

Abstract
Systemic lupus erythematosus (SLE) is a chronic, heterogeneous autoimmune disease short of effective therapeutic agents. A multitude of studies of SLE in the last decade have accentuated a central role of the interferon alpha (IFN-α) pathway in SLE pathogenesis. We report here a candidate therapeutic neutralizing antibody, AIA22, with a different binding epitope and discrepant neutralizing profile from the anti-multiple IFN-α subtype antibodies currently in clinical trials. AIA22 specifically interacts with multiple IFN-α subtypes, binds to the type I IFN receptor 2 (IFNAR2) recognition region of IFN-α (considered a novel antigen epitope), and effectively neutralizes the activity of almost all of the IFN-α subtypes (with the exception of IFN-α7) both in vitro and in vivo. Concurrently, structural modeling and computational design yielded a mutational antibody of AIA22, AIAmut, which exhibited substantially improved neutralizing activity to multiple IFN-α subtypes.
Keywords: antigen epitope, computational design, interferon alpha, monoclonal antibody, neutralizing activity, systemic lupus erythematosus
Abbreviations
- SLE
systemic lupus erythematosus
- IC
immune complex
- IFN
interferon
- IFNAR
type I interferon receptor
- PBMC
peripheral blood mononuclear cell
- PD
pharmacodynamic
- scFv
single-chain variable fragment
- PDB
protein data bank
- IgG
immunoglobulin G
- SPR
surface plasma resonance
- ka
association rate constant
- kd
disassociation rate constant
- KD
equilibrium disassociation constant
- CDR
complementary determining region
- AHC
anti-human-IgG conjugated biosensor
- Fab
antigen binding fragment
- Adv
adenoviral vector
- VEGF
vascular endothelial growth factor
- TNF
tumor necrosis factor
- ADCC
antibody-dependent cell-mediated cytotoxicity
- WB
whole blood
- OPD
o-phenylenediamine dihydrochloride
- OD
optical density
- DEPC
diethyl pyrocarbonate
- RT-PCR
reverse transcription polymerase chain reaction
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease typically characterized by the occurrence of many different autoantibodies that induce inflammation and damage of vital organs and tissues by the formation of immune complexes (ICs) with corresponding autoantigens. The etiopathogenesis of SLE has been studied intensively for many years.1 Genetic and epigenetic factors, environmental triggers, sex hormones, immunoregulatory factors, and stochastic events are commonly believed to expound a predisposition for the development of SLE.2 However, these factors fail to fully explain the etiology and pathogenesis of this disease.3 Complex pathogenesis, multisystem involvement, and clinical heterogeneity render SLE a difficult disease both in its treatment and diagnosis.4 Recently, SLE patients have been routinely treated with non-targeted chemotherapeutic drugs, including the antimalarial agents chloroquine and hydroxychloroquine, steroids, or immunosuppressive drugs, resulting in an overall increase in survival.4,5 However, due to the progression of SLE in a substantial percentage of patients and the side effects of recent standard therapy, further research is needed to better characterize the pathogenetic mechanisms of SLE, identify specific therapeutic targets, and develop effective and nontoxic novel agents.
Interferons (IFNs) are a family of mammalian cytokines that exhibit similar biological effects, including antiviral, antiproliferative, and immunomodulatory activities. They are classified as type I, II, or III based on their chromosomal location, protein sequence, structure, receptor recognition, and physicochemical properties. Human type I IFNs consist of 6 distinct classes (IFN-α, IFN-β, IFN-ε, IFN-κ, IFN-ω, and IFN-υ), and share the same cellular receptor (type I IFN receptor), which is composed of 2 subunits commonly termed IFNAR1 and IFNAR2.6 In humans, IFN-α consists of at least 12 subtypes that share nearly 85% amino acid homology.7,8 It remains unclear why there are so many different type I IFNs including multiple IFN-α subtypes. A variety of studies suggest they possess an overlapping, but also unique, set of biological activities.6
Early functional studies of IFN-α focused on their therapeutic applications and effects on immunocytes, and several recent studies have revealed their role in the pathogenesis of human disease, especially in SLE.9-13 Studies both in mice and humans have demonstrated the correlation between IFN-α and disease activity and severity of SLE. The implication of IFNs in the development of SLE was first observed in their therapeutic applications in cancer and viral infections, which induce autoantibody formation in 4–19% of patients and a variety of SLE symptoms in 0.15–0.70%.9 Elevated levels of IFN-α (particularly IFN-α2) were also detected in the serum of some SLE patients.10,11 Microarray studies that investigated IFN-induced gene expression profiles (so-called IFN signature) in peripheral blood mononuclear cells (PBMCs) of SLE patients have further supported the idea that IFN-α is involved in disease pathogenesis.10-13 Additionally, type I IFN receptor deficiency in lupus-prone NZM 2328 mice has been shown to decrease dendritic cell numbers and activation, which protects the mice from lupus disease.14 These findings have provided additional rationale for IFN-α blocking strategies for human SLE therapy.
Clinical trials in patients with SLE have recently been conducted using either monoclonal antibodies against IFN-α (sifalimumab, rontalizumab [discontinued after completing Phase 2 trials], AGS-009) or IFNAR (MEDI-546), or a therapeutic vaccine (IFN-α kinoid) that induces host polyclonal antibodies against IFN-α. These agents have shown a positive pharmacodynamic (PD) effect with respect to inhibition of type I IFN signature and some promising signs in clinical efficacy.15 The monoclonal antibodies that target multiple IFN-α subtypes bind to amino acid residues of IFN-α that are involved in IFNAR1 (IFNAR1 recognition region), but not to those involved in IFNAR2, which is the receptor subunit with higher affinity to ligand. Notably sifalimumab, a therapeutic antibody developed by MedImmune LLC, is reported to display high affinity and extensive neutralizing activity to multiple IFN-α subtypes. The Phase 2b clinical study of sifalimumab, which enrolled 835 patients with moderate/severe systemic lupus erythematosus (SLE or lupus), met its primary endpoint and showed clinically important improvements in organ-specific outcome measures (joint, skin) and patient-reported outcomes.
Here, we describes a novel monoclonal antibody acquired from a human single-chain antibody (scFv) phage library, which has an extensive neutralizing potency to multiple IFN-α subtypes, but has disparate binding epitopes (IFNAR2 recognition region) from the anti-IFN-α monoclonal antibodies currently in clinical trials. Structural modeling of the antibody-antigen complex revealed the interface residues involved in antigen binding and provided the details of the recognition mechanism; subsequent computational design yielded a variant antibody with extensively improved neutralizing activities to multiple IFN-α subtypes. Furthermore, the inhibitory effects of the novel monoclonal antibodies on IFN-α signature in IFN-α adenovirus treated mice have also been demonstrated.
