

Summary
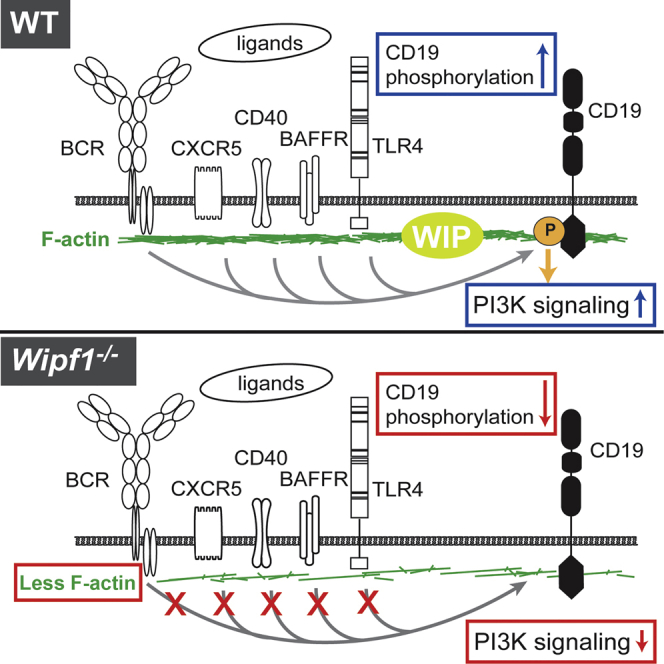
Humans with Wiskott-Aldrich syndrome display a progressive immunological disorder associated with compromised Wiskott-Aldrich Syndrome Interacting Protein (WIP) function. Mice deficient in WIP recapitulate such an immunodeficiency that has been attributed to T cell dysfunction; however, any contribution of B cells is as yet undefined. Here we have shown that WIP deficiency resulted in defects in B cell homing, chemotaxis, survival, and differentiation, ultimately leading to diminished germinal center formation and antibody production. Furthermore, in the absence of WIP, several receptors, namely the BCR, BAFFR, CXCR4, CXCR5, CD40, and TLR4, were impaired in promoting CD19 co-receptor activation and subsequent PI3 kinase (PI3K) signaling. The underlying mechanism was due to a distortion in the actin and tetraspanin networks that lead to altered CD19 cell surface dynamics. In conclusion, our findings suggest that, by regulating the cortical actin cytoskeleton, WIP influences the function of CD19 as a general hub for PI3K signaling.
Graphical Abstract
Highlights
-
•
WIP deficiency leads to faulty cortical actin, altering CD19 dynamics and function
-
•
CD19 is a common adaptor used by multiple receptors for PI3K signaling
-
•
CD19 malfunction results in PI3K activation defects by BCR, CD40, BAFFR, and CXCR5
-
•
The absence of WIP in B cells is sufficient to cause immunodeficiency in vivo
Batista and colleagues report that WIP deficiency leads to distorted cortical actin, causing CD19 malfunction. Defective CD19 activation affects multiple receptors, suggesting that CD19 functions as a hub of PI3K signaling. Therefore, the absence of WIP in B cells is sufficient to trigger immunodeficiency in vivo, mimicking Wiskott-Aldrich syndrome.
Introduction
Naive B cells express a B cell receptor (BCR) composed of the nonsignaling membrane immunoglobulins (Ig) IgM and IgD, which recognize extracellular antigen, and the associated signaling transmembrane components Ig-α and Ig-β, containing immunoreceptor tyrosine-based activation motifs (ITAMs) (Reth, 1989; Weiss and Littman, 1994). Cognate antigen recognition by the BCR initiates rapid phosphorylation of the ITAMs, which form a signaling platform for the tyrosine kinases Lyn and Syk. Subsequently both kinases recruit and phosphorylate several adaptor proteins and the co-receptor CD19. CD19 further recruits other molecules such as the Vav adaptor protein, Bruton’s tyrosine kinase (Btk), the PI3 kinase (PI3K) subunit p85α and Lyn itself via its cytosolic domain thus lowering the threshold of B cell activation (Carter and Fearon, 1992). In addition, CD19 plays a prominent role in PI3K pathway activation after BCR ligation (Otero et al., 2001).
To initiate the signaling cascade, early antigen recognition requires alteration of the actin cytoskeleton, enabling the spreading and contraction of B cells across the surface of antigen-presenting cells (Fleire et al., 2006). Antigen-induced BCR signaling leads to an early rapid wave of actin depolymerization that is dependent on the amount of BCR cross-linking (Hao and August, 2005). This radical reorganization of the actin cytoskeleton removes barriers to BCR diffusion and modifies BCR dynamics at the cell surface. In addition, TLR signaling has been shown to influence actin organization, thereby increasing BCR mobility and facilitating BCR signaling (Freeman et al., 2015). Furthermore, changes in actin organization alone increases BCR diffusion and triggers signaling similar to BCR crosslinking (Treanor et al., 2010). This ligand-independent signal not only requires increased BCR diffusion but also an immobilized co-receptor CD19, held in place by its association with the tetraspanin molecule CD81 (Mattila et al., 2013). In this context, we believe CD19 provides a mechanism for signal amplification via the PI3K pathway; however, the molecular linkage between CD19, the PI3K pathway, and the actin cytoskeleton is incompletely defined.
The PI3K pathway is one of the main signaling pathways regulating B cell homeostasis, survival, differentiation, and class-switch recombination. In addition to the BCR, several other receptors have been shown to activate the PI3K pathway in B cells, including chemokine and cytokine receptors, Toll-like receptors and receptors of the tumor necrosis factor (TNF) family, namely BAFFR and CD40 (Arron et al., 2001; Patke et al., 2006). Recently, it has been suggested that CD19 also mediates PI3K signaling in response to BAFFR or CD40 stimulation (Hojer et al., 2014; Jellusova et al., 2013) and raises the question of a general involvement of CD19 in PI3K activation in B cells.
WIP, the Wiskott-Aldrich syndrome protein (WASP) interacting protein, which is encoded by the Wipf1 gene, plays a key regulatory role in remodeling of the actin cytoskeleton. WIP binding to WASP, a central activator of the Arp2/3 complex, protects it from degradation and regulates its cellular distribution (Fried et al., 2014). WIP promotes actin polymerization independently of WASP by binding and stabilizing actin filaments (Martinez-Quiles et al., 2001; Ramesh et al., 1997). Furthermore, WIP associates with the adaptor molecules Nck and Grb2, thus potentially linking the actin network to signaling cascades (Antón et al., 1998; Barda-Saad et al., 2005; Donnelly et al., 2013; Moreau et al., 2000).
In humans, mutations in the WIP binding site of WASP (Stewart et al., 1999) or in WIP itself (Lanzi et al., 2012) results in the development of the immunodeficiency Wiskott-Aldrich syndrome (WAS). WAS is an X-linked disorder associated with eczema, increased susceptibility to infections and heightened risk of autoimmune disorders (Thrasher, 2009). Mice deficient in WIP display severe lymphopenia, increased spleen size but reduced numbers of B cells, lack of marginal zone B cells and a severe T cell dysfunction thus mimicking WAS (Curcio et al., 2007). WIP-deficient B cells exhibit defects in their actin network and increased proliferation in response to BCR stimulation in vitro (Antón et al., 2002); however, the role of WIP in B cell activation remains to be studied.
Here, we have shown that WIP deficiency in B cells resulted in defects in homing, chemotaxis, survival, and differentiation, ultimately leading to a reduction in germinal centers and antibody production in response to infection or immunization. These defects were the result of an impaired CD19 activation and PI3K signaling in WIP-deficient B cells after triggering a variety of receptors, namely BCR, BAFFR, CXCR4, CXCR5, CD40, and TLR4. Addressing the underlying mechanism, we found a distorted actin cytoskeleton and CD81-tetraspanin network in WIP-deficient B cells, which resulted in altered cell surface mobility of CD19. On the basis of these findings, it appears that WIP, by controlling actin-dependent receptor dynamics, influences CD19 in its function as a general hub for PI3K signaling.
Results
B Cell-Specific WIP Deficiency Impairs Mouse Immune Responses to Viral Infection or Immunization
To establish whether the absence of WIP exclusively in B cells has any effect in altering humoral immune responses, we generated mixed bone marrow (BM) chimeras by adoptively transferring a mix of WIP-deficient BM (from now on Wipf1−/− cells referring to its standard gene symbol) and BM from mice that lack B cells (JHT mice) into lethally irradiated JHT recipients. In this setting, all newly generated B cells will be Wipf1−/− in an environment containing mainly WT cells.
