

Abstract
Epratuzumab has demonstrated therapeutic activity in patients with non-Hodgkin lymphoma, acute lymphoblastic leukemia, systemic lupus erythematosus, and Sjögren's syndrome, but its mechanism of affecting normal and malignant B cells remains incompletely understood. We reported previously that epratuzumab displayed in vitro cytotoxicity to CD22-expressing Burkitt lymphoma cell lines (Daudi and Ramos) only when immobilized on plates or combined with a crosslinking antibody plus a suboptimal amount of anti-IgM (1 μg/mL). Herein, we show that, in the absence of additional anti-IgM ligation, extensive crosslinking of CD22 by plate-immobilized epratuzumab induced intracellular changes in Daudi cells similar to ligating B-cell antigen receptor with a sufficiently high amount of anti-IgM (10 μg/mL). Specifically, either treatment led to phosphorylation of CD22, CD79a and CD79b, along with their translocation to lipid rafts, both of which were essential for effecting caspase-dependent apoptosis. Moreover, such immobilization induced stabilization of F-actin, phosphorylation of Lyn, ERKs and JNKs, generation of reactive oxygen species (ROS), decrease in mitochondria membrane potential (Δψm), upregulation of pro-apoptotic Bax, and downregulation of anti-apoptotic Bcl-xl and Mcl-1. The physiological relevance of immobilized epratuzumab was implicated by noting that several of its in vitro effects, including apoptosis, drop in Δψm, and generation of ROS, could be observed with soluble epratuzumab in Daudi cells co-cultivated with human umbilical vein endothelial cells. These results suggest that the in vivo mechanism of non-ligand-blocking epratuzumab may, in part, involve the unmasking of CD22 to facilitate the trans-interaction of B cells with vascular endothelium.
Keywords: BCR, CD22, epratuzumab, human B-cell lymphoma, HUV-EC, immobilized
Abbreviations
- Anti-IgM
F(ab’)2 fragment of affinity-purified goat anti-human IgM, Fc5μ fragment
- BCR
B-cell antigen receptor
- BSA
bovine serum albumin
- CM-H2DCF-DA
2′,7′-dichlorodihydrofluorescein diacetate
- Δψm
mitochondria membrane potential
- DNP
2,4-dinitrophenyl
- EC
endothelial cells
- ERKs
extracellular signal-regulated kinases
- FBS
fetal bovine serum
- FITC-DNase I
fluorescein isothiocyanate-conjugated DNase I
- 488-annexin V
Alexa Fluor 488-conjugated annexin V
- GAH
F(ab′)2 fragment of affinity-purified goat anti-human IgG Fcγ fragment-specific
- HUV-EC
human umbilical vein endothelial cells
- ITIM
immunoreceptor tyrosine-based inhibition motif
- JNKs
c-Jun N-terminal kinases
- JP
jasplakinolide
- LatB
latrunculin B
- Lyn
Lck/Yes novel tyrosine kinase
- MAP kinases
mitogen-activated protein kinases
- mIgM
membrane IgM
- MTS
(3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PLCγ2
phospholipase C, isotype gamma 2
- Rhodamine-anti-IgG
rhodamine-conjugated F(ab′)2 fragment of affinity-purified goat anti-human IgG, F(ab′)2 fragment-specific
- ROS
reactive oxygen species
- 7-AAD
7-aminoactinomycin D
- Syk
spleen tyrosine kinase
- TMRE
/tetramethylrhodamine/ethyl ester
Introduction
Epratuzumab,1 a humanized monoclonal antibody specific for human CD22 (hCD22), is currently under clinical investigation for the treatment of non-Hodgkin lymphoma (NHL),2,3 pediatric and adult acute lymphoblastic leukemia,4,5 and systemic lupus erythematosus (SLE),6-8 and it has shown promise in patients with primary Sjögren's syndrome in a Phase 1/2 study.9 CD22, also known as Siglec-2,10 is a type-I transmembrane glycoprotein whose expression in humans can be detected in the cytoplasm of pro-B and pre-B cells, as well as on the surface of IgM+ B cells upon the appearance of IgD, but not on terminally-differentiated plasma cells.11 Structurally, CD22 in its extracellular region comprises 7 immunoglobulin-like domains, of which the 2 N-terminal domains are involved in ligand binding.12 The cytoplasmic tail of CD22 contains 6 conserved tyrosine (Y) residues,13 4 of which (Y762, Y796, Y822, and Y842) in