Results
IFN-α1b and IFN-α2b are chosen from all of the IFN-α subtypes as the antigen for panning
The span of polymorphisms of IFN-α renders it an onerous choice for selecting suitable subtypes to screen antibodies with high affinity and extensive neutralizing potency to IFN-α subtypes from the fully synthetic phage display library. Semi-quantitative studies have demonstrated that IFNAR2 is mainly responsible for the high affinity interaction between ligand and receptor, while IFNAR1 (required for the stability of the ternary compound of IFNAR1/IFN-α/IFNAR2 and the further initiation of the signaling cascade) displays almost no binding to IFN-α.6,16 Accordingly, the IFNAR2 recognition region was considered to be the preferred binding epitope of further neutralizing antibodies. The crystal structure of the IFN-α2/IFNAR2 binary complex at resolution of 2.0Å (PDB:3S9D) revealed the recognition mode and interaction residues between IFN-α2 and IFNAR2.17 The amino acid sequence alignment of the 12 IFN-α subtypes was then investigated, and the residues that interacted with IFNAR2 were labeled as described in the Figure S1A legend. Additionally, the crystal structure PDB:3UX9 describes a high specificity antibody that interacts only with IFN-α1b, but not IFN-α2b whose interaction residues on IFN-α1b partially overlap the IFNAR2 recognition region.18 The analysis of these residues indicates that it may be beneficial to avoid interaction with residues 26, 27, 30, and 31 of IFN-α for future antibodies with extensive IFN-α subtype-neutralizing activity. IFN-α1 and IFN-α13 show significant differences with other subtypes at these 4 residue sites, which are graphically described by the alignment of the structure of IFN-α2 (PDB:3S9D) and IFN-α1b (PDB:3UX9) (Fig. S1B–E). Ultimately, IFN-α1b and IFN-α2b were employed to screen target antibodies blocking the interaction between IFN-α and IFNAR2 and neutralizing multiple human IFN-α subtypes effectively from the phage display library.
AIA22 specifically recognizes recombinant IFN-α with high affinity
A human single-chain antibody (scFv) phage library was screened with immunotubes coated with IFN-α1b and IFN-α2b in turn to select scFvs that bind to multiple human IFN-α subtypes. The three rounds of panning with progressively stringent washing conditions resulted in about 43 positive clones with specific genes (Table S1 and Fig. S2), which were then cloned into full-length human IgG1 expression vectors. From 23 antibodies that expressed well in the FreeStyle™ 293-F system, one candidate, AIA22, was selected for its specific binding activity to both IFN-α1b and IFN-α2b. The binding specificity of AIA22 to IFN-α was approximated using ELISA. The result (Fig. 1) revealed that AIA22 specifically recognizes IFN-α, including IFN-α1b and IFN-α2b.
Figure 1.
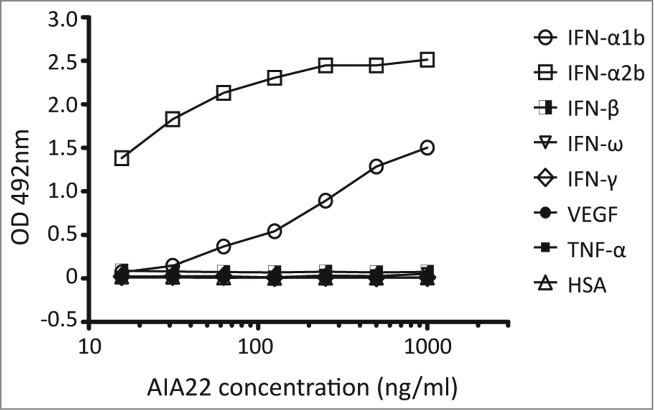
Binding activity of AIA22 to different antigens. All the antigens were coated at 5 μg/ml. The optical density (OD) at 492 nm reduced in a dose-dependent manner with decreasing amounts of AIA22 when binding to IFN-α1b and IFN-α2b. No cross-reactivity was observed when AIA22 was incubated with other type I IFN (IFN-β, IFN-ω), type II IFN (IFN-γ), cytokines (VEGF, TNF), and human serum albumin (HSA).
The affinity constant of AIA22 to recombinant IFN-α1b and IFN-α2b were examined through surface plasma resonance (SPR) analysis using the BIAcore™ T200 system. The results are shown in Table 1. Apparently, sifalimumab forms stronger interactions with IFN-α2b and IFN-α1b than AIA22. It should be noted that the binding mechanism of AIA22 to IFN-α1b (Fig. S3D) does not give a straightforward relationship between affinity and kinetics. We determined the affinity constant of AIA22 to IFN-α1b from analysis of the binding level at steady state (Fig. S4A, B ). The results (Table 1) show that the affinity of AIA22 to IFN-α1b is about 1/600th of that to IFN-α2b. According to the sensorgrams (Figs. S3D and S4A), AIA22 displays a sharply decreased disassociation phase when binding to IFN-α1b that may result from disharmonious recognition of the binding interface.
Table 1.
Affinity constant of AIA22 and sifalimumab
| Mean ± Standard error |
|||||
|---|---|---|---|---|---|
| Monoclonal antibody | IFN-α subtype | ka(104 M−1s−1) | kd(10−4 s−1) | KD (10−9 M) | Chi2 (RU2) |
| AIA22 | IFN-α2b | 200.67±41.28 | 6.49±0.56 | 0.33±0.05a | 0.24±0.07 |
| IFN-α1b | NA | NA | 210.90±6.13b | 0.15±0.13 | |
| Sifalimumab | IFN-α2b | 47.83±0.29 | 0.58±0.13 | 0.12±0.03a | 0.27±0.04 |
| IFN-α1b | 23.67±5.68 | 20.08±2.41 | 8.69±0.91a | 0.27±0.02 | |
ka, association rate constant; kd, disassociation rate constant; KD, equilibrium disassociation constant; NA, not available. All of the values are determined by BIAcore™ T200 system and represent the mean ± standard error from 3 separate assays.
The value is derived from kinetics measurements by kd/ka (1:1 binding).
The value is derived from steady-state measurements (1:1 binding).
Structural prediction of AIA22's recognition mode to IFN-α2b and IFN-α1b
To clarify the recognition mode of AIA22 to IFN-α1b, we initially built a structure model of AIA22 and its complex with IFN-α2b (Fig. S5). Interestingly, the AIA22/IFN-α2b complex structure revealed that the binding epitope of AIA22 on IFN-α2b partially overlaps the IFNAR2 recognition region (Fig. 2A and 2B), which is a potentially novel antigen epitope different from that of neutralizing antibodies sifalimumab and AGS-009. The analysis of the predicted interaction interface also indicated that the light chain residues Ser31a, Asn31b, Tyr32, Asp50, Asn52, Lys66, and the heavy chain residues Tyr95, Ser97, Tyr99 make direct interaction with IFN-α2b by forming extensive hydrogen-bonds (or π interaction networks) (Fig. 2C). This indicates that the complementary-determining region (CDR) L1, L2 and CDR H3 play an important role in AIA22's binding to IFN-α2b. Glu96 of CDR L3 is also a key residue that maintains the stability between light and heavy chains (Fig. 2C). To demonstrate these interactions, we substituted these sites with alanine and detected the relative affinity to IFN-α compared with AIA22 by ELISA.19 According to the results (Table S3), the alanine replacement of light chain residues Tyr32, Asp50, Glu96 and heavy chain residue Tyr95 resulted in considerable loss of binding activity to IFN-α2b, which was in accordance with the prediction of AIA22/IFN-α2b complex model. Accordingly, we considered the AIA22/IFN-α2b complex model plausible, and next built the AIA22/IFN-α1b complex structure by using a structural superposition method. The predicted AIA22/IFN-α1b interaction interface is shown in Figure 2D. The key residues of interaction were also demonstrated by the results of alanine scanning experiments (Table S3). Additionally, the analysis of AIA22/IFN-α1b interaction revealed that residue Tyr99 of AIA22 heavy chain appeared to account for the low affinity to IFN-α1b due to its strong clash with the backbone of Leu30 and Met31 on IFN-α1b (Fig. 2D).