Wipf1−/− and wild-type (WT) chimeric mice were challenged with a low dose (1 × 104 PFU) of Vaccinia virus intra footpad. Eight days later, using flow cytometry, the generation of germinal center (GC) B cells was measured as a percentage of GL7+CD95+ of B220+ B cells. We observed a robust formation of GC B cells in the draining lymph node of WT chimeras, with 16% of B cells staining positive for GC markers. This percentage was significantly diminished to 9% when B cells were deficient in WIP (Figure 1A). We also observed a marked diminution in the percentage of T follicular helper cells (CD4+CXCR5+PD1+) from 13% to 7% in Wipf1−/− chimeras (Figure 1A), suggesting a defective interaction between Wipf1−/− B cells and T cells.
Figure 1.

B Cell-Specific WIP Deficiency Compromises Humoral Immune Responses
JHT-WT or JHT-Wipf1−/− mixed BM chimeric mice were infected intra footpad with Vaccinia virus (A) or immunized intra-peritoneally with NP-KLH in alum (B–D).
(A) Analysis of immune responses in popliteal lymph nodes at day 8 post-infection by flow cytometry. GC cells (B220+CD95+GL7+) or Tfh cells (CD4+CD44+CD62L−PD-1+CXCR5+) are shown. Quantifications (right column) indicate the percentage of GC B cells or Tfh cells per lymph node analyzed (mean ± SEM).
(B and C) Analysis of splenic NP-specific GC cells (NP+B220+CD95+GL7+) at day 13 post-immunization by flow cytometry. Immunohistochemistry showing IgD+ B cell follicles, Bcl-6 expressing GC cells and TCR-β+ T cell areas in frozen spleen sections. Graph indicates quantifications of GC cells in spleen sections (mean ± SEM). Scale bar, 150 μm.
(D) ELISA of NP-specific IgM, IgG, IgG1, and IgG3 antibodies in the sera of immunized chimeras at indicated time points (mean ± SEM).
Data are representative of two independent experiments (eight mice each).
See also Figure S1.
Similar to our observations after Vaccinia virus infection, B cell responses were severely impaired when Wipf1−/− chimeric mice were challenged with the T cell-dependent antigen NP-KLH. Here, the GC B cell population was reduced from 14.1% to 4.1% in Wipf1−/− chimeras (Figure 1B). Furthermore, using confocal microscopy, we detected smaller Bcl-6+ GC in frozen sections of spleens from immunized Wipf1−/− chimeras, with substantially reduced numbers of Wipf1−/− GC cells compared to WT GC cells (an average of 22 Wipf1−/− cells compared to an average of 970 WT cells) (Figure 1C). Quantifying NP-specific antibody titers in the serum, we found that Wipf1−/− chimeras had about one-fifth of the NP-specific IgM antibody concentration compared to WT chimeras. Notably, the production of NP-specific IgG antibody titers was severely delayed and reduced with IgG1 and IgG3 antibodies titers being diminished by at least one-fourth in Wipf1−/− chimeric mice (Figure 1D). The substantial reduction in the percentage of GC B cells and the differences in antibody production were unlikely to be due to a reduction of the percentage of circulating B cells in Wipf1−/− chimeras to about half that of the WT chimeras (Figures S1A and S1B), but mainly indicate an intrinsic differentiation defect in the absence of WIP. Furthermore, we observed a similarly impaired GC B cell response of Wipf1−/− cells when we challenged chimeras reconstituted with a mixture of 50% CD45.2 Wipf1−/− BM and 50% CD45.1 WT BM with NP-KLH in alum (Figure S1C).
Collectively, these in vivo experiments suggest that B cell-specific WIP deficiency renders mice unable to respond effectively to viral infection or immunization.
Wipf1−/− B Cells Show Impaired Homing and Migration
During analysis of the mixed chimeras, we found the expected ratio of approx. 50:50 (50% CD45.1+ WT cells versus 50% CD45.2+ Wipf1−/− cells) in the B cell compartment of the BM and the blood (Figures 2A and 2B). However, we observed a ratio of approx. 70:30 of CD45.1+ WT B cells versus CD45.2+ Wipf1−/− B cells in the spleen and lymph node (Figure 2B). A similar difference in the percentages of cells homing to spleen and lymph nodes was observed when fluorescently labeled CD45.1+ WT and CD45.2+ Wipf1−/− B cells were adoptively transferred into WT recipients (Figure S2A). Together, these results indicate that Wipf1−/− B cells might be deficient in their capacity to migrate and/or to sense chemokines.
Figure 2.

WIP Deficiency Affects B Cell Homing and Chemotaxis
(A) Flow cytometric analysis of BM mixture (B220+CD45.1+ WT and B220+CD45.2+Wipf1−/− BM in a mixture of 1:1) before transfer into irradiated recipients.
(B) Flow cytometric analysis of WT cells (B220+CD45.1+) and Wipf1−/− cells (B220+CD45.2+) in indicated organs of mixed BM chimeras (upper row). Graphs (±SEM) indicate the percentage of B220+CD45.1+ WT and B220+CD45.2+Wipf1−/− cells in respective organs. Data are representative of three independent experiments (n ≥ 4 mice).
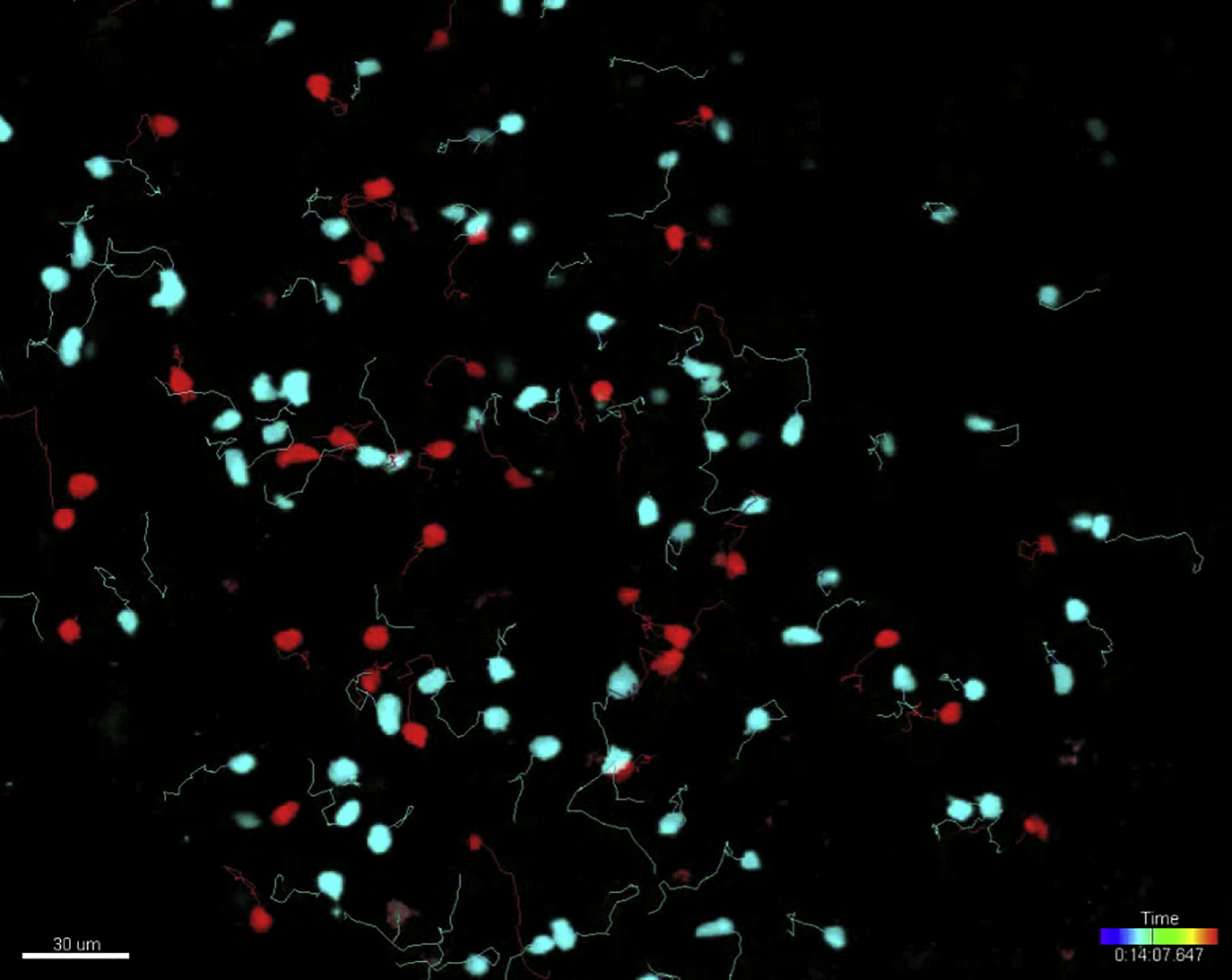
(C) Representative two-photon microscopy images 24 hr after adoptive transfer of labeled WT (cyan) and Wipf1−/− (red) B cells. Left image is a whole fixed popliteal lymph node (scale bar, 150 μm). B cell follicles are highlighted in gray for illustrative purposes. Middle and right images are examples of the visualization (including representative tracks) of homeostatic movement (scale bars, 30 μm). Smaller images indicate two representative cells, highlighted by arrows at different times. Graphs (±SEM) on the bottom right indicate the quantification of the average speed over the span of the movie and the mean displacement length. Data are representative of three independent experiments.
