

Abstract
Diabetic foot ulcers (DFUs) are a serious complication of diabetes. Previous exposure to hyperglycemic conditions accelerates a decline in cellular function through metabolic memory despite normalization of glycemic control. Persistent, hyperglycemia-induced epigenetic patterns are considered a central mechanism that activates metabolic memory; however, this has not been investigated in patient-derived fibroblasts from DFUs. We generated a cohort of patient-derived lines from DFU fibroblasts (DFUF), and site- and age-matched diabetic foot fibroblasts (DFF) and non-diabetic foot fibroblasts (NFF) to investigate global and genome-wide DNA methylation patterns using liquid chromatography/mass spectrometry and the Illumina Infinium HumanMethylation450K array. DFFs and DFUFs demonstrated significantly lower global DNA methylation compared to NFFs (p = 0.03). Hierarchical clustering of differentially methylated probes (DMPs, p = 0.05) showed that DFFs and DFUFs cluster together and separately from NFFs. Twenty-five percent of the same probes were identified as DMPs when individually comparing DFF and DFUF to NFF. Functional annotation identified enrichment of DMPs associated with genes critical to wound repair, including angiogenesis (p = 0.07) and extracellular matrix assembly (p = 0.035). Identification of sustained DNA methylation patterns in patient-derived fibroblasts after prolonged passage in normoglycemic conditions demonstrates persistent metabolic memory. These findings suggest that epigenetic-related metabolic memory may also underlie differences in wound healing phenotypes and can potentially identify therapeutic targets.
Keywords: diabetes, diabetic foot ulcer, DNA methylation, fibroblast, metabolic memory, wound healing, epigenetics
Abbreviations
- ANOVA
Analysis of Variance
- BMP
Bone Morphogenic Protein
- COL4A1
Collagen 4A1
- DAVID
Database for Annotation, Visualization, and Integrative Discovery
- DCCT
Diabetes Control and Complications Trial
- DFF
Diabetic Foot Fibroblast
- DFU
Diabetic Foot Ulcer
- DFUF
Diabetic Foot Ulcer Fibroblast
- DHS
DNase Hypersensitive Site
- DMP
Differentially Methylated Probe
- dNTPs
deoxynucleotide
- ECM
Extracellular Matrix
- EDIC
Epidemiology of Diabetes Interventions and Complications
- ENCODE
Encyclopedia of DNA Elements
- FGF1
Fibroblast Growth Factor 1
- HbA1c
Hemoglobin A1c
- NFF
Non-diabetic Foot Fibroblast
- NHLF
Normal Human Lung Fibroblast
- PLAU
Plasminogen Activator Urokinase
- SNP
Single Nucleotide Polymorphism
- TFBS
Transcription Factor Binding Site
- TGFb
Transforming Growth Factor b
- TNFa
Tumor Necrosis Factor a
- TSS
Transcription Start Site
- UTR
Untranslated Region
Introduction
A constellation of disease complications arise from chronic hyperglycemia and associated low-grade inflammation, contributing to the morbidity and mortality of diabetes. There is strong epidemiological evidence that previous exposure to hyperglycemia drives the progression of diabetic complications through a mechanism called “metabolic memory.”1 Metabolic memory in diabetes is defined as a persistent decline in cellular function in response to previous exposure to hyperglycemia despite normalization of glycemic control.1,2 This was well established by the Diabetes Control and Complications Trial (DCCT), where subjects treated with intensive insulin therapy for many years maintained superior glycemic control and developed fewer diabetic complications. The persistence of these outcomes was identified in the observational Epidemiology of Diabetes Interventions and Complications (EDIC) follow-up trial, in which all subjects from DCCT were treated with long-term, intensive insulin therapy.3 Subjects demonstrated indistinguishable hemoglobin A1c (HbA1c) levels; however, individuals initially treated with conventional insulin therapy displayed accelerated progression of diabetic complications, including hypertension, retinopathy, neuropathy, and microvascular complications, such as nephropathy.4,5 These findings, which have been further substantiated at the 20-year follow up EDIC reports,3 demonstrate the impact of persistent metabolic memory and the urgent need for strict glucoregulatory control in the management of diabetic patients.
It is now well established that a central mechanism underlying metabolic memory is the persistence of hyperglycemia-associated epigenetic patterns that sustain and drive the progression of disease phenotypes, despite long-term normalization of glycemic control. Unlike other molecular markers of diabetic status, such as the presence of glycated proteins, heritable transmission of epigenetic patterns across mitotic cell division can maintain hyperglycemia-driven epigenetic patterns despite removal of the glycemic insult.6,7 This metabolic memory has been demonstrated at the level of genome-wide DNA methylation,8,9 defined histone modifications,6,8,10,11 microRNA expression patterns12,13 and specific modulation of epigenetic regulatory machinery in animal and human studies12 across a range of diabetes-relevant tissues, including retinal, renal and vascular cells.2 Collectively, these studies have demonstrated gene expression and epigenetic profiles that are enriched in diabetes-relevant pathways, including metabolism and inflammation, and correlated with impaired cellular function and clinical complications. While a role for metabolic memory in diabetic complications has been studied in numerous cell types, including vascular smooth muscle cells,6,12 endothelial cells,8 glomerular mesangial cells,10 and retinal cells,11 no previous studies14 have focused on the role of epigenetic metabolic memory in establishing a diabetic phenotype specifically in fibroblasts derived from chronic, non-healing diabetic foot ulcers (DFUs).
Diabetic foot ulceration is a serious complication that significantly impairs the quality of patient life. Existing therapies, such as growth factor treatment15 and bioactive dressings harboring naïve fibroblasts that do not integrate into host tissues,16-18 have high failure rates and require repeated applications.17,18 Furthermore, there is limited understanding as to which diabetic patients are susceptible to the development of chronic, non-healing DFUs19 and why DFU treatments are effective in some patients and not in others. This lack of response to therapy demonstrates the urgent need to develop new insights into the risk profile of diabetic patients who may develop these complications in ways that can lead to next generation treatments for these chronic wounds.
As a mediator of crosstalk between multiple cell types critical to normal wound repair, fibroblasts are an essential cell type in wound healing due to their production of extracellular matrix (ECM), secretion of cytokines, support of re-epithelialization, and activation of angiogenesis.20 Previous studies have demonstrated that fibroblasts are altered in many types of chronic wounds,21,22 but their role in initiating and maintaining the non-healing, diabetic environment remains unclear.23,24 In particular, the epigenetic profile associated with metabolic memory in fibroblasts resident in diabetic ulcers or in patients at risk for developing them has not been studied and may reveal new targets that underlie compromised tissue regeneration and repair. To date, there are no studies that have characterized the epigenetic profile of a large human cohort of fibroblasts derived from DFUs when compared to site-, age-, and disease-matched, and normal-matched fibroblasts.
The aim of our current study was to characterize the DNA methylation signature of diabetic fibroblasts harvested directly from diabetic wounds and diabetic patients to determine whether the diabetic microenvironment alters epigenetic patterns. Differences in DNA methylation detected after prolonged culture of these diabetic, patient-derived fibroblast cell lines in normoglycemic conditions strongly suggest retained metabolic memory that is associated with poor wound healing outcomes in these diabetic patients.
Results
Generation of patient-derived cell lines
We generated a cohort of 12 patient-derived foot fibroblast cell lines from 3 groups of study participants including (1) Diabetic Foot Ulcer Fibroblasts (DFUF), (2) Diabetic Foot Fibroblasts (DFF) from diabetic patients without foot ulcers, and (3) Non-diabetic Foot Fibroblasts (NFF) from non-diabetic subjects without foot ulcers. The subject demographics and biopsy characteristics of the 4 patient-derived cell lines per group are presented in Table 1. No significant differences in age or gender were observed between patient groups. Mean diabetes duration, blood glucose, and hemoglobin A1c are presented per group.
Table 1.
– Clinical characteristics
| Cell line | Age | Sex | Diabetes | Specimen origin | Neuropathy | Blood glucose (mg/dl) | HbA1c (%, mmol/mol) | Diabetes duration (yrs) |
|---|---|---|---|---|---|---|---|---|
| DFUF1 | 52 | M | Type 2 | Plantar – ulcer | Y | 197+/−88.7 | 7.7+/−1.6, 61+/−18 | 14.3+/−1.2, |
| DFUF2 | 59 | F | Type 2 | Plantar – ulcer | Y | |||
| DFUF3 | 50 | M | Type 2 | 2nd metatarsal head – ulcer | Y | |||
| DFUF4 | 52 | F | Type 2 | Ankle – ulcer | N | |||
| DFF5 | 50 | M | Type 2 | R 5th toe | Y | 129+/−23.8 | 7.0+/−0.72, 53+/−8.2 | 2.29+/−2.4 |
| DFF6 | 49 | M | Type 2 | 2nd digit | Y | |||
| DFF7 | 67 | M | Type 2 | 2nd digit | Y | |||
| DFF8 | 68 | F | Type 2 | 2nd digit | Y | |||
| NFF9 | 57 | F | NA | Hammertoe | N | 90+/−49 | 5.4+/−0.28, 36+/−3.5 | NA |
| NFF10 | 39 | F | NA | R foot excision cyst | N | |||
| NFF11 | 67 | M | NA | 5th digit | N | |||
| NFF12 | 63 | F | NA | 3rd and 4th digit | N |
Donor characteristics of primary foot fibroblast cell lines. Primary foot fibroblast cell lines were derived from non-diabetic subjects, diabetic subjects with foot ulcers and diabetic subjects without foot ulcers. No differences in age or sex were observed between the 3 groups. We did not statistically compare blood glucose, HbA1c, or diabetes duration between groups because we did not have complete data of these parameters for all subjects, with 10 subjects’ blood glucose measures, 8 subjects’ HbA1c measures and 5 diabetic subjects’ disease duration data, therefore we present only summary statistics for these parameters. DFUF = diabetic foot ulcer fibroblast, DFF = diabetic foot fibroblast, NFF = non-diabetic foot fibroblast.
Global DNA methylation
To gain a top-down view of global epigenomic patterns, we first investigated global DNA methylation by liquid chromatography/mass spectrometry (LC/MS). Both DFF and DFUF samples displayed significantly lower percent DNA methylation compared to NFF (mean difference −0.32%; p = 0.03; Fig. 1). We observed a trend for lower percent DNA methylation in DFUF compared to DFF, though differences were not significant (p > 0 .05). This demonstrates that there are substantive changes in global DNA methylation in response to diabetes, suggesting major alterations in genomic integrity and gene expression.
Figure 1.

Diabetes is associated with lower global DNA methylation. Regardless of ulcer status, diabetic fibroblasts demonstrate significantly lower DNA methylation compared to non-diabetic fibroblasts (p = 0.03, mean difference −0.32%) by 2-way ANOVA for diabetes and ulcer status.
Genome-wide DNA methylation array
Three 2-group contrasts were performed with the data, comparing DFUF vs. NFF, DFF vs. NFF, and DFUF vs. DFF (Supplementary Table). One 3-group contrast was performed comparing DFUF, DFF, and NFF to each other. All 2-group contrasts identified a similar number of differentially methylated probes (DMPs), range 172–190, and 375 DMPs were identified in the 3-group comparison (Fig. 2a). Hierarchical clustering analysis of DMPs in the 3-group comparison demonstrated that DFUFs and DFFs cluster together and separately from NFFs, visualized as a heat map (Fig. 2b). Comparison of all the DMPs identified in the 3 2-group contrasts demonstrated that DFUF vs. NFF and DFF vs. NFF identified 45 of the same sites as being differentially methylated, which represents 25% of all the DMPs identified in these contrasts (Fig. 2c). Shared diabetic DMPs demonstrated a similar methylation β value in DFFs and DFUFs [mean absolute (βDFF-βDFUF) of 0.122, SD = 0.099] with the same direction of difference compared to NFFs (Table 2). Taken together, these observations support that we have identified a consistent diabetic epigenotype in foot-derived fibroblasts, regardless of patient ulcer status. Three probes were identified to be differentially methylated across all 3 contrasts; however, there was no consistent pattern in the direction of methylation differences between the groups or evident biological relevance of the associated genes in our context (Supplementary Table).
Figure 2.
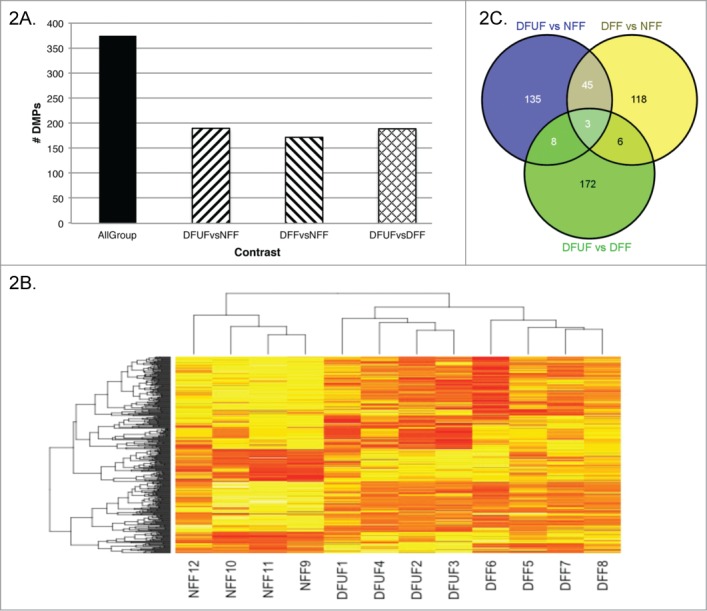
Diabetic fibroblasts demonstrate similar DNA methylation patterns. Differentially methylated probes (DMPs) identified in a 3-group comparison and each 2-group comparison (A). Overall, DFFs and DFUFs cluster together and separately from NFFs based on Euclidean hierarchical clustering of DMPs identified in the all-group analysis (B). Of the DMPs identified in the 2-group contrasts, 25% of DFF vs. NFF and DFUF vs. NFF identified differential methylation at the same probe (C).
Table 2.
Annotation of common diabetic DMPs
| Probe | Gene | Location | p value | NFFβ | DFFβ | DFUFβ |
|---|---|---|---|---|---|---|
| cg00945409 | ZMIZ1 (−91127); RPS24 (+944148) | chr10:80737665 | 0.057 | 0.81 +/−0.08 | 0.57 +/−0.21 | 0.45, +/−0.25 |
| cg01230320 | NT5C1B−RDH14 (−779135); OSR1 (+8392) | chr2:19549980 | 0.057 | 0.92 +/−0.18 | 0.56 +/−0.16 | 0.59 +/−0.16 |
| cg01632562 | SUPT3H (−284090); CLIC5 (+418326) | chr6:45629759 | 0.057 | 0.5 +/−0.12 | 0.23 +/−0.08 | 0.28 +/−0.09 |
| cg02100397 | FGF22 (+6965); RNF126 (+16343) | chr19:646890 | 0.057 | 0.88 +/−0.15 | 0.3 +/−0.26 | 0.53 +/−0.32 |
| cg02138953 | PLAU (−4583) | chr10:75666279 | 0.057 | 0.65 +/−0.03 | 0.41 +/−0.14 | 0.35 +/−0.20 |
| cg02368820 | ARHGEF16 (−318646); PRDM16 (+66760) | chr1:3052501 | 0.057 | 0.27 +/−0.29 | 0.71 +/−0.05 | 0.66 +/−0.14 |
| cg02799905 | NRP2 (−204998); PARD3B (+931711) | chr2:206342226 | 0.057 | 0.68 +/−0.12 | 0.43 +/−0.15 | 0.45 +/−0.15 |
| cg03217995 | HOXA7 (−7135); HOXA9 (+1719) | chr7:27203430 | 0.057 | 0.36 +/−0.05 | 0.56 +/−0.13 | 0.58 +/−0.12 |
| cg03301058 | GABRR1 (−80361); GABRR2 (+17111) | chr6:90007856 | 0.057 | 0.6 +/−0.17 | 0.35 +/−0.11 | 0.34 +/−0.18 |
| cg03859028 | TTC23 (−159475); MEF2A (−156844) | chr15:99949289 | 0.057 | 0.17 +/−0.11 | 0.42 +/−0.02 | 0.4 +/−0.10 |
| cg04478875 | SPRY4 (−319155); FGF1 (+41879) | chr5:142023774 | 0.057 | 0.68 +/−0.04 | 0.34 +/−0.07 | 0.4 +/−0.12 |
| cg04500819 | GORAB (−124288); METTL11B (+261788) | chr1:170376975 | 0.057 | 0.81 +/−0.03 | 0.56 +/−0.17 | 0.5 +/−0.13 |
| cg04888234 | FCRLA (−1183) | chr1:161675579 | 0.057 | 0.85 +/−0.18 | 0.49 +/−0.18 | 0.48 +/−0.03 |
| cg05949913 | CTGF (−285040); MOXD1 (+165107) | chr6:132557557 | 0.057 | 0.65 +/−0.09 | 0.38 +/−0.10 | 0.42 +/−0.1 |
| cg06486129 | C21orf33 (+19917); ICOSLG (+87424) | chr21:45573410 | 0.057 | 0.14 +/−0.05 | 0.43 +/−0.10 | 0.36 +/−0.11 |
| cg06766860 | GALNT9 (+41922); NOC4L (+234991) | chr12:132863983 | 0.057 | 0.44 +/−0.28 | 0.74 +/−0.04 | 0.66 +/−0.19 |
| cg07891658 | ZNF469 (−497148); BANP (+11694) | chr16:87996731 | 0.057 | 0.27 +/−0.03 | 0.48 +/−0.15 | 0.55 +/−0.20 |
| cg08797704 | CDH5 (−707920); CDH11 (−536687) | chr16:65692605 | 0.057 | 0.45 +/−0.08 | 0.2 +/−0.09 | 0.16 +/−0.02 |
| cg09259081 | KIAA1609 (−602) | chr16:84538889 | 0.057 | 0.32 +/−0.05 | 0.53 +/−0.14 | 0.55 +/−0.11 |
| cg10270430 | MLN (−252570); GRM4 (+77274) | chr6:34024362 | 0.057 | 0.19 +/−0.05 | 0.4 +/−0.10 | 0.4 +/−0.09 |
| cg10590622 | PDHA2 (−294) | chr4:96760945 | 0.057 | 0.61 +/−0.24 | 0.15 +/−0.01 | 0.14 +/−0.19 |
| cg10776061 | MAN2B1 (+9201); ZNF791 (+46659) | chr19:12768390 | 0.057 | 0.54 +/−0.14 | 0.3 +/−0.25 | 0.29 +/−0.23 |
| cg11728145 | PXDN (+90101); TPO (+240958) | chr2:1658190 | 0.057 | 0.72 +/−0.08 | 0.44 +/−0.20 | 0.43 +/−0.22 |
| cg12214399 | SPATA18 (+293068); USP46 (+314842) | chr4:53210660 | 0.057 | 0.14 +/−0.24 | 0.59 +/−0.12 | 0.55 +/−0.21 |
| cg13205848 | NTF3 (+134226); ANO2 (+379893) | chr12:5675505 | 0.057 | 0.75 +/−0.03 | 0.51 +/−0.13 | 0.52 +/−0.19 |
| cg14223671 | GNG13 (−7249); LMF1 (+163003) | chr16:857981 | 0.057 | 0.08 +/−0.08 | 0.3 +/−0.16 | 0.29 +/−0.09 |
| cg14447606 | CYP26B1 (+4635); DYSF (+676497) | chr2:72370328 | 0.057 | 0.11 +/−0.01 | 0.32 +/−0.14 | 0.44 +/−0.20 |
| cg14646613 | KLF4 (−160662) | chr9:110412708 | 0.057 | 0.79 +−0.06 | 0.49 +/−0.24 | 0.39 +/−0.18 |
| cg14895374 | HMBOX1 (+182571); KIF13B (+190129) | chr8:28930481 | 0.057 | 0.71 +/−0.06 | 0.49 +/−0.22 | 0.42 +/−0.11 |
| cg15752756 | HLA−DQB1 (−16) | chr6:32634481 | 0.057 | 0.45 +/−0.05 | 0.23 +/−0.04 | 0.17 +/−0.05 |
| cg16112880 | TMEM9 (−114) | chr1:201123745 | 0.057 | 0.14 +/−0.02 | 0.42 +/−0.32 | 0.67 +/−0.14 |
| cg17013691 | TRIO (+236495); ANKH (+491564) | chr5:14380323 | 0.057 | 0.83 +/−0.13 | 0.55 +/−0.02 | 0.62 +/−0.11 |
| cg18004235 | OSR1 (−249959); TTC32 (+293414) | chr2:19808330 | 0.057 | 0.73 +/−0.12 | 0.48 +/−0.05 | 0.49 +/−0.17 |
| cg20321086 | CLVS1 (−148318); CHD7 (+460884) | chr8:62052207 | 0.057 | 0.47 +/−0.14 | 0.23 +/−0.03 | 0.25 +/−0.02 |
| cg20895691 | ATAD2B (+508386) | chr2:23641550 | 0.057 | 0.55 +/−0.08 | 0.32 +/−0.09 | 0.26 +/−0.10 |
| cg23763647 | AKR1E2 (+289) | chr10:4868690 | 0.057 | 0.29 +/−0.20 | 0.07 +/−0.03 | 0.05 +/−0.01 |
| cg24284539 | CCDC3 (+44105); CAMK1D (+608017) | chr10:12999599 | 0.057 | 0.74 +/−0.07 | 0.45 +/−0.26 | 0.47 +/−0.24 |
| cg25570222 | SIX2 (−568090); SRBD1 (+33802) | chr2:45804631 | 0.057 | 0.66 +/−0.13 | 0.88 +/−0.04 | 0.91 +/−0.08 |
| cg25638870 | NOX4 (−65) | chr11:89224717 | 0.057 | 0.18 +/−0.14 | 0.43 +/−0.12 | 0.42 +/−0.02 |
| cg27333018 | ZNF57 (−3382) | chr19:2897514 | 0.057 | 0.69 +/−0.11 | 0.48 +/−0.05 | 0.47 +/−0.04 |
| cg00945409 | ZMIZ1 (−91127); RPS24 (+944148) | chr10:80737665 | 0.057 | 0.81 +/−0.08 | 0.57 +/−0.21 | 0.45, +/−0.25 |
| cg01230320 | NT5C1B−RDH14 (−779135); OSR1 (+8392) | chr2:19549980 | 0.057 | 0.92 +/−0.18 | 0.56 +/−0.16 | 0.59 +/−0.16 |
| cg01632562 | SUPT3H (−284090); CLIC5 (+418326) | chr6:45629759 | 0.057 | 0.5 +/−0.12 | 0.23 +/−0.08 | 0.28 +/−0.09 |
| cg02100397 | FGF22 (+6965); RNF126 (+16343) | chr19:646890 | 0.057 | 0.88 +/−0.15 | 0.3 +/−0.26 | 0.53 +/−0.32 |
| cg02138953 | PLAU (−4583) | chr10:75666279 | 0.057 | 0.65 +/−0.03 | 0.41 +/−0.14 | 0.35 +/−0.20 |
Shared DMPs have a similar methylation status between diabetic fibroblasts. Each DMP that was common to both DFF vs NFF and DFUF vs NFF are listed with their Illumina CpG identifier, probe chromosomal location, gene associations and CpG island relation. All 2 group comparisons that yielded the common diabetic DMPs had p values of 0.057. Mean β values per group, which reflect the methylation status, are listed for DFUF, DFF and NFF demonstrating similar methylation values for diabetic fibroblasts compared to non-diabetic fibroblasts.
Characterization of shared diabetic DMPs
Annotation of common diabetic DMPs to genomic regions identified that differential methylation was localized to both proximal and distal gene regions, with the greatest number of DMPs within gene bodies and DHSs (Fig. 3a). Due to the complicated nature of genome structure, one individual probe site may fit the criteria of several genomic region categories. Overlap between our diabetic DMP sites and histone modification peaks identified in Normal Human Lung Fibroblast (NHLF) ChIP-seq ENCODE data was explored through EpiExplorer and demonstrated substantial association with activating histone marks H3K4me1/2/3, H3K9ac, and H3K27ac compared to the randomly generated control data set (Fig. 3b). This suggested that our diabetic differential methylation may be associated with sites of active gene expression.
Figure 3.

Genomic context of common diabetic differentially methylation loci. Diabetic methylation localizes to proximal and distal regulatory regions and within gene bodies. (A). Common diabetic DMPs annotated to gene-centric and CpG island-centric regions based on the Illumina annotation, to DnaseI hypersensitive sites (DHS), insulators and enhancer regions through association with ENCODE data.68,69 (B). Co-localization of diabetic DMPs compared to permutation-generated control sites with histone modifications in Normal Human Lung Fibroblasts, derived from the EpiExplorer database. (C). Localization of diabetic DMPs, compared to control sites, to transcription factor binding sites (TFBS) across all tissues, derived from the EpiExplorer database.
We explored overlap between our diabetic DMPs and TFBS identified across multiple cell types in the ENCODE dataset (Fig. 3c). Evidence is surmounting that DNA methylation can modify the binding affinity of transcription factors to DNA to fine-tune gene expression regulation.25 Although further confirmation would be required in our samples for appropriate tissue and biological context, this analysis identified localization of diabetic DMPs to TFBSs of transcription factors that are known to regulate gene expression programs relevant to diabetes and wound healing, namely MAX, Sp1, and YY1.
We used a 2-pronged approach to associate our diabetic DMPs with genes to generate our list presented in Table 2. First, we used a proximal approach in which a DMP is associated with a gene if it falls within 1500bp upstream of the TSS through to the 3’UTR. Second, we employed a distal approach in which a DMP is associated with a gene if it falls within a DHS that is associated with expression changes of that gene. This gene annotation method enabled us to estimate gene associations based on both proximity and public data repositories.
Using the gene list presented in Table 2, we performed further bioinformatics analyses to gain biological insight into common diabetic differential methylation. Our DAVID functional annotation clustering identified 3 clusters that may offer biological insight with enrichment of genes related to oxidation/reduction, myofibril function and angiogenesis/extracellular matrix functions (Table 3).
Table 3.
- Functional annotation clustering of diabetic DMPs
| Term | p value | Genes | Fold Enrichment |
|---|---|---|---|
| Annotation Cluster #1 – Enrichment Score: 1.45 | |||
| oxidoreductase | 0.004 | NOX4, PXDN, AKR1E2, CYP26B1, PDHA2, MOXD1 | 5.40 |
| GO:0055114∼oxidation reduction | 0.008 | NOX4, PXDN, AKR1E2, CYP26B1, PDHA2, MOXD1 | 4.54 |
| GO:0020037∼heme binding | 0.030 | NOX4, PXDN, CYP26B1 | 10.73 |
| GO:0046906∼tetrapyrrole binding | 0.033 | NOX4, PXDN, CYP26B1 | 10.06 |
| GO:0005506∼iron ion binding | 0.150 | NOX4, PXDN, CYP26B1 | 4.22 |
| endoplasmic reticulum | 0.401 | NOX4, CYP26B1, MOXD1 | 2.13 |
| Annotation Cluster #2 – Enrichment Score: 1.28 | |||
| GO:0030016∼myofibril | 0.026 | CACNA1C, CACNA1S, VCL | 11.52 |
| GO:0044449∼contractile fiber part | 0.027 | CACNA1C, CACNA1S, VCL | 11.31 |
| GO:0043292∼contractile fiber | 0.031 | CACNA1C, CACNA1S, VCL | 10.56 |
| disease mutation | 0.367 | COL4A1, MAN2B1, CACNA1C, CACNA1S, VCL | 1.59 |
| Annotation Cluster #3 – Enrichment Score: 1.18 | |||
| angiogenesis | 0.007 | COL4A2, COL4A1, FGF1 | 23.01 |
| GO:0005578∼proteinaceous extracellular matrix | 0.035 | COL4A2, COL4A1, NAV2, FGF1 | 5.33 |
| GO:0031012∼extracellular matrix | 0.042 | COL4A2, COL4A1, NAV2, FGF1 | 4.94 |
| GO:0005576∼extracellular region | 0.074 | PXDN, COL4A2, COL4A1, NTF3, NAV2, FCRLA, FGF1, PLAU, VCL | 1.91 |
| hsa05200:Pathways in cancer | 0.276 | COL4A2, COL4A1, FGF1 | 2.74 |
| GO:0044421∼extracellular region part | 0.373 | COL4A2, COL4A1, NAV2, FGF1 | 1.78 |
Differentially methylated diabetic genes are enriched in functions associated with redox reactions, contraction, extracellular matrix and angiogenesis. DAVID functional annotation clustering of genes associated with diabetic DMPs demonstrates an enrichment of biologically relevant functions within the context of diabetes and wound healing. p-values of individual annotation terms are generated from a modified Fisher exact test and cluster enrichment scores are calculated as the geometric mean of all the enrichment p-values of each annotation term in the group72.
To gain additional insight into our genes associated with diabetic differential methylation, we uploaded our list into Ingenuity Pathway Analysis (IPA) to explore whether it generates any interaction networks, deemed significant if the negative log p value of the Fisher exact test is greater than 2. We identified a network titled “cell to cell signaling and interaction, tissue development, cancer” with a significant score of 8, displaying an interaction network with wound-healing relevant nodes including TGFβ, Myc, and TNFα (Fig. 4).
Figure 4.

Interaction network of differentially methylated diabetic genes. Ingenuity Pathway Analysis (IPA) was used to generate an interaction network using all the genes in our list and any first, second or third order interactions with other genes based on literature findings with specificity to the dermis, epidermis, and skin. Gray indicates genes that are differentially methylated in diabetic fibroblasts, while white represents genes that were not identified to be differentially methylated but interact with genes in our list.
Discussion
In this study we have identified a diabetic epigenotype using a cohort of 12 patient-derived cell lines, including diabetic foot ulcer fibroblasts (DFUF), which demonstrate characteristic features of metabolic memory based on their persistent expression of a diabetic phenotype upon prolonged cell culture. We have used a cohort of 12 fibroblast derived cell lines, including 8 cell lines from diabetic patients. Previous investigations of fibroblast cell lines from chronic wounds have been limited by using only a small number of fibroblast cell lines, and most of these studies have studied fibroblasts from venous ulcers and not from diabetic foot ulcers (DFUs).26-28 This is because fibroblasts from DFUs have been difficult to culture, as they have shown both an impaired responsiveness to growth factors and elevated numbers of senescent cells.29 Thus, we report one of the largest cohorts of fibroblasts from diabetic patients that are site-matched, to healthy control fibroblasts. This is the first report of a comprehensive DNA methylation analysis at the global and genome-wide levels using patient-derived fibroblasts within the context of diabetic wound healing. Identification of a persistent diabetic wound healing-related epigenotype in a cohort of patient fibroblasts contributes to a growing literature demonstrating that epigenetically-driven, metabolic memory directs cellular phenotypes that lead to complications associated with diabetes,1,2,6,8,9,14,30 adding further urgency to the need for strict glycemic control in diabetic therapy.
In order to further our understanding of foot ulcer development and wound repair incompetency in diabetic patients who may be at risk for their development, mechanisms underlying cellular alterations both before and after DFU development are needed. While our understanding of the pathophysiology of neuropathy and ischemia leading to DFUs has increased in recent years,31-33 insights into underlying alterations in fibroblasts in the chronic wound environment in general, and their participation in metabolic memory in particular, require further study. One of the challenges in the treatment of DFUs is the unpredictable variability in patient response to therapy, leading to treatment failures and limb amputation.19,34 This is partially due to limitations linked to the isolation and growth of fibroblasts in primary culture that has complicated their study in vitro. For example, while numerous studies have examined the behavior of primary fibroblasts derived from chronic wounds,26–28 fibroblasts from DFUs exhibit a spectrum of biological properties that are dependent on their site of origin within the wound.29 This heterogeneity is further complicated by the fact that fibroblasts isolated and cultured from the superficial dermis contain varied subpopulations that each have distinct phenotypes and proliferation kinetics.35 In spite of this, our findings extend previous studies on chronic wound fibroblasts26–28 by showing that DFU- and DFF-derived fibroblasts maintain their distinct phenotype and epigenetic profile in culture. Previous human studies investigating the role of fibroblasts in DFUs have primarily used immortalized cell lines following extensive passaging isolated from tissues that are not from the dermal foot region,36-39 which are less predictive of in vivo patient outcomes. It has previously been shown that tissue and context specificity is critical when studying epigenetic profiles because patterns have been shown to vary greatly in the same cell type between different tissue microenvironments,40,41 emphasizing the need to evaluate epigenetic patterns in cells derived from context-relevant tissues. By profiling epigenetic signatures using well-characterized, patient-derived fibroblasts, we have found evidence that these cell lines have biological relevance to their in vivo, biological microenvironment and offer opportunities to gain insights into mechanisms predisposing diabetic patients to the development of chronic foot ulcers. This supports the utility of primary fibroblast cultures as a tool to study chronic wound pathogenesis and to test potential therapeutic agents.
This is the first report of epigenetic metabolic memory in a cohort of patient-derived fibroblast cell lines derived from DFUs. In our study, DFFs and DFUFs were derived from diabetic patients with hyperglycemia and subsequently cultured through 4 passages in normoglycemic conditions. Therefore, the diabetic epigenotype that we have identified represents a hyperglycemia-associated DNA methylation pattern that is persistent despite multiple cell culture passages under normoglycemic conditions. Our finding that the diabetic epigenotype we identified was associated with hyperglycemia is supported by previous studies showing persistent epigenetic changes in endothelial cells from diabetic patients.42 This builds upon previous observations in animal studies performed with vascular smooth muscle cells derived from diabetic mice showing maintenance of altered histone modification patterns following ex vivo culturing in normoglycemic conditions.6 Zebrafish models of diabetic wound healing have demonstrated persistent, hyperglycemia-induced gene expression and DNA methylation patterns that are associated with impaired healing and tissue regeneration in response to a wounding insult.9,30 Previous studies in monozygotic twins (MZ) discordant for Type 1 diabetes have demonstrated differences in gene expression profiles in dermal fibroblasts,7 which has been attributed to hyperglycemia-driven epigenetic patterns; however, this was not investigated directly at the epigenetic level. Our study extends the identification of epigenetic metabolic memory from these animal models of diabetic wound healing9,30 and free-living humans7 to include cell culture studies on human fibroblasts in the context of diabetic foot ulceration. Though further confirmatory studies are needed, the decrease in global DNA methylation observed is similar to decreases observed in cancer studies,43 thus supporting clinical significance of our findings.
The most striking finding in our study was the consistent DNA methylation pattern identified in both DFFs and DFUFs when they were compared to NFFs. Importantly, this identifies a consistent diabetic epigenotype of metabolic memory regardless of ulcer status across multiple primary fibroblast cell lines. Our study is novel in that we are comparing cells derived from non-diabetic patients to cells derived from diabetic patients with and without ulceration, whereas previous epigenetic metabolic memory studies have only compared diabetic to non-diabetic tissue6,9,12,13,30 or hyperglycemia vs. normoglycemia.8,10,11 Whether the metabolic memory that we have identified in our study directly predisposes diabetic patients to foot ulceration in response to a wounding insult will require further study. Previous reports in the literature have demonstrated drifts in DNA methylation patterns over the course of the aging process44 that could potentially confound their interpretation in clinical studies. However, we identified a consistent methylation profile despite a range of ages in our patient cohort. This illustrates that the patient's diabetic history may be a more critical determinant in methylation than other factors such as age. Furthermore, it is possible that the prolonged exposure of DFUF to a sustained inflammatory environment when compared to DFF (Table 1) may impact their cellular and molecular phenotype. For example, it is known that mouse models of DFU are typified by a failure to shift from an M1 inflammatory macrophage to an M2 reparative macrophage responses leading to poor healing.45-47 This suggests that a longer duration of diabetes in DFUF compared to DFF may result in other inflammatory complications that are not unique to diabetes, but rather are common to other complex diseases such as atherosclerosis. However, at the level of DNA methylation, our analysis identifies consistent patterns despite the differing diabetes duration and exposure to inflammatory conditions between DFUF and DFF.
The identified metabolic memory epigenetic patterns seen in fibroblast cell lines derived from diabetic patients are localized to genes implicated in diabetic pathogenesis and wound healing.19 Functional annotation identified enrichment of gene clusters implicated in angiogenesis, ECM regulation, and myofibril function. In addition, the interaction network generated using literature specific to skin through Ingenuity Pathway Analysis (IPA) identified interactions with genes that are key regulators of normal wound healing and are altered in non-healing skin. For example, Type I collagen and fibronectin play important roles in the development of a provisional ECM that is essential for normal wound healing.33 The interaction network identified several notable nodes, including integrin α 3 and laminin 5, which play central roles in directing re-epithelialization, and c-myc, which is associated with a non-healing epithelial phenotype in chronic, non-healing wounds.32 We identified differential methylation associated with COL4A1, which is biologically important as this protein directly interacts with BMPs to modulate TGFβ regulation of myofibroblast differentiation required for wound contraction.48 Differential methylation associated with the pro-angiogenic cytokine FGF1 may underlie impaired angiogenesis characteristic of diabetic patients19 and contribute to poor wound healing outcomes. PLAU is a critical regulator in tissue remodeling and angiogenesis, with PLAU-deficient mice displaying delayed wound healing, decreased keratinocyte migration required for re-epithelialization, and impaired angiogenesis.49 Taken together, this supports that the identified epigenetic profile includes genes and interactions that are implicated in wound healing and repair.
Most of our gene specific findings are novel, as they differ from previous reports investigating genome wide DNA methylation profiles within the context of diabetes. For example, Hidalgo et al. identified significant associations between DNA methylation and clinical markers of diabetes in CD4 cells that were localized to ABCG1.50 We did not find differential methylation of ABCG1 in any of our contrasts, suggesting that these findings are likely to be specific for specific cell and tissue types.40 However, other studies have identified significant differences in the KCNQ1 gene, which we found to be differentially methylated in NFF vs. DFUF contrast, when comparing diabetic to non-diabetic subjects in pancreatic β cells,51 in adipose tissue of MZ twins discordant for type 2 diabetes,52 and in placenta and cord blood in mothers with gestational diabetes.53
Interpreting genome-wide epigenomic data relies on analyzing the interdependency of DNA methylation and histone modification patterns that cooperatively determine transcriptional activity.54,55 Previous studies in the context of cancer56 and cell differentiation57 have identified DNA methylation patterns and histone modifications that interact to drive gene expression and functional outcomes. In this study, we have identified the epigenomic landscape at the level of DNA methylation and we have associated this pattern with histone modifications by comparing them to other fibroblast lines and conserved transcription factor binding sites (TFBS) to gain insight into these cooperative, higher levels of epigenomic regulation. Although subject to further studies, our analyses suggest that diabetic DNA methylation patterns co-localize with activating histone modifications and previously identified TFBSs, suggesting that differentially methylated regions are highly regulated at multiple levels. Our present study is limited in that we have do not have corresponding gene expression analyses; however, we have interpreted the potential functional effects of our DNA methylation patterns to gene expression based on localization and anticipate that future studies will incorporate integrated analyses of histone modifications and methylation profiles that will inform more directed analyses of transcriptional activation.
We have presented the first evidence of an epigenetic pattern of metabolic memory in fibroblasts within the context of DFUs. By investigating a cohort of multiple cell lines derived from diabetic patients and site-matched controls, we have identified common epigenetic profiles that are consistent across diabetic patients that reflect the in vivo conditions. This is the case even when control diabetic cells were sampled from an adjacent site on the foot. Identification of these DNA methylation patterns in cell lines derived from diabetic patients after prolonged passage in normoglycemic conditions demonstrates the presence of a persistent, metabolic memory in these cells. Identification of genes in our epigenetic profile that are linked to angiogenesis, ECM production and myofibril contraction indicate that hyperglycemia-driven, metabolic memory is targeting functions critical to wound healing. Additional studies will be needed to establish if this epigenetic-related, metabolic memory may predispose diabetic patients to impaired healing in response to a wound insult and will lead to identification of novel therapies directed to these targets.
Research Design and Methods
Collection of patient-derived cell lines
We collected de-identified discarded skin specimens from 21 to 80 year-old subjects who underwent elective foot surgery at the Foot Center and Vascular Surgery clinic at the Joslin/Beth Israel Deaconess Medical Center. Specimens were representative of 3 patient groups: Diabetic Foot Ulcer Fibroblasts (DFUF) from individuals with an active foot ulcer, Diabetic non-ulcerated Foot Fibroblasts (DFF) from diabetic individuals without a foot ulcer and Non-diabetic Foot Fibroblasts (NFF) from healthy, non-diabetic subjects. All groups were matched for age and gender (Table 1).In addition, the diabetic groups were matched for duration of diabetes and glycemic control as defined by the levels of HbA1c. The discarded skin specimens were collected at the time of the surgical procedure in the operating room. Only the skin specimens that were determined by the operating surgeon to be discarded specimens that required no further evaluation were collected and used for this study. All procedures were approved by the Beth Israel Deaconess Medical Center Institutional Review Board (IRB).
Fibroblasts were isolated from patient biopsies as previously described.58 In brief, biopsies were treated in dispase overnight at 4°C and then centrifuged to collect any released cells followed by removal of the epidermis from the dermis the following day. Subsequently, the dermis was cut into small pieces and added to the cell pellet, then treated with collagenase and hyalurondiase in DMEM-F12 (Invitrogen, #11330–057) for one hour at 37°C with stirring. The cell suspension was mixed with red blood cell lysis buffer for 2 minutes, centrifuged, and then cells were collected and plated with the biopsy pieces on tissue culture plastic. Fibroblasts were expanded and second passage stocks were frozen in liquid nitrogen. Fibroblasts were grown in 1g/L glucose DMEM (Invitrogen, #11885–092), 10% FBS (ThermoScientific, HyClone #SH30071.03), HEPES (Sigma, #4034) and Pen/Strep/Fung (Invitrogen, #15240–062). Cells were passaged after reaching confluence. Lines were routinely checked for mycoplasma contamination with MycoAlert® Mycoplasma detection kin (Lonza #LT07–218, Rockland, ME) and PCR (Sigma, #MP0025). Flow cytometry analysis was performed to confirm fibroblast identity (data not shown).
DNA extraction
Cells were collected as cell pellets after 4 passages in normoglycemic culture media. DNA was extracted using the Qiagen DNeasy kit (Qiagen, #S407992) per manufacturer's instructions. DNA quality was confirmed by a clean band observed on agarose gel electrophoresis (data not shown) and 260/280 >1.8 as measured by Nanodrop.
LC/MS assay
Global genomic DNA methylation was investigated using the electrospray ionization LC/MS method as previously described.59 Briefly, 500ng of genomic DNA was digested with nuclease P1 (Sigma-Aldrich #N8630), venom phosphodiesterase I (Sigma-Aldrich, #P-3243) and alkaline phosphatase (Sigma-Aldrich #P-4252) to yield individual nucleotides. Isotope labeled [15N3]2’-deoxycytidine and (methyl-d3, ring-6-d1)-5-methyl-2’-deoxycytidine internal standards were added to the digested DNA, then samples were run in the electrospray LC/MS. To determine percent methylation, we first calculated the mass of sample methylated cytosine by multiplying the ratio of the known mass of the labeled methylcytosine internal standard (ISmC) to ISmC peak area by the peak area of the unlabeled methylcytosine. We calculated the mass of the sample unmethylated cytosine by multiplying the ratio of the known mass of the labeled cytosine internal standard (ISC) to ISC peak area by the peak area of the unlabeled cytosine. Percent methylated cytosine was calculated as the mass of methylated cytosine divided by the sum of the masses of methylated cytosine plus unmethylated cytosine.
DNA methylation array
The Illumina Infinium HumanMethylation450K BeadChip array (Illumina, #WG-314–1003) was used to investigate genome-wide DNA methylation at 485,577 loci across the genome, covering 99% of RefSeq genes at single-base resolution.60 The Methylation450K array uses 2 different probe types with unique chemistries enabling interrogation of both high and low density CpG regions in the genome. Briefly, high-quality genomic DNA was sent to the Microarray Facility at the Yale Center for Genome Analysis (New Haven, CT) and samples were sodium bisulfite treated, amplified, fragmented, and hybridized to the array. Subsequently, fluorescently labeled dNTPs were incorporated onto the probe through single base extension enabling determination of the methylation status at the single loci. All procedures were performed per manufacturer's instructions.
Statistical Analysis
The Methylation450K array was analyzed using the minfi package61 through R/Bioconductor.62 Briefly, 2 Illumina IDAT files per sample were read into RStudio63 with fluorescence data per probe in Cy3 and Cy5 fluorescence channels. All minfi quality control analyses were performed, including determining whether any probes yielded a detection p value >0 .01 in >50 % of samples. All analyses supported that all arrays were of good quality and did not require any specific array or subset of probes to be filtered out due to poor quality. Arrays were normalized using Subset quantile Within Array Normalization (SWAN) through minfi, which is a normalization method designed specifically to address the technical and biological variability in the Methylation450K arrays due to the type I and type II probes.64 Following normalization, all probes containing SNPs and probes mapped to sex chromosomes were filtered out to prevent bias due to unknown genetic background and mixed gender of samples, respectively, yielding 470,870 probes for subsequent analysis. Beta values per probe were calculated as β = M/(M+U+α), where M and U are the methylated and unmethylated signals and α is an arbitrary offset of 100. Three 2-group contrasts were performed with the data, namely DFUF vs. NFF, DFF vs. NFF, DFUF vs. DFF. Differentially Methylated Probes (DMPs) in the 2-group contrasts were identified as probes with a β value difference of > = 0.2 between groups and p < 0.06 in the Mann-Whitney test. We used p < 0 .06 as our threshold in these analyses because we identified a large number of probes at the p = 0.0596 level and deemed that this to be equivalent to the canonical threshold of p = 0.05. As we expected to find fewer, more subtle differences in our biological model compared to previous cancer epigenomic studies,65 we generated our DMP lists using an uncorrected p value and then identified similarities in the lists between contrasts as an additional level of statistical robustness. One analysis was performed using all 3 groups, defining DMPs as probes with a β value difference of p > = 0.2 and p < 0 .05 in the Wilcoxon rank sum test. We used the subset of probes identified from the 3-group analysis for hierarchical clustering using the Euclidean method. Based on the individual CpG resolution of the Methylation450K array and the findings from Bibikova et al.60 that a β value difference of > = 0.2 can be detected with 99%, subsequent site-specific validation is outside the scope of this analysis, similar to other reports using this array in the literature.66,67
All other statistical analyses were performed in RStudio.63 Two-way ANOVAs by diabetes and ulcer status were used to analyze LC/MS global DNA methylation.
Bioinformatic Analysis
DMPs were annotated to gene-centric and CpG island-centric regions using the provided Illumina annotation, enabling association of DMPs to 1500 or 200 base pairs upstream of Transcription Start Sites (TSS1500, TSS200 respectively), 5’ untranslated region (5’UTR), first exon, gene body, 3’UTR, CpG island and north or south CpG island shore or shelf. The genomic region annotation was expanded by associating DMP locations with publicly available databases including Regulatory Elements Database68 (http://dnase.genome.duke.edu/) to identify DNase Hypersensitive Sites (DHSs) and the EpiExplorer69 (http://epiexplorer.mpi-inf.mpg.de/) database to identify insulators, enhancers, histone modification patterns and Transcription Factor Binding Sites (TFBSs). All EpiExplorer analyses compared our set of DMP-loci with a randomly generated control set of loci to gain insight into enrichments within our set.69
We used 2 annotation methods to associate DMPs with genes based on localization. First, DMPs were associated with genes by using the provided Illumina annotation for probes that fall within 1500bp upstream of the gene to the 3’UTR. Second, since it is well documented that methylation sites located far away from genes can influence gene expression,70 we expanded on our gene annotation by identifying DMPs that fall within a DHS site that is associated with changes in expression of genes and subsequently associated our DMPs with those genes as has been previously performed by others.68,71
We explored our list of genes associated with DMPs by using the functional annotation table and functional annotation clustering analyses in DAVID72 with medium stringency parameters and by using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA) to generate interaction networks based on literature in dermal and epidermal tissues.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
L.K.P. conceived and designed the DNA methylation portion of the study, wrote the manuscript, collected and assembled data, analyzed and interpreted data. A.G.M collected data, assembled data, analyzed data and interpreted data. A.S. collected data. B.G. contributed to the interpretation of data. L.K.I. assisted in the analysis and interpretation of the data. A.V. and D.J.M. conceived and designed the study, obtained financial support and interpreted data. J.A.G. conceived and designed the study, obtained financial support, interpreted data and wrote manuscript. The authors would like to acknowledge Dr. Sang-Woon Choi and Dr. Stephanie Tammen for their assistance in the genomic DNA methylation LC/MS assay and Judith Edwards for her assistance.
J.A.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by NIH grant #DK098055–06A1.
Supplementary Material
References
- 1.Villeneuve LM, Reddy MA, Natarajan R. Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol 2011; 38:451-9; PMID:21309809; http://dx.doi.org/ 10.1111/j.1440-1681.2011.05497.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Intine RV, Sarras MP. Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Current diabetes reports 2012; 12:551-9; PMID:22760445; http://dx.doi.org/ 10.1007/s11892-012-0302-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan DM. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 2014; 37:9-16; PMID:24356592; http://dx.doi.org/ 10.2337/dc13-2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 2003; 290:2159-67; PMID:14570951; http://dx.doi.org/ 10.1001/jama.290.16.2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002; 287:2563-9; PMID:12020338; http://dx.doi.org/ 10.1001/jama.287.19.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A 2008; 105:9047-52; PMID:18579779; http://dx.doi.org/ 10.1073/pnas.0803623105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caramori ML, Kim Y, Moore JH, Rich SS, Mychaleckyj JC, Kikyo N, Mauer M. Gene expression differences in skin fibroblasts in identical twins discordant for type 1 diabetes. Diabetes 2012; 61:739-44; PMID:22315306; http://dx.doi.org/ 10.2337/db11-0617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, et al. . Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011; 21:1601-15; PMID:21890681; http://dx.doi.org/ 10.1101/gr.116095.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsen AS, Sarras MP, Leontovich A, Intine RV. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 2012; 61:485-91; PMID:22228713; http://dx.doi.org/ 10.2337/db11-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao C, Chen G, Liu L, Li X, He J, Jiang L, Zhu J, Xu Y. Impact of high glucose and proteasome inhibitor MG132 on histone H2A and H2B ubiquitination in rat glomerular mesangial cells. J Diabetes Res 2013; 2013:589474; PMID:23738337; http://dx.doi.org/ 10.1155/2013/589474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Q, Kowluru RA. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest Ophth Vis Sci 2013; 54:244-50; http://dx.doi.org/ 10.1167/iovs.12-10854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natarajan R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 2010; 59:2904-15; PMID:20699419; http://dx.doi.org/ 10.2337/db10-0208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madhyastha R, Madhyastha H, Nakajima Y, Omura S, Maruyama M. MicroRNA signature in diabetic wound healing: promotive role of miR-21 in fibroblast migration. Int Wound J 2012; 9:355-61; PMID:22067035; http://dx.doi.org/ 10.1111/j.1742-481X.2011.00890.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafehi H, El-Osta A, Karagiannis TC. Epigenetic mechanisms in the pathogenesis of diabetic foot ulcers. J Diabetes Complicat 2012; 26:554-61; PMID:22739801; http://dx.doi.org/ 10.1016/j.jdiacomp.2012.05.015 [DOI] [PubMed] [Google Scholar]
- 15.Falanga V, Eaglstein WH, Bucalo B, Katz MH, Harris B, Carson P. Topical use of human recombinant epidermal growth factor (h-EGF) in venous ulcers. J Dermatol Sur Oncol 1992; 18:604-6; http://dx.doi.org/ 10.1111/j.1524-4725.1992.tb03514.x [DOI] [PubMed] [Google Scholar]
- 16.Dinh TL, Veves A. The efficacy of Apligraf in the treatment of diabetic foot ulcers. Plast Rec Sur 2006; 117:152S-7S; discussion 8S-9S. [DOI] [PubMed] [Google Scholar]
- 17.Mustoe TA, O'Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Rec Sur 2006; 117:35S-41S. [DOI] [PubMed] [Google Scholar]
- 18.Ehrenreich M, Ruszczak Z. Update on tissue-engineered biological dressings. Tissue Eng 2006; 12:2407-24; PMID:16995775 [DOI] [PubMed] [Google Scholar]
- 19.Dinh T, Tecilazich F, Kafanas A, Doupis J, Gnardellis C, Leal E, Tellechea A, Pradhan L, Lyons TE, Giurini JM, et al. . Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 2012; 61:2937-47; PMID:22688339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999; 341:738-46; PMID:10471461 [DOI] [PubMed] [Google Scholar]
- 21.Stanley AC, Park HY, Phillips TJ, Russakovsky V, Menzoian JO. Reduced growth of dermal fibroblasts from chronic venous ulcers can be stimulated with growth factors. J Vasc Surg 1997; 26:994-9; discussion 9-1001; . [DOI] [PubMed] [Google Scholar]
- 22.Mendez MV, Stanley A, Park HY, Shon K, Phillips T, Menzoian JO. Fibroblasts cultured from venous ulcers display cellular characteristics of senescence. J Vasc Surg 1998; 28:876-83; PMID:9808856 [DOI] [PubMed] [Google Scholar]
- 23.Hehenberger K, Kratz G, Hansson A, Brismar K. Fibroblasts derived from human chronic diabetic wounds have a decreased proliferation rate, which is recovered by the addition of heparin. J dermatol Sci 1998; 16:144-51; PMID:9459127 [DOI] [PubMed] [Google Scholar]
- 24.Loot MA, Kenter SB, Au FL, van Galen WJ, Middelkoop E, Bos JD, Mekkes JR. Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. Eur J Cell Biol 2002; 81:153-60; PMID:11998867 [DOI] [PubMed] [Google Scholar]
- 25.Chen CC, Xiao S, Xie D, Cao X, Song CX, Wang T, He C, Zhong S. Understanding variation in transcription factor binding by modeling transcription factor genome-epigenome interactions. PLoS Comput Biol 2013; 9:e1003367; PMID:24339764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harding KG, Morris HL, Patel GK. Science, medicine and the future: healing chronic wounds. BMJ 2002; 324:160-3; PMID:11799036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook H, Davies KJ, Harding KG, Thomas DW. Defective extracellular matrix reorganization by chronic wound fibroblasts is associated with alterations in TIMP-1, TIMP-2, and MMP-2 activity. J Invest Dermatol 2000; 115:225-33; PMID:10951240 [DOI] [PubMed] [Google Scholar]
- 28.Kim BC, Kim HT, Park SH, Cha JS, Yufit T, Kim SJ, Falanga V. Fibroblasts from chronic wounds show altered TGF-β-signaling and decreased TGF-β Type II receptor expression. J Cell Physiol 2003; 195:331-6; PMID:12704642 [DOI] [PubMed] [Google Scholar]
- 29.Brem H, Golinko MS, Stojadinovic O, Kodra A, Diegelmann RF, Vukelic S, Entero H, Coppock DL, Tomic-Canic M. Primary cultured fibroblasts derived from patients with chronic wounds: a methodology to produce human cell lines and test putative growth factor therapy such as GMCSF. J Translat Med 2008; 6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarras MP Jr., Leontovich AA, Olsen AS, Intine RV. Impaired tissue regeneration corresponds with altered expression of developmental genes that persists in the metabolic memory state of diabetic zebrafish. Wound Repair Regen: Off Pub Wound Heal Soc Eur Tissue Repair Soc 2013; 21:320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falanga V. Chronic wounds: pathophysiologic and experimental considerations. J Invest Dermatol 1993; 100:721-5; PMID:8491995; http://dx.doi.org/ 10.1111/1523-1747.ep12472373 [DOI] [PubMed] [Google Scholar]
- 32.Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, Merchant A, Galiano RD, Tomic-Canic M. Molecular pathogenesis of chronic wounds: the role of β-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol 2005; 167:59-69; PMID:15972952; http://dx.doi.org/ 10.1016/S0002-9440(10)62953-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008; 453:314-21; PMID:18480812; http://dx.doi.org/ 10.1038/nature07039 [DOI] [PubMed] [Google Scholar]
- 34.Lantis JC, Marston WA, Farber A, Kirsner RS, Zhang Y, Lee TD, Cargill DI, Slade HB. The influence of patient and wound variables on healing of venous leg ulcers in a randomized controlled trial of growth-arrested allogeneic keratinocytes and fibroblasts. J Vasc Surg 2013; 58:433-9; PMID:23588112; http://dx.doi.org/ 10.1016/j.jvs.2012.12.055 [DOI] [PubMed] [Google Scholar]
- 35.Sorrell JM, Caplan AI. Fibroblasts-a diverse population at the center of it all. Int Rev Cell Mol Biol 2009; 276:161-214; PMID:19584013; http://dx.doi.org/ 10.1016/S1937-6448(09)76004-6 [DOI] [PubMed] [Google Scholar]
- 36.Sekhejane PR, Houreld NN, Abrahamse H. Irradiation at 636 nm positively affects diabetic wounded and hypoxic cells in vitro. Photomed Laser Sur 2011; 29:521-30; http://dx.doi.org/ 10.1089/pho.2010.2877 [DOI] [PubMed] [Google Scholar]
- 37.Loughlin DT, Artlett CM. Modification of collagen by 3-deoxyglucosone alters wound healing through differential regulation of p38 MAP kinase. PLoS ONE 2011; 6:e18676; PMID:21573155; http://dx.doi.org/ 10.1371/journal.pone.0018676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendoza-Naranjo A, Cormie P, Serrano AE, Wang CM, Thrasivoulou C, Sutcliffe JE, Gilmartin DJ, Tsui J, Serena TE, Phillips AR, et al. . Overexpression of the gap junction protein Cx43 as found in diabetic foot ulcers can retard fibroblast migration. Cell Biol Int 2012; 36:661-7; PMID:22455314; http://dx.doi.org/ 10.1042/CBI20110628 [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q, Wei F, Fong CC, Yu WK, Chen Y, Koon CM, Lau KM, Leung PC, Lau CB, Fung KP, et al. . Transcriptional profiling of human skin fibroblast cell line Hs27 induced by herbal formula Astragali Radix and Rehmanniae Radix. Journal of ethnopharmacology 2011; 138:668-75; PMID:22075453; http://dx.doi.org/ 10.1016/j.jep.2011.08.080 [DOI] [PubMed] [Google Scholar]
- 40.Maruyama R, Choudhury S, Kowalczyk A, Bessarabova M, Beresford-Smith B, Conway T, Kaspi A, Wu Z, Nikolskaya T, Merino VF, et al. . Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet 2011; 7:e1001369; PMID:21533021; http://dx.doi.org/ 10.1371/journal.pgen.1001369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gehrke S, Brueckner B, Schepky A, Klein J, Iwen A, Bosch TC, Wenck H, Winnefeld M, Hagemann S. Epigenetic regulation of depot-specific gene expression in adipose tissue. PLoS ONE 2013; 8:e82516; PMID:24340035; http://dx.doi.org/ 10.1371/journal.pone.0082516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008; 205:2409-17; PMID:18809715; http://dx.doi.org/ 10.1084/jem.20081188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friso S, Udali S, Guarini P, Pellegrini C, Pattini P, Moruzzi S, Girelli D, Pizzolo F, Martinelli N, Corrocher R, et al. . Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk. Cancer Epidemiol Biomarkers Prev 2013; 22:348-55; PMID:23300023; http://dx.doi.org/ 10.1158/1055-9965.EPI-12-0859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. . Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A 2012; 109(26):10522-7; PMID:22689993; http://dx.doi.org/ 10.1073/pnas.1120658109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8:958-69; PMID:19029990; http://dx.doi.org/ 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, et al. . An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest 2011; 121:985-97; PMID:21317534; http://dx.doi.org/ 10.1172/JCI44490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol 2013; 183:1352-63; PMID:24091222; http://dx.doi.org/ 10.1016/j.ajpath.2013.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet 2012; 21:R97-110; PMID:22914737; http://dx.doi.org/ 10.1093/hmg/dds346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juncker-Jensen A, Lund LR. Phenotypic overlap between MMP-13 and the plasminogen activation system during wound healing in mice. PLoS ONE 2011; 6:e16954; PMID:21326869; http://dx.doi.org/ 10.1371/journal.pone.0016954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hidalgo B, Irvin MR, Sha J, Zhi D, Aslibekyan S, Absher D, Tiwari HK, Kabagambe EK, Ordovas JM, Arnett DK. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes 2014; 63:801-7; PMID:24170695; http://dx.doi.org/ 10.2337/db13-1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, Kirkpatrick CL, Wollheim CB, Eliasson L, Ronn T, et al. . Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 2014; 10:e1004160; PMID:24603685; http://dx.doi.org/ 10.1371/journal.pgen.1004160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, Poulsen P, Ribel-Madsen R, Pedersen NL, Almgren P, et al. . Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014; 63:2962-76; PMID:24812430; http://dx.doi.org/ 10.2337/db13-1459 [DOI] [PubMed] [Google Scholar]
- 53.Ruchat SM, Houde AA, Voisin G, St-Pierre J, Perron P, Baillargeon JP, Gaudet D, Hivert MF, Brisson D, Bouchard L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013; 8:935-43; PMID:23975224; http://dx.doi.org/ 10.4161/epi.25578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295-304; PMID:19308066; http://dx.doi.org/ 10.1038/nrg2540 [DOI] [PubMed] [Google Scholar]
- 55.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010; 143:470-84; PMID:21029866; http://dx.doi.org/ 10.1016/j.cell.2010.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li D, Bi FF, Cao JM, Cao C, Liu B, Yang Q. Regulation of DNA methyltransferase 1 transcription in BRCA1-mutated breast cancer: a novel crosstalk between E2F1 motif hypermethylation and loss of histone H3 lysine 9 acetylation. Mol Cancer 2014; 13:26; PMID:24502362; http://dx.doi.org/ 10.1186/1476-4598-13-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luu PL, Scholer HR, Arauzo-Bravo MJ. Disclosing the crosstalk among DNA methylation, transcription factors, and histone marks in human pluripotent cells through discovery of DNA methylation motifs. Genome Res 2013; 23:2013-29; PMID:24149073; http://dx.doi.org/ 10.1101/gr.155960.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Normand J, Karasek MA. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev Biol Anim 1995; 31:447-55; PMID:8589888; http://dx.doi.org/ 10.1007/BF02634257 [DOI] [PubMed] [Google Scholar]
- 59.Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem 2002; 74:4526-31; PMID:12236365; http://dx.doi.org/ 10.1021/ac020050h [DOI] [PubMed] [Google Scholar]
- 60.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. . High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288-95; PMID:21839163; http://dx.doi.org/ 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 61.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA Methylation microarrays. Bioinformatics 2014; PMID:24478339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. . Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80; PMID:15461798; http://dx.doi.org/ 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: 2012, http://www.R-project.org [Google Scholar]
- 64.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; PMID:22703947; http://dx.doi.org/ 10.1186/gb-2012-13-6-r44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaneda A, Matsusaka K, Sakai E, Funata S. DNA methylation accumulation and its predetermination of future cancer phenotypes. J Biochem 2014; 156:63-72; PMID:24962701; http://dx.doi.org/ 10.1093/jb/mvu038 [DOI] [PubMed] [Google Scholar]
- 66.Kippler M, Engstrom K, Mlakar SJ, Bottai M, Ahmed S, Hossain MB, Raqib R, Vahter M, Broberg K. Sex-specific effects of early life cadmium exposure on DNA methylation and implications for birth weight. Epigenetics 2013; 8:494-503; PMID:23644563; http://dx.doi.org/ 10.4161/epi.24401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, Yang BT, Lang S, Parikh H, Wessman Y, et al. . Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012; 61:3322-32; PMID:23028138; http://dx.doi.org/ 10.2337/db11-1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, Furey TS. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 2013; 23:777-88; PMID:23482648; http://dx.doi.org/ 10.1101/gr.152140.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halachev K, Bast H, Albrecht F, Lengauer T, Bock C. EpiExplorer: live exploration and global analysis of large epigenomic data sets. Genome Biol 2012; 13:R96; PMID:23034089; http://dx.doi.org/ 10.1186/gb-2012-13-10-r96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/ 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 71.Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, Haroutunian V, Zhu J, Donovan MJ, Sharp AJ, et al. . Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nature Neurosci 2014; 17:121-30; PMID:24270187; http://dx.doi.org/ 10.1038/nn.3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.