hCD22 are considered to be localized within the immunoreceptor tyrosine-based inhibition motifs (ITIM).14,15 These tyrosine residues also fit in the specific internalization motifs of YXXø (where ø denotes a hydrophobic residue),16 which mediate the recruitment of CD22 to clathrin-coated pits and are required for the internalization of CD22.17 Whereas endocytosed CD22 has been documented to be degraded intracellularly,18 a recent report contended that CD22 instead is constitutively recycled back to the cell surface.19 Functionally, CD22 recognizes α2,6-linked sialic acids on glycoconjugates in both cis (on the same cell)20 and trans (on different cells)21 interactions, and modulates B cells via interaction with CD79a and CD79b, the signaling components of the BCR complex.22 Crosslinking the BCR with cognate antigens or appropriate antibodies against membrane immunoglobulin (mIg) on the cell surface induces translocation of the aggregated BCR complex to lipid rafts,23 where CD79a, CD79b and CD22, among others, are phosphorylated by Lyn,24 which in turn triggers various downstream signaling pathways, culminating in proliferation, survival, or death.25 Research on anti-CD22 antibodies, which can be either blocking or nonblocking,26 has also led to intriguing observations that CD22 may positively26-28 or negatively29 affect BCR-mediated signaling pathways, with the ultimate outcome depending on the characteristics of the B cells (differentiation stage, expression of BCR isotype, and being malignant, abnormal, or normal). Thus, fully understanding the role of CD22 in B-cell malignancies, as well as B-cell-implicated autoimmune diseases, is of considerable importance for improving CD22-targeted therapies.
As a single agent, epratuzumab was well-tolerated, depleting circulating B cells transiently in NHL patients,3 and by an average of 35% at 18 weeks in SLE patients.30 When evaluated in vitro, epratuzumab displayed modest antibody-dependent cellular cytotoxicity, but no complement-dependent cytotoxicity,31 and was shown to inhibit the proliferation of B cells from patients with SLE, but not from normal donors, under all culture conditions.32 Additional studies with B cells from SLE patients indicated that (i) binding of epratuzumab was particularly enhanced on CD27− compared to CD27+ B cells;33 (ii) a decrease of CD62L and β7 integrin and an increase of β1 integrin in the cell surface expression, as well as an enhanced migration toward CXCL12, were noted for CD27− B cells preferentially;33 and (iii) the in vivo effects of epratuzumab in SLE patients included an immediate and sustained (up to 18 weeks) decrease in CD22 surface expression on circulating CD27− and CD27+ B cells.33 Pre-incubation with F(ab′)2 of epratuzumab was recently reported to inhibit calcium mobilization and phosphorylation of Syk and PLCγ2 in normal human B cells after BCR stimulation,34 and the ability of epratuzumab-based agents to effectively mediate Fc-dependent trogocytosis of multiple B-cell surface markers by FcγR-bearing cells, was established.35,36
In cell lines and xenografts of human Burkitt lymphoma, soluble epratuzumab, although capable of phosphorylating CD2237 and translocating CD22 to lipid rafts,38 as demonstrated in vitro, was not cytotoxic or cytostatic,31,38 and displayed only minimal toxicity even when crosslinked by goat anti-human IgG Fcγ (GAH).31 On the other hand, in vitro cytotoxicity of epratuzumab comparable to that achievable with anti-IgM (10 μg/mL) could be consistently demonstrated in Ramos and D1–1, a subclone of Daudi selected for a high expression of membrane IgM (mIgM), when the antibody was immobilized to plastic plates, or added in combination with suboptimal amounts of anti-IgM (for example, 1 μg/mL or less) along with GAH.31 Despite all of this knowledge, how epratuzumab kills or modulates normal and malignant B cells in patients, and inhibits the growth of lymphoma lines in vitro upon immobilization, remains poorly understood.