(D) Chemotactic response of purified WT and Wipf1−/− B cells to CXCL12 (left graph) or CXCL13 (right graph) using transwell plates. Graphs (±SEM) show percentages of migrating cells normalized to the starting input number of B cells set to 100%. Data are pooled from four independent experiments.
(E) Immunoblot of splenic WT or Wipf1−/− B cells treated with CXCL12 or CXCL13 and probed with antibodies as indicated.
(F) Quantifications of intensity of proteins normalized by densitometry to total Erk or tubulin and to the signal in unstimulated WT cells at t = 0. Data are representative of at least three independent experiments.
We next investigated the steady-state migration of naive B cells in vivo. For this, fluorescently labeled WT and Wipf1−/− B cells were adoptively transferred into WT recipients. After 24 hr, both cell types were found in similar regions of explanted popliteal and inguinal lymph nodes using two-photon microscopy, indicating that both cell types are able to localize to B cell follicles (Figure 2C). While WT B cells moved with an average speed of 6.2 ± 0.07 μm/min, Wipf1−/− B cells were significantly slower with an average speed of 5.1 ± 0.1 μm/min (Figure 2C; Movie S1). In addition, Wipf1−/− B cells showed 30% less displacement during the acquisition period compared to WT B cells. These results support our hypothesis that WIP might regulate cell migration in vivo.
Homing of B cells to follicles in secondary lymphoid organs involves migration mediated by the chemokines CXCL12 and CXCL13 (Okada et al., 2002). We found WIP to be slightly phosphorylated after stimulating naive B cells with CXCL12 as measured by flow cytometry (Figure S2B), likely indicating an involvement of WIP in chemokine signaling. We next evaluated the B cell chemotactic response to a gradient of these chemokines in vitro using transwell plates. Here, Wipf1−/− B cells displayed a significant reduction in the percentage of migratory B cells to both, CXCL12 and CXCL13 compared to WT cells (Figure 2D). This reduction was not due to altered expression of CXCR4 (the receptor binding CXCL12) and CXCR5 (the receptor binding CXCL13) on the B cell surface (Figure S2C). Examining signaling after CXCR4 stimulation, Wipf1−/− B cells displayed a severely diminished phosphorylation of Akt kinase, used as an indicator for PI3K signaling, when compared to WT B cells (Figures 2E and 2F). A similar reduced phosphorylation of Akt was observed in Wipf1−/− WEHI-231 B cells. Re-expression of WIP in Wipf1−/− WEHI B cells restored Akt phosphorylation to amounts similar to those in WT WEHI cells (Figure S2E). Furthermore, splenic Wipf1−/− B cells showed a 50% reduction in phosphorylation of the transcription factor Foxo and a 80% reduction in phosphorylation of p70S6K compared to the phosphorylation in WT cells, both downstream targets of the PI3K signaling pathway (Figures 2E and 2F; Figure S2D). Wipf1−/− B cells stimulated with CXCL13 similarly displayed severely diminished activation of the PI3K/Akt signaling pathway when compared to WT cells (Figures 2E and 2F; Figure S2D).
Together, these results demonstrate an impaired chemokine receptor-induced PI3K signaling in Wipf1−/− B cells. In line with this, we also found reduced chemokine-sensing and migration of these cells in vitro and in vivo, which likely contributes to a decreased humoral immune response in Wipf1−/− mice.
BCR Signaling Requires WIP for PI3K Pathway Activation
To address whether WIP was recruited to active sites of BCR signaling, we expressed human GFP-WIP in WEHI-231 B cells. These cells were settled on planar lipid bilayers containing Alexa Fluor 633-labeled antibody against the BCR (anti-BCR) to mimic the interaction of B cells with antigen-presenting cells (Fleire et al., 2006). Immediately after B cells contacted the membrane, GFP-WIP was recruited to sites of antigen aggregation (Figure 3A and Movie S2). In parallel, we found strong phosphorylation of WIP at residue S488 already 3 min after BCR cross-linking of naive WT B cells, as detected by flow cytometry (Figure 3B) or immunoblot (Figure S3A). These findings demonstrate that during early events of BCR signaling, WIP is recruited to signalosomes. This recruitment is concomitant with an increase in phosphorylation of the protein.
Figure 3.

WIP Is Required for Efficient BCR-Induced Activation of the PI3K Pathway
(A) TIRF microscopy of WEHI-231 B cells expressing GFP-WIP imaged after spreading over a planar lipid bilayer labeled with anti-κ-chain antibody linked to Alexa Fluor 633 (left) and quantification of fluorescent intensities along the white diagonal line (right, colors match labels above images). Scale bar, 3 μm.
(B) Flow cytometric analysis of WIP phosphorylation after anti-κ-chain stimulation of WT B cells. Grey shaded histogram represents the unstimulated WT control. Graph shows the mean fluorescent intensity (MFI) normalized to the MFI of Wipf1−/− cells at indicated time points.
(C) Phalloidin staining indicating intracellular amount of F-actin in fixed WT or Wipf1−/− B cells settled on coverslips coated with anti-κ-chain antibody for 10 min. Scale bar, 3 μm. Quantification (right graph) shows F-actin foci per cell (mean ± SEM). Data are representative of three independent experiments.
(D and E) Immunoblot of splenic WT or Wipf1−/− B cells stimulated with anti-κ-chain antibody and probed with antibodies as indicated. Quantifications of intensity of proteins normalized by densitometry to total Erk or tubulin and to the signal in unstimulated WT cells at t = 0. Data are representative of at least three independent experiments.
(F) Graph show mean (±SEM) percentage of live WT or Wipf1−/− B cells cultured in medium or in the presence of BAFF at different time points, normalized to the starting input number of live B cells set to 100%. Results pooled from three independent experiments with one mouse each.
(G) Immunoblot of splenic WT or Wipf1−/− B cells stimulated with BAFF, probed with antibodies against p-Akt and Erk. Quantifications of intensity of proteins normalized by densitometry to total Erk and to the signal in unstimulated WT cells at t = 0.
(D and G) Experiments were performed simultaneously with experiments in Figure 5A and therefore results can be directly compared. The loading control measurements (Erk for anti-κ-chain and BAFF stimulation) were part of both experiments.
We next investigated whether ablation of WIP influences the quantity of filamentous actin (F-actin) in activated B cells. We found that Wipf1−/− B cells exhibited a lower amount of F-actin when B cells were allowed to spread on coverslips coated with immobilized antigen. Such difference was mainly manifested by the absence of visible F-actin foci measured by total internal reflection microscopy (TIRFM) (with an average of 6.8 F-actin foci per WT cell compared to an average of 0.8 F-actin foci in Wipf1−/− cells), but resulted neither in the incapacity to spread nor in a defect of F-actin polymerization at the leading edge (Figure 3C).
We next tested the capacity of Wipf1−/− cells to internalize antigen. B cells were loaded with fluorescent microspheres containing anti-κ-chain antibody on ice, washed to remove unbound microspheres, and then incubated at 37°C to allow BCR internalization. Wipf1−/− B cells internalized less particulate antigen compared to WT B cells at all time-points analyzed (Figure S3B), indicating that WIP-mediated reorganization of the actin cytoskeleton might be necessary for BCR-mediated antigen internalization.
We went on to measure BCR-induced Ca2+ mobilization in naive splenic B cells by flow cytometry and found it to be increased in Wipf1−/− B cells relative to that of WT B cells (Figure S3D). This was not due to increased BCR expression on the surface of Wipf1−/− B cells (Figure S3C). Next, we stimulated both cell types with a soluble anti-κ-chain antibody and measured the BCR-induced phosphorylation of Syk and Erk kinases, as well as the PI3K subunit p85, Akt and Foxo. Both WT and Wipf1−/− B cells displayed robust phosphorylation of Syk and Erk after BCR cross-linking. Of note, naive Wipf1−/− B cells expressed higher basal phosphorylation of Syk and Erk (Figures 3D and 3E). However, Wipf1−/− B cells showed an at least 50% reduction of phosphorylation of p85, Akt and Foxo compared to WT cells (Figures 3D and 3E). Similar results for Erk and Akt phosphorylation after stimulation were obtained using flow cytometry (Figure S3E).
