by just the lowercase letter c. The shift, slide and rise parameters are presented in Ångström, and the tilt, roll and twist in degrees. The average values are depicted by the dotted lines. The corresponding average values for each parameter and their standard deviations are present in Table S4.
The central A-T regions (AA/TT, AT-AT, TT-AA) are almost identical in every parameter, between all structures. They display a smaller range of values than any of the other bp steps, meaning that they only sample a limited region of conformational space, in accordance to the stiffness previously reported for TA bps.25 However, there are differences in the steps flanking the modified base pair compared to the same bases in the WT. This is specifically seen in the twist parameter for the 3-DNA structure (Fig. S1, Table S2), which defines the overall flexibility of the oligomer, and is associated with the winding/unwinding of the helix.30 The methylated structure displays an average twist value that is ∼3° lower for the 5-mCG-GC and CG-5-mCG steps comparing to the corresponding steps in the unmethylated structure. This local unwinding has been reported for other modified bases, e.g., in 2 crystal structures with N6-methylated adenine residues, these bps were unwound by roughly 2° in comparison to the unmodified DNA or in O6-alkylated guanine lesions, the guanine dimers showed strong unwinding (5 to 6°).31 For the 9-DNA structure (Fig. S2, Table S3), we observe a smaller variation in the twist value. However, there is a ∼2–3° difference in the roll for the GA-T5-mC and T5-mC-GA steps. The 3,9-DNA structure (Fig. 3, Table S4) presents differences in both the twist and roll angles. It presents similar differences to the sum of the 3-DNA and the 9-DNA structures, but also shows ∼2 to 3° differences in the roll of the G5-mC-5-mCG steps and differences of ∼2 to 3° in the tilt for the GA-T5-mC and T5-mC-GA.
Regarding the local base pair parameters, most of them show no differences between the WT and modified DNA on this timescale, except for the buckle and the propeller twist, which show small differences near the modification sites. The differences are smaller than those observed for the base step parameters. Figs. S3-S5 and Tables S5-S7 show the obtained values of the local base parameters for 3*100ns simulations of the WT and methylated sequences at position 3, 9 and both 3 and 9 (see Fig. 2). Also, Figs. S6-S9 and Tables S8-S11 show the values for the base step parameters and local parameters for the hydroxymethylated structures at position 3 and 9.
The 3-DNA and 9-DNA structures are different with regard to the twist angle. Specifically, the 3-DNA structure shows significant local unwinding near the methylation position, whereas the 9-DNA structure behaves mostly as the unmodified structure with regard to this parameter (Fig. S1 and S2, and Tables S2 and S3). The atomic level explanation for this relies on the relative position of the methyl groups at the major groove of DNA. The 3-DNA has the methyl group of the modified nucleotide in one strand, whereas the methyl groups of the thymine bases 7 and 8 are on the other strand. Although these residues are too far away to directly interact, there is a change in the hydration patterns that leads to localized untwisting of the DNA, since the methyl group in the modified base pair creates a hydrophobic region on the DNA (the corresponding RDF of which is shown in Fig. 4), followed by a more hydrated region (Fig. 5). Note that the methyl hydrogens are still able to interact with water molecules; however the water molecules are further away from the base. This is visible in the RDF of the H41 hydrogen (one of the amine hydrogen atoms), which in the unmodified structure has a much higher probability to interact with a water molecule than for the methylated sequence. The DNA molecule seems to unwind in order to accommodate the change in solvation.
Figure 4.

Top: Radial distribution functions for the added cytosine methyl group (red) in the 3-DNA sequence and the corresponding hydrogen in the unmodified base (black). Integration of the first peak for the 3-DNA structure results in 10 water molecules, whereas the first peak for unmethylated corresponds to 7 water molecules. The difference is caused by the water molecule's CH…O dipole-dipole interactions with the methyl group. Bottom: Radial distribution functions for one of the amino hydrogens (atom H41) in the methylated (red) and unmodified (black) cytosine in the 3-DNA sequence. Compared to the methylated sequence, the WT amino hydrogen has a much higher probability of interacting with a water molecule.
Figure 5.