In this study, we further explored the molecular events associated with the cytotoxicity of epratuzumab in vitro, examining epratuzumab presented in soluble or immobilized form in various combinations, as summarized in Table 1. We show in Daudi and D1–1 cells that epratuzumab by non-covalent adsorption on microtiter plates (the Dried-I format, Table 1) induces phosphorylation of CD22, CD79a and CD79b, as well as their translocation to lipid rafts, which are instrumental for cell death via caspase-dependent apoptosis. Such plate-immobilized epratuzumab likewise induced substantial apoptosis and growth inhibition in Ramos cells. Treatment of D1–1 cells with the Dried-I format also revealed multiple intracellular changes that included sustained phosphorylation of ERK and JNK MAP kinases, decrease in Δψm, generation of ROS, activation of caspases, as well as upregulation and downregulation of pro- and anti-apoptotic proteins, respectively. Interestingly, several of the in vitro effects observed with the Dried-I format, including apoptosis, drop in Δψm, and generation of ROS, could be observed with the Dried-II format (Table 1) comprising co-cultivation of Daudi cells over a monolayer of human umbilical vein endothelial cells (HUV-EC) in the presence of soluble epratuzumab (20 μg/mL). These findings indicate a physiological relevance of immobilized epratuzumab, and suggest that the in vivo mechanism of non-ligand-blocking epratuzumab may, in part, involve the unmasking of CD22 to facilitate the trans-interaction of B cells with vascular endothelium.
Table 1.
Invitro conditions to evaluate the cytotoxicity of epratuzumab against CD22-expressing B cells
| Conditions | Format |
|---|---|
| Target cells + epratuzumab IgG immobilized onto microtiter wells overnight | Dried-I |
| Target cells + labetuzumab IgG immobilized onto microtiter wells overnight | Isotype control for Dried-I |
| Target cells + epratuzumab IgG over a monolayer of HUV-EC | Dried-II |
| Target cells + labetuzumab IgG over a monolayer of HUV-EC | Isotype control for Dried-II |
| Target cells + epratuzumab IgG or F(ab’)2 in solution | Wet-I |
| Target cells + epratuzumab IgG + GAH in solution | Wet-IIA |
| Target cells + epratuzumab IgG + anti-IgM (1 μg/mL) in solution | Wet-IIB |
| Target cells + epratuzumab IgG + GAH + anti-IgM (1 μg/mL) in solution | Wet-III |
| Target cells + epratuzumab IgG conjugated to polystyrene beads | Particulate-I |
| Target cells + epratuzumab IgG bound to Protein A-Sepharose | Particulate-II |
| Target cells + anti-IgM (10 μg/mL) in solution | Positive control |
Results
Inhibition of proliferation and induction of apoptosis
To evaluate the effect on cell proliferation, varying amounts of epratuzumab were coated on non-tissue-culture, U-bottom plates, and the results of the MTS cell viability assay indicate that at 5 μg/mL, immobilized epratuzumab of the Dried-I format could inhibit about 60% proliferation of D1–1 cells compared to untreated cells (P < 0.005), with little change found at higher concentrations of 10 and 20 μg/mL (Fig. 1A). In Ramos cells, which express a lower level of CD22 than D1–1, epratuzumab achieved about 45% growth-inhibition when coated at 10 μg/mL compared to untreated cells (P < 0.005). Immobilized labetuzumab (anti-CEACAM5), serving as an isotype control of the Dried-I format, did not induce appreciable growth-inhibition in either cell line (Fig. 1A). Soluble epratuzumab (the Wet-I format), even at the highest concentration (20 μg/mL) tested, did not induce growth-inhibition in both cell lines (Fig. 1B), indicating the requirement for immobilization.
Figure 1.