We next investigated whether WIP deficiency influences B cell survival. In addition to BCR signaling, the BAFFR pathway is critical for survival of B cells. We detected slightly lower expression of BAFFR on the surface of Wipf1−/− B cells compared to WT cells by flow cytometry (Figure S3F). Cultivating purified splenic B cells from Wipf1−/− or WT mice in either unconditioned medium or in the presence of BAFF, we found reduced survival in the absence of WIP. In cultures with BAFF, only 40% of Wipf1−/− B cells were alive at day 3 compared to 60% of WT B cells (Figure 3F). In line with the impaired survival, phosphorylation of Akt was impaired in Wipf1−/− B cells after short-term stimulation with BAFF (Figure 3G). These results suggest a role of WIP in BAFFR-induced PI3K activation and survival.
Collectively, these results demonstrate that, in the absence of WIP, the actin cytoskeleton after BCR stimulation is altered. Furthermore, WIP deficiency led to impaired PI3K signaling after BCR cross-linking, which eventually results in a reduced BAFFR-triggered survival.
Formation of Plasma Cells In Vitro Depends on WIP Mediated PI3K Signaling
We went on to measure phosphorylation of WIP in response to CD40 and Toll-like receptor-4 (TLR4) stimulation by flow cytometry and immunoblot, respectively, and found WIP to be phosphorylated, again indicating a direct contribution of WIP in response to those stimuli (Figures S4A and S4B). We next analyzed the activation status of Akt and Foxo after triggering CD40 and TLR4 in WT and Wipf1−/− B cells by immunoblot. We observed robust phosphorylation of Akt and Foxo in WT B cells, but phosphorylation of both was strongly reduced in Wipf1−/− B cells (Figures 4A and 4B), similar to our results after BCR and chemokine receptor stimulation. Furthermore, we measured reduced phosphorylation of Akt after CD40 stimulation of Wipf1−/− WEHI-231 B cells by flow cytometry, which was restored to the amount of phosphorylation in WT cells after re-expression of WIP (Figure S4F). Thus, WIP deficiency might indeed lead to a general impaired PI3K and Akt activation.
Figure 4.

WIP Regulates PI3K Signaling and Mediates Class-Switching and Plasma Cell Formation In Vitro
(A and B) Immunoblot of splenic WT or Wipf1−/− B cells stimulated with CD40L (A) or LPS (B), probed with antibodies as indicated. Quantifications of intensity of proteins normalized by densitometry to Erk or tubulin, and to the signal in unstimulated WT cells at t = 0. This experiment and experiment in Figure S4D were performed simultaneously. The loading control measurements were part of both experiments.
(C and D) CTV labeled splenic WT and Wipf1−/− B cells were stimulated for 4 days with CD40L (C) or LPS (D) and IL-4 and IL-5. Flow cytometry measurements of proliferation (CTV dilution, upper row), class-switch recombination (IgG1+ cells, middle row) and plasma cell differentiation (CD138hi cells, lower row). Graphs on the right of (C) and (D) indicate the percentages of class-switched and PCs normalized to total live cells. Data are pooled from at least three experiments (mean ± SEM).
See also Figure S4.
We next analyzed whether the defect in PI3K/Akt signaling was reflected in the capability of Wipf1−/− B cells to class-switch and differentiate into plasma cells (PCs). Splenic WT and Wipf1−/− B cells were cultivated with either CD40L or LPS in the presence or absence of interleukin-4 (IL-4) and IL-5 in vitro. After four days of culture, both, Wipf1−/− and WT B cells proliferated extensively to CD40L stimulation as indicated by the dilution of CTV (Figure 4C). However, in response to LPS or LPS in the presence of IL-4 and IL-5, Wipf1−/− B cells showed a marked increase in the percentage of proliferating cells of 80% versus 50% when compared to WT B cells (Figure 4D). This correlated with an increase in Erk phosphorylation as detected by immunoblot (Figures S4D and S4E). Furthermore, Wipf1−/− B cells showed a 2–3 times higher percentage of class-switched cells compared to WT cells in both stimulatory conditions (Figures 4C and 4D). The proportion of Wipf1−/− PCs (evaluated by CD138 expression) was substantially diminished to less than half (Figures 4C and 4D). Taken together, these results indicate that WIP is required for PI3K pathway activation and thus regulates class-switch recombination and plasma cell formation.
WIP Regulates PI3K Activation via Phosphorylation of CD19
Having identified an essential function for WIP in regulating PI3K/Akt signaling in response to stimulation of chemokine receptors, BAFFR, cross-linking of the BCR or the co-receptor CD40 or TLR4, we wondered whether the co-receptor CD19 itself might be involved in all the signaling pathways we analyzed. As expected, triggering of the BCR, BAFFR, and CD40 induced robust phosphorylation of CD19 (Figures 5A and 5B). Triggering of TLR4, CXCR4, and CXCR5 also induced low but consistent phosphorylation of CD19 (Figures 5A and 5B). Furthermore, we noted that, in B cells lacking WIP, basal CD19 phosphorylation was reduced, likely contributing to overall reduced CD19 phosphorylation after stimulation. We found that triggering CD40, TLR4, and CXCR4 induced phosphorylation of Src family kinases and that this phosphorylation was again reduced in the absence of WIP (Figure S5A). Together, these results indicate that CD19 is not only a critical component of the BCR, BAFFR, and CD40 pathway but might also be relevant for PI3K signaling after TLR4, CXCR4, and CXCR5 stimulation. In addition, WIP is involved in these signaling pathways possibly influencing the phosphorylation of CD19.
Figure 5.

WIP Regulates PI3K Activation via Phosphorylation of CD19
(A) Immunoblot of splenic WT or Wipf1−/− B cells stimulated with (from top to bottom) anti-κ-chain antibody, BAFF, CC40L, LPS, CXCL12, or CXCL13 and probed with antibodies as indicated. Experiments were performed simultaneously with experiments in Figure 3 (anti-κ-chain and BAFF), Figure 4 (LPS) and Figure S2D (CXCL12 and CXCL13). Single loading control measurements are shown in figures.
(B) Quantifications of intensity of proteins stimulated as in (A) normalized by densitometry to Erk or tubulin and to the signal in unstimulated WT cells at t = 0.
(C–F) CTV labeled splenic WT and Cd19−/− B cells were stimulated for 4 days with CD40L and IL-4 and IL-5. Flow cytometry measurements of (C) proliferation (CTV dilution), (D) class-switch recombination (IgG1+ cells, upper row) and plasma cell differentiation (CD138hi cells, lower row). Graphs on the right of (D) indicate the percentages of class-switched and PCs normalized to total live cells. Data are pooled from at least three experiments (mean ± SEM).
(E and F) Immunoblot of splenic WT or Cd19−/− B cells stimulated with CD40L and probed with antibodies as indicated. Quantifications of intensity of proteins normalized by densitometry to actin and to the signal in unstimulated WT cells at t = 0.
(G) Chemotactic response of purified WT and Cd19−/− B cells to CXCL13 using transwell plates. Graphs (±SEM) show percentages of migrating cells normalized to the starting input number of B cells set to 100%. Data are pooled from three independent experiments.
(H) Immunoblot of splenic WT or Cd19−/− B cells stimulated with CXCL13 and probed with antibodies as indicated. Quantifications of intensity of proteins normalized by densitometry to Erk and to the signal in unstimulated WT cells at t = 0.
See also Figure S5.
Cd19−/− mice have been repeatedly used to study the role of the PI3K/Akt pathway in B cell function (Depoil et al., 2008; Engel et al., 1995; Fujimoto et al., 1999; Rickert et al., 1995). We used B cells from Cd19−/− mice to compare results of experiments performed under similar conditions. Of note, BCR cross-linking of Cd19−/− B cells induced a robust phosphorylation of WIP (Figure S5E), suggesting that WIP activation might be independent of CD19 signaling. We went on to investigate the effect of CD19 deficiency on plasma cell formation in vitro. Splenic purified B cells from Cd19−/− and WT mice were cultivated with either LPS or CD40L in the presence or absence of IL-4 and IL-5 as described before. After 4 days in culture, both, Cd19−/− and WT B cells proliferated extensively (Figure 5C and Figure S5C). In similarity to what has been observed in cultures of Wipf1−/− B cells, the proportion of Cd19−/− B cells that underwent class-switch recombination was about 2-fold higher compared to WT cells when stimulated with CD40L (Figure 5D). However, class-switch recombination of Cd19−/− B cells in response to LPS was comparable to WT cells (Figure S5B). Furthermore, Cd19−/− B cells demonstrated a complete block in differentiation into CD138-positive PCs (Figure 5D). In line with these results, and similar to results obtained with Wipf1−/− B cells, Cd19−/− B cells showed reduced Akt and Foxo phosphorylation after CD40L or LPS stimulation (Figures 5E and 5F and Figure S5D).