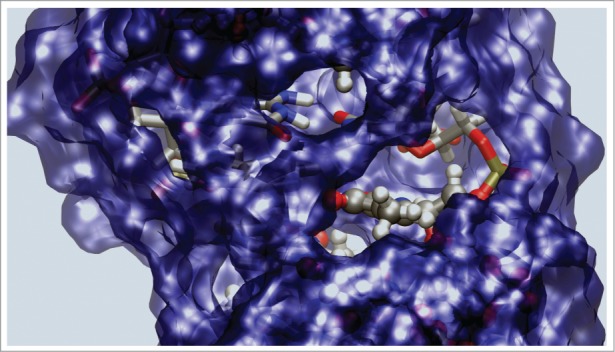
Surface representation of the water molecules within 3.5 Å from the DNA. This figure shows both the hydrophobic region created by the extra methyl group, and the hydrophobic regions already present and created by the 2 methyl groups of the thymine bases.
This effect is not observed for the 9-DNA structure, since both the methylated base and the 2 thymine bases are in the same strand, and then the twisting of the structure would not lead to any proximity between the hydrophobic regions (Fig. 2). The opposite scenario occurs with regard to the roll and tilt parameters, where we observe differences in the 9-DNA structure and in 3,9-DNA structures, but not in the 3-DNA structure. This local bending can also be a consequence of the proximity of the methyl groups of the modified cytosine bases and of the thymine bases lying in the major groove (Fig. 2).
The central part of the dodecamer is an A-tract with 4 A/T base pairs, a sequence that has its own intrinsic properties including a fully methylated major groove.32,33 We have previously shown that continuous major groove methylation affects both the structure and dynamics of dsDNA by improving base stacking and probably also by modulating water binding.34-36 Thus, it might be of significance that methylation of d(CGCGAATTCGCG)2 at position 9 creates a 6 bases long spine of major groove methyl groups, that double-methylation at positions 3 and 9 provides 8 consecutive methyl groups, and that the methyl group afforded by single methylation at position 3 remains isolated (Fig. 2).
For the hydroxymethylated structures, we observe similar profiles as for the methylated structures. Furthermore, our simulations also allow us to probe the preferred orientation of the hydroxymethyl group: DFT predicts a slight preference for the OH group to point in 3’ over 5’ and an optimized C6-C5-C5A-O5 torsion angle of 118.4°, while our MD simulations show this angle to vary between 85 and 120° over 100 ns of simulation. This is in good agreement with the experimental and theoretical results of Renciuk et al.,12 who reported values between 72 and 133° using X-ray diffraction methods.
These results indicate that DNA modifications can increase DNA flexibility and alter the twisting angle of the structure if the methylation is in a different strand than a neighboring thymine. Large twist fluctuations for CG-CG steps have been reported before23 and have been shown to be sequence dependent. Finally, if the methylation is in the same strand as a neighboring thymine we observe differences in roll and tilt, which are related to DNA bending. From our analysis it seems that these small chemical modifications (methylation and hydroxymethylation of cytosines) lead to increased structural diversity in the roll, tilt and twist parameters, which are related to unwinding and bending of the DNA. This in principle can modulate the affinity of the DNA to proteins and hence have a role in protein-DNA recognition.
For the 20 bp structure we found that methylation leads again to modifications in the twist and roll/tilt parameters near the modified sites (Fig. 6 and Table S12). However, now the pattern is more complicated than before, since now there are more neighboring thymine residues, which leads to changes between the modified and unmodified sequences further away from the central methylated CpG. Direct comparison of these sequences with protein DNA complexes is not possible, since in all the crystals of protein-methylated DNA complexes the methylated nucleotide is flipped.37-39 However, indirect readout or shape (topography) recognition is a well-known mechanism for DNA recognition.40 This was further corroborated by Hansen et al., who showed that DNA shape is under evolutionary selection pressure.40,41 Consequently, it is feasible that the differences in DNA shape upon methylation observed by us can influence protein-DNA recognition and help to clarify the structural and mechanistic details of epigenetics.
Figure 6.