Figure 2.
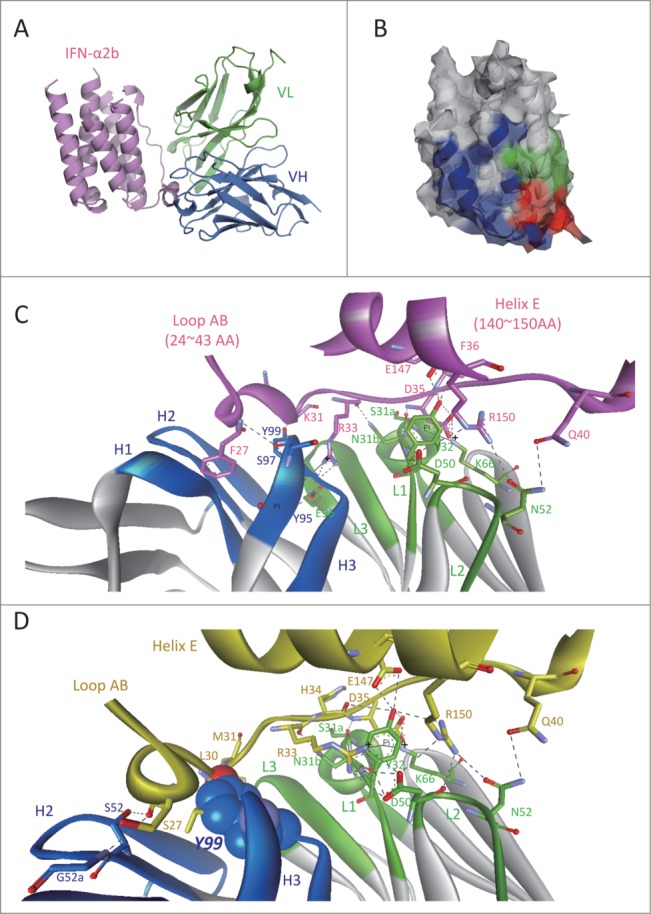
Predicted interaction mechanism of AIA22 with IFN-α2b and IFN-α1b. (A) The AIA22/IFN-α2b complex model. Variable region of AIA22 light chain (VL): green, variable region of heavy chain (VH): marine, IFN-α2b: pink. (B) The recognition region mapping of AIA22 (Green), IFNAR2 (Blue), and their overlapping region (Red) on IFN-α2b surface. (C) The predicted interaction interface between AIA22 and IFN-α2b. H1∼H3 and L1∼L3 represent the 6 CDR regions of heavy chain and light chain respectively. IFN-α2b and its residues are labeled as pink. The dotted lines between resides indicate hydrogen-bonds and solid lines indicate π interactions. (D) The predicted interaction interface between AIA22 and IFN-α1b. IFN-α1b and its residues were labeled as yellow. The atoms of Tyr99 (CDR H3) are displayed as CPK scale to clearly reveal its clash with IFN-α1b backbone residues Leu30 and Met31.
AIA22 binds to the IFNAR2 recognition region of IFN-α2b, which is different from sifalimumab
The structural modeling predicted the overlapping of AIA22's binding epitope with IFNAR2 recognition region. To identify the binding region of AIA22 on the IFN-α surface experimentally, a series of competitive binding assays were performed by using the forteBio Octet QKe system, which is an application of biolayer interferometry (BLI) technology. The interface between the molecular layer on the fiber (biosensor) and the solution will change with the addition of bound molecules that are monitored by the Octet system in wavelength shift approach over time. Figure 3A shows the interaction course of a sandwich-like binding assay. Purified AIA22 was captured on the interface of the anti-human-IgG conjugated (AHC) biosensor to saturation as the first layer of the “sandwich,” while AIA22 (control), sifalimumab, or IFNAR2-hFc (R&D, 4015-AB) individually acted as the top layer. The intermediate layer was occupied by IFN-α2b or buffer, as shown in the legend box of Figure 3A. The results showed that sifalimumab could interact with IFN-α2b immobilized on AIA22 (NO. I curve), while the IFNAR2 did not (NO. II curve). This indicated that AIA22 might share a proximal binding region on IFN-α2b with IFNAR2. In reverse, both AIA22 and IFNAR2 could interact with IFN-α2b immobilized on sifalimumab (Fig. 3B). Furthermore, we performed a competitive binding assay (Fig. 3). In Figure 3C, the binding response reduced in a dose-dependent manner to increasing amounts of AIA22-Fab (Fab region of AIA22) (Table S4), indicating that AIA22-Fab can compete against IFNAR2 for the interaction with IFN-α2b. An overlapping binding course was then executed, and results showed that sifalimumab also interacted with IFN-α2b immobilized on IFNAR2 in a dose-dependent manner (Fig. 3D) and identified that the binding epitope of sifalimumab is different from the IFNAR2 recognition region. To summarize, AIA22, but not sifalimumab, overlapped the IFNAR2 recognition region of IFN-α partially, which confirmed the prediction of the molecular modeling results. With IFNAR2 responsible for high affinity binding,6,22 we predict effective blocking of IFN-α/IFNAR interaction by AIA22.
Figure 3.

For figure legend, see next page.