Again similar to results with Wipf1−/− B cells, we found that the chemotaxis of Cd19−/− B cells toward a gradient of CXCL13 using transwell plates was significantly reduced compared to WT B cells (Figure 5G). In addition, Cd19−/− B cells displayed diminished phosphorylation of Akt and Foxo in response to CXCL13 stimulation compared to WT cells (Figure 5H). These experiments demonstrate that Cd19−/− B cells recapitulate many of the phenotypes observed in Wipf1−/− B cells, providing strong evidence that WIP is involved in regulating the PI3K pathway via CD19 activation.
WIP Regulates BCR and CD19 Diffusion on the B Cell Surface
How could WIP affect CD19 activation? We noticed that while CD19 expression was similar on the surface of WT and Wipf1−/− B cells, expression of the tetraspanin protein CD81 was reduced on Wipf1−/− B cells (Figure 6A). No substantial alterations in the expression of other members of the tetraspanin family were detected in the absence of WIP (data not shown). A reduction in CD81 was also observed in two independent Wipf1−/− cell lines generated by employing the CRISPR/Cas9 deletion system. Transient re-expression of WIP in Wipf1−/− A20 cells increased CD81 expression (Figures S6A–S6D) further supporting the notion that WIP is required to maintain CD81 expression.
Figure 6.

WIP Regulates BCR and CD19 Diffusion on the B Cell Surface
(A) Flow cytometric analysis of CD19 and CD81 on the surface of WT (blue line) or Wipf1−/− (red line) B cells. Grey shaded histogram indicates the isotype control. Graph (mean ± SEM) on the right shows the MFI of CD81 expression on splenic B cells of 4 WT and 4 Wipf1−/− mice.
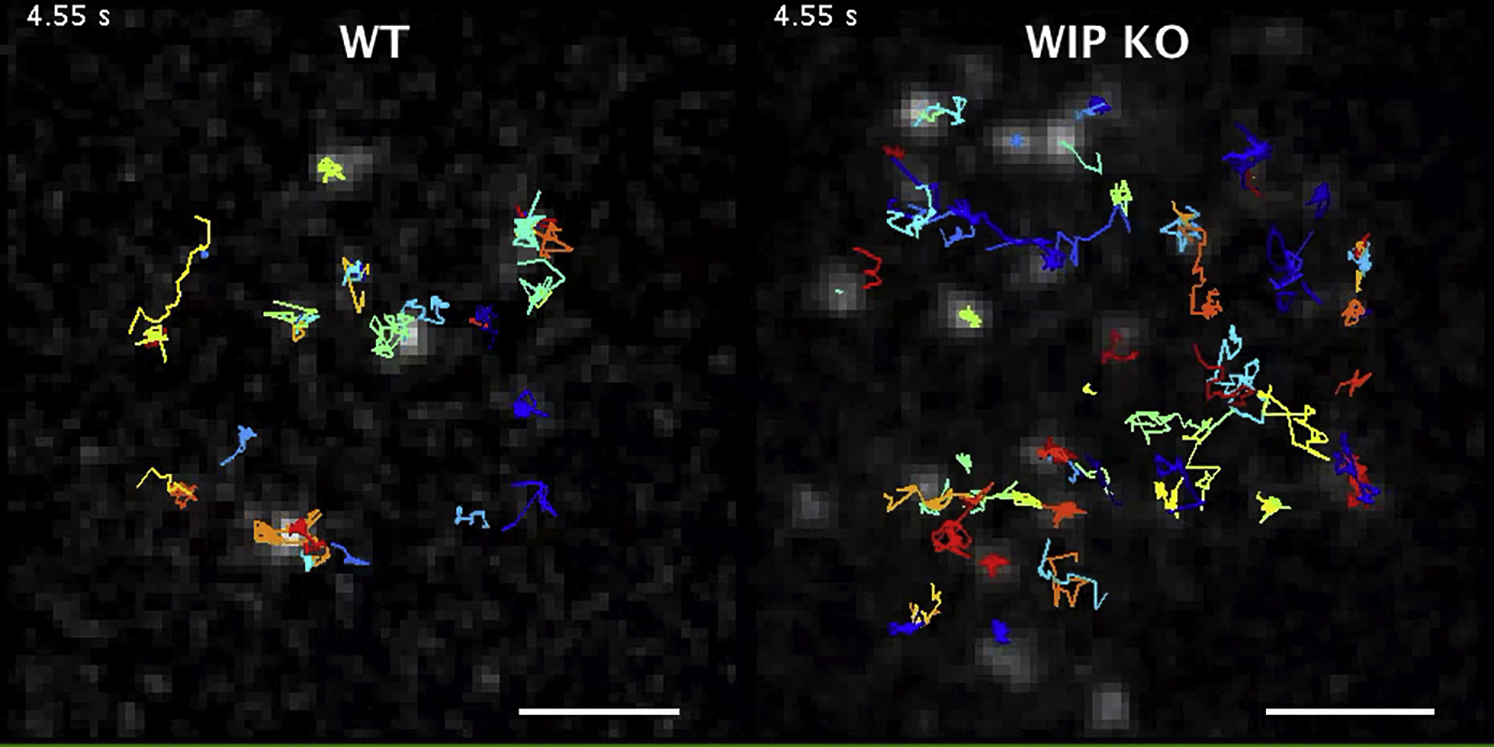
(B) Single-particle tracking (SPT) of CD19 on WT or Wipf1−/− B cells settled on nonstimulatory coverslips. Shown is the diffusion coefficient (D) in μm2/s. The median of 300 representative cells is given as value and indicated by black bars. 1,000–2,000 tracks were analyzed with a minimum of 20 cells from 3 experiments.
(C) Trajectories of CD19 in WT (left) and Wipf1−/− cells (right) showing diffusion of single particles over 10 s and magnified regions from white rectangles. Scale bar, 2 μm.
(D) Analysis of the CD19 diffusion coefficient plotted in a logarithmic histogram and fitted to two distinct populations exhibiting slower (light gray) and faster (dark gray) diffusion on WT (left graph), Wipf1−/− (middle graph) and a comparison of the CD19 diffusion on WT and Wipf1−/− B cells (right graph).
(E) Flow cytometric analysis of F-actin by intracellular phalloidin staining of resting WT (blue line) or Wipf1−/− (red line) B cells.
(F) SPT of IgM and IgD on WT or Wipf1−/− B cells settled on nonstimulatory coverslips. Shown is the diffusion coefficient (D) in μm2/s. 1,000–2,000 tracks were analyzed with a minimum of 20 cells from 2 experiments. The median of 300 representative cells is given as value and indicated by black bars.
(G) Ratiometric intracellular Ca2+ flux of WT and Wipf1−/− B cells, stimulated with Latranculin A and analyzed by flow cytometry. Results are presented as fluorescent emission at 450 nm relative to that at 530 nm. Data are representative of three independent experiments.
(H) Immunoblot of splenic WT or Wipf1−/− B cells stimulated with Latrunculin A and probed with antibodies as indicated. Quantifications of intensity of proteins normalized by densitometry to Erk or tubulin and to the signal in unstimulated WT cells at t = 0.
In resting B cells, CD81 stabilizes and immobilizes CD19 on the cell surface (Mattila et al., 2013). To investigate whether WIP deficiency alters CD19 dynamics, we performed single-particle tracking of CD19 by incubating primary B cells with limited amounts of fluorescently labeled anti-CD19 Fab fragments followed by analysis with TIRFM. While CD19 was relatively immobile on the surface of resting WT cells, CD19 particles on the surface of Wipf1−/− B cells showed an accelerated diffusion, with the median diffusion coefficient increasing from 0.008 μm2/s on WT B cells to 0.03 μm2/s on Wipf1−/− B cells (Figures 6B and 6C and Movie S3). A more detailed analysis revealed two different populations of CD19 in WT cells, with a slower diffusion population centered around D = 0.003 μm2/s and a population retaining higher mobility (centered around D = 0.032 μm2/s). CD19 diffusion of both, the slower and higher populations accelerated on Wipf1−/− B cells with the slower diffusion population centered around D = 0.010 μm2/s and the higher diffusion population centered around D = 0.060 μm2/s) (Figure 6D). These results indicate that the reduced CD81 amount indeed correlate with an increase in CD19 dynamics, likely leading to a misplacement of the co-receptor.
We went on to analyze the impact of WIP deficiency on BCR dynamics on resting B cells. While CD19 is immobilized by the tetraspanin network, BCR mobility is regulated by the cortical actin cytoskeleton. Measuring phalloidin staining by flow cytometry, we observed a reduced amount of F-actin in naive Wipf1−/− B cells when compared to WT B cells (Figure 6E). Given this reduction in F-actin in steady state and the impaired actin polymerization after BCR stimulation, we wondered whether the diffusion of the BCR was altered in the absence of WIP. As previously shown (Mattila et al., 2013), we observed a very low mobility of a large proportion of IgD particles in resting WT B cells, with a median diffusion coefficient of 0.008 μm2/s. Notably, on Wipf1−/− B cells, we detected a 2-times accelerated diffusion of IgD particles, with a median diffusion coefficient of 0.015 μm2/s (Figure 6F, right graph). Although IgM is already quite mobile on resting WT B cells, we detected a slight but consistent increase also in IgM diffusion on Wipf1−/− B cells (Figure 6F, left graph). The diffusion of CD40 was also accelerated on Wipf1−/− B cells (Figure S6F). From these results, we conclude that the diminished amount of F-actin in Wipf1−/− B cells is reflected in higher mobility of B cell receptors.