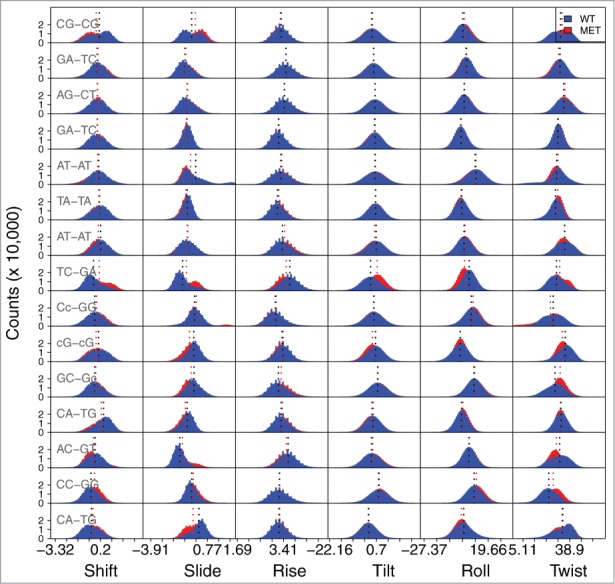
Frequency of values for the local base parameters for the d(5’- CCGAGATATCCGCACCAACG-3’)2 and the corresponding methylated sequence. The base step values for the 2 last bps at the 5′and 3′ termini are not shown. Due to space constraints on the figure, the 5-mC modification is represented by just the lowercase letter c. The shift, slide and rise parameters are expressed in Ångström, and the tilt, roll and twist parameters in degrees. The average values are depicted by the dotted lines. The values for the average and standard deviations of each parameter are presented in Table S12.
Discussion
The regulation of gene expression by DNA methylation is of great importance, since it is required for maintaining genomic stability in cells. Changes in methylation patterns can give rise to dreadful consequences for the cell (and, therefore by extension, the organism), and are involved in many human diseases, including cancer. Studies of DNA methylation have provided insightful information about the factors underlying the establishment and maintenance of methylation in mammalian cells,42 but corresponding work aimed at obtaining a fundamental understanding of how the intrinsic behavior of DNA is influenced by such modifications has been limited.
The use of theoretical methods to describe complex biomolecules such as DNA is becoming increasingly common,19,25,43-46 with theory providing further insight into the interpretation of the experimental data. One example of this is the work of Schulten and co-workers on DNA strand separation.8,47 These authors combined single molecule force microscopy experiments with steered MD simulations and showed that cytosine methylation and hydroxymethylation alter the mechanical properties of DNA, thus facilitating strand separation.8,47 However, although they identified this effect, they did not determine its underlying mechanism. Another important example is a recent study of methylation in CpG-rich sequences, which showed that methylation lowers the energy difference between the B- and Z-DNA conformations for these highly repetitive sequences.48 However, the authors again did not find any apparent significant differences in structure that accounted for this energy difference.
In the present work, we report the structural dynamics of 5-methylcytosine and 5-hydroxymethylcytosine DNA sequences. Overall, the employment of force field and molecular dynamics approaches appears to provide a better representation of the experimental data to when compared to the static description provided by hybrid QM/MM approaches when compared to a static description. The flexibility and deformability of native DNA has been extensively studied,16,25,46,49 and it is well established that both features are sequence-dependent, with base composition and the presence of specific sequences substantially contributing to the malleability of the double helix.16,50 In the present work, the different conformations that DNA can adopt were evaluated by measuring relevant base pair parameters during our simulations. Previous work has shown that the AA/TT and GA/TC steps are considered to be the ones with less variability.15 The obtained data from our MD simulations concerning all the modified structures are consistent with this observation as far as the AA/TT step is concerned, which shows no variability at all between the different structures. However, the 5mCG-CG step appears to be a variable feature between structures, especially as far as the twisting angle is concerned with 3′GA. The CA/T5m-C roll and tilt parameters are a variable feature with 3′T. This strongly suggests that the cytosine modifications do in fact have an effect on the conformational state of the steps as well as on the overall structure.
These differences in the conformational flexibility of DNA were even more pronounced when we created a symmetrically methylated CpG dinucleotide-containing structure. From our point of view, the study of this structure was of extreme importance, since it contains a suitable amount of methylated cytosines to properly represent cellular patterns of methylation. The two consecutive methylations affected the step conformations in the same way as the combination of the single methylations, except for the roll parameter. In this last case, the differences due to the unmodified structure are more pronounced than in the 9-DNA structure. We do not believe that the observed differences are due to end-effects. Otherwise, we would not observe consistent results for the different types of structural modifications examined (i.e., methylated and hydroxymethylated DNA).
The comparison of results between the 2 doubly modified structures, with different positions of modifications, confirms that DNA conformational flexibility depends on the presence of particular sequences, and that the neighboring steps influence the conformation adopted by each step. Thus, static structures do not provide a good description of such a flexible molecule as DNA, and a proper description of the behavior of such systems must take their flexibility into account. Techniques that report static structures, such as purely QM optimization or X-ray crystallography12 may therefore miss key differences in the base-pair helical parameters between non-modified and modified structures. In this study, we show unprecedented results demonstrating that modified structures have the propensity to adopt different conformations, which can hypothetically have an impact on recognition of modified cytosines by important proteins, such as DNMTs and MeCPs. Finally, this augmentation in flexibility may be an aid for chromatin condensation, which is a process also connected to methylated cytosines and gene silencing.1,3,5,42
Methods
Initial system setup
Atomic coordinates of the wild-type, methylated and hydroxymethylated DNA dodecamers examined in this work, based on Dickerson's well-studied self-complementary d(5’-CGCGAATTCGCG-3’)2 sequence,27 were obtained from X-ray structures deposited in the Protein Data Bank12 (PDB IDs: 1BNA, 4GJU, 4GLG, 4GLH and 4GLC). The 4GJU structure is methylated in the position 3 of each strand and the 4GLG structure is methylated in each position 9. We also constructed an additional structure methylated in positions 3 and 9 (4 methylations), this structure mimics 2 symmetrical methylated CpG dinucleotides, as it occurs in the cells. For simplicity, in the text, we refer to the 4GJU structure as 3-DNA, and 4GLG structure as 9-DNA, and the combined structure 3,9-DNA. We also analyzed an additional sequence (5’-CCGAGATATCCGCACCAACG-3′)′and the corresponding methylated one (5’-CCGAGATATCmCGCACCAACG-3’), which were initially modeled using the X3DNA program.
Calculation of point charges
All QM calculations were performed with the Gaussian09 simulation package,51-53 with HF as well as the 6–31G(d,p) basis set.54-56 Charges for the methylated and hydroxymethylated bases were obtained using the restrained electrostatic potentials method (RESP), in accordance with the traditional AMBER charge derivation methodology57 (Tables S13 and S14).
Molecular dynamics simulations
All molecular dynamics (MD) simulations were performed using the GROMACS simulation package58 and the amber parmbsc0 force field.59 This force field was specifically chosen because it has an improved description of the α/γ-concerted rotation in nucleic acids compared to simple parm99.60 For a more detailed discussion on available force fields for nucleic acids please consult Sponer et al.61 The DNA models were enclosed in a cubic box with a volume of 68.47 nm3 (2.403 nm × 2.901 nm × 4.496 nm) containing TIP3P water molecules62 as well as Na+ and Cl− counter ions to create a neutral electrostatic environment. Electrostatic interactions were accounted for by using the fast Smooth Particle-Mesh Ewald (SPME) method,63 with a 10Å cutoff being employed to both electrostatic and van der Waals interactions.
We prepared our system by performing an initial brief energy minimization with 200 steps of the full simulation system, followed by 2 500ps equilibration runs. The first equilibration run was performed in a canonical ensemble with the standard velocity rescaling thermostat64, and the second equilibration run was conducted in the isothermal-isobaric ensemble using the Berendsen thermostat.65 The temperature and pressure coupling during the simulations were 300K and 1 atm, respectively, and periodic boundary conditions (PBC) were used. Subsequent production runs were performed with no restraints and in an isothermal-isobaric ensemble, using the Parrinello-Rahman barostat.66 All simulations were performed using a 2 fs stepsize. We have sampled each structure for 100 ns based on previous work by Pérez et al.67 While it is possible that this is still inadequate simulation time to obtain fully convergent results on these systems, these authors found that a shorter 100 ns simulation had a self-similarity index of 0.92 to 0.99 to a very long simulation of 1.2 μs, so it could correctly describe the DNA flexibility.67 Finally, we repeated each simulation 3 times, with different initial velocities. The combined total simulation time for all our systems was therefore 2.1 μs (for a full list of simulations, see Table S1).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the Swedish National Infrastructure for Computing (SNIC) for generous allocation of computer time (SNIC grant 025/12–10). This work was supported by calculations performed on the Triolith and Tintin clusters at the National Supercomputing Center in Linköping and UPPMAX in Uppsala respectively. JAP is grateful for access to Advanced Research Computing at Cardiff (ARCCA) facilities.
Funding
This work was supported by the Swedish Research Council (Vetenskapsrådet, 2010–5026) and funding from the Sven and Ebba Christina Hagbergs Foundation.
References
- 1. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128:669-81; PMID:17320505; http://dx.doi.org/ 10.1016/j.cell.2007.01.033 [DOI] [PubMed] [Google Scholar]
- 2. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333:1300-3; PMID:21778364; http://dx.doi.org/ 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robertson KD. DNA methylation and chromatin - unraveling the tangled web. Oncogene 2002; 21:5361-79; PMID:12154399; http://dx.doi.org/ 10.1038/sj.onc.1205609 [DOI] [PubMed] [Google Scholar]
- 4. Soubry A, Hoyo C, Jirtle RL, Murphy SK. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. Bioessays 2014; 36:359-71; PMID:24431278; http://dx.doi.org/ 10.1002/bies.201300113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005; 6:597-610; PMID:16136652; http://dx.doi.org/ 10.1038/nrg1655 [DOI] [PubMed] [Google Scholar]
- 6. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007; 8:286-98; PMID:17339880; http://dx.doi.org/ 10.1038/nrg2005 [DOI] [PubMed] [Google Scholar]
- 7. Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321:209-13; PMID:2423876; http://dx.doi.org/ 10.1038/321209a0 [DOI] [PubMed] [Google Scholar]
- 8. Severin PMD, Zou X, Schulten K, Gaub HE. Effects of cytosine hydroxymethylation on DNA strand separation. Biophys J 2013; 104:208-15; PMID:23332073; http://dx.doi.org/ 10.1016/j.bpj.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010; 468:839-43; PMID:21057493; http://dx.doi.org/ 10.1038/nature09586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu H, Zhang Y. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells. Cell Cycle 2011; 10:2428-36; PMID:21750410; http://dx.doi.org/ 10.4161/cc.10.15.16930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clark SJ, Harrison J, Molloy PL. Sp1 binding is inhibited by (m)Cp(m)CpG methylation. Gene 1997; 195:67-71; PMID:9300822; http://dx.doi.org/ 10.1016/S0378-1119(97)00164-9 [DOI] [PubMed] [Google Scholar]
- 12. Renciuk D, Blacque O, Vorlickova M, Spingler B. Crystal structures of B-DNA dodecamer containing the epigenetic modifications 5-hydroxymethylcytosine or 5-methylcytosine. Nucleic Acids Res 2013; 41:9891-900; PMID:23963698; http://dx.doi.org/ 10.1093/nar/gkt738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banyay M, Gräslund A. Structural effects of cytosine methylation on DNA sugar pucker studied by FTIR. J Mol Biol 2002; 324:667-76; PMID:12460569; http://dx.doi.org/ 10.1016/S0022-2836(02)01104-X [DOI] [PubMed] [Google Scholar]
- 14. Maehigashi T, Hsiao C, Woods KK, Moulaei T, Hud NV, Williams LD. B-DNA structure is intrinsically polymorphic: even at the level of base pair positions. Nucleic Acids Res 2012; 40:3714-22; PMID:22180536; http://dx.doi.org/ 10.1093/nar/gkr1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Travers AA. The structural basis of DNA flexibility. Philos Trans A Math Phys Eng Sci 2004; 362:1423-38; PMID:15306459; http://dx.doi.org/ 10.1098/rsta.2004.1390 [DOI] [PubMed] [Google Scholar]
- 16. Fujii S, Kono H, Takenaka S, Go N, Sarai A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res 2007; 35:6063-74; PMID:17766249; http://dx.doi.org/ 10.1093/nar/gkm627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Portella G, Battistini F, Orozco M. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput Biol 2013; 9:e1003354; PMID:24278005; http://dx.doi.org/ 10.1371/journal.pcbi.1003354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang S-Y, Yang X-L, Yao L-F, Wang H-B, Sun C-K. Effect of CpG methylation on DNA binding protein: molecular dynamics simulations of the homeodomain PITX2 bound to the methylated DNA. J Mol Graph Model 2011; 29:920-7; PMID:21498098; http://dx.doi.org/ 10.1016/j.jmgm.2011.03.003 [DOI] [PubMed] [Google Scholar]
- 19. Yusufaly TI, Li Y, Olson WK. Five-Methylation of cytosine in CG:CG base-pair steps: a physicochemical mechanism for the epigenetic control of DNA nanomechanics. J Phys Chem B 2013; 117:16436-42; PMID:24313757; http://dx.doi.org/ 10.1021/jp409887t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poater J, Swart M, Bickelhaupt FM, Fonseca Guerra C. B-DNA structure and stability: the role of hydrogen bonding, π-π stacking interactions, twist-angle, and solvation. Org Biomol Chem 2014; 12:4691-700; http://dx.doi.org/ 10.1039/c4ob00427b [DOI] [PubMed] [Google Scholar]
- 21. Barone G, Fonseca Guerra C, Bickelhaupt FM. B-DNA structure and stability as function of nucleic acid composition: dispersion-corrected DFT study of dinucleoside monophosphate single and double strands. ChemistryOpen 2013; 2:186-93; PMID:24551565; http://dx.doi.org/ 10.1002/open.201300019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garrec J, Patel C, Rothlisberger U, Dumont E. Insights into intrastrand cross-link lesions of DNA from QM/MM molecular dynamics simulations. J Am Chem Soc 2012; 134:2111-9; PMID:22200321; http://dx.doi.org/ 10.1021/ja2084042 [DOI] [PubMed] [Google Scholar]
- 23. Lavery R, Zakrzewska K, Beveridge D, Bishop TC, Case DA, Cheatham T, Dixit S, Jayaram B, Lankas F, Laughton C, et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res 2010; 38:299-313; PMID:19850719; http://dx.doi.org/ 10.1093/nar/gkp834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pérez A, Luque FJ, Orozco M. Frontiers in molecular dynamics simulations of DNA. Acc Chem Res 2012; 45:196-205 [DOI] [PubMed] [Google Scholar]
- 25. Lankas F, Sponer J, Langowski J, Cheatham TE. DNA basepair step deformability inferred from molecular dynamics simulations. Biophys J 2003; 85:2872-83; PMID:14581192; http://dx.doi.org/ 10.1016/S0006-3495(03)74710-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dixit SB, Beveridge DL, Case DA, Cheatham TE, Giudice E, Lankas F, Lavery R, Maddocks JH, Osman R, Sklenar H, et al. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys J 2005; 89:3721-40; PMID:16169978; http://dx.doi.org/ 10.1529/biophysj.105.067397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE. Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci USA 1981; 78:2179-83; http://dx.doi.org/ 10.1073/pnas.78.4.2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou X, Ma W, Solov’yov IA, Chipot C, Schulten K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012; 40:2747-2758; PMID:22110028; http://dx.doi.org/ 10.1093/nar/gkr1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zgarbová M, Otyepka M, Sponer J, Lankas F, Jurečka P. Base pair fraying in molecular dynamics simulations of DNA and RNA. J Chem Theory Comput. 2014; 10:3177-89; http://dx.doi.org/ 10.1021/ct500120v [DOI] [PubMed] [Google Scholar]
- 30. Gardiner EJ, Hunter CA, Packer MJ, Palmer DS, Willett P. Sequence-dependent DNA Structure: A database of octamer structural parameters. J Mol Biol 2003; 332:1025-35; PMID:14499606; http://dx.doi.org/ 10.1016/j.jmb.2003.08.006 [DOI] [PubMed] [Google Scholar]
- 31. Gorin AA, Zhurkin VB, Wilma K. B-DNA twisting correlates with base-pair morphology. J Mol Biol 1995; 247:34-48; PMID:7897660; http://dx.doi.org/ 10.1006/jmbi.1994.0120 [DOI] [PubMed] [Google Scholar]
- 32. Sandström K, Wärmländer S, Gräslund A, Leijon M. A-tract DNA disfavours triplex formation. J Mol Biol 2002; 315:737-48; PMID:11812143; http://dx.doi.org/ 10.1006/jmbi.2001.5249 [DOI] [PubMed] [Google Scholar]
- 33. Sandström K, Wärmländer S, Bergman J, Engqvist R, Leijon M, Gräslund A. The influence of intercalator binding on DNA triplex stability: correlation with effects on A-tract duplex structure. J Mol Recognit 2004; 17:277-85; PMID:15227636; http://dx.doi.org/ 10.1002/jmr.665 [DOI] [PubMed] [Google Scholar]
- 34. Wärmländer S, Sponer JE, Sponer J, Leijon M. The influence of the thymine C5 methyl group on spontaneous base pair breathing in DNA. J Biol Chem 2002; 277:28491-7; http://dx.doi.org/ 10.1074/jbc.M202989200 [DOI] [PubMed] [Google Scholar]
- 35. Wärmländer S, Sen A, Leijon M. Imino proton exchange in DNA catalyzed by ammonia and trimethylamine: evidence for a secondary long-lived open state of the base pair. Biochemistry 2000; 39:607-15; http://dx.doi.org/ 10.1021/bi991863b [DOI] [PubMed] [Google Scholar]
- 36. Lankas F, Cheatham TE, Spackova N, Hobza P, Langowski J, Sponer J. Critical effect of the N2 amino group on structure, dynamics, and elasticity of DNA polypurine tracts. Biophys J 2002; 82:2592-609; PMID:11964246; http://dx.doi.org/ 10.1016/S0006-3495(02)75601-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hashimoto H, Horton JR, Zhang X, Cheng X. UHRF1, a modular multi-domain protein, regulates replication-coupled crosstalk between DNA methylation and histone modifications. Epigenetics 2009; 4:8-14; PMID:19077538; http://dx.doi.org/ 10.4161/epi.4.1.7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rechkoblit O, Delaney JC, Essigmann JM, Patel DJ. Implications for damage recognition during Dpo4-mediated mutagenic bypass of m1G and m3C lesions. Structure 2011; 19:821-32; PMID:21645853; http://dx.doi.org/ 10.1016/j.str.2011.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wojciechowski M, Czapinska H, Bochtler M. CpG underrepresentation and the bacterial CpG-specific DNA methyltransferase M.MpeI. Proc Natl Acad Sci USA 2013; 110:105-10; http://dx.doi.org/ 10.1073/pnas.1207986110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. Origins of specificity in protein-DNA recognition. Annu Rev Biochem 2010; 79:233-69; PMID:20334529; http://dx.doi.org/ 10.1146/annurev-biochem-060408-091030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parker SCJ, Hansen L, Abaan HO, Tullius TD, Margulies EH. Local DNA topography correlates with functional noncoding regions of the human genome. Science 2009; 324:389-92; PMID:19286520; http://dx.doi.org/ 10.1126/science.1169050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ooi SKT, O’Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci 2009; 122:2787-91; PMID:19657014; http://dx.doi.org/ 10.1242/jcs.015123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Araúzo-Bravo MJ, Fujii S, Kono H, Ahmad S, Sarai A. Sequence-dependent conformational energy of DNA derived from molecular dynamics simulations: toward understanding the indirect readout mechanism in protein-DNA recognition. J Am Chem Soc 2005; 127:16074-89; PMID:16287294; http://dx.doi.org/ 10.1021/ja053241l [DOI] [PubMed] [Google Scholar]
- 44. Cheatham TE, Kollman PA. Molecular dynamics simulations highlight the structural differences among DNA:DNA, RNA:RNA, and DNA:RNA hybrid duplexes. J Am Chem Soc 1997; 119:4805-25; http://dx.doi.org/ 10.1021/ja963641w [DOI] [Google Scholar]
- 45. Xiao S, Zhu H, Wang L, Liang H. DNA conformational flexibility study using phosphate backbone neutralization model. Soft Matter 2014; 10;1045-55; PMID:24983118; http://dx.doi.org/ 10.1039/c3sm52345d [DOI] [PubMed] [Google Scholar]
- 46. Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci USA 1998; 95:11163-8; http://dx.doi.org/ 10.1073/pnas.95.19.11163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Severin PMD, Zou X, Gaub HE, Schulten K. Cytosine methylation alters DNA mechanical properties. Nucleic Acids Res 2011; 39:8740-51; PMID:21775342; http://dx.doi.org/ 10.1093/nar/gkr578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Temiz NA, Donohue DE, Bacolla A, Luke BT, Collins JR. The role of methylation in the intrinsic dynamics of B- and Z-DNA. PLoS ONE 2012; 7:e35558; PMID:22530050; http://dx.doi.org/ 10.1371/journal.pone.0035558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lankas F, Sponer J, Langowski J, Cheatham TE. DNA deformability at the base pair level. J Am Chem Soc 2004; 126:4124-5; PMID:15053599; http://dx.doi.org/ 10.1021/ja0390449 [DOI] [PubMed] [Google Scholar]
- 50. Araúzo-Bravo MJ, Fujii S, Kono H, Ahmad S, Sarai A. Sequence-dependent conformational energy of DNA derived from molecular dynamics simulations: toward understanding the indirect readout mechanism in protein-DNA recognition. J Am Chem Soc 2005; 127:16074-89; PMID:16287294; http://dx.doi.org/ 10.1021/ja053241l [DOI] [PubMed] [Google Scholar]
- 51. Zheng G, Lu X-J, Olson WK. Web 3DNA-a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res 2009; 37:W240-6; PMID:19474339; http://dx.doi.org/ 10.1093/nar/gkp358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu X-J, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res 2003; 31:5108-21; PMID:12930962; http://dx.doi.org/ 10.1093/nar/gkg680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT, 2009. [Google Scholar]
- 54. Ditchfield R, Hehre WJ, Pople JA. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J Chem Phys 1971; 54:724-8; http://dx.doi.org/ 10.1063/1.1674902 [DOI] [Google Scholar]
- 55. Hariharan PC, Pople JA. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret Chim Acta 1973; 28:213-22; http://dx.doi.org/ 10.1007/BF00533485 [DOI] [Google Scholar]
- 56. Clark T, Chandrasekhar J, Spitznagel GNW, Schleyer PVR. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J Comput Chem 1983; 4:294-301; http://dx.doi.org/ 10.1002/jcc.540040303 [DOI] [Google Scholar]
- 57. Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J Comput Chem 1995; 16:1357-77; http://dx.doi.org/ 10.1002/jcc.540161106 [DOI] [Google Scholar]
- 58. Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013; 29:845-54; PMID:23407358; http://dx.doi.org/ 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/gamma conformers. Biophys J 2007; 92:3817-29; http://dx.doi.org/ 10.1529/biophysj.106.097782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem 2000; 21:1049-74; http://dx.doi.org/ 10.1002/1096-987X(200009)21:12%3c1049::AID-JCC3%3e3.0.CO;2-F [DOI] [Google Scholar]
- 61. Sponer J, Banáš P, Jurečka P, Zgarbová M, Kührová P, Havrila M, Krepl M, Stadlbauer P, Otyepka M. Molecular dynamics simulations of nucleic acids. From tetranucleotides to the ribosome. J Phys Chem Lett 2014; 5:1771-82; [DOI] [PubMed] [Google Scholar]
- 62. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983; 79:926; http://dx.doi.org/ 10.1063/1.445869 [DOI] [Google Scholar]
- 63. Essmann U, Perera L, Berkowitz ML. A smooth particle mesh Ewald method. J Chem Phys 1995; 103:8577; http://dx.doi.org/ 10.1063/1.470117 [DOI] [Google Scholar]
- 64. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007; 126:014101; PMID:17212484; http://dx.doi.org/ 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 65. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81:3684; http://dx.doi.org/ 10.1063/1.448118 [DOI] [Google Scholar]
- 66. Parrinello M. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys 1981; 52:7182; http://dx.doi.org/ 10.1063/1.328693 [DOI] [Google Scholar]
- 67. Pérez A, Luque FJ, Orozco M. Dynamics of B-DNA on the microsecond time scale. J Am Chem Soc 2007; 129:14739-45; http://dx.doi.org/ 10.1021/ja0753546 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.