AIAmut derived from AIA22 has an improved affinity to IFN-α1b
The results of structural prediction clearly revealed the possible improper recognition mechanism of AIA22/IFN-α1b interaction by complex modeling, and was confirmed by alanine scanning of the CDR region and competitive assays experimentally. We emphasized Tyr99 of the heavy chain and the neighboring interface residues (e.g., Ser28, Val33, Ser34, Glu96 from the light chain; Tyr95, Tyr96, Thr100 from the heavy chain), employed mutations of these sites to all other natural amino acids, and estimated the changes in the binding free energy of the protein-to-protein complexes. The variants showing a decrease in binding free energy (ΔΔG < 0) were selected and a visual inspection was also executed for the variants with anomalous ΔΔG value. The selected variants were used in the binding activity analysis by ELISA (some variants with apparently decreased binding activity to IFN-α1b or IFN-α2b are given in Table S5).19 Additionally, according to the results of alanine scanning, the mutational residues with remarkably improved affinity were combined with the positive variant sites yielded from computational design. The identification results of these variants are shown in Table 2. To isolate candidate antibodies with extensive neutralizing potency to multiple IFN-α subtypes, we selected mutants according to their neutralizing activities to different IFN-α subtypes. The variant L-S34A + H-32G96W (the combined mutation of L-S34A, H-Y32G, and H-Y96W) displayed extensively improved neutralizing activities to multiple IFN-α subtypes and was termed AIAmut. The affinities of AIAmut to IFN-α1b and IFN-α2b were also determined (Table S6, Figs. S3 and S4). Apparently, AIAmut forms stronger interaction with IFN-α2b and IFN-α1b than AIA22.
Table 2.
The evaluation results of variants derived from AIA22
| ΔΔG (kcal/mol) ** |
Relative Affinity*** |
Relative Neutralizing Activity**** |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variants*IFN-α: | 1b | 2b | 1b | 2b | 1b | 2b | 5 | 6 | 8 | 14 | 17 | 21 |
| L-S34A | −0.3 | 0.24 | 0.4 | 0.78 | 0.88 | 1.24 | – | – | 3.85 | 5.22 | – | – |
| H-Y32A | −0.305 | −0.115 | 0.07 | 0.88 | – | 1.15 | 3.42 | >10 | – | 8.9 | 0.83 | – |
| H-Y32G | −0.045 | −0.17 | 0.44 | 2.52 | 1.03 | 1.12 | 0.73 | – | – | 0.99 | 1.07 | – |
| H-A33G | −8.53 | 0.04 | 0.06 | 0.76 | 0.33 | 0.91 | – | >10 | >10 | 2.53 | 2.22 | >10 |
| H-Y96W | −0.02 | 0 | 0.13 | 2.15 | 0.87 | 1.05 | – | >10 | – | 2.23 | 0.51 | 1.67 |
| H-Y99V | −13.44 | −55.425 | 0.56 | 0.87 | 0.48 | 5.86 | 7.71 | – | – | – | – | – |
| L-S34A+H-A33G | −1.925 | 0.52 | 0.18 | 1.01 | – | 4.34 | – | >10 | >10 | 7.48 | >20 | – |
| L-S34A+H-Y96W | −0.735 | 0.395 | 0.27 | 1.58 | 0.49 | 1.85 | – | >10 | 6.02 | >20 | 1.25 | – |
| H-32A96W | −0.65 | −0.075 | 0.32 | 7.33 | – | 0.91 | 3.15 | ˜7.28 | – | – | 2.36 | – |
| H-32G96W | −0.36 | −0.18 | 0.22 | 3.56 | 1.09 | 0.88 | 2.37 | 2.89 | – | 7.98 | 0.93 | – |
| H-33G96W | −5.68 | −0.14 | 0.21 | 1.08 | 0.13 | 1.35 | – | 1.22 | 1.05 | 0.87 | 2.15 | 2.23 |
| L-S34A+H-32A96W | −1.065 | 0.105 | 0.29 | 2.02 | – | 2.77 | – | 1.58 | – | – | – | – |
| L-S34A+H-32G96W | −0.05 | −0.14 | 0.21 | 1.15 | 0.15 | 0.38 | 0.19 | 2.11 | 1.01 | 8.53 | 0.62 | ˜0.01 |
| L-S34A+H-33G96W | −0.06 | 0.23 | 0.19 | 2.39 | 0.13 | 1.71 | – | 11.34 | 7.59 | 7.33 | 1.98 | – |
| H-32G96W99V | 0.37 | 0.66 | 0.86 | 0.99 | 0.22 | 2.15 | 9.63 | – | – | – | – | – |
The “+” symbol represents the combination of light and heavy chain. For multi-site mutation of the same chain, the initial AA is not shown in the name.
ΔΔG (= ΔGVariants − ΔGAIA22) was estimated by FoldX based on the AIA22/IFN-α complex model. ΔG represents the binding free energy between antibody (AIA22 or variants) and corresponding IFN-α subtype.
The value of KDVariants/KDAIA22 (KD was derived from the same ELISA assay with Table S3).
The value of EC50Variants/EC50AIA22 (EC50 was derived from proliferation assay of Daudi cell).
The horizontal short line (–) indicates the value was not determined.
Both AIA22 and AIAmut show effective neutralizing activity to multiple IFN-α subtypes in vitro
IFNs exert a series of biological activities, many of which are measurable in isolated cell culture systems. Notably, Daudi cells (Burkitt's lymphoma-derived B cell line) have been demonstrated to be highly susceptible to the antiproliferation of IFN-α.23,24 Neutralizing antibodies blocking IFN-α binding to its receptors have the ability to restore proliferation. First, we examined the dose-response curves of the antiproliferation activity of multiple IFN-α subtypes and determined the critical concentrations of different subtypes, where IFN-α displayed 100% suppression of cell proliferation (Table 3). Next, the cell proliferation assay was performed and the neutralization of 12 IFN-α subtypes by the monoclonal antibodies was examined (Table 3). According to the results obtained, AIAmut displayed stronger neutralizing potency to IFN-α subtypes than AIA22. The neutralizing activities of AIAmut to IFN-α1b, 2a, 2b, 6, and 21 were considerably improved compared with AIA22. Compared with sifalimumab, AIA22 and AIAmut exhibited different neutralizing profiles of the 12 IFN-α subtypes. Sifalimumab performed the most effective blocking to IFN-α6, while AIA22 and AIAmut showed better neutralizing activity to IFN-α2. AIAmut displays superior neutralizing potency compared with sifalimumab to 7 of the 12 IFN-α subtypes, but inferior neutralizing activity to 4 of them. Interestingly, with a remarkably lower affinity to IFN-α2b, AIA22 exhibited better neutralizing effects than sifalimumab. Taking the antigen epitope into consideration, we can conclude that the binding to IFNAR2 recognition region may provide AIA22 (and AIAmut) with more effective neutralizing activity to IFN-α.
Table 3.
Neutralizing activities of monoclonal antibodies to multiple IFN-α subtypes
| Neutralizing Activity (EC50, nM) |
||||
|---|---|---|---|---|
| IFN | Critical Concentration (IU/ml, pM) | AIA22 | AIAmut | Sifalimumab |
| IFN-α1b | 60, 36 | 335.90 | 49.66 | 18.71 |
| IFN-α2b | 0.5*, 25 | 0.63 | 0.24 | 1.80 |
| IFN-α4b | 150, 10.29 | 94.65 | 26.37 | 100.30 |
| IFN-α5 | 100, 17.9 | 82.3 | 15.3 | 49.7 |
| IFN-α6 | 200, 10.5 | 1.34 | 2.83 | 0.05 |
| IFN-α7 | 200, 20.7 | NA | NA | 100.2 |
| IFN-α8 | 70, 10 | 24.6 | 29.6 | ∼4.17§ |
| IFN-α10 | 150, 51 | 335.4 | 76.85 | 188.4 |
| IFN-α14 | 60, 25.4 | 28.4 | 246.4 | ∼2.0§ |
| IFN-α16 | 30, 45 | 109.3 | 23.89 | 41.21 |
| IFN-α17 | 100, 20.6 | 116.5 | 72.8 | 194.7 |
| IFN-α21 | 100, 30 | 79.93 | ∼5.0§ | 44.43 |
The concentration unit is ng/ml.