In view of the impaired CD19 phosphorylation and the altered dynamics of CD19, we next analyzed whether Wipf1−/− B cells were able to react to the disruption of the actin cytoskeleton through the actin monomer-sequestering agent Latrunculin A (LatA). Compared to WT cells, Wipf1−/− B cells displayed a markedly delayed calcium response to LatA treatment together with a severely impaired capability to phosphorylate the co-receptor CD19 and the signaling molecules Akt, Syk, and Erk (Figures 6G and 6H). These results show that in the absence of WIP, ligand-independent signaling via CD19 triggered by actin cytoskeleton disruption is distorted, again hinting at the regulatory effect of WIP in mediating CD19 activation. We thus suggest that in the absence of WIP, the actin cytoskeleton but also the CD81 tetraspanin network is disturbed, leading to an increased diffusion of both, the BCR and CD19 on resting cells. This possibly causes an altered organization of B cell surface receptors, leading to a misplacement of the receptors and impaired signaling.
Discussion
Here, we have shown that WIP functions as a regulator of CD19 activation and PI3K/Akt signaling in B cells. WIP deficiency resulted in defects in homing, chemotaxis, survival and differentiation, highlighting an essential role for WIP in B cell function. Furthermore, in the absence of WIP, B cells displayed distortions in the actin cytoskeleton and the CD81-tetraspanin network, which resulted in altered cell surface mobility of the BCR and CD19. These changes in surface receptor dynamics resulted in reduced phosphorylation of CD19 after triggering a variety of receptors and thus place CD19 at the heart of PI3K signaling in B cells.
Challenging mixed BM chimeras with either Vaccinia virus or NP-KLH revealed that WIP deficiency compromised GC formation and antibody production, thereby hampering humoral immune responses. Similarly, the small Rho GTPase Cdc42, a known regulator of actin remodeling and interaction partner of WASP and N-WASP, controls GC formation and antibody production in B cells (Burbage et al., 2015). In contrast, B cells deficient for WASP mount a normal antibody response to T-dependent immunization (Recher et al., 2012; Snapper et al., 1998). This can be partly explained by compensatory effects of the ubiquitously expressed protein N-WASP (Westerberg et al., 2012). We therefore suggest that the impaired capability of our mixed BM chimeras to mount a humoral immune response is due to the absence of WIP in B cells.
We have further shown that WIP regulated chemotaxis of B cells toward the chemokines CXCL12 and CXCL13, mediating B cell homing to lymph nodes and spleen. In support of our findings, WIP functions in F-actin-rich lamellopodium formation and amoeboid motility in response to CXCL13 in vitro (Banon-Rodriguez et al., 2013). T cells lacking the actin-binding domain of WIP displayed decreased cellular F-actin content, impaired chemotaxis and defective homing to lymph nodes although having normal WASP expression (Gallego et al. 2006; Massaad et al., 2014). These findings strongly argue for a crucial role for WIP in regulating actin-cytoskeleton-mediated migration.
Early signaling events following BCR stimulation by membrane-bound antigen trigger the formation of antigen microclusters (Fleire et al., 2006). We found here that WIP was recruited to microclusters and phosphorylated at the serine residue S488 immediately after BCR engagement, suggesting that WIP participates in the early events after antigen recognition. How phosphorylation regulates WIP function in B cells is unclear to date. In T cells, S488 of WIP is phosphorylated in a PKCΘ-dependent manner in response to T cell receptor (TCR) activation, mediating the recruitment of WASP to the immunological synapse (Fried et al., 2014; Sasahara et al., 2002). In contrast, tyrosine phosphorylation of WIP by Btk triggers the release of WASP from the WIP–WASP complex, resulting in WASP degradation and termination of actin filament generation (Vijayakumar et al., 2015). Phosphorylation and activation of WIP in B cells might thus regulate actin dynamics. Indeed, in the absence of WIP, we found lower amounts of F-actin in resting B cells and less F-actin foci when B cells spread on antigen-coated coverslips. Similar F-actin foci were recently described as being a WASP-generated fraction of the total synaptic F-actin in T cells (Kumari et al., 2015). We suggest that, in B cells, WIP plays a central role in cytoskeletal actin dynamics that is triggered by the engagement of a variety of receptors, having shown that BCR crosslinking, CXCL12, CD40, and TLR4 signaling all induce the phosphorylation of WIP.
A high percentage of WAS patients develop autoimmune diseases. Similarly, B cell-intrinsic deletion of WASP is sufficient to induce autoantibody production and antibody deposition in the kidneys of mice (Becker-Herman et al., 2011; Recher et al., 2012). In the absence of the WASP homolog N-WASP, B cells show delayed BCR internalization, enhanced tyrosine phosphorylation and higher Ca2+ flux (Liu et al., 2013). WIP interacts with N-WASP but is not required for its stability (de la Fuente et al., 2007). In Wipf1−/− B cells, N-WASP activation upon BCR stimulation was only mildly impaired (data not shown), suggesting that the negative regulatory role of WIP is probably independent of N-WASP function. We found that Wipf1−/− B cells displayed an increased Ca2+ flux after BCR crosslinking, enhanced proliferation, a delayed and reduced BCR internalization, and increased basal phosphorylation of Syk and Erk. These findings are in line with a previous report suggesting that blocking of BCR endocytosis leads to hyperphosphorylation of Syk and Erk and lower phosphorylation of the PI3K pathway (Chaturvedi et al., 2011). We suggest that the higher basal Syk activation in Wipf1−/− B cells might explain the partially hyperreactive phenotype, because Syk is both essential to and sufficient for antigen-induced Ca2+ flux after BCR clustering by multimeric ligands (Mukherjee et al., 2013; Takata et al., 1994).
In B cells, CD19 contributes to BAFF- and CD40-mediated survival, consistent with CD19 phosphorylation and Akt activation (Hojer et al., 2014; Jellusova et al., 2013). Here, we extend this model by showing that triggering a variety of receptors, namely the BCR, BAFFR, CXCR4, CXCR5, CD40, and TLR, led to the phosphorylation of CD19. We thus suggest a critical role for CD19 as a general “hub” of PI3K signaling. The precise molecular mechanism by which CD19 is phosphorylated remains unclear. Recent data demonstrated that BAFFR stimulation triggers the phosphorylation of the Igα chain and Syk, potentially leading to CD19 activation (Schweighoffer et al., 2013). Furthermore, in a model of CD40 overexpression, CD19 phosphorylation is likely mediated by the Src family kinase Lyn (Hojer et al., 2014). We indeed detected Lyn phosphorylation after triggering of most of the receptors studied. We moreover have demonstrated here that CD19 phosphorylation is dependent on WIP. Wipf1−/− B cells were unable to efficiently phosphorylate CD19. Concomitantly, activation of the PI3K/Akt pathway was impaired, leading to multiple defects in homing, survival, class-switching, and antibody production. Supporting our hypothesis, many of the defects seen in Wipf1−/− B cells were recapitulated in B cells lacking CD19.
Dissecting the molecular mechanism of how WIP might mediate the function of CD19, we found that in the absence of WIP, BCR and CD19 diffusion is enhanced. We recently showed that in the plasma membrane of resting B cells, the BCR is immobilized by the actin cytoskeleton (Treanor et al., 2010) and CD19 is kept relatively immobile by the CD81-tetraspanin network (Mattila et al., 2013). Actin cytoskeleton disruption increases BCR diffusion, leading to signal induction. This ligand-independent signal not only requires an increased BCR diffusion but also an immobilized co-receptor CD19. In the absence of CD81, CD19 mobility on the B cell surface increases, failing to promote BCR signaling induced by cytoskeleton disruption. CD19 mobility increased even more by treating Cd81−/− cells with LatA (Mattila et al., 2013). We concluded that CD19 diffusion correlates with CD81 expression and in the absence of CD81, CD19 diffusion is partially controlled by the actin cytoskeleton. We now propose that in the absence of WIP, altered dynamics of the actin cytoskeleton together with a diminished expression of CD81 induce a faster diffusion of CD19. This might cause a dysregulated function of CD19 because both its diffusion and organization might be altered. In line with this suggestion, ligand-independent actin cytoskeleton disruption induced a delayed Ca2+ flux and almost absent signaling in B cells lacking WIP.
A growing body of evidence suggests a crucial role for changes in actin dynamics in mediating signal integration from a multitude of surface receptors. On the basis of our findings, we propose that WIP, and possibly other actin regulators, are essential to control correct organization of the actin cytoskeleton and receptor mobility. This might be especially critical in the case of a low amount of signaling (e.g., monovalent antigen) or tonic signaling requiring signal amplification via CD19. Altered receptor dynamics, such as seen in the absence of WIP, might hamper receptor crosstalk and limit CD19 in its function as a general hub for PI3K signaling, thereby influencing critical survival and differentiation signals for mature B cells.
Experimental Procedures
For an extended description of experimental procedures, see the Supplemental Information.
Mice
Wipf1−/− mice (Antón et al., 2002) were a kind gift from Raif Geha (Boston’s Children Hospital); Igh-Jtm1Dhu mice (BALB/c JHT; Chen et al., 1993) were purchased from Taconic Bioscience; CBy.SJL(B6)-Ptprca/J mice (BALB/c CD45.1) were purchased from The Jackson Laboratory. BALB/c WT and C57BL/6 WT mice, CD19cre+/+ (referred to as Cd19−/−) (Rickert et al., 1995) mice were obtained from the breeding facility at Cancer Research UK (CRUK). Gene targeted mice were bred and maintained at the animal facility of CRUK. Radiation chimeras were generated using standard protocols. Mixed chimeras were generated by injecting a mixture of 80% of Balb/c JHT with 20% WT or Wipf1−/− BM into Balb/c JHT or Balb/c CD45.1 mice or by injecting a 1:1 mixture of Balb/c CD45.1 WT and CD45.2 Wipf1−/− into CD45.1 mice. All experiments were approved by the Animal Ethics Committee of Cancer Research UK and the UK Home Office.
Immunization and Infection
For infection, mice were injected with a low dose (1 × 104 PFU) of Vaccinia virus WR strain (VV) intra footpad and mice sacrificed 8 days later. Draining popliteal lymph nodes were analyzed by flow cytometry. For immunization, mice were injected i.p. with 50 μg NP33-KLH (Biosearch Technology) in 4 mg Alum (ThermoScientific).
Immunoblotting
Splenic naive B were purified using negative B cell isolation kits yielding enriched populations of about 95%–98% (Miltenyi). Purified splenic B cells were stimulated with 3 μg/mL anti-κ antibody (HB558, ATCC), 200 ng/ml BAFF (Peprotech), 2.5 ng/ml CXCL12, 0.5 μg/mL CXCL13, 0.5 μg/mL CD40L (all R&D systems), 5 μg/mL LPS, or 1 μM Latranculin A (Calbiochem). Lysates were prepared using lysis buffer with 1% NP-40. Immunoblotting was performed using standard procedures. Blot densitometry analysis was performed using the ImageJ software.
Generation of Wipf1−/− Cell Lines
WIP was deleted in A20 cells and WEHI-231 cells using CRISPR/Cas9 system (Cong et al., 2013; Ran et al., 2013). Briefly, custom-designed plasmids expressing the gRNA and Cas9-GFP protein in a single vector (U6gRNA-Cas9-2A-GFP) were bought from Sigma-Aldrich. The following target sites within the coding region of WIP were used: CACTGGCGCCGGCTCCCTTTGG and AGGGAGCCGGCGCCAGTGCTGG. The cell lines were transiently transfected with the desired vector for 12 hr followed by single-cell sorting of GFP-positive cells into a 96-well plate containing complete B cell medium. After 2 weeks, the individual colonies were screened for WIP deletion by immunoblotting.
Single Particle Tracking
Single particle tracking was performed and analyzed as previously described (Mattila et al., 2013; Treanor et al., 2010). Briefly, primary naive B cells were labeled with limiting dilutions of Atto633 or Atto488 conjugated anti-CD19 Fab (1D3), anti-IgD Fab (11-26c), or anti-IgM Fab (Jackson ImmunoResearch) for 10 min on ice and washed with PBS. Cells were then resuspended in imaging buffer and allowed to settle on anti-MHCII-coated coverslips in Focht Chamber System 2 (FCS2) chambers (Bioptechs) at 37°C for 2 min prior to imaging with TIRFM for 15 min. Single particle tracking data was analyzed and confinement size was calculated as described (Mattila et al., 2013; Treanor et al., 2010).
Experimental Data and Statistical Analysis
All statistical comparisons were carried out using the nonparametric two-tailed Student’s t test and p values were calculated using PRISM software. Statistically significant differences are indicated on the figures as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Author Contributions
S.J.K. designed and performed the experiments, analyzed the data, and wrote the paper. F.G., M.B., S.A., and B.F. performed and analyzed experiments. R.G. provided Wipf1−/− mice. M.W. provided the GFP-WIP construct. A.B. assisted with microscopy and data analysis. F.D.B. and S.J.K. conceived of the study and prepared the manuscript.
Acknowledgments
We thank the biological resource unit (LIF Laboratories) for animal husbandry. We thank all members of the Lymphocyte Interaction Laboratory, Beatriz Montaner for managing the animal colony, Julia Coleman for critical reading of the manuscript, and Jan Sodenkamp and Sabine Zoellner for help with graphics. All the work presented here was supported by The Francis Crick Institute, the German Research Foundation (DFG) grant Ke1737/1-1 (S.J.K.), the Institute Pasteur-Fondazione Cenci Bolognetti (F.G.), a long-term EMBO fellowship (S.A.), and a Royal Society Wolfson Research Merit Award (F.D.B).
Published: October 6, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes six figures, Supplemental Experimental Procedures, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.09.004.
Supplemental Information
{kind=link}
{kind=link}
{kind=link}
References
- Antón I.M., Lu W., Mayer B.J., Ramesh N., Geha R.S. The Wiskott-Aldrich syndrome protein-interacting protein (WIP) binds to the adaptor protein Nck. J. Biol. Chem. 1998;273:20992–20995. doi: 10.1074/jbc.273.33.20992. [DOI] [PubMed] [Google Scholar]
- Antón I.M., de la Fuente M.A., Sims T.N., Freeman S., Ramesh N., Hartwig J.H., Dustin M.L., Geha R.S. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity. 2002;16:193–204. doi: 10.1016/s1074-7613(02)00268-6. [DOI] [PubMed] [Google Scholar]
- Arron J.R., Vologodskaia M., Wong B.R., Naramura M., Kim N., Gu H., Choi Y. A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine (trance) and CD40L-mediated Akt activation. J. Biol. Chem. 2001;276:30011–30017. doi: 10.1074/jbc.M100414200. [DOI] [PubMed] [Google Scholar]
- Banon-Rodriguez I., Saez de Guinoa J., Bernardini A., Ragazzini C., Fernandez E., Carrasco Y.R., Jones G.E., Wandosell F., Anton I.M. WIP regulates persistence of cell migration and ruffle formation in both mesenchymal and amoeboid modes of motility. PLoS ONE. 2013;8:e70364. doi: 10.1371/journal.pone.0070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barda-Saad M., Braiman A., Titerence R., Bunnell S.C., Barr V.A., Samelson L.E. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat. Immunol. 2005;6:80–89. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- Becker-Herman S., Meyer-Bahlburg A., Schwartz M.A., Jackson S.W., Hudkins K.L., Liu C., Sather B.D., Khim S., Liggitt D., Song W. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J. Exp. Med. 2011;208:2033–2042. doi: 10.1084/jem.20110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbage M., Keppler S.J., Gasparrini F., Martínez-Martín N., Gaya M., Feest C., Domart M.-C., Brakebusch C., Collinson L., Bruckbauer A., Batista F.D. Cdc42 is a key regulator of B cell differentiation and is required for antiviral humoral immunity. J. Exp. Med. 2015;212:53–72. doi: 10.1084/jem.20141143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter R.H., Fearon D.T. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–107. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- Chaturvedi A., Martz R., Dorward D., Waisberg M., Pierce S.K. Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat. Immunol. 2011;12:1119–1126. doi: 10.1038/ni.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Trounstine M., Alt F.W., Young F., Kurahara C., Loring J.F., Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int. Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio C., Pannellini T., Lanzardo S., Forni G., Musiani P., Antón I.M. WIP null mice display a progressive immunological disorder that resembles Wiskott-Aldrich syndrome. J. Pathol. 2007;211:67–75. doi: 10.1002/path.2088. [DOI] [PubMed] [Google Scholar]
- de la Fuente M.A., Sasahara Y., Calamito M., Antón I.M., Elkhal A., Gallego M.D., Suresh K., Siminovitch K., Ochs H.D., Anderson K.C. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP) Proc. Natl. Acad. Sci. USA. 2007;104:926–931. doi: 10.1073/pnas.0610275104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depoil D., Fleire S., Treanor B.L., Weber M., Harwood N.E., Marchbank K.L., Tybulewicz V.L.J., Batista F.D. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat. Immunol. 2008;9:63–72. doi: 10.1038/ni1547. [DOI] [PubMed] [Google Scholar]
- Donnelly S.K., Weisswange I., Zettl M., Way M. WIP Provides an Essential Link between Nck and N-WASP during Arp2/3-Dependent Actin Polymerization. Current Biology. 2013;23:999–1006. doi: 10.1016/j.cub.2013.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P., Zhou L.J., Ord D.C., Sato S., Koller B., Tedder T.F. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Fleire S.J., Goldman J.P., Carrasco Y.R., Weber M., Bray D., Batista F.D. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- Freeman S.A., Jaumouille V., Choi K., Hsu B.E., Wong H.S., Abraham L., Graves M.L., Coombs D., Roskelley C.D., Das R. Toll-like receptor ligands sensitize B-cell receptor signalling by reducing actin-dependent spatial confinement of the receptor. Nature Communications. 2015;6:1–16. doi: 10.1038/ncomms7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried S., Matalon O., Noy E., Barda-Saad M. WIP: more than a WASp-interacting protein. J. Leukoc. Biol. 2014;96:713–727. doi: 10.1189/jlb.2RU0314-162R. [DOI] [PubMed] [Google Scholar]
- Fujimoto M., Poe J.C., Jansen P.J., Sato S., Tedder T.F. CD19 amplifies B lymphocyte signal transduction by regulating Src-family protein tyrosine kinase activation. J. Immunol. 1999;162:7088–7094. [PubMed] [Google Scholar]
- Gallego M.D., de la Fuente M.A., Anton I.M., Snapper S., Fuhlbrigge R., Geha R.S. WIP and WASP play complementary roles in T cell homing and chemotaxis to SDF-1alpha. Int. Immunol. 2006;18:221–232. doi: 10.1093/intimm/dxh310. [DOI] [PubMed] [Google Scholar]
- Hao S., August A. Actin depolymerization transduces the strength of B-cell receptor stimulation. Mol. Biol. Cell. 2005;16:2275–2284. doi: 10.1091/mbc.E04-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojer C., Frankenberger S., Strobl L.J., Feicht S., Djermanovic K., Jagdhuber F., Hömig-Hölzel C., Ferch U., Ruland J., Rajewsky K., Zimber-Strobl U. B-cell expansion and lymphomagenesis induced by chronic CD40 signaling is strictly dependent on CD19. Cancer Res. 2014;74:4318–4328. doi: 10.1158/0008-5472.CAN-13-3274. [DOI] [PubMed] [Google Scholar]
- Jellusova J., Miletic Ana V., Cato Matthew H., Lin W.-W., Hu Y., Bishop Gail A., Shlomchik Mark J., Rickert Robert C. Context-Specific BAFF-R Signaling by the NFκB and PI3K Pathways. Cell Reports. 2013;27:1022–1035. doi: 10.1016/j.celrep.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S., Depoil D., Martinelli R., Judokusumo E., Carmona G., Gertler F.B., Kam L.C., Carman C.V., Burkhardt J.K., Irvine D.J., Dustin M.L. Actin foci facilitate activation of the phospholipase C-γ in primary T lymphocytes via the WASP pathway. eLife. 2015;4:1–31. doi: 10.7554/eLife.04953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzi G., Moratto D., Vairo D., Masneri S., Delmonte O., Paganini T., Parolini S., Tabellini G., Mazza C., Savoldi G. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J. Exp. Med. 2012;209:29–34. doi: 10.1084/jem.20110896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Bai X., Wu J., Sharma S., Upadhyaya A., Dahlberg C.I.M., Westerberg L.S., Snapper S.B., Zhao X., Song W. N-wasp is essential for the negative regulation of B cell receptor signaling. PLoS Biol. 2013;11:e1001704. doi: 10.1371/journal.pbio.1001704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Quiles N., Rohatgi R., Antón I.M., Medina M., Saville S.P., Miki H., Yamaguchi H., Takenawa T., Hartwig J.H., Geha R.S., Ramesh N. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat. Cell Biol. 2001;3:484–491. doi: 10.1038/35074551. [DOI] [PubMed] [Google Scholar]
- Massaad M.J., Oyoshi M.K., Kane J., Koduru S., Alcaide P., Nakamura F., Ramesh N., Luscinskas F.W., Hartwig J., Geha R.S. Binding of WIP to actin is essential for T cell actin cytoskeleton integrity and tissue homing. Mol. Cell. Biol. 2014;34:4343–4354. doi: 10.1128/MCB.00533-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila P.K., Feest C., Depoil D., Treanor B., Montaner B., Otipoby K.L., Carter R., Justement L.B., Bruckbauer A., Batista F.D. The Actin and Tetraspanin Networks Organize Receptor Nanoclusters to Regulate B Cell Receptor-Mediated Signaling. Immunity. 2013;38:461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Moreau V., Frischknecht F., Reckmann I., Vincentelli R., Rabut G., Stewart D., Way M. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat. Cell Biol. 2000;2:441–448. doi: 10.1038/35017080. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Zhu J., Zikherman J., Parameswaran R., Kadlecek T.A., Wang Q., Au-Yeung B., Ploegh H., Kuriyan J., Das J., Weiss A. Monovalent and multivalent ligation of the B cell receptor exhibit differential dependence upon Syk and Src family kinases. Sci. Signal. 2013;6:ra1. doi: 10.1126/scisignal.2003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T., Ngo V.N., Ekland E.H., Förster R., Lipp M., Littman D.R., Cyster J.G. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J. Exp. Med. 2002;196:65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero D.C., Omori S.A., Rickert R.C. Cd19-dependent activation of Akt kinase in B-lymphocytes. J. Biol. Chem. 2001;276:1474–1478. doi: 10.1074/jbc.M003918200. [DOI] [PubMed] [Google Scholar]
- Patke A., Mecklenbräuker I., Erdjument-Bromage H., Tempst P., Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J. Exp. Med. 2006;203:2551–2562. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N., Antón I.M., Hartwig J.H., Geha R.S. WIP, a protein associated with wiskott-aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc. Natl. Acad. Sci. USA. 1997;94:14671–14676. doi: 10.1073/pnas.94.26.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recher M., Burns S.O., de la Fuente M.A., Volpi S., Dahlberg C., Walter J.E., Moffitt K., Mathew D., Honke N., Lang P.A. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. 2012;119:2819–2828. doi: 10.1182/blood-2011-09-379412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. doi: 10.1038/338383b0. [DOI] [PubMed] [Google Scholar]
- Rickert R.C., Rajewsky K., Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- Sasahara Y., Rachid R., Byrne M.J., de la Fuente M.A., Abraham R.T., Ramesh N., Geha R.S. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol. Cell. 2002;10:1269–1281. doi: 10.1016/s1097-2765(02)00728-1. [DOI] [PubMed] [Google Scholar]
- Schweighoffer E., Vanes L., Nys J., Cantrell D., McCleary S., Smithers N., Tybulewicz V.L. The BAFF Receptor Transduces Survival Signals by Co-opting the B Cell Receptor Signaling Pathway. Immunity. 2013;38:475–488. doi: 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper S.B., Rosen F.S., Mizoguchi E., Cohen P., Khan W., Liu C.H., Hagemann T.L., Kwan S.P., Ferrini R., Davidson L. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9:81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- Stewart D.M., Tian L., Nelson D.L. Mutations that cause the Wiskott-Aldrich syndrome impair the interaction of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein. J. Immunol. 1999;162:5019–5024. [PubMed] [Google Scholar]
- Takata M., Sabe H., Hata A., Inazu T., Homma Y., Nukada T., Yamamura H., Kurosaki T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 1994;13:1341–1349. doi: 10.1002/j.1460-2075.1994.tb06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrasher, A.J. (2009). New insights into the biology of Wiskott-Aldrich syndrome (WAS). Hematology: the Education Program of the American Society of Hematology. 132–138. [DOI] [PubMed]
- Treanor B., Depoil D., Gonzalez-Granja A., Barral P., Weber M., Dushek O., Bruckbauer A., Batista F.D. The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity. 2010;32:187–199. doi: 10.1016/j.immuni.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakumar V., Monypenny J., Chen X.J., Machesky L.M., Lilla S., Thrasher A.J., Antón I.M., Calle Y., Jones G.E. Tyrosine phosphorylation of WIP releases bound WASP and impairs podosome assembly in macrophages. J. Cell Sci. 2015;128:251–265. doi: 10.1242/jcs.154880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A., Littman D.R. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Westerberg L.S., Dahlberg C., Baptista M., Moran C.J., Detre C., Keszei M., Eston M.A., Alt F.W., Terhorst C., Notarangelo L.D., Snapper S.B. Wiskott-Aldrich syndrome protein (WASP) and N-WASP are critical for peripheral B-cell development and function. Blood. 2012;119:3966–3974. doi: 10.1182/blood-2010-09-308197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.