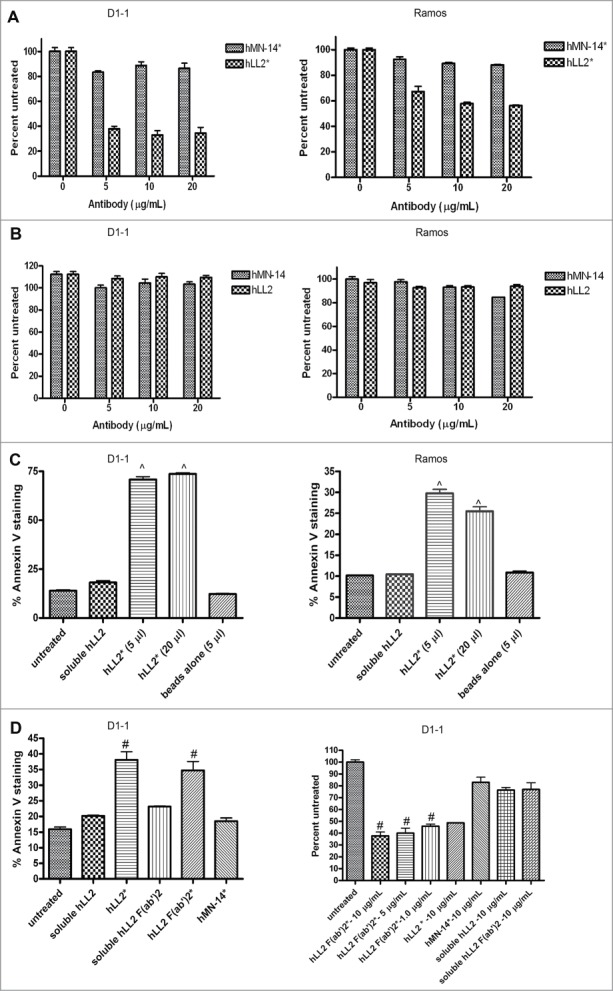
Evaluation of growth-inhibition and apoptosis in D1–1 and Ramos cells. Cell viability determined by the MTS assay after 4-day incubation for (A) the Dried-I format of epratuzumab (hLL2*) or labetuzumab (hMN-14*) and (B) the Wet-I format of epratuzumab (hLL2) or labetuzumab (hMN-14). Apoptosis as determine by Annexin V staining (C) following the indicated treatments of D1–1 and Ramos cells for 24 and 48 h, respectively. (D) Plate-immobilized F(ab’)2 of epratuzumab (hLL2 F(ab’)2*) effectively induced apoptosis (left panel) and inhibited proliferation (right panel) in D1–1 cells as determined by the annexin V assay at 24 h and the MTS assay after 4 days, respectively. Error bars represent standard deviation (SD), where n = 3. Significant differences compared to untreated or nonspecific antibody are indicated with ^ (P < 0.005) and # (P < 0.05).
Evidence that immobilization of epratuzumab was required to induce apoptosis was provided by the Particulate-I format (Table 1) of bead-conjugated epratuzumab (Fig. 1C), which, at both 5- and 20-μL doses, caused about 75% apoptosis in D1–1 cells following a 24-h incubation, as compared to approximately 20% (P < 0.005) for the 3 controls (cells with no treatment, cells treated with soluble epratuzumab, and cells treated with unconjugated beads). The same particulate epratuzumab also resulted in about 30% apoptosis in Ramos cells, which was significant (P < 0.005) compared with the 3 controls (10% apoptosis). Similar results were obtained with the Dried-I format of epratuzumab F(ab’)2 in D1–1 cells, as shown in Figure 1D for apoptosis (left panel; P < 0.05 vs. controls) and growth inhibition (right panel; P < 0.025 vs. controls), indicating a lack of Fc involvement in the cytotoxicity of plate-immobilized epratuzumab.
