

Abstract
AIM: To study the effect of cyclin G2 on proliferation of gastric adenocarcinoma cell line-SGC-7901 cell in vitro.
METHODS: By use of cation lipofectamine transfection reagent, the pIRES-G2 and pIRESneo plasmids were transferred into SGC-7901cell line. Anticlones were selected by G418. Positive clones were observed and counted using Giemsa staining. Cell proliferative ability was assayed by MTT.
RESULTS: (1) The clone number of pIRES-G2 group decreased, clone volume reduced. The number of cell clones in pIRESneo group was 87 ± 3, that of pIRES-G2 group was 53 ± 4, occupying 60.1% of pIRESneo group, there was significant difference obviously (P < 0.01, t = 15.45). (2) The average absorbance of clone cell obtained by stable transfection of pIRES-G2 at 570 nm was 1.6966 ± 0.2125, the average absorbance of clone cell obtained by stable transfection of pIRESneo at 570 nm was 2.1182 ± 0.3675, there was significant difference between them (P < 0.01, t = 3.412).
CONCLUSION: Cyclin G2 can inhibit SGC-7901cell proliferative ability obviously, it may be a negative regulator in cell cycle regulation.
INTRODUCTION
Cell cycle regulation is the core part in cell proliferation, which has a close relationship with cell carcinogenesis. In recent years, the study of cell cycle regulation is the foreland and hotspot field in cell biology and genetics. At present, research on the formation, activation, inactivation of cyclin-CDK (cyclin-dependent kinase) holo-enzyme complex is the main molecular basis of cell cycle regulation[1,2]. All the 12 type different cyclins found in mammalian cells contain a 100 amino-acid homologous region named cyclin box, which is the molecular structure marker of cyclins[3]. Cyclin G is a new member of cyclin family, which includes cyclin G1 and cyclin G2. Although the identity of amino acid sequence and nucleotide sequence of cDNA of cyclin G1 and cyclin G2 was 53% and 60% respectively, their biological function and distribution in tissue and organ differ greatly[4,5]. Research shows that cyclin G1 is located mainly in nucleus, whose expression is induced by the damage of DNA and depends on the expression level of p53[6,7]. Cyclin G2 is located mainly in cytoplasm, whose expression can be induced by the VHL protein and to some degree is independent of p53[8]. We have known that cyclin G1 is a positive regulator in cell cycle regulation. But we still do not know if cyclin G2 is connected with CDKs and with which kind of CDKs. The expression of cyclin G2 induced by cancer -suppressing gene hints that it may act as a negative regulator in cell proliferation[9]. Moreover, we purified oral squamous carcinoma cell by laser capture micro-dissection, and found that 4 cases showed low expression of cyclin G2 in 5 cases in the gene expression pedigree research with gene chip analysis[10]. Thus, it is much more likely for cyclin G2 to have the negative regulating effect on the cell proliferation.
In this article, we transfected pIRES-G2 into SGC-7901 cell and studied the effect of cyclin G2 expression on the cell proliferation in vitro.
MATERIALS AND METHODS
Materials
The eukaryotic plasmid expression vector contains the whole length of cyclin G2 and selective marker gene, neo was constructed by the Medical Gene Group of China Medical University[11]. pIRESneo vector was purchased from Clontech. E. Coli JM109 competent cell was the product of TaKaRa, plasmid extraction and purification kits were from Qiagen. Lipofectamine PlusTM and G418 were the products of Invitrogen. MTT was from Huamei. SABC kit and DAB kit were from Boster.
The human gastric adenocarcinoma cell line SGC-7901(Cell Biology Laboratory of China Medical University) was grown in DMEM (Gibco) supplemented with 100 mL/L heat-inactivated fetal calf serum (Hyclone), 100 U/mL of penicillin sodium and 100 μg/mL streptomycin sulfate, cultured at 37 °C, with 50 mL/L CO2.
Methods
Plasmid amplification reaction and evaluation: E. Coli JM109 competent cells were transformed by pIRES-G2 plasmid, cultured in LB medium supplemented with Amp+ (100 μg/ mL). The positive clones were selected and cultured. Abundant plasmids were produced and purified by alkaline lysis method, identified by 10 g/L agarose gel electrophoresis after restriction endogenous enzyme digestion. The concentration and purity were determined by ultraviolet scanning spectrophotometer.
Gene transfection: Exponentially growing SGC-7901 cells were seeded using 2 mL non-antibiotics culture medium into 6-well plates. After 24 h when cells grew to 60%-80% confluence transfection began. Cells were transiently transfected according to the protocol of Lipofectamine PlusTM. Briefly, the plasmid DNA was premixed with the Plus reagent by diluting 1 μg DNA into 100 μL transfection DMEM (serum-free, non-antibiotics) and adding 6 μL Plus reagents in polystyrene tubes. Solutions were combined, gently mixed, and incubated for 15 min at room temperature to allow formation of DNA/Plus complexes. Meanwhile, 4 μL Lipofectamine reagents were diluted in 100 μL transfection DMEM. The DNA/Plus reagent was mixed with the diluted Lipofectamine reagent and incubated for 15 min again at room temperature. The growth medium was removed from the cells and replaced with 800 μL transfection DMEM. The complexes were added to the cells and incubated for 3 h at 37 °C, with 50 mL/L CO2. Then 1 mL of non-antibiotics DMEM containing 200 mL/L FCS was added to it. Transfection of the pIRESneo vector into SGC-7901cell was the same.
Cell clone: At 24 h after gene transfection, cells were trypsin digested and transferred into 100 mm culture dish, and cultured for another 2 wk in G418 (400 μg/ mL) selective culture medium. The clone formation and cell morphology were observed under an inverted microscope (Olympus IX70). Images were acquired using a cool charge-coulped device (Spot) and transferred into computer. Cells were fixed in methanol/ice acetic acid (3:1) and stained in 50-100 g/L Giemsa. Cell clones were observed and counted.
Immunocytochemistry staining for exogenous cyclin G2 expression: Cell transfection and G418 selection were done as above. After fourteen days, cell clones could be seen in pIRES-G2 and pIRESneo group. After PBS washing, the cell clones were circled, separated with sterile tip and cultured in G418 (250 μg/mL) selective culture medium for another 2 wk. The stably transfected cell lines were obtained after passage by 2-3 generations. Cells were seeded into 6-well culture plates containing coverslips allowed to grow until they were 60%-80% confluence and coverslips were picked up, washed with PBS, then fixed in ice cold acetone for 30 min at room temperature, washed 3 times (5 min each) in PBS. Cytochemistry staining was preformed by the avidin-biotin peroxidase complex method using the SABC kit as follows: cells were treated to remove endogenous peroxidase activity, blocked with rabbit serum for 30 min at room temperature, and then incubated in primary goat anti-cyclin G2 polyclonal antibody (American Santa Cruz Company) for 1 h at 37 °C or overnight at 4 °C, washed 3 times with PBS, then incubated with a rabbit-anti-goat secondary antiboby conjugated to horseradish peroxidase for 1 h. Following PBS washing, the coverslips were incubated with SABC elite reagent for 30 min at room temperature and washed 3 times in PBS. The bound horseradish peroxidase complexes were developed using DAB kit. The coverslips were counterstained with hematoxylin, for colored by 10 mL/L hydrochloric acid ethanol, dehydrated by graded ethanol, and enveloped by neutral resin, negative contrast was made by using PBS instead of primary antibody.
MTT assay: The stably transfected cells were seeded into 24-well culture plates and cultured for 24 h at 37 °C, with 50 mL/L CO2. Cells were seeded into 96-well culture plates at 4000-6000 cells/well. After 48 h culture, the culture solution was poured out and washed twice with PBS. 150 μL (0.5 mg/mL) MTT was added at 37 °C for 4 h, the culture medium solution was poured out and 150 μL/each well DMSO was added again, shaken for 10 min in horizontal shaker. The absorbance at 570 nm was read by enzyme-labelling analysis equipment. The average absorbance reflected the cell proliferative ability. Data showed in mean ± SD format and statistic significance was analyzed by t test.
RESULTS
Plasmid appraisal
The cyclin G2 fragment was inserted between the BamHI and BstXI restriction sites, the restriction enzyme digestion and gel electrophoresis results were the same as predicted.
Selection of the stable transfection cell line and exogenous cyclin G2 gene expression
After 14 d of selection by G418 in transfected cells, several cell clones could be seen in pIRES-G2 and pIRESneo group. After PBS washing, the cell clones were circled, and the larger single cell clones were separated with sterile tip, cultured in G418 (250 μg/ mL) selective culture medium for another 2 wk, the stable transfection cell lines were obtained after passage by 2-3 generations.
Immunocytochemistry staining result showed that a large number of gray positive granules appeared in SGC-7901cell cytoplasma after pIRES-G2 transfection, while pIRESneo cell cytoplasma only appeared blue and no positive granule was seen in negative contrast cell.
The effect of cyclin G2 overexpression on the colony-formation of SGC-7901 cell
Observing the stably transfected SGC-7901 cell obtained by G418 selection under microscope, the clone number of pIRES-G2 group decreased, clone volume reduced and the cells in clone changed irregularly, many granules and bubbles appeared in cytoplasm. The 5 repeated experimental results were the same. The number of pIRESneo group was 87 ± 3, that of pIRES-G2 group was 53 ± 4, occuping 60.1% of pIRESneo group, there was significant difference obviously (P < 0.01, t = 15.45) (Figures 1 and 2).
Figure 1.
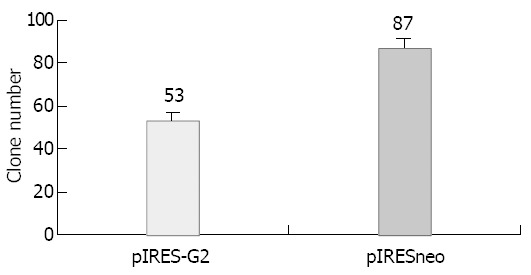
The positive clone number obtained with pIRES-G2 and pIRESneo plasmids after stable transfection in SGC-7901 cell line.
Figure 2.
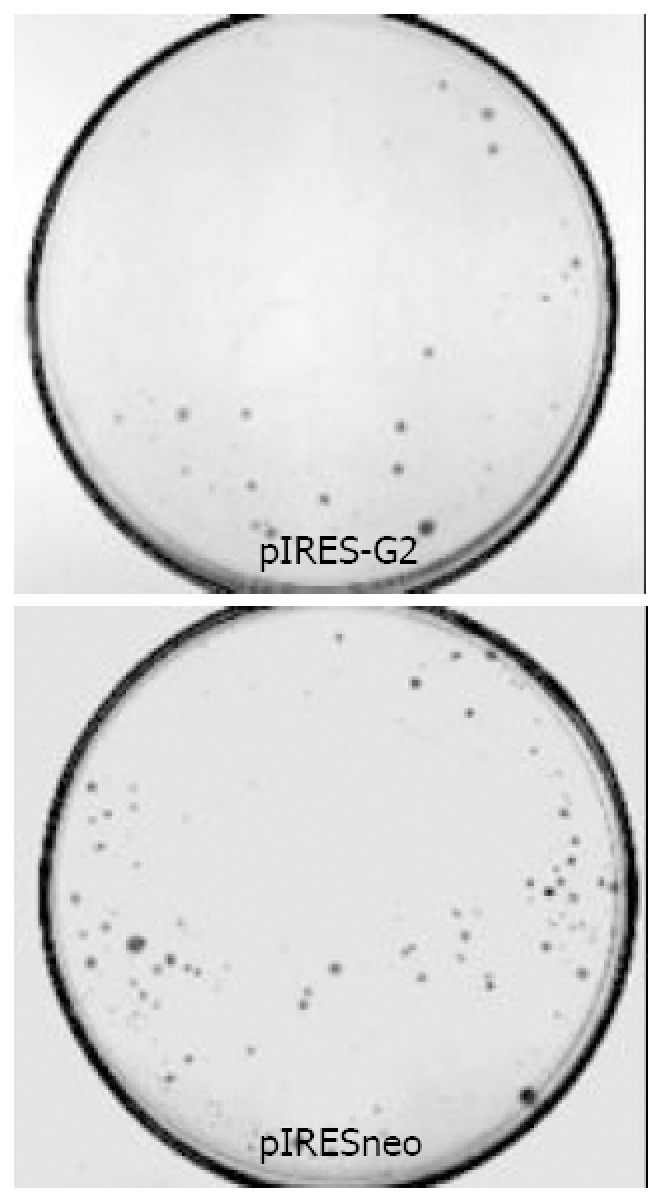
Effect of cyclin G2 overexpression on the colony-formation ability of SGC-7901.
Effects of cyclin G2 overexpression on SGC-7901 proliferative ability
By MTT assay analysis the proliferative ability change of stably transfected SGC-7901 cell obtained by G418 selection was as following: the average absorbance of clone cell obtained by stably transfected pIRES-G2 at 570 nm was 1.6966 ± 0.2125, the average absorbance of clone cell obtained by stably transfected pIRESneo at 570 nm was 2.1182 ± 0.3675, there was statistical difference between them (P < 0.01, t = 3.412) (Table 1) .
Table 1.
The proliferative ability compared between 2 clone-cell
| Group | Well number | Average absorbancee |
| pIRESneo | 96 | 2.1182±0.3675b |
| pIRES-G2 | 96 | 1.6966±0.2125 |
P < 0.01, t = 3.412 vs pIRES-G2.
DISCUSSION
There are many kind of cyclins and CDKs in mammalin cells, which combine with each other forming active cyclin-CDK complex as a triggering factor in the regulation of cell cycle. The expression level of cyclins shows phase specificity, which is obviously regulated by transcription and the rate of protein hydrolysis. Cyclin G, a recent addition to the cyclin family, has homology to fisson yeast Cig1, B-type cyclins, and human cyclin A and I. The molecular structure of cyclin G differs from other cyclins. NH2 terminus lacks a “destruction box” sequence controlling the ubiquitin-dependent degradation mitotic cyclins, while contains an epidermal growth factor receptor (EGF-R/ ErbB) like autophosphorylation motif in its carboxyl terminus[12]. The structure suggested that a role for cyclin G in signal transduction. Cyclin G has not yet been matched with a CDK binding partner, and its biologic function is still elusive. Cyclin G is the only known cyclin that is transcriptionally activated by the p53 tumor suppressor gene. Cyclins G1 and G2 share the highest sequence identity and similarity to the fission yeast B-type cyclins Cig1 and Cig2 respectively, while cyclin G2 exhibits high sequence identity to the S-phase cyclin Clb-5 of budding yeast. Cloning and sequencing a partial cDNA sequence revealed the cyclin G1 transcript does not encode motifs which resemble known degradation signals. In contrast, the cyclin G2 protein contains a prototypic protein destabilizing (PEST) rich sequences which may be responsible for its potential regulated degradation in cell cycle. The cyclin G1 mRNA expression does not fluctuate with cell cycle phase, whereas the expression of cyclin G2 mRNA oscillated with cell cycle and reached peak in the mid-late S phase. In contrast to cyclin G1, cyclin G2 mRNA weakly expressed in skeletal muscles and heart. It showed high expression in cerebrum, thymus, spleen, prostate gland and kidney[13]. Notably cyclin G2 transcripts are abundant in tissue rich in either terminally differentiated cells or cells reacting to growth inhibitory signals and apoptosis[5]. Cyclin G1 is a positive regulator and prompts cell proliferation, so far, neither the physiological role nor the biochemical function of cyclin G2 has been defined. The mRNA expression of cyclin G1 and cyclin G2 can be induced by DNA-damage drug actinomycin-D, which has the p53 dependency for cyclin G1 but the p53 independency for cyclin G2[14]. Cyclin G2 can be induced by negative growth regulators such as TGF-β1 and dexamethasone. In the growth inhibition state of B cell, the transcription level of cyclin G2 is up-regulated. Re-conducting the VHL cancer-suppressing gene into renal cancer cells with VHL gene defect can induce cyclin G2 expression. The newest research also showed that lack of cyclin G2 plays an important role in the malignant transformation of papillary carcinoma of the thyroid. It may play an adjuvant role in the transformation of follicular adenoma to carcinoma[15]. So cyclin G2 most likely appears to be a negative regulator of the cell cycle.
The result showed that compared with the control group, the clones formed by transfected pIRES-G2 group were smaller and fewer, cells became wrinkled and irregular, which proved that the high expression of cyclin G2 inhibited the cell proliferation. In this research we had the gene transfection experiment through cloning cyclin G2 cDNA into bicistronic eukaryotic expression vector pIRESneo, the 2 open reading frame (ORF) of inserted gene cyclin G2 and selective label gene neo were separated by the inserted brain cardiomyoditis virus IRES[16,17]. So the recombinant plasmid pIRES-G2 in host cells only produced a single transcriptional template, which made the 2 proteins to be translated simultaneously to ensure all positive cells after G418 selection express target genes, overcame the shortcoming that these 2 genes could not be transcribed and translated at the same time when using other eukaryotic expression vectors[18,19]. The positive clones formed by G418 selection in the experimental group of pIRES-G2 should have the expression of cyclin G2. Thus, the difference of clones between the experimental and control groups was only caused by the high expression of cyclin G2. The MTT assay also testified that the average absorbance of pIRES-G2 group cell was lower than that of pIRESneo group, it showed that the proliferative ability decreased after pIRES-G2 transfection. Because by MTT assay, the cell numbers in pIRES-G2 group and pIRESneo group were the same and they also grew at the exponentially growing state, thus the average absorbance difference between them was merely caused by the difference of transfected genes. From this we could draw the conclusion that cyclin G2 overexpression makes the proliferative ability decrease.
Our experiment indicates that cyclin G2 could inhibit the SGC-7901 cell proliferation. Unlike other cyclins, it may be a negative regulating factor of cell cycle.
Footnotes
Supported by A grant for Distinguished Young Teachers of Higher Education of the Ministry of Education of the Teaching and Research Encouragement Plan
Edited by Zhu LH, Xu FM
References
- 1.Hunter T, Pines J. Cyclins and cancer. Cell. 1991;66:1071–1074. doi: 10.1016/0092-8674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- 2.Pines J, Hunter T. Cyclin-dependent kinases: a new cell cycle motif? Trends Cell Biol. 1991;1:117–121. doi: 10.1016/0962-8924(91)90116-q. [DOI] [PubMed] [Google Scholar]
- 3.Pines J. Cyclins and cyclin-dependent kinases: theme and variations. Adv Cancer Res. 1995;66:181–212. doi: 10.1016/s0065-230x(08)60254-7. [DOI] [PubMed] [Google Scholar]
- 4.Draetta GF. Mammalian G1 cyclins. Curr Opin Cell Biol. 1994;6:842–846. doi: 10.1016/0955-0674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 5.Horne MC, Goolsby GL, Donaldson KL, Tran D, Neubauer M, Wahl AF. Cyclin G1 and cyclin G2 comprise a new family of cyclins with contrasting tissue-specific and cell cycle-regulated expression. J Biol Chem. 1996;271:6050–6061. doi: 10.1074/jbc.271.11.6050. [DOI] [PubMed] [Google Scholar]
- 6.Zauberman A, Lupo A, Oren M. Identification of p53 target genes through immune selection of genomic DNA: the cyclin G gene contains two distinct p53 binding sites. Oncogene. 1995;10:2361–2366. [PubMed] [Google Scholar]
- 7.Kimura SH, Ikawa M, Ito A, Okabe M, Nojima H. Cyclin G1 is involved in G2/M arrest in response to DNA damage and in growth control after damage recovery. Oncogene. 2001;20:3290–3300. doi: 10.1038/sj.onc.1204270. [DOI] [PubMed] [Google Scholar]
- 8.Wykoff CC, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Identification of novel hypoxia dependent and independent target genes of the von Hippel-Lindau (VHL) tumour suppressor by mRNA differential expression profiling. Oncogene. 2000;19:6297–6305. doi: 10.1038/sj.onc.1204012. [DOI] [PubMed] [Google Scholar]
- 9.Horne MC, Donaldson KL, Goolsby GL, Tran D, Mulheisen M, Hell JW, Wahl AF. Cyclin G2 is up-regulated during growth inhibition and B cell antigen receptor-mediated cell cycle arrest. J Biol Chem. 1997;272:12650–12661. doi: 10.1074/jbc.272.19.12650. [DOI] [PubMed] [Google Scholar]
- 10.Alevizos I, Mahadevappa M, Zhang X, Ohyama H, Kohno Y, Posner M, Gallagher GT, Varvares M, Cohen D, Kim D, et al. Oral cancer in vivo gene expression profiling assisted by laser capture microdissection and microarray analysis. Oncogene. 2001;20:6196–6204. doi: 10.1038/sj.onc.1204685. [DOI] [PubMed] [Google Scholar]
- 11.Tian YL, Liu FR, Liu J, Jiang L, Luo Y, Zhang X. [Ectopic expression of cyclin G2 inhibits cell proliferation in HeLa cancer cell line] Ai Zheng. 2002;21:577–581. [PubMed] [Google Scholar]
- 12.Tamura K, Kanaoka Y, Jinno S, Nagata A, Ogiso Y, Shimizu K, Hayakawa T, Nojima H, Okayama H. Cyclin G: a new mammalian cyclin with homology to fission yeast Cig1. Oncogene. 1993;8:2113–2118. [PubMed] [Google Scholar]
- 13.Jensen MR, Audolfsson T, Keck CL, Zimonjic DB, Thorgeirsson SS. Gene structure and chromosomal localization of mouse cyclin G2 (Ccng2) Gene. 1999;230:171–180. doi: 10.1016/s0378-1119(99)00057-8. [DOI] [PubMed] [Google Scholar]
- 14.Bates S, Rowan S, Vousden KH. Characterisation of human cyclin G1 and G2: DNA damage inducible genes. Oncogene. 1996;13:1103–1109. [PubMed] [Google Scholar]
- 15.Ito Y, Yoshida H, Uruno T, Nakano K, Takamura Y, Miya A, Kobayashi K, Yokozawa T, Matsuzuka F, Kuma K, et al. Decreased expression of cyclin G2 is significantly linked to the malignant transformation of papillary carcinoma of the thyroid. Anticancer Res. 2003;23:2335–2338. [PubMed] [Google Scholar]
- 16.Rees S, Coote J, Stables J, Goodson S, Harris S, Lee MG. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques. 1996;20:102–104, 106, 108-110. doi: 10.2144/96201st05. [DOI] [PubMed] [Google Scholar]
- 17.Gaines P, Wojchowski DM. pIRES-CD4t, a dicistronic expression vector for MACS- or FACS-based selection of transfected cells. Biotechniques. 1999;26:683–688. doi: 10.2144/99264st04. [DOI] [PubMed] [Google Scholar]
- 18.Felgner PL. Improvements in cationic liposomes for in vivo gene transfer. Hum Gene Ther. 1996;7:1791–1793. doi: 10.1089/hum.1996.7.15-1791. [DOI] [PubMed] [Google Scholar]
- 19.Bennett CF, Chiang MY, Chan H, Shoemaker JE, Mirabelli CK. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Mol Pharmacol. 1992;41:1023–1033. [PubMed] [Google Scholar]