The top and bottom value was not reached. NA = no activity.
Both AIA22 and AIAmut inhibit IFN-α inducible genes expression in Adv-mIFN-α treated mice
Prolonged mouse IFN-α administration in Balb/c mice using adenovirus-mediated gene transfer technology was performed to evaluate the neutralizing activity of AIA22 and AIAmut in vivo by detecting the expression level of type I IFN inducible genes. As shown in Figure 4A, all the groups that received Adv-mIFN-α (recombinant adenovirus vector containing the mouse IFN-α cDNA) injection exhibited significantly improved blood concentrations of mIFN-α after 24 hours, and the subsequent administration of equivalent neutralizing antibodies also made the serum human IgG levels reach a comparative level between groups (Fig. 4B). The elevated serum IFN-α levels resulting from Adv-mIFN-α administration markedly increased the expression of the type I IFN inducible genes IRF-7, IP-10, ISG-15, and Mx-1 in peripheral blood cells. The over expressions of these inducible genes were also significantly inhibited by the injection of a large amount of AIA22 or AIAmut (Fig. 4 C–F).
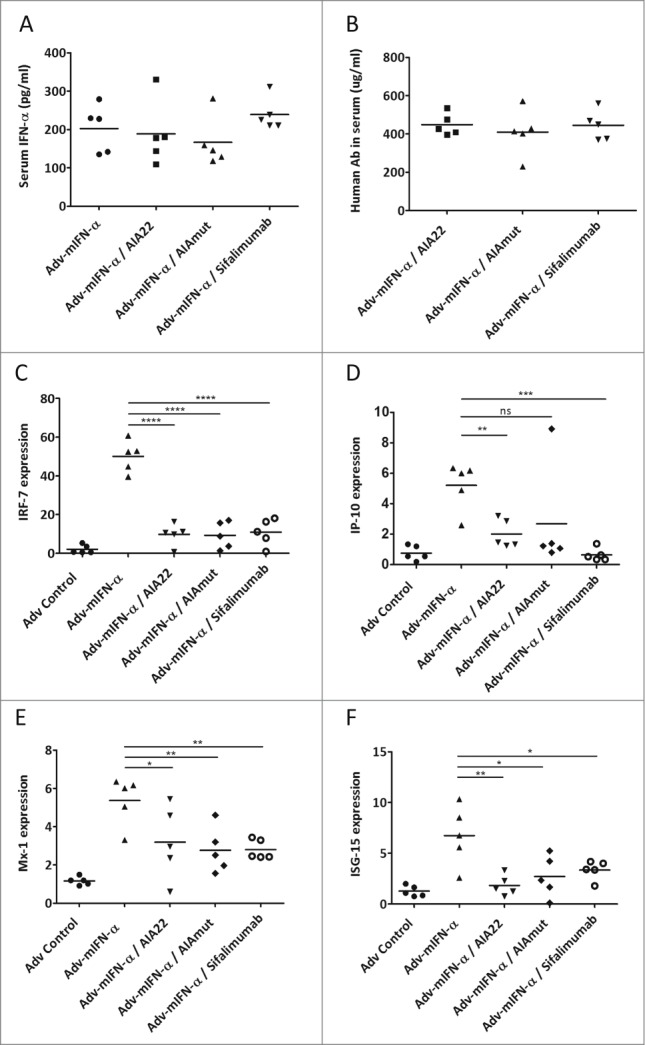
Discussion
Many studies10-13,20-22 have revealed a connection between the increased IFN-α levels (in both circulation and local tissues) and disease activity and severity of SLE. The investigation into the genetics of SLE 12 has found significant overexpression of mRNAs of almost all IFN-α subtypes in the WB of SLE patients, making it important that IFN-α-targeted therapeutic agents neutralize extensive IFN-α subtypes. Additionally, the possibility that treatment with IFNAR-targeted agents may provide greater neutralization than IFN-α-targeted strategies should be considered. It should be noted, however, that use of canonical anti-IFNAR antibodies may result in undesired antibody-dependent cell-mediated cytotoxicity (ADCC), and the thorough blocking of type I IFN signaling could increase the risk of infection. Taking all these factors into account, we directed our endeavors toward the development of antibodies that target multiple IFN-α subtypes for SLE therapy.
As described in results, the IFNAR2 recognition region is preferred as the binding epitope of anti-multiple IFN-α subtype antibodies. As expected, AIA22 and its derived antibody AIAmut were shown to bind to the IFNAR2 recognition region of IFN-α. It must be noted that blocking of IFNAR1 recognition region can also stop the effects of IFN-α because of its requirement for the further initiation of the signaling cascade.23,24 For example, sifalimumab, which has a binding epitope that overlaps the IFNAR1 recognition region of IFN-α, can also neutralize the activity of multiple IFN-α subtypes (Table 3). Interestingly, AIA22 displays a slightly higher neutralizing potency to IFN-α2b (Table 3) in spite of its considerably lower affinity to IFN-α2b over sifalimumab (Table 1). It is very likely that the blocking advantage of AIA22 over sifalimumab can be attributed to its recognition region of IFN-α. Concurrently, when considering the low binding affinity between IFNAR1 and IFN-α, it can be expected that the binding of AIA22 and AIAmut to IFNAR2 recognition region of IFN-α may effectively avoid the ADCC mediated by IFN-α (Fig. S6). Additionally, the retaining of sifalimumab/IFN-α/IFNAR2 complexes on the cell surface partially antagonizes IFN-β and IFN-ω-induced signaling pathways, which would be avoided by the recognition mode of AIA22 and AIAmut (Fig. S6).
It has been reported that SLE patients with lower levels of IFN-I signature responded better than groups with higher levels in a Phase 2 trial of rontalizumab (Genentech).25 This indicates a necessity for the improvement of the neutralizing ability for anti-IFN-α antibodies. Generally, the neutralizing activity of an antibody displays a positive correlation with its affinity. In this study, the structural modeling of the AIA22/IFN-α complex confirmed by the alanine scanning and the affinity maturation based on computational design resulted in a variant antibody, AIAmut that exhibited an extensively increased neutralizing potency to IFN-α subtypes (Tables 2, 3). Alongside the neutralizing activity, AIAmut also showed higher affinity to both IFN-α1b and IFN-α2b over AIA22 (Table 1 and Table S6). Here, we must emphasize the discrepancy seen in the neutralizing profiles of IFN-α subtypes among AIA22, AIAmut, and sifalimumab and note that AIA22 and AIAmut may have novel therapeutic effects.
As previously reported, Adv-mIFN-α treatment of preautoimmune (NZB×NZW)F1 mice,26 a classic mouse model of spontaneous lupus, results in a rapid and severe disease with all characteristics of SLE. In this work (Fig. 4), the inhibitory effects of AIA22 and AIAmut on IFN-α signature in Adv-mIFN-α treated Balb/c mice have been demonstrated. According to the results, we anticipate a promising PD effect of AIA22 and AIAmut in Adv-mIFN-α-treated (NZB×NZW)F1 mice; such experiments are planned for the future.
In conclusion, we developed a novel candidate therapeutic antibody AIA22 that specifically interacts with multiple IFN-α subtypes and effectively neutralizes their activities in vitro. Different from the anti-IFN-α antibodies in clinical trials (especially sifalimumab), AIA22 binds to the IFNAR2 recognition region of IFN-α, which is considered to be a novel antigen epitope. Concurrently, structural modeling and computational design yielded a mutational antibody AIAmut, which exhibited remarkably improved neutralizing activity. Both AIA22 and AIAmut employ quite different neutralizing profile of IFN-α subtypes from sifalimumab, and may yield novel therapeutic effects. Subsequently, the inhibitory of IFN-α signature by AIA22 and AIAmut in Adv-mIFN-α treated Balb/c mice predicts a promising PD effect and warrants further development of AIA22 and AIAmut.
Materials and methods
Generation of monoclonal antibodies
As previously described,27 the phage display single-chain variable fragment (scFv) library was exposed to 3-round selections in immunotubes (Nunc, 443990) coated with recombinant IFN-α2b (ProSpec-Tany, CYT-205) and IFN-α1b (gifted from Professor Xiaojie Dong) in turn at a decreasing concentration range of 20 μg/ml, 5 μg/ml, or 1 μg/ml (Table S1). The variable regions of light or heavy chain genes of positive scFv antibodies were isolated for sequencing and then cloned into expression vectors pABG and pABL, respectively (both constructed in our lab). FreeStyle™ 293-F cells (Invitrogen) were cotransfected with equal parts of both vectors for simultaneous expression and were cultured according to the manufacturer's instructions. Antibodies secreted in the supernatant were purified using HiTrap™ rProtein A FF (GE healthcare, 17-5079-01/17-5080-01).
Specificity and binding activity ELISAs
Ninety-six-well microtiter plates (Costar, 9018) were coated with recombinant IFN-α1b, IFN-α2b, IFN-β (Abnova, P3623), IFN-ω (gifted from Professor Wei Chen), IFN-γ, or control antigens in carbonate buffer (pH9.6) and incubated overnight at 4°C. They were then blocked with phosphate-buffered saline (PBS) supplemented with 5% (M/V) fat-free powdered milk (Amresco, M203-10G-10PK) for one hour at room temperature (RT). Serial dilutions of purified AIA22 (15.625–1000 ng/ml) in a dilution buffer of PBS containing 5% fat-free powdered milk and 0.1% Tween20 were incubated for one hour at 37°C. After washing, HRP-conjugated goat anti-human IgG (Sigma-Aldrich, A0170) was added and incubated for 30 minutes at 37°C. Plates were developed with o-phenylenediamine dihydrochloride (OPD) substrate. The optical density (OD) was detected at 492 nanometers (nm) with 630 nm as a reference by using a microplate reader (Thermo Multiskan MK3). The binding activities of AIA22 and mutant antibodies were also compared following a coating of IFN-α2b or IFN-α1b at 1 μg/ml.
Affinity Determination by BIAcore Analysis
The interaction affinity between antibodies and recombinant IFN-α2b were determined by calculation from kinetic constants using BIAcore™ T200 systems. Multi-cycle kinetics was used for kinetic analysis experiments. Purified antibodies at 1.0 μg/ml were first captured on a CM5 chip (GE healthcare, 10226578) handled with the Human Antibody Capture Kit (GE healthcare, BR-1008-39). IFN-α2b with a concentration from 1.2 nanomolar (nM) to 120 nM in HBS-EP+ running buffer were passed over the chip at a rate of 30 μl/min and 25°C. The two minute association time was followed by a 10 to 40 minute dissociation period. The regeneration step consisted of chip flushing with the regeneration buffer for 30 seconds at the rate of 30 μl/min. Repeated measurements were taken at 7.68 nM and 2 baseline binding readings of the buffer to the chip with corresponding captured monoclonal antibody were also detected to correct for system bias. The experimental data were fitted to 1:1 binding model using BIAcore™ T200 evaluation software.
Considering the lack of a straightforward relationship between the affinity of AIA22 (and AIAmut) to IFN-α1b and corresponding kinetics, we examined the binding affinity by measurement of steady-state binding levels using BIAcore™ T200 systems. Purified antibodies at 0.4 μg/ml were first captured. Solutions of IFN-α1b at concentrations from 3 nM to 750 nM in HBS-EP+ running buffer were passed over the chip at a rate of 30 μl/min and 25°C. The seven minute association time was followed by a 15 minute dissociation period. Repeated measurements were taken at 19.2 nM, and 2 baseline binding readings of the buffer to the chip with corresponding captured monoclonal antibody were detected. The steady-state binding data were analyzed by fitting the curve of binding level against concentration using BIAcore™ T200 evaluation software.
All reagents and analysis measures used above were obtained from GE Healthcare, USA.
Molecular modeling, docking and superposition
A scFv homology model for AIA22 was built using Discovery Studio (Accelrys) according to the structure of PDB:3UX9, which describes an antibody sharing a high sequence similarity with AIA22. The CDR loops were refined by using Loop Refinement (MODELER) (a built-in module of Discovery Studio). The conformations of IFN-α1b and IFN-α2b were taken from the crystal structures PDB:3UX9 (2.8angstrom [Å] resolution) and PDB:3S9D (2.0 Å resolution), respectively. The ZDOCK algorithm28 was applied to the docking simulations of AIA22 and IFN-α2b. Two thousand docked poses generated in a global docking search were scored and clustered (ZDock Score and ZRank Score). The poses with higher ZDock score and lower ZRank score were identified (Fig. S5) and were chosen for refinement by RDOCK.29 The top refined pose was selected for further analysis. The complex structure of AIA22 and IFN-α1b was procured by using a structural superposition method with Align Structures (MODELER). Molecular structure illustrations were generated using PyMOL Molecular Graphics Software (www.pymol.org) or Discovery Studio.