Further experiments in Daudi cells demonstrated that the in vitro cytotoxicity of epratuzumab, as determined by the MTS assay, could be observed dose-dependently with the Dried-I or the Wet-III format (Fig. 2A, right panel), but not with the Wet-I or the Wet-IIB format (Fig. 2A, left panel), and confirmed that the Dried-I format induced apoptosis comparable to the positive control of anti-IgM as determined by the Annexin V assay (Fig. 2B). More importantly, we have discovered that the Dried-II format, which employed plates coated with a monolayer of HUV-EC, was capable of inducing apoptosis in Daudi cells in the presence of soluble epratuzumab to a similar extent (∼50%), when compared with the Dried-I format (Fig. 2C).
Figure 2.
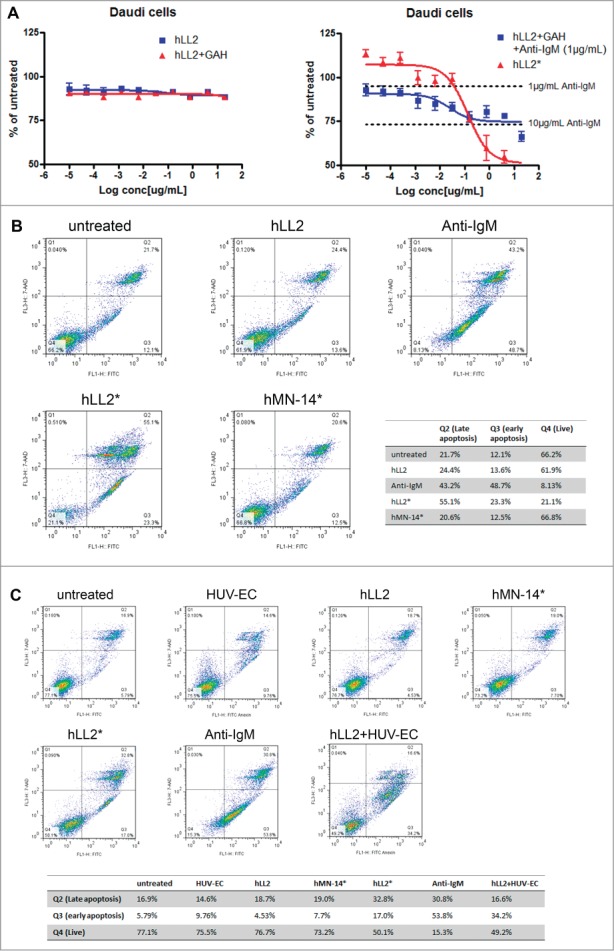
Cytotoxicity of epratuzumab in various formats to Daudi cells. (A) Epratuzumab presented as the Dried-I (hLL2*) or Wet-III (hLL2 + GAH + anti-IgM) format (right panel), but not the Wet-I (hLL2) or Wet-IIB (hLL2 + GAH) format (left panel), induced dose-dependent cytotoxicity in Daudi cells, as measured by the MTS assay. (B) The Dried-I format of epratuzumab (hLL2*) induced apoptosis comparable to the positive control (anti-IgM) as determined by the Annexin V assay. (C) The Dried-I format (hLL2*) and the Dried-II format (hLL2 + HUV-EC), in which soluble epratuzumab was added to a monolayer of HUV-EC, induced apoptosis in Daudi cells to a similar extent (∼50%).
Phosphorylation of CD22, CD79a and CD79b
To elucidate the differential effect induced on D1–1 or Ramos cells by soluble (in various Wet-based formats) and immobilized (the Dried-I format) epratuzumab, we evaluated their roles in phosphorylating CD22, CD79a, and CD79b, and compared the results with those of anti-IgM. As shown in Fig. 3A (left panel) for D1–1 cells, soluble anti-IgM at 10 μg/mL induced phosphorylation of CD22, CD79a and CD79b, while soluble epratuzumab (lane: hLL2/Wet-I) induced notable phosphorylation of CD22 and some CD79b, but not CD79a. In contrast, Fig. 3A (right panel) shows immobilized epratuzumab (lane: hLL2*/Dried-I), and immobilized anti-IgM (lane: anti-IgM*) as well, induced phosphorylation of CD22, CD79a and CD79b to a similar extent. However, whereas the Wet-III format of epratuzumab (Fig. 3B, lane 7), comprising a mixture of epratuzumab (7.5 μg/mL), GAH (10 μg/mL) and anti-IgM (1 μg/mL), induced the phosphorylation of CD22, CD79a, and CD79b as soluble anti-IgM at 10 μg/mL (Fig. 3B, lane 8), omitting one or 2 components from the Wet-III format (Fig. 3B, lanes 2–5), or the provision of only a very small amount of epratuzumab (10 ng/mL) to GAH and anti-IgM (Fig. 3B, lane 6), failed to induce phosphorylation of all 3 molcules. These results correlate the observed cytotoxicity of anti-IgM (10 μg/mL) and epratuzumab presented in the Dried-I or Wet-III format with their ability to simultaneously phosphorylate CD22, CD79a, and CD79b in target cells.
Figure 3.
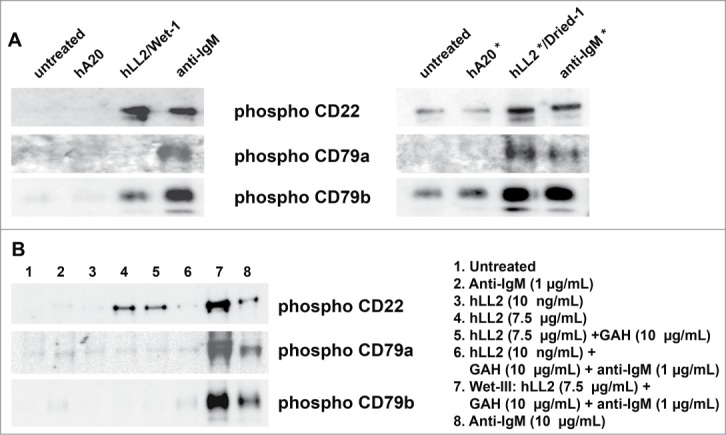
Phosphorylation of CD79a, CD79b, and CD22. Western blot analyses of phosphorylated CD79a, CD79b, and CD22 in D1–1 cells treated for 2 h with (A) the Wet-I format of soluble antibodies (left panel) or the Dried-I format of immobilized antibodies (right panel), and (B) various formats of soluble epratuzumab, including Wet-I (lane 4; hLL2, 7.5 μg/mL), Wet-IIA (lane 5; hLL2, 7.5 μg/mL; GAH, 10 μg/mL), and Wet-III (lane 7; hLL2, 7.5 μg/mL; GAH, 10 μg/mL; anti-IgM, 1 μg/mL). In lane 6, the amounts of GAH and anti-IgM were the same as those in lane 7, but the concentration of epratuzumab was too low (10 ng/mL) to induce a notable effect.
Translocation of CD22 and CD79 to lipid rafts
Treatment of Daudi cells with anti-IgM (10 μg/mL) or epratuzumab either in the Dried-I format (Fig. 4A, left panel; sample: hLL2*) or the Wet-I format (Fig. 4A, left panel; sample: hLL2) all resulted in the redistribution of CD22 to the lipid rafts (fractions 3–6, Fig. S1). However, redistribution of CD79b to lipid rafts (Fig. 4A, right panel) was observed only with anti-IgM or the Dried-I format (sample: hLL2*), but not with the Wet-I format (sample: hLL2). Additional experiments also revealed that only CD22, not CD79a or CD79b, could be detected in lipid rafts from cells treated with soluble epratuzumab either in the Wet-I (Fig. 4B, lane 2) or the Wet-IIA format (Fig. 4B, lane 5). These results confirm the ability of soluble epratuzumab (the Wet-I format) to stabilize the localization of CD22 in lipid rafts,38 and suggest that the cytotoxicity of epratuzumab requires the concurrent translocation of CD22, CD79a, and CD79b to lipid rafts.
Figure 4.
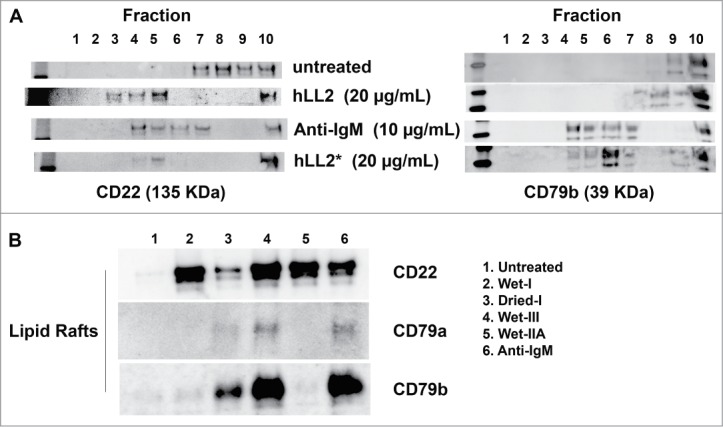
Translocation of CD79a, CD79b, and CD22 to the lipid rafts. (A) left panel, CD22 was detected in the lipid rafts (fractions 3–6) following treatment of D1–1 cells with anti-IgM (10 μg/mL) and both the Wet-I (hLL2, 20 μg/mL) and Dried-I (hLL2*, 20 μg/mL) formats of epratuzumab; right panel, CD79b was translocated to the lipid rafts (fractions 4–7) by either anti-IgM (10 μg/mL) or the Dried-I format of epratuzumab (hLL2*, 20 μg/mL), but not soluble epratuzumab (hLL2). (B) Anti-IgM (lane 6) and epratuzumab of the Dried-I (lane 3) or Wet-III (lane 4) format, but not the Wet-I (lane 2) or Wet-IIA (lane 5) format, induced redistribution of CD22, CD79a, and CD79b to the lipid rafts.
Activation of BCR-mediated signals and modulation of MAP kinases
The Dried-I format of epratuzumab induced in D1–1 cells rapid and prolonged phosphorylation of Lyn, Syk, and PLCγ2, as shown in Figure 5A. Changes of intracellular signals induced by the Dried-I format also included a rapid (detectable within 30 min) and continuous (over a period of 24 h) activation of both ERKs and JNKs (Fig. 5B). A functional role of JNK was established by showing SP600125, a known inhibitor of JNK, given at low doses (2.5 and 5 nM) to D1–1 cells 2 h before treatment with plate-immobilized epratuzumab, could effectively prevent apoptosis when determined at 24 h (Fig. 5C).
Figure 5.
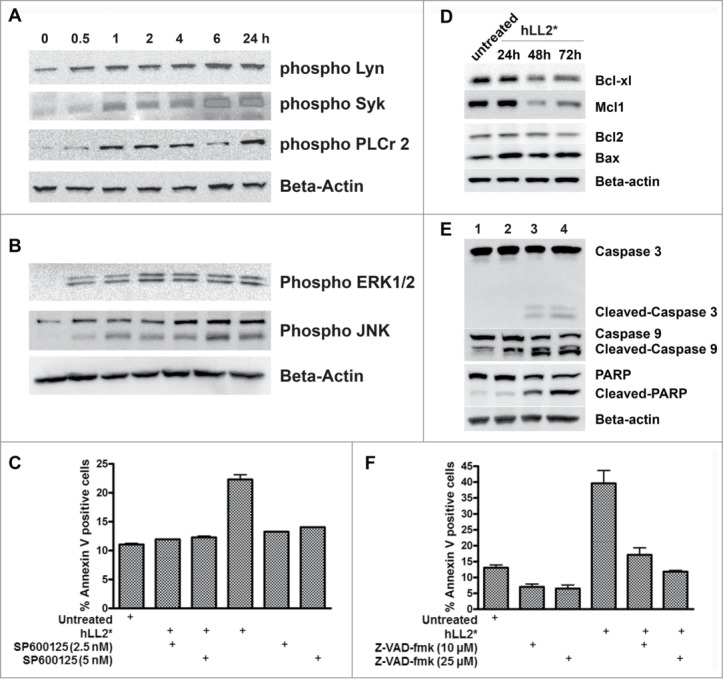
Phosphorