

Abstract
Non-enzymatic glycation is a challenging post-translational modification to characterize due to the structural heterogeneity it generates in proteins. Glycation has become increasingly recognized as an important product quality attribute to monitor, particularly for the biotechnology sector, which produces recombinant proteins under conditions that are amenable to protein glycation. The elucidation of sites of glycation can be problematic using conventional collision-induced dissociation (CID)-based mass spectrometry because of the predominance of neutral loss ions. A method to characterize glycation using an IgG1 monoclonal antibody (mAb) as a model is reported here. The sugars present on this mAb were derivatized using sodium borohydride chemistry to stabilize the linkage and identified using CID-based MS2 mass spectrometry and spectral search engines. Quantification of specific glycation sites was then done using a targeted MS1 based approach, which allowed the identification of a glycation hot spot in the heavy chain complementarity-determining region 3 of the mAb. This targeted approach provided a path forward to developing a structural understanding of the propensity of sites to become glycated on mAbs. Through structural analysis we propose a model in which the number and 3-dimensional distances of carboxylic acid amino acyl residues create a favorable environment for glycation to occur.
Keywords: glycation, targeted mass spectrometry, boronate affinity chromatography, structural modeling
Abbreviations
- Å
angstroms
- BA
boronate affinity chromatography
- CDR3
complementary-determining region 3
- CEX
cation exchange chromatography
- CID
collision induced dissociation
- CV
coefficient of variation
- Da
daltons
- EIC
extracted ion chromatogram
- HC-CDR3
heavy chain complementary determining region 3
- HPLC
high performance liquid chromatography
- LC-MS2
liquid chromatography coupled with tandem mass spectrometry
- mAb
monoclonal antibody
- MS1
a mass to charge ratio survey scan
- MS2
tandem mass spectrometry - selected ions from MS1 are fragmented and fragment ion mass measured
- UPLC
ultrahigh performance liquid chromatography
Introduction
Interactions between sugars and amino acids are well known to occur. For example, it has been known for over a century that, when heat is applied, reaction of these molecules causes the browning of food. Since the first description of the interaction between reducing sugars and amino acids by Malliard in 1912,1 much of the additional work on the mechanistic underpinnings of these reactions has been food-focused. It is now widely recognized, however, that this reaction has important consequences in biotechnology. Glycation is a non-enzymatic modification in which the primary amine of a protein, most often the epsilon amino group of the exposed lysine (K) residues, reacts with the aldehyde group of a reducing sugar to form a Schiff base derivative of the protein. This derivative can then spontaneously undergo an Amadori rearrangement, forming a stable, covalent bond between the sugar and protein (for a review see ref. 2).
Glycation can have profound effects on protein structure, function and stability.3 Once glycated, proteins can undergo further oxidation of the Amadori compounds to generate a complex set of end products, collectively termed advanced glycation end (AGE) products.4 These products underlie, or are involved in, a host of pathological conditions including diabetes, osteoarthritis and aging,5 and as such, the study of non-enzymatic glycation is an active area of research.
Our interest in glycation is rooted in biologic drug manufacturing. Recombinant antibody production represents a substantial portion of biotechnology activity. Antibodies can undergo a host of modifications, from amino acid inter-conversion through glycation (for reviews see refs. 6–8). The exposure of these recombinant proteins to excipients, such as the sugars in the growth medium, makes them susceptible to glycation as a post-translational modification.9 Factors controlling glycation levels include a host of cell culture conditions, e.g., temperature, pH, exposure time, reactant concentrations (both amines and sugars), and glucose feed schedules.10 At first glance, the probability for glycation on these recombinant proteins appears high. For monoclonal antibody (mAb) production, the sugars are in molar excess during cell culture, often by several orders of magnitude, relative to the recombinant protein. In spite of this disproportionate molar excess, the extent of glycation is generally limited to only 1 to 2 glycation sites. These glycated residues are present in less than 20% of the total recombinant protein pool. Factors mediating glycation may include a reduction in the molar excess of sugars by the number of primary amines generally found on a mAb (more than 100), as well as additional reactive amines found in other compounds and free amino acids in cell culture.10 Nonetheless, changes in process that increase, decrease or change the type of sugar, for example galactose rather than glucose feeds, can have significant effects on protein glycation during production.10,11 As molecules advance through later stages of development, the analytical comparability burden associated with addressing molecule glycation increases, highlighting the need for rapid and definitive methods for glycation site identification.
The extent of glycation on a mAb can be measured by different methodologies, including boronate affinity chromatography and mass spectrometry-based top-down intact mass analysis.2 Beyond the extent of glycation, the specific sites of glycation are of paramount importance, particularly in the context of functional protein domains. A number of methods have been employed to characterize glycation sites, generally some form of bottom-up mass spectrometric application.10,12 This approach has demonstrated varying degrees of success, but tends to be distinguished by the predominance of neutral loss ions that occur during collision-induced dissociation (CID). The underlying peptide background then undergoes limited fragmentation, precluding identification of the glycated site. Other fragmentation strategies have also been employed including CID using MS3 and multistage activation approaches,13,14 electron transfer dissociation (ETD),15-17 and higher energy collisional dissociation (HCD).18
Given that CID is the most prevalent of the dissociation methods available to researchers, a method that relies upon derivatization of the glycated peptide to prevent neutral loss was developed, facilitating elucidation of the peptide backbone using CID. In this study glycation on a therapeutic IgG1 mAb was characterized. A high degree of glycation on this mAb was identified, enriching in the acidic fraction when the mAb was subjected to cation exchange chromatography (CEX). Sites of glycation were mapped by derivatizing the glycated species and analyzing the resulting peptides by MS2 mass spectrometry and spectral search engines. Glycation sites were quantified using high resolution, high mass accuracy MS1 mass spectrometry and software that facilitated a targeted, quantitative assay. This straightforward approach allowed for the characterization of several glycation sites, including a dominant site in the heavy chain complementarity-determining region 3 (CDR3) of the mAb, which enriches in the acidic portion of the CEX fraction. Finally, through structural modeling, we propose a mechanism where glycation in the heavy chain CDR3 increases due to the number of nucleophilic amino acid residues in proximity to the glycated lysine.
Results
The mAb is glycated and the glycation enriches in the acidic CEX fraction
Mass spectrometry-based, intact mass analysis is a rapid means of identifying alterations to a protein, including the addition or removal of amino acids and sugars. This approach was used to assay for any significant changes between in the mAb between the early development and late stage manufacturing processes. It was found that the deglycosylated mAb, both early and late versions, show shifts in mass of 162 Da, indicative of the addition of a reduced sugar, likely non-enzymatic protein glycation (Fig. 1A). The levels of glycation detected increase slightly from 15% in the early stage process to 18% in the late stage process molecule.
Figure 1.
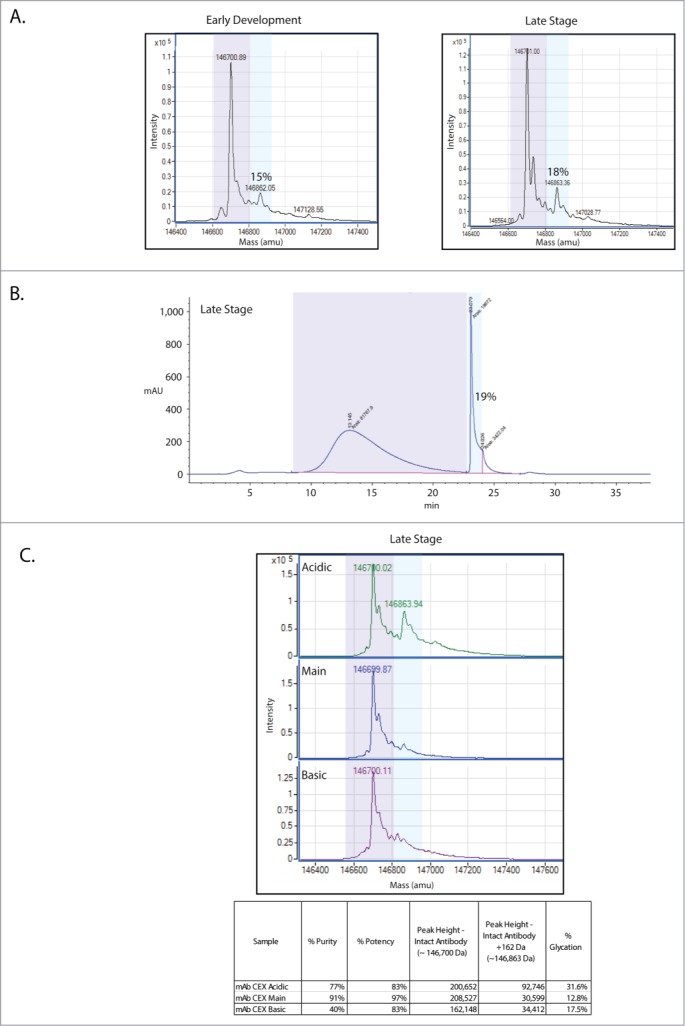
This mAb is glycated and the glycation enriches in the acidic CEX fraction. (A) Intact mass analysis of the deglycosylated mAb shows that glycation increases from early development (15% glycation) to late stage (18%) production processes. The glycated species is indicated by light blue shading while the non-glycated population is indicated by light purple shading. (B) Boronate affinity estimates of glycation levels are in agreement with the intact mass analysis at 19%. (C) Intact mass analysis of the CEX fractions shows that glycation is enriched in the acidic fraction. Estimates of purity, potency, peak height and glycation level are shown for each fraction in the table.
In order to quantify total glycation levels via an orthogonal means, a boronate affinity chromatography method for the mAb was employed (Fig. 1B). Sugars, including those bound to proteins, can be captured by boronate affinity chromatography.19,20 The boronate affinity method works via formation of esters between boronate ligands and the cis-diols that are present on sugars.21 The only major requirements for formation of these boronate esters are that the 2 hydroxyl groups of a diol be adjacent and coplanar. As seen in Figure 1B, the estimate of 19% glycation from this chromatographic approach agrees with the estimates of glycation levels on the late stage version of the mAb made from mass spectrometry-based intact mass analyses.
As part of the routine characterization of this mAb, the protein produced during the late stage process was subjected to CEX, separating the molecule into acidic, main and basic forms.22 These CEX fractions were then tested for potency using an assay that measures the presence of phosphorylated target protein (Fig. 1C). This mAb acts to block a receptor, thus inhibiting the phosphorylation and activation of the target protein. Both the acidic and the basic fractions show reductions in the ability to inhibit phosphorylation of the target protein, with potencies near 83% (3% coefficient of variation (CV)). The main fraction maintains potency at 97% (3% CV), near reference standard levels. These CEX fractions were individually subjected to mass spectrometric intact mass analysis (Fig. 1C). Interestingly, this analysis shows an enrichment of glycation in the acidic fraction. We then sought to further characterize the glycation enriching in the CEX acidic fraction to determine if there is a relationship between the increase in glycation and the decrease in potency in this fraction.
In an orthogonal approach to map glycation sites at a broad level, the mAb was digested with FabRICATOR, a proteolytic enzyme that cleaves IgGs at the hinge region, resulting in 2 F(ab) fragments.23,24 After reduction of the digested antibody, the masses of the 3 resulting fragments (Fc, Fd, and Light Chain; Fig. 2A) were analyzed by mass spectrometry. This analysis demonstrated that 71.5% of glycation within the acidic CEX fraction is found on the Fd fragment, and furthermore this glycation accounts for 68.4% of glycation on the Fd fragments between the acidic, main, and basic fractions. These data demonstrate that the majority of glycation on the mAb is found on the Fd region and the single glycation site that was identified by peptide mapping on the light chain, is not the primary reservoir of glycation on this mAb.
Figure 2.

Characterization of the mAb Glycation by MS2 peptide mapping and FabRICATOR-Intact Mass analysis. (A) A cartoon of cleavage fragments generated by treatment with FabRICATOR and reduction (left panel). Intact mass analysis of the FabRICATOR fragments (right panel) shows glycation (light blue shading) enriching in the Fd portion of the Acidic CEX fraction. (B) Searches of the MS2 data using the Sequest search engine identify one peptide in the light chain with an XCorr score of 2.93. The glycated lysine residue is shown by the bold K. Inset shows a typical neutral loss peak that arises in the MS2 spectra precluding identification of the ion series. Labeled spectra were overlaid with equivalent intensity, thicker lines, to enhance visibility.
Derivatization chemistry and mass spectrometric search algorithms facilitate the identification of glycation sites by peptide mapping
In order to characterize the glycation on the mAb, we attempted to determine the specific sites of glycation. To this end, the mAb CEX fractions were subjected to LC-MS2-based peptide mapping. While the peptide mapping resulted in 100% coverage of both the heavy and light chains, no new peaks arose in the 214 nm or 280 nm chromatographic traces relative to the late stage process molecule (data not shown). The mass spectrometric data were searched with the Sequest and X!Tandem search algorithms using the mass of glycation as a variable modification. A lone higher probability glycated peptide was identified by the Sequest search engine on the light chain (Fig. 2B), with a cross correlation (XCorr) value of 2.93. This peptide was not identified by the X!Tandem search engine (data not shown). Visual inspection of this spectrum reveals a significant amount of noise with a number of unassigned ions. The MS2 spectra for the low probability glycated peptide identifications were also of poor quality, with large neutral loss peaks that appear to preclude species identification (Fig. 2B inset).
Given both the poor quality of the spectra from the glycated peptides and the results of the FabRICATOR analysis demonstrating that the bulk of glycation likely resides on the Fd fragment (Fig. 2B), a means to enhance identification of glycation sites by CID mass spectrometry was sought. A sodium borohydride derivatization procedure to convert the double bond present in the Amadori moiety to a single bond was employed, thus stabilizing the bond between the sugar and the lysine residues20,25 (Fig. 3A). This stabilization of this bond facilitates collision-induced dissociation without the generation of neutral loss peaks, allowing for the unambiguous assignment of the MS2 spectral series (Fig. 3B and 3B inset). The mAb was derivatized, subjected to peptide mapping, and the resulting spectra searched with Proteome Discoverer using the Sequest search engine and the Tran-Proteomic Pipeline using the X!Tandem search engine. For both searches, derivatized hexose was used as a variable modification; reduction of the double bond via derivatization increases the mass of the hexose by 2 Da. After manual inspection of the spectra, peptides that scored well by either search engine (Prob ≥ 0.9 by TPP or XCorr ≥ 3 by Sequest) were included. This led to a final tally of 7 unique peptides identified after derivatization (Table 1). Importantly, 2 of the 5 glycated peptides identified in the heavy chain reside in the Fd portion, consistent with the results from the FabRICATOR–Intact Mass analysis. The glycated residue K99 (Peptide 1, Table 1) is located within the CDR3, while K222 is located in the Fd side of the “hinge” region of the mAb (Fig. 3B dashed white line).
Figure 3.

Sodium borohydride derivatization facilitates the identification of MS2 ion series. (A) Mechanism of action of NaBH derivatization, where the double bond between the reducing sugar and the nitrogen of the lysine residue is reduced. (B) MS2 spectra of the derivatized peptide showing the differences between derivatized and underivatized (inset) samples. Labeled spectra were overlaid with equivalent intensity, thicker lines, to enhance visibility. (C) A cartoon of sites of glycation identified by derivatization of glycation and spectral searching for peptides with derivatized glycan mass shifts. Approximate sites of glycation are shown by red arrows and the numbers correspond to numbers identified in Table 2.
Table 1.
Identification of glycated peptides in the late stage production mAb. Sites of glycation are shown by lower case (k). Chain indicates heavy chain (HC) or light chain (LC). The numbers for the TPP (Trans Proteomic Pipeline) and PD (Proteome Discoverer) columns indicate the number of times the respective peptide was identified. Prob is the Protein Prophet probability while XCorr refers to the Sequest search engine's XCorr value
| Filtered | Number | Peptide | Chain | Position | TPP | Prob | PD | Xcorr |
|---|---|---|---|---|---|---|---|---|
| 1 | DkXXXYXDXWGX | HC | 98–109 | 2 | 0.9984 | 2 | 3.25 | |
| 2 | XCDkXXXCPPCPAPXXXXGPSVFLFPPKPK | 219–248 | 1 | 0.9879 | 0 | 0 | ||
| 3 | XXXcPPcPAPXXXXGPSVFLFPPkPK | 223–248 | 2 | 0.9985 | 2 | 3.99 | ||
| 4 | FNWYVDGVEVHNAKTkPR | 275–292 | 1 | 0.9845 | 0 | 0 | ||
| 5 | VSNkXLPAPIEK | 323–334 | 1 | 0.9871 | 1 | 3.15 | ||
| 6 | DSTYSLSSTLTLSkADYEK | LC | 176–194 | 1 | 0.9512 | 2 | 3.4 | |
| 7 | VYACEVTHQGLSSPVTkSFNR | 197–217 | 1 | 0.9789 | 0 | 0 |
This approach was then used to analyze the CEX fractions previously assayed without derivatization. Acidic, main and basic fractions from CEX chromatography were subjected to sodium borohydride derivatization and analyzed by peptide mapping as described in the Materials and Methods section. In total, 14 glycated peptides were identified (Table 2-I). After excluding peptides that did not score well in either X!Tandem or Sequest searches, and manual inspection of the MS2 spectra, 8 high-confidence glycated peptides were identified (Table 2-II). Based on the number of “hits” observed for each peptide, there appeared to be an overall enrichment of glycation in the acidic fraction, consistent with the intact mass and FabRICATOR-reduced intact mass analyses of the CEX fractions. The two glycated peptides located in the Fd region, identified in the late-stage process molecule derivatized peptide map described above, also enriched in the acidic fraction. Taken together, these data reveal an enrichment of glycated peptides in the acidic CEX fraction, and identify additional glycation sites localized within the Fc and light chain of the mAb.
Table 2.
Identification of glycated peptides in CEX fractions of the mAb. (I) All identified sites of glycation identified in the acidic, main and basic fractions. Values colored blue indicate enrichment for that peptide in the respective CEX fraction. Values colored purple were only identified in the indicated fractions. (II) Curated peptides from I. Also shown is the position of the peptides in the primary sequence of the mAb
| ACIDIC |
MAIN |
BASIC |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALL | Number | Peptide | Chain | TPP | Prob | PD | Xcorr | TPP | Prob | PD | Xcorr | TPP | Prob | PD | Xcorr | |
| 1 | DkXXXYXDXWGX | HC | 5 | 0.99838 | 5 | 3.875 | 2 | 0.9974 | 2 | 2.99 | 2 | 0.99805 | 2 | 2.99 | ||
| 2 | FNWYVDGVEVHNAKTkPR | 0 | 0 | 0 | 0 | 2 | 0.1202 | 3 | 1.59 | |||||||
| 3 | kXXXYXDXWGX | 0 | 3 | 2.315 | 0 | 2 | 1.755 | 0 | 2 | 1.765 | ||||||
| 4 | PPGk | 0 | 0 | 0 | 0 | 0 | 1 | 0.72 | ||||||||
| 5 | XCDkXXXCPPCPAPXXXXGPSVFLFPPKPK | 3 | 0.999933 | 3 | 4.755 | 0 | 0 | 0 | 0 | |||||||
| 6 | XXXCPPCPAPXXXXGPSVFLFPPKPk | 4 | 0.44915 | 8 | 4.06 | 0 | 0 | 0 | 0 | |||||||
| 7 | VSNkXLPAPIEK | 2 | 0.9616 | 2 | 3.045 | 2 | 0.9105 | 2 | 3.21 | 2 | 0.9673 | 2 | 3.1 | |||
| 8 | VTISVXTSkNQFSLK | 1 | 0.9915 | 1 | 2.64 | 1 | 0.0639 | 1 | 1.37 | 0 | 0 | |||||
| 9 | ADYEkHK | LC | 0 | 2 | 1.67 | 0 | 0 | 0 | 0 | |||||||
| 10 | DSTYSLSSTLTLSkADYEK | 2 | 0.6894 | 4 | 3.995 | 1 | 0.4724 | 2 | 2.81 | 0 | 0 | |||||
| 11 | HkVYACEVTHQ | 0 | 2 | 1.915 | 0 | 0 | 0 | 0 | ||||||||
| 12 | HkVYACEVTHQGLSSPVTK | 1 | 0.9994 | 2 | 4.05 | 0 | 0 | 0 | 0 | |||||||
| 13 | TFGQGTXXEIkR | 0 | 0 | 0 | 0 | 0 | 1 | 1.17 | ||||||||
| 14 | VYACEVTHQGLSSPVTkSFNR | 2 | 0.9974 | 2 | 3.515 | 1 | 0.9962 | 1 | 2.67 | 1 | 0.9991 | 1 | 2.68 | |||
| ACIDIC | MAIN | BASIC | ||||||||||||||
| Filtered | Number | Peptide | Chain | Position | TPP | Prob | PD | Xcorr | TPP | Prob | PD | Xcorr | TPP | Prob | PD | Xcorr |
| 1 | VTISVETSkNQFSLK | HC | 67–81 | 1 | 0.9915 | 1 | 2.64 | 1 | 0.0639 | 1 | 1.37 | 0 | 0 | |||
| 2 | DkXXXYXDXWGX | 98–109 | 5 | 0.99838 | 5 | 3.875 | 2 | 0.9974 | 2 | 2.99 | 2 | 0.99805 | 2 | 2.99 | ||
| 3 | XCDkXXXCPPCPAPXXXXGPSVFLFPPKPK | 219–248 | 3 | 0.999933 | 3 | 4.755 | 0 | 0 | 0 | 0 | ||||||
| 4 | XXXCPPCPAPXXXXGPSVFLFPPKPk | 223–248 | 4 | 0.44915 | 8 | 4.06 | 0 | 0 | 0 | 0 | ||||||
| 5 | VSNkXLPAPIEK | 323–334 | 2 | 0.9616 | 2 | 3.045 | 2 | 0.9105 | 2 | 3.21 | 2 | 0.9673 | 2 | 3.1 | ||
| 6 | DSTYSLSSTLTLSkADYEK | LC | 176–194 | 2 | 0.6894 | 4 | 3.995 | 1 | 0.4724 | 2 | 2.81 | 0 | 0 | |||
| 7 | HkVYACEVTHQGLSSPVTK | 195–213 | 1 | 0.9994 | 2 | 4.05 | 0 | 0 | 0 | 0 | ||||||
| 8 | VYACEVTHQGLSSPVTkSFNR | 197–217 | 2 | 0.9974 | 2 | 3.515 | 1 | 0.9962 | 1 | 2.67 | 1 | 0.9991 | 1 | 2.68 | ||
Quantification of site specific glycation by targeted MS1
With the sites of glycation identified, we next sought to quantify individual sites to determine if the amount of glycation detected on each site could be stratified. In our first approach, the 214 nm and 280 nm chromatographic trace of the tryptic digest was analyzed as it eluted from the reversed phase column. Using this method, no peaks that could clearly be assigned to the derivatized form of the peptide or specifically enriching in the acidic fraction could be identified, likely due to low abundance or co-elution.
In a second approach, the extracted ion chromatograms were used to assign areas to ion peaks that were identified in sufficient isolation (defined here as 4 times greater than any other ion in the MS1 scan). While relative quantities for some peptides can be determined, others are more problematic, again due to low abundance or co-elution with other ion species. A further drawback of this method is the assumption that the glycated species, in which the glycated lysine residue is not cleaved by trypsin, has similar ionization properties to the 2 parent ions that are generated in the absence of glycation. Using this approach, an estimate for only one peptide, located in the CDR3 of the Fd portion of the heavy chain, was possible. This peptide shows 4% glycation relative to the parent peptide in the Acidic CEX fraction (data not shown).
The third approach taken to quantify specific sites of glycation was to analyze a tryptic digest of the mAb using a high resolution, high mass accuracy Thermo Exactive Plus™ mass spectrometer in combination with the Pinpoint™ software package. This approach is MS1-based. Our MS1-based analyses allowed quantification of the previously detected glycated peptides (Table 3 MS2 & MS1), as well as peptides that were only identified in the MS1 experiment (Table 3 MS1 only). The peptide containing the glycated lysine residue (K99) is located in the Fd portion of the heavy chain, and accounts for the most significantly glycated peptide (6.62%). The quantities of the remaining glycated peptides each accounts for less than 1% of the glycated peptide population. The sum of all the identified glycated species in the Fd, Fc and Light Chain domains of the Acidic CEX fraction of the mAb shows remarkable agreement with the FabRICATOR-reduced intact mass analysis (10.3% for MS1 versus 11.6% for FabRICATOR). Interestingly, we note that glycation at K99 increases from early development to the late-stage production processes.
Table 3.
Targeted MS1 quantification of glycated peptides in CEX fractions of the mAb. Shown are the peptides identified with the glycated lysine shown by lower case (k). Carboxymethyl cysteine is shown in lower case (c). HC indicates heavy chain while LC indicates lower chain. Those peptides mapping to the Fd portion of the heavy chain are shaded in blue. BA refers to identification of the peptide by Boronate Affinity enrichment of the glycated mAb and subsequent derivatization and peptide mapping. The starred sequence refers to the fact that this peptide was not identified by derivatization and peptide mapping
| % Area |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chain | MS Type | Peptide | Sequence | BA | Domain | Retention Time (min) | Acidic | Main | Basic |
| HC | MS2 & MS1 | 1 | VTISVXTSkNQFSLK | Yes | Fd | 23.22 | 0.12 | 0.04 | 0.04 |
| 2 | DkXXXYXDXWGX | Yes | Fd | 50.77 | 6.62 | 0.65 | 1.44 | ||
| 3 | XcDkXXXcPPcPAPXXXXGPSVFLFPPKPK | Yes | Fd | 44.76 | 0.18 | 0.11 | 0.09 | ||
| 4 | XXXCPPCPAPXXXXGPSVFLFPPkPK | Yes | Fc | 45.62 | 0.18 | 0.09 | 0.09 | ||
| 5 | XXXcPPcPAPXXXXGPSVFLFPPkPKDTLMISR | No | Fc | 47.67 | 0.28 | 0.19 | 0.19 | ||
| 6 | FNWYVDGVEVHNAkTKPR | Yes | Fc | 20.55 | 0.23 | 0.13 | 0.11 | ||
| 7 | VSNkXLPAPIEK | Yes | Fc | 12.45 | 0.33 | 0.22 | 0.24 | ||
| MS1 only | 8 | NQFSLkLXSVTAADTAVYYcAR | No | Fd | 11.04 | 0.23 | 0.28 | 0.20 | |
| 9 | GTLVTVSSASTkGPSVFPLAPXSX | No | Fd | 32.43 | 0.03 | 0.02 | 0.01 | ||
| 10 | GPSVFPLAPXSkSTSXXTAALGcLVK | Yes | Fd | 38.65 | 0.12 | 0.07 | 0.06 | ||
| 11 | VVSVLTVXHQDWLNXkEYK | Yes | Fc | 42.54 | 0.07 | 0.05 | 0.06 | ||
| Fd Total | 7.30 | 1.17 | 1.85 | ||||||
| Fc/2 Total | 1.08 | 0.69 | 0.68 | ||||||
| LC | MS2 & MS1 | 12 | VQWkVDNALQSGNSQESVTEQDSK | No | 19.73 | 0.43 | 0.29 | 0.27 | |
| 13 | HkVYAcEVTHQGLSSPVTK | Yes | 13.28 | 0.12 | 0.07 | 0.05 | |||
| 14 | DSTYSLSSTLTLSkADYEK | Yes | 27.93 | 0.47 | 0.30 | 0.27 | |||
| 15 | VYAcEVTHQGLSSPVTkSFNR | Yes | 21.98 | 0.16 | 0.09 | 0.07 | |||
| MS1 only | 16 | XFGQGTkVEIK | Yes | 12.62 | 0.60 | 0.39 | 0.44 | ||
| 17 | EAkVQWK | No | 9.82 | 0.05 | 0.03 | 0.03 | |||
| 18 | VDNALQSGNSQESVTEQDSkDSTYSLSSTLTLSK | No | 28.82 | 0.04 | 0.03 | 0.03 | |||
| LC Total | 1.88 | 1.21 | 1.17 | ||||||
| Fraction Totals | 10.26 | 3.07 | 3.70 | ||||||
Seven glycated peptides were only identified by targeted MS1 (Table 3). We attempted to confirm the glycation status of these peptides by enriching for glycated mAbs from the pool of mAbs by boronate affinity chromatography (Fig. 1B). The glycated fraction was derivatized and peptide mapped, as done previously, and the resulting MS2 spectra searched using the Sequest search engine. After manual inspection and elimination of low probability peptides, the glycation status of 3 of the glycated peptide only identified by targeted MS1 was confirmed (Table 3). Six previously unobserved glycated peptides were also confirmed; however, after elimination of low probability identifications and noisy spectra by manual inspection, only one of these peptides appears to be a bona fide glycated peptide. This peptide has the same glycated lysine as Peptide 4 (Table 3), and is part of the hinge region on the heavy chain. The confirmation of 3 of the glycated peptides only identified by targeted MS1 speaks to the sensitivity of the targeted MS1 approach, in which no glycation enrichment or derivatization was performed.
With respect to the peptides only identified by the MS1 analysis, they exist at very low levels, but, admittedly, at levels similar to other peptides detected by MS2. In the absence of MS2 data or an orthogonal data type, we cannot determine if these peptides are bona fide glycated peptides or represent artifacts inherent to our method.
Forced glycation of this mAb increases glycation of the heavy chain complementary determining region 3 and reduces biological potency
As described above, potency analysis of the CEX fractions revealed that the more heavily glycated acidic fraction shows a drop in potency to 83%. In order to further define the relationship between glycation and potency, the mAb in high levels of glucose was incubated over a 6 day time course. Over this time window of forced glycation, the majority of the antibody population became multiply glycated by intact mass analysis (Fig. 4A), and glycation levels increased significantly compared to when the mAb was incubated with the non-reducing sugar sorbitol. Both the glucose- and sorbitol-incubated samples were tested for potency. As seen in Figure 4B, the forced glycated sample decreased in relative potency to ∼82% by day 6. This level was similar to that observed for the acidic CEX fraction (∼83%), which was approximately 30% glycated (Fig. 1C).
Figure 4.

(See previous page). Forced glycation of the mAb reveals a decrease in potency and an increase in glycation of K99. (A) Intact mass of the forced glycated mAb. On the top is the mAb incubated with 5% glucose while below is the mAb incubated with 5% sorbitol, a negative control. (B) Potency, expressed here as the relative potency of the mAb in glucose over the mAb in sorbitol. By day 6, potency has decreased to ˜82%. (C) Base Peak and survey MS1 scans of the Base Peak area for the glycated form of the peptide. Note that in the late stage process molecule, the +2 and +3 forms of the ions co-elute with another ion species (the 726 m/z species). The sum of the relative intensity of the 613 m/z (+3) and 919 m/z (+2) ions is estimated to be 36% of the 726 m/z ion. This estimate is used to adjust the estimate of the area for the glycated form.
The forced glycated sample was sodium borohydride derivatized and subjected to MS2-based peptide mapping. By analyzing the area of the glycated peptide in the extracted ion chromatogram and comparing it to the nonglycated form of the peptide, an increase in glycation at the K99 hot spot was observed (Fig. 4C). We estimate that glycation at K99 increases from less than 4% to 34% relative to the parent ion. While we cannot eliminate the effects of the additional glycation sites and levels of glycation at those sites, we do observe an inverse correlation between glycation at K99 and potency of this mAb.
Structural analysis reveals a putative glycation hotspot mechanism
Finally, an understanding of the physical properties that govern the propensity of sites to become glycated was sought. Inspection of the primary sequence of glycated peptides reveals that there are 43 lysine residues on this mAb. Of these 43, 8 have appreciable levels of glycation, though clearly K99 is the dominant glycation site. Structural modeling predicts that the vast majority of lysines on the mAb are solvent exposed (Fig. 5A). The lack of obvious differences in solvent exposure between glycated and non-glycated lysines indicates that proximal amino acids are involved in promoting or inhibiting glycation. The presence of a carboxylic acid has been proposed to act as an internal catalyst for glycation, with the oxygen atom acting as a nucleophile to propagate the conversion of the Schiff base to the more stable Amadori product.26,27 As seen in Figure 5B, K99 has 2 carboxylic acids, D105 on the heavy chain and E61 on the light chain, in proximity (at 2.5Å and 2.6Å, respectively), and a third carboxylic acid at D98 on the heavy chain at 11.6Å, a distance still close enough to nucleate glycation.27 Further structural analysis reveals that, of the glycated lysine residues identified in the targeted MS1 dataset, K99 is the only lysine where 2 carboxylic acids residues are within 4Å, with a third carboxylic acid also able to act as an effector (Fig. 5C). We propose that K99 is preferentially glycated over other lysine residues because of this relative surplus of catalytic opportunities to stabilize glycation.
Figure 5.

Structural modeling of this mAb reveals a putative mechanism for mediation of non-enzymatic glycation. (A) A three dimensional ribbon model of the mAb. The heavy chain is shown in green while the light chain is shown in gray. Lysine residues are shown in blue, while glycated lysine residues are shown in burgundy. (B) Two views of the K99 glycation hot spot. Two residues on the heavy chain, D98 and D105, and one residue on the light chain, E61, are able to exert an effect on K99. The red coloring on D98, D105 and E61 shows the position of oxygen atoms in the carboxylic acid.
Discussion
In this study, we report evidence that a therapeutic mAb undergoes non-enzymatic glycation that increases between early development and late-stage processes. The presence of glycation was first identified this mAb through top down intact-molecule mass spectrometry, a relatively straightforward approach that identifies the presence of glycation and provides quantification of glycation on the whole molecule. This approach does not provide information on the site of glycation, although, when employed in combination with cleavage of the molecule using the FabRICATOR enzyme, we were able to broadly localize a significant amount of glycation to the Fd region. The extent of glycation on the whole molecule was confirmed by an orthogonal approach, boronate affinity chromatography. While boronate affinity chromatography is a relatively straightforward and quantitative means of quantifying overall glycation levels, this method also does not provide information on sites of glycation. The sites of glycation were mapped, obtaining high quality MS2 spectra by derivatizing the glycation sites using sodium borohydride and using search spectral search algorithms to identify derivatized-hexose modified versions of peptides. This approach is more labor intensive than the intact mass and boronate affinity approaches described above, and, while providing site specific sites of glycation, the quantification of the glycated peptides was challenging, likely due to the low abundance of these peptides and the relative lower sensitivity of the LTQ mass spectrometer compared to the Exactive Plus mass spectrometer used for the MS1 approach. Quantification of specific glycation levels was achieved using a targeted MS1 approach on a high resolution, high mass accuracy mass spectrometer and Pinpoint spectral analysis software. This method provided highly sensitive quantification of the glycated peptides without prior derivatization of the hexoses, and these peptides can be confirmed by derivatization and MS2 analysis. The predominant glycation site is at K99 in the CDR3 of the Fd portion of the heavy chain. Finally, through structural modeling, we postulate that the glycation hotspot at K99 is mediated by the presence of 3 carboxylic acid containing residues; these residues act to catalyze the conversion of a Schiff base to the more stable Amadori product.
The methods presented herein to identify sites of glycation by derivatization and CID MS2 mass spectrometry, and to estimate site-specific quantities of glycation by targeted MS1, are a part of a larger effort in recombinant protein production. Increasingly, there is a shift away from the use of a multitude of different assessments, e.g., chromatographic separations, toward the capture of relevant product quality attributes by mass spectrometry and targeted, computationally assisted analyses. This will be particularly relevant in the biosimilar space where these high resolution analyses can assess similarity of complex post-translational modifications in a rapid and comprehensive manner.28,29
Glycation derivatization can be completed in very few steps and essentially adds only a one hour incubation to the entire protocol. This can easily be executed by analysts as part of routine peptide mapping protein characterization. The MS2-based approach does not appear to impede the generation of a complete and highly sensitive peptide map in the context of this mAb (data not shown). This method could be employed when process changes occur, in particular changes in sugar in a given medium. By mapping the sites of glycation, production teams can assess if potentially important domains are being glycated. One advantage of the derivatization approach is the ability to differentiate glycation from 2 different samples using differential stable isotope labeling during derivatization.25 Alternatively, others have used isotopically labeled sugars to identify sites with a propensity to become glycated, identifying the sites by MS2 analyses.12 These sites are then searched for glycation in the native peptide map.
Once a suitable glycation site assessment has been established by glycation derivatization and MS2-based peptide mapping, the targeted MS1 approach described herein could be employed to monitor changes in glycation during production in a near real-time analytical context, immediately assessing glycation, among other attributes, in response to process changes. The targeted MS1 approach can also be used to monitor glycation during Quality Control (QC), though further exploration of reproducibility and limits of detection still needs to be defined to meet the rigorous needs of a QC environment. Since the MS1 approach obviates the need for derivatization, no additional sample processing is required.
In the example presented in this study, during production of the mAb, among other changes, maximum glucose feed levels were increased between early development and late stage processes from 7 g/L to 11 g/L. An increase in glycation levels on the mAb from 15% in the early development process to 18% in the late stage process was also observed (Fig. 1B), presumably in response to the higher levels of glucose. By the targeted MS1 method we observe an increase in K99, the glycation hotspot on the mAb, between the 2 processes. Through forced glycation experiments, an inverse correlation between HC-CDR3 glycation and mAb potency is also observed. Therefore, there is likely a direct correlation between a change in process and potency. Further experimentation testing the relationship between glycation levels and process glucose levels would clarify the nature of this correlation.
Factors that promote the formation of aldemines, here Schiff bases, or the conversion of aldemines to ketones, in this case an Amadori product, are thought to be key mediators of protein glycation.30,31 Zhang and coworkers27 assessed the glycation levels of a series of mAbs that became variably glycated and found a correlation between glycation levels and the presence of an aspartic acid residue (D) within 11 Å of the glycated lysine. Though the distance of hydrogen bonding between N and O is approximately 3 Å, the authors propose that side chain flexibility can greatly reduce the 11 Å distance between the D and K side chains in their model. The presence of a D residue within interaction distance of the site of glycation is at the heart of the glycation mechanism; the carboxylic side chains on D and glutamic acid (E) residues are able to promote the conversion of aldemines to ketones.32 Structural analysis of this mAb provides an intuitive hypothesis for why a hot spot is generated at K99. There are 2 carboxylic acid-containing amino acids within 3 Å of K99 and another within 12 Å. Analysis of the glycated lysine residues identified by targeted MS1 (Fig. 5C) finds that K99 is located in a loop region in the heavy chain (Fig. 5B), and, as such, has considerable flexibility. Given side chain flexibility in addition to loop flexibility, it is possible that there are 3 sites able to catalyze conversion of the Schiff base to the Amadori product. We note that several other K residues have more than one potential catalytic residue that may be close enough to interact, depending on localized flexibilities. Our current hypothesis is that the bias toward K99 glycation results from the combined catalytic effects of at least 2 nucleophiles that are within 3 Å, that stabilize Amadori products at a much faster rate than at other sites. Mutagenesis experiments removing one of the nucleophiles would be a means of testing this hypothesis directly.
Our study demonstrates that this therapeutic mAb is glycated and links glycation levels to the production process and potency. The characterization of this mAb is an excellent example of the power of these methods to provide highly resolved analytical data, which informs our process changes and facilitates robust analytical comparability throughout development. Ultimately these data will inform structural models and protein design principles.
Materials and Methods
Intact mass analysis
Intact mass analysis was performed on an Agilent 6210 Time-of-Flight instrument equipped with electrospray ionization and an Agilent HP1100 chromatography system.
The mAb was deglycosylated with PNGase F (New England Biolabs) according to the manufacturer's instructions. Following deglycosylation the sample was diluted to 1 mg/mL. 5 µg of the mAb was loaded onto a size exclusion column (Waters Acquity UPLC BEH 200 SEC 1.7 µm, 4.6 mm × 150 mm) using an isocratic gradient of 0.1% formic acid and 15% acetonitrile at 0.4 mL/min, separating the mAb from buffer components. The total run time was 10 minutes and data was collected on the mass spectrometer between 1 and 4.3 minutes. The gas temperature was set to 350°C while the capillary voltage was set to 4000 V.
The resulting spectra were deconvoluted using Agilent's Mass Hunter software with a mass window of 140,000 – 150,000 Da.
CEX fractionation
The mAb was fractionated using semi-preparative CEX. A semi-preparative-CE-HPLC column, Dionex ProPac WCX-10 22 mm × 250 mm was employed on an Agilent HP1200 HPLC. The flow rate was 3 mL/min and the species were eluted over a 30 minute gradient. Acidic, main, and basic peak fractions were collected on a fraction collector over multiple cycles and subsequently pooled. The fraction pools were concentrated, buffer exchanged into formulation buffer, and then re-analyzed by analytical CE-HPLC to confirm identity and purity. This analytical CEX is highly resolving and would not be applicable at the process scale.
Boronate affinity
Boronate affinity chromatography was performed on an Agilent HP1100 system with a fraction collector using a Tosoh TSK-gel Boronate-5PW, 7.5 mm ID × 7.5 cm column. The mobile phase consisted of 100 mM HEPES pH 8.5, 200 mM NaCl, and 25 mM Tris 8.5. Elution was achieved over 5 minutes with the mobile phase plus the addition of 500 mM sorbitol. Total run time was 38 minutes with elution starting at 19 minutes.
Potency assay
Cells expressing human versions of the cognate receptors were incubated with varying concentrations of a mAb Reference Standard (0.26–1000 ng/mL) or test samples for 75 minutes at 37°C. 0.5 ng/mL of ligand was added to the cells and incubated at 25°C for 45 minutes. Cell lysates were generated using the lysis buffer supplied with the AlphaScreen® SureFire® kit as per the manufacturer's instructions. This assay detects phosphorylated target using a biotinylated anti-target antibody and an anti-phospho-target as well as 2 bead types: acceptor beads conjugated with Protein A and a hydrogel containing thioxene and donor beads coated with a hydrogel that contains phthalocyanine, a photosensitizer, and streptavidin. The biotinylated anti-target antibody in the reaction buffer is captured by the streptavidin-coated donor bead, which in turn captures the endogenous target protein in the cell lysate. The second antibody (anti-phospho target) is captured by the Protein A-conjugated acceptor bead, but only recognizes a specific phosphorylated form of the target protein in the cell lysates. The two beads are brought into proximity in the presence of the phosphorylated target protein. When a laser is applied to the complex, ambient oxygen is converted to singlet oxygen by the donor bead. An energy transfer to the acceptor bead occurs, resulting in light production (luminescence), which is measured in a plate reader equipped for AlphaScreen® signal detection.
Fabricator antibody digestion
Samples (60 µg) were incubated with 60 µl of the FabRICATOR enzyme (QED Biosciences) and the supplied reaction buffer in a total volume of 30 µl. Samples were then incubated at 37°C for 30 minutes, digesting the mAb into Fc/2 and Fab’2 fragments. For reduced intact mass analyses, the resulting digest was reduced in 4 M guanidine hydrochloride, 50 mM Tris pH 8.3, and 50 mM dithiothreitol (DTT) at 55°C for 30 minutes, which generates Fd (HC variable and CH1 region), LC, and Fc/2 fragments.
Sodium borohydride derivatization
The mAb was buffer exchanged into 50 mM Tris pH 7.5 using a Biospin column (Biorad) and diluted to a working concentration of 10 mg/mL. A freshly prepared stock of 100 mM sodium borohydride in 0.2 N NaOH was diluted 1:5 in water and mixed at a 1:1 ratio with the diluted, buffer exchanged mAb. This mixture was incubated at room temperature for 1 hour and the reaction stopped by acidification using 8 µl of 1.0 N HCl. After five minutes of incubation, the mixture was neutralized by the addition of an equal volume of 100 mM Tris 7.5. The pH was checked and adjusted to pH 7.5 if necessary.
Peptide mapping
The mAb was denatured with 5.5 M guanidine hydrochloride in 0.1 M Tris 8.3 and reduced with 10 mM DTT at 55°C for 30 minutes. After cooling to room temperature the protein was alkylated with 22 mM iodoacetic acid for 15 minutes, and this reaction quenched by the further addition of DTT (0.8 mM). Reduced and alkylated mAb was then buffer exchanged into 50 mM Tris 7.5 on a Biospin 6 size exclusion column. Deionized urea was added to a final concentration of 0.1 M and the sample then digested with trypsin at 1:40 for 4 hours. The reaction was stopped by the acidification of the sample (0.2% Cf) with trifluoroacetic acid.
For the derivatized MS2 analysis, samples were analyzed on a LC-MS2 mass spectrometry on a Thermo-Fisher LTQ-Velos ion trap coupled in-line to a Waters Acquity UPLC. Trypsin digest cleavage products were separated by reversed phase chromatography using an Agilent SB-C3 column in 0.1% trifluoroacetic acid with a gradient of acetonitrile. Eluted peaks were detected at 215 nm and were introduced into the mass spectrometer using a heated electrospray ionization source. The mass spectrometer was run in data-dependent acquisition mode with dynamic exclusion.
For the targeted MS1 analysis, samples were analyzed on an Exactive Plus coupled in-line to a Thermo Accela UPLC. Tryptic peptides were separated by reversed phase chromatography using an Agilent SB-C18 PRHO 1.8 µm column in 0.1% formic acid with a gradient of acetonitrile. Eluted peaks were detected at 214 nm and introduced into the mass spectrometer using a heated electrospray ionization source.
Searches
MS2 mass spectrometric spectral files generated on the LTQ Velos were processed using either the Trans Proteomic Pipeline with X! tandem33 as the search engine or Proteome Discoverer, using Sequest34 as the search engine. Searches were done for the mAb protein sequence, the NCBI Chinese Hamster Ovary reference proteome (GenBank Assembly ID GCA_000223135.1 consisting of 24240 entries), and trypsin and keratin sequence data sets, as well as a reverse decoy database. Searches also included the static modification of cysteine carboxymethylation (58.005 Da) and variable modifications of methionine oxidation (15.995 Da) and glycated, derivatized lysine (164.073 Da). Semi-tryptic fragments with up to 2 missed cleavages were allowed. The precursor mass tolerance for the Sequest search was 3 Da and the fragment mass tolerance was 0.6 Da. For the X! tandem searches, the precursor mass tolerance was from −2 to 4 Da and fragment mass tolerance was 0.4 Da. X! tandem search results were validated using PeptideProphet35 while Sequest search results were validated using Percolator36 in Proteome Discoverer.
Mass spectrometric spectral files generated on the Thermo Exactive Plus™ were processed using the Pinpoint Software package. Samples were analyzed using the screening tool function. This allowed for the targeted identification of glycated species based on the following criteria: the peptide shows a variable modification of 162 Da for the addition of hexose over the predicted mass of the parent ion, 4 isotopic masses matching the theoretical isotopic masses are identified, the non-glycated parent peptide elutes at or near the glycated form of the peptide, and the mass of the glycated ion is within 5 ppm of the theoretical mass. Additional parameters included the static modification of cysteine residues by carboxymethylation and tryptic cleavage with one missed cleavage. Sensitivity was set to allow for entities of less than 1% intensity. A dot product score of greater than 95% was also used as a selection criterion. The dot product score in this assessment is a correlation score between the experimental isotopic distribution acquired for a peptide and the theoretical isotopic distribution for that peptide, and is used as a measure of confidence in the identification of a given peptide. The presence of multiple charges states for a given ion was also used as a selection criterion, but in the absence of multiple charge states, the spectra were manually inspected to ensure the correct charge states and isotopic distributions were being assigned by the software.
Forced glycation
The mAb was diluted to 14 mg/mL in a 1 ml total volume of 20 mM NaPO4 pH 6.5 with 5% (w/v) glucose or sorbitol (a non-reducing sugar control). Samples were incubated at 37°C in a sealed cryotube over a period of 6 d and sampled on days 0, 2, 4, and 6.
For potency testing of the forced glycated samples, the results were normalized to the reference standard for the mAb, and the final result given as Glucose Potency over Sorbitol Potency to control for the effects of forced glycation.
Structural modeling
Structural homology models of the mAb were generated in-house using the Molecular Operating Environment (MOE – Chemical Computing Group, Montreal, Canada). MOE utilizes an in-house database of antibody structures that is composed on both publically deposited and Amgen proprietary structures to generate a knowledge-based homology model. To build the Fab domains, appropriate framework and loop templates were identified, grafted into the destination framework and followed with energy minimization to correct for stained geometries. Pymol (www.pymol.org) was used for visualization and distance measurements.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Lowell Brady for helpful discussions and insights regarding sodium borohydride derivatization and boronate affinity capture of glycated proteins. We also thank Dr. Richard Rogers, Dr. Alain Balland, and Jennifer Kerr for their work on the general approach for the targeted MS1 method used in this study.
References
- 1.Maillard L. Action des acides amines sure les sucres; formation des melanoidines par voie methodique. Comptes Rendus Hebdomadaires Des Seances De L'academie Des Sciences 1912; 154:66-8 [Google Scholar]
- 2.Priego Capote F, Sanchez JC. Strategies for proteomic analysis of non-enzymatically glycated proteins. Mass Spectr Rev 2009; 28:135-46; PMID:18949816; http://dx.doi.org/ 10.1002/mas.20187 [DOI] [PubMed] [Google Scholar]
- 3.Baynes JW, Watkins NG, Fisher CI, Hull CJ, Patrick JS, Ahmed MU, Dunn JA, Thorpe SR. The Amadori product on protein: structure and reactions. Prog Clin Biol Res 1989; 304:43-67; PMID:2675036 [PubMed] [Google Scholar]
- 4.Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia 2001; 44:129-46; PMID:11270668; http://dx.doi.org/ 10.1007/s001250051591 [DOI] [PubMed] [Google Scholar]
- 5.Frye EB, Degenhardt TP, Thorpe SR, Baynes JW. Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem 1998; 273:18714-9; PMID:9668043; http://dx.doi.org/ 10.1074/jbc.273.30.18714 [DOI] [PubMed] [Google Scholar]
- 6.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715-36; PMID:23134362; http://dx.doi.org/ 10.1021/ac3032355 [DOI] [PubMed] [Google Scholar]
- 7.Haberger M, Bomans K, Diepold K, Hook M, Gassner J, Schlothauer T, Zwick A, Spick C, Kepert JF, Hienz B, et al.. Assessment of chemical modifications of sites in the CDRs of recombinant antibodies: susceptibility vs. functionality of critical quality attributes. mAbs 2014; 6:327-39; PMID:24441081; http://dx.doi.org/ 10.4161/mabs.27876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Ponniah G, Zhang HM, Nowak C, Neill A, Gonzalez-Lopez N, Patel R, Cheng G, Kita AZ, Andrien B. In vitro and in vivo modifications of recombinant and human IgG antibodies. mAbs 2014; 6:1145-54; PMID:25517300; http://dx.doi.org/ 10.4161/mabs.29883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller AK, Hambly DM, Kerwin BA, Treuheit MJ, Gadgil HS. Characterization of site-specific glycation during process development of a human therapeutic monoclonal antibody. J Pharm Sci 2011; 100:2543-50; PMID:21287557; http://dx.doi.org/ 10.1002/jps.22504 [DOI] [PubMed] [Google Scholar]
- 10.Quan C, Alcala E, Petkovska I, Matthews D, Canova-Davis E, Taticek R, Ma S. A study in glycation of a therapeutic recombinant humanized monoclonal antibody: where it is, how it got there, and how it affects charge-based behavior. Anal Biochem 2008; 373:179-91; PMID:18158144; http://dx.doi.org/ 10.1016/j.ab.2007.09.027 [DOI] [PubMed] [Google Scholar]
- 11.Bunn HF, Higgins PJ. Reaction of monosaccharides with proteins: possible evolutionary significance. Science 1981; 213:222-4; PMID:12192669; http://dx.doi.org/ 10.1126/science.12192669 [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Zhang T, Jiang L, Hewitt D, Huang Y, Kao YH, Katta V. Rapid identification of low level glycation sites in recombinant antibodies by isotopic labeling with 13C6-reducing sugars. Anal Chem 2012; 84:2313-20; PMID:22324758; http://dx.doi.org/ 10.1021/ac202995x [DOI] [PubMed] [Google Scholar]
- 13.Hoffman MD, Sniatynski MJ, Rogalski JC, Le Blanc JC, Kast J. Multiple neutral loss monitoring (MNM): a multiplexed method for post-translational modification screening. J Am Soc Mass Spectr 2006; 17:307-17; PMID:16443369; http://dx.doi.org/ 10.1016/j.jasms.2005.11.002 [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Petyuk VA, Schepmoes AA, Orton DJ, Monroe ME, Yang F, Smith RD, Metz TO. Analysis of non-enzymatically glycated peptides: neutral-loss-triggered MS(3) versus multi-stage activation tandem mass spectrometry. Rapid Commun Mass Spectr 2008; 22:3027-34; PMID:18763275; http://dx.doi.org/ 10.1002/rcm.3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, Frolov A, Tang N, Hoffmann R, van de Goor T, Metz TO, Smith RD. Application of electron transfer dissociation mass spectrometry in analyses of non-enzymatically glycated peptides. Rapid Commun Mass Spectr 2007; 21:661-6; PMID:17279487; http://dx.doi.org/ 10.1002/rcm.2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Qian WJ, Knyushko TV, Clauss TR, Purvine SO, Moore RJ, Sacksteder CA, Chin MH, Smith DJ, Camp DG, 2nd, et al.. A method for selective enrichment and analysis of nitrotyrosine-containing peptides in complex proteome samples. J Proteome Res 2007; 6:2257-68; PMID:17497906; http://dx.doi.org/ 10.1021/pr0606934 [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Tang N, Brock JW, Mottaz HM, Ames JM, Baynes JW, Smith RD, Metz TO. Enrichment and analysis of nonenzymatically glycated peptides: boronate affinity chromatography coupled with electron-transfer dissociation mass spectrometry. J Proteome Res 2007; 6:2323-30; PMID:17488106; http://dx.doi.org/ 10.1021/pr070112q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Priego-Capote F, Scherl A, Muller M, Waridel P, Lisacek F, Sanchez JC. Glycation isotopic labeling with 13C-reducing sugars for quantitative analysis of glycated proteins in human plasma. Mol Cell Proteomics 2010; 9:579-92; PMID:NOT_FOUND; http://dx.doi.org/ 10.1074/mcp.M900439-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brena BM, Batista-Viera F, Ryden L, Porath J. Selective adsorption of immunoglobulins and glucosylated proteins on phenylboronate-agarose. J Chromatogr 1992; 604:109-15; PMID:1639919; http://dx.doi.org/ 10.1016/0021-9673(92)85535-2 [DOI] [PubMed] [Google Scholar]
- 20.Brady LJ, Martinez T, Balland A. Characterization of nonenzymatic glycation on a monoclonal antibody. Anal Chem 2007; 79:9403-13; PMID:17985928; http://dx.doi.org/ 10.1021/ac7017469 [DOI] [PubMed] [Google Scholar]
- 21.Liu X-C, Scouten W. Boronate Affinity Chromatography In: Bailon P, Ehrlich G, Fung W-J, Berthold W, eds. Affinity Chromatography: Methods and Protocols. Totowa, NJ: Humana Press, 2000:119-28. [DOI] [PubMed] [Google Scholar]
- 22.Fekete S, Beck A, Fekete J, Guillarme D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part I: salt gradient approach. J Pharma Biomed Anal 2015; 102:33-44; PMID:25240157; http://dx.doi.org/ 10.1016/j.jpba.2014.08.035 [DOI] [PubMed] [Google Scholar]
- 23.von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. The EMBO J 2002; 21:1607-15; PMID:11927545; http://dx.doi.org/ 10.1093/emboj/21.7.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.An Y, Zhang Y, Mueller HM, Shameem M, Chen X. A new tool for monoclonal antibody analysis: application of IdeS proteolysis in IgG domain-specific characterization. mAbs 2014; 6:879-93; PMID:24927271; http://dx.doi.org/ 10.4161/mabs.28762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Ponniah G, Neill A, Patel R, Andrien B. Identification and comparative quantitation of glycation by stable isotope labeling and LC-MS. J Chromatogr B Anal Technol Biomed Life Sci 2014; 958:90-5; PMID:24705536; http://dx.doi.org/ 10.1016/j.jchromb.2014.03.021 [DOI] [PubMed] [Google Scholar]
- 26.Venkatraman J, Aggarwal K, Balaram P. Helical peptide models for protein glycation: proximity effects in catalysis of the Amadori rearrangement. Chem Biol 2001; 8:611-25; PMID:11451663; http://dx.doi.org/ 10.1016/S1074-5521(01)00036-9 [DOI] [PubMed] [Google Scholar]
- 27.Zhang B, Yang Y, Yuk I, Pai R, McKay P, Eigenbrot C, Dennis M, Katta V, Francissen KC. Unveiling a glycation hot spot in a recombinant humanized monoclonal antibody. Anal Chem 2008; 80:2379-90; PMID:18307322; http://dx.doi.org/ 10.1021/ac701810q [DOI] [PubMed] [Google Scholar]
- 28.Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, Van Dorsselaer A, Sanglier-Cianférani S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. TrAC Trends Anal Chem 2013; 48:81-95; PMID:NOT_FOUND; http://dx.doi.org/ 10.1016/j.trac.2013.02.014 [DOI] [Google Scholar]
- 29.Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013; 27:495-507; PMID:23649935; http://dx.doi.org/ 10.1007/s40259-013-0036-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feeney RE, Blankenhorn G, Dixon HB. Carbonyl-amine reactions in protein chemistry. Adv Protein Chem 1975; 29:135-203; PMID:237412 [DOI] [PubMed] [Google Scholar]
- 31.Acharya AS, Roy RP, Dorai B. Aldimine to ketoamine isomerization (Amadori rearrangement) potential at the individual nonenzymic glycation sites of hemoglobin A: preferential inhibition of glycation by nucleophiles at sites of low isomerization potential. J Protein Chem 1991; 10:345-58; PMID:1910466; http://dx.doi.org/ 10.1007/BF01025633 [DOI] [PubMed] [Google Scholar]
- 32.Isbell HS, Frush HL. Mutarotation, Hydrolysis, and Rearrangement Reactions of Glycosylamines. J Organic Chem 1958; 23:1309-19; PMID:NOT_FOUND; http://dx.doi.org/ 10.1021/jo01103a019 [DOI] [Google Scholar]
- 33.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004; 20:1466-7; PMID:14976030; http://dx.doi.org/ 10.1093/bioinformatics/bth092 [DOI] [PubMed] [Google Scholar]
- 34.Eng JK, McCormack AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectr 1994; 5:976-89; PMID:24226387; http://dx.doi.org/ 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- 35.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 2002; 74:5383-92; PMID:12403597; http://dx.doi.org/ 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- 36.Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res 2009; 8:3176-81; PMID: 19338334; http://dx.doi.org/ 10.1021/pr800982s [DOI] [PMC free article] [PubMed] [Google Scholar]