Competitive binding assay
The competitive binding experiment was conducted to determine the binding region of AIA22 on the IFN-α surface experimentally. A sandwich-like binding assay was designed using forteBIO® Octet QKe System (Pall ForteBio Corporation, USA). Purified AIA22 in HBS-EP buffer at 200 nM was loaded on an individual anti-human-IgG Conjugated (AHC) biosensor followed by a one minute wash at 1000 rpm in HBS-EP buffer. Afterward, a 10 minute association was performed in the sample plate with 100 nM IFN-α2b or HBS-EP buffer control. This was followed with a 2–10 minute disassociation in the HBS-EP buffer plate, which resulted in a decrease of binding that did not surpass more than 10% of the peak value of binding response. A re-association step was then executed in a new sample plate containing either purified antibodies or recombinant proteins at 100 nM. Background binding of IFN-α2b to sensor was performed for quality control by detecting the binding to biosensor loaded with control human IgG. The reverse experiment with sifalimumab captured on AHC biosensors was performed in the same manner. The results were analyzed with Octet Date Analysis Software (Pall ForteBio).
Computational design of mutants using FoldX
The computational design of high-affinity AIA22 mutants to IFN-α1b was performed using FoldX version 3.0 (Center de Regulació Gen∫mica, Barcelona E).30 The interface residues (in particular the collision site and its adjacent sites) were identified according to the complex structure model, after which the mutation of the specific residue to all other 19 naturally occurring amino acids were scanned using the mutate residue function in FoldX. Changes in the binding free energy (ΔΔG, kcal/mol) of protein-to-protein complexes due to residue mutations were estimated, and the amino acid substitutions showing a notable decrease in binding free energy were selected. Mutagenesis experiments of AIA22 were carried out with Quik Change Site-Directed Mutagenesis (Stratagene, 200518) in accordance with the manufacturer's instructions. Mutants were expressed, purified, and tested as described in previous sections.
A detailed description of the protein design algorithm FoldX is available at http://foldx.crg.es/
Cell culture and neutralizing activity
Daudi cells (TCHu140) were purchased from the Committee on Type Culture Collection of Chinese Academy of Science (Shanghai, China) and cultured in RPMI-1640 medium (Gibco, 31800-022) supplemented with 10% FBS (Gibco, 10099-141), 1/100 Penicillin-Streptomycin solution (Gibco, 15140-122), and 0.11 g/L sodium pyruvate at 37°C in a 5% CO2 humidified incubator unless otherwise indicated. Daudi cells at logarithmic phase were inoculated in 96-well plates (Costar, 3599) to a final density of 1 × 104 cells/well and were grown with a serial titration of each IFN-α subtype (PBL Biomedical Labs, 11002-1) for 72 hours to examine cytotoxicity curves. Cell viability was detected by the addition of CCK-8 (Dojindo, CK04) at an OD of 450 nm after a 2–3 hour incubation period and was calculated relative to Daudi proliferation in the absence of IFN-α (100% cytoactive) and in the presentence of IFN-α alone without Daudi cells inoculated (0% cytoactive). The critical concentration of different subtypes (where Daudi cells displayed the lowest proliferation) was determined, and the neutralizing activity was assayed at EC50 with the determined critical concentration as the final concentration. A serial titration of AIA22 or mutant antibodies mixed either with or without a certain IFN-α was added to Daudi cells in a 96-well plate at a final density of 1 × 104 cells/well. Following a 72-hour conditioned incubation, relative cell viability was measured using CCK-8. The neutralizing activity percentage was calculated by setting the OD 450nm in the absence of the monoclonal antibodies and IFN-α as ‘Ao’, in the presence of IFN-α and monoclonal antibodies at different concentration as ‘As’, and in the presence of IFN-αalone as ‘Ab’. The neutralizing percentage of IFN-α equation was as follows:
The EC50 of the monoclonal antibodies to different IFN-α subtypes was derived with GraphPad Prism software using non-linear regression, dose-response stimulation, and variable slope fit.
Mice and treatment
Female BALB/c mice (6–8 weeks old) were purchased from Beijing Laboratory Animal Center and housed in a pathogen free facility in barrier cages. These mice were treated with a single intravenous injection in the vena caudalis of 1×108 Adv-mIFN-α particles (HanBio, Shanghai, China), recombinant adenovirus vector containing the mouse IFN-α subtype 5 cDNA. Controls received the same amount of control Adv particles. Twenty-four hours after Adv treatment, these mice received a single intraperitoneal injection of AIA22, AIAmut, sifalimumab, or isotype human IgG at 1 mg/mouse. Blood serum was collected before injection of antibodies and at 24 hours after the injection. Mice were sacrificed on the fourth day and PBMCs were harvested for RNA extraction using TRIzol regent (Life Technologies, 15596-026) according to the manufacturer's instructions.
The study was approved by the Animal Ethics Committee of the Academy of Military Medical Sciences (AMMS) and performed in accordance with the guidelines of the National Institute of Biological Sciences Guide for the care and use of laboratory animals.
RT-PCR and Quantitative PCR
Total RNA was extracted and resuspended in diethyl pyrocarbonate (DEPC, Amresco, E147) treated water. The RT-PCR was performed using TransScript All-in-One First-Strand cDNA Synthesis SuperMix for PCR (TransGene Biotech, AT321) concordant with the manufacturer's instructions. Gene expression was relatively quantified by real-time PCR, and SYBR® Green Real-time PCR Master Mix (Applied Biosystems, 4367659) was used in compliance with provided instructions. Amplification conditions were performed in succession as follows: 95°C for 10 minutes, 45 cycles at 94°C for 15 seconds, 60°C for 25 seconds, and 72°C for 25 seconds using the ABI PRISM® 7500 Fast RT-PCR system. A standard melt curve stage followed. Primers (Table S7) were synthesized by Sangon Biotech (Shanghai, China) and consistent with ones reported before.31,32 Transcripts were quantified using the comparative (2−ΔΔCt) method.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Wei Chen and Dr. Xiaojie Dong (Beijing Institute of Biotechnology, Beijing, China) for the gift of IFN-ω and IFN-α1b. We are also grateful to Dr. Long Liang for his support of computational design workstation. We further thank Drs. Jun Wu, Chunjie Liu, Zhixin Yang, and Jian Wang for their expert technical help and Xiaoyan Yu, Ph.D., for her editorial assistance in the preparation of the manuscript.
Funding
This work was funded by National Science and Technology Major Project of China (2012ZX09301003).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Ronnblom L. Potential role of IFN alpha in adult lupus. Arthritis Res Ther 2010; 12 Suppl 1:S3; PMID:20392290; http://dx.doi.org/ 10.1186/ar2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2008; 358:929-39; PMID:18305268; http://dx.doi.org/ 10.1056/NEJMra071297 [DOI] [PubMed] [Google Scholar]
- 3.Eisenberg R. Why can't we find a new treatment for SLE? J Autoimmun 2009; 32:223-30; PMID:19329279; http://dx.doi.org/ 10.1016/j.jaut.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong W, Lahita RG. Novel treatments for systemic lupus erythematosus. Ther Adv Musculoskelet Dis 2011; 3:255-66; PMID:22870484; http://dx.doi.org/ 10.1177/1759720X11415456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anolik JH, Aringer M. New treatments for SLE: cell-depleting and anti-cytokine therapies. Best Pract Res Clin Rheumatol 2005; 19:859-78; PMID:16150407; http://dx.doi.org/ 10.1016/j.berh.2005.05.006 [DOI] [PubMed] [Google Scholar]
- 6.Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, Pestka S, Schreiber G. Binding and activity of all human alpha interferon subtypes. Cytokine 2011; 56:282-9; PMID:21856167; http://dx.doi.org/ 10.1016/j.cyto.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 7.Krause CD, Pestka S. Evolution of the Class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther 2005; 106:299-346; PMID:15922016; http://dx.doi.org/ 10.1016/j.pharmthera.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 8.Weissmann C, Weber H. The Interferon Genes. Progress in Nucleic Acid Research and Molecular Biology 1986; 33:251-300; PMID:3025923 [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006; 25:383-92; PMID:16979570; http://dx.doi.org/ 10.1016/j.immuni.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 10.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003; 197:711-23; PMID:12642603; http://dx.doi.org/ 10.1084/jem.20021553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al.. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci 2003; 100:2610-5; PMID:12604793; http://dx.doi.org/ 10.1073/pnas.0337679100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao Y, Higgs BW, Morehouse C, de Los Reyes M, Trigona W, Brohawn P, White W, Zhang J, White B, Coyle AJ, et al.. Development of potential pharmacodynamic and diagnostic markers for Anti-IFN-alpha monoclonal antibody trials in systemic lupus erythematosus. Hum Genomics Proteomics 2009; 2009; pii: 374312; PMID:20948567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum 2006; 54:1906-16; PMID:16736505; http://dx.doi.org/ 10.1002/art.21890 [DOI] [PubMed] [Google Scholar]
- 14.Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, et al.. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 2009; 183:6021-9; PMID:19812195; http://dx.doi.org/ 10.4049/jimmunol.0803872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin Immunol 2013; 148:303-12; PMID:23566912; http://dx.doi.org/ 10.1016/j.clim.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 16.Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem 2008; 283:32925-36; PMID:18801736; http://dx.doi.org/ 10.1074/jbc.M806019200 [DOI] [PubMed] [Google Scholar]
- 17.Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, et al.. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011; 146:621-32; PMID:21854986; http://dx.doi.org/ 10.1016/j.cell.2011.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouyang S, Gong B, Li JZ, Zhao LX, Wu W, Zhang FS, Sun L, Wang SJ, Pan M, Li C, et al.. Structural insights into a human anti-IFN antibody exerting therapeutic potential for systemic lupus erythematosus. J Mol Med (Berl) 2012; 90:837-46; PMID:22307521; http://dx.doi.org/ 10.1007/s00109-012-0866-3 [DOI] [PubMed] [Google Scholar]
- 19.Smith K, Crowe SR, Garman L, Guthridge CJ, Muther JJ, McKee E, Zheng NY, Farris AD, Guthridge JM, Wilson PC, et al.. Human monoclonal antibodies generated following vaccination with AVA provide neutralization by blocking furin cleavage but not by preventing oligomerization. Vaccine 2012; 30:4276-83; PMID:22425791; http://dx.doi.org/ 10.1016/j.vaccine.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol 2006; 18:676-82; PMID:17011763; http://dx.doi.org/ 10.1016/j.coi.2006.09.014 [DOI] [PubMed] [Google Scholar]
- 21.Becker-Merok A, Ostli-Eilersten G, Lester S, Nossent J. Circulating interferon- 2 levels are increased in the majority of patients with systemic lupus erythematosus and are associated with disease activity and multiple cytokine activation. Lupus 2012; 22:155-63; PMID:23213068; http://dx.doi.org/ 10.1177/0961203312468964 [DOI] [PubMed] [Google Scholar]
- 22.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 2005; 23:307-36; PMID:15771573; http://dx.doi.org/ 10.1146/annurev.immunol.23.021704.115843 [DOI] [PubMed] [Google Scholar]
- 23.Strunk JJ, Gregor I, Becker Y, Li Z, Gavutis M, Jaks E, Lamken P, Walz T, Enderlein J, Piehler J. Ligand binding induces a conformational change in ifnar1 that is propagated to its membrane-proximal domain. J Mol Biol 2008; 377:725-39; PMID:18294654; http://dx.doi.org/ 10.1016/j.jmb.2008.01.017 [DOI] [PubMed] [Google Scholar]
- 24.Lamken P, Gavutis M, Peters I, Van der Heyden J, Uze G, Piehler J. Functional cartography of the ectodomain of the type I interferon receptor subunit ifnar1. J Mol Biol 2005; 350:476-88; PMID:15946680; http://dx.doi.org/ 10.1016/j.jmb.2005.05.008 [DOI] [PubMed] [Google Scholar]
- 25.Kalunian K, Merrill JT, Maciuca R, Ouyang WJ, McBride JM, Townsend MJ, Park E, Li J, Wei X, Morimoto A, et al.. Efficacy and Safety of Rontalizumab (Anti-Interferon Alpha) in SLE Subjects with Restricted Immunosuppressant Use: Results of A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study. Arthritis Rheum 2012; 64:S1111-S; http://dx.doi.org/ 10.1002/art.34669 [DOI] [Google Scholar]
- 26.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-alpha Induces Early Lethal Lupus in Preautoimmune (New Zealand Black x New Zealand White)F1 but Not in BALB/c Mice. J Immunol 2005; 174:2499-506; PMID:15728455; http://dx.doi.org/ 10.4049/jimmunol.174.5.2499 [DOI] [PubMed] [Google Scholar]
- 27.Yu R, Wang S, Yu YZ, Du WS, Yang F, Yu WY, Sun ZW. Neutralizing antibodies of botulinum neurotoxin serotype A screened from a fully synthetic human antibody phage display library. J Biomol Screen 2009; 14:991-8; PMID:19726786; http://dx.doi.org/ 10.1177/1087057109343206 [DOI] [PubMed] [Google Scholar]
- 28.Chen R, Weng Z. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins 2002; 47:281-94; PMID:11948782; http://dx.doi.org/ 10.1002/prot.10092 [DOI] [PubMed] [Google Scholar]
- 29.Li LCR, Weng ZP. RDOCK: Refinement of rigid-body protein docking predictions. Proteins 2003; 53:693-707; PMID:14579360; http://dx.doi.org/ 10.1002/prot.10460 [DOI] [PubMed] [Google Scholar]
- 30.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res 2005; 33:W382-8; PMID:15980494; http://dx.doi.org/ 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, Sobel E, Kuroda Y, Akaogi J, Satoh M, et al.. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am J Pathol 2006; 168:1227-40; PMID:16565497; http://dx.doi.org/ 10.2353/ajpath.2006.050125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, Reeves WH. A Novel Type I IFN-Producing Cell Subset in Murine Lupus. J Immunol 2008; 180:5101-8; PMID:18354236; http://dx.doi.org/ 10.4049/jimmunol.180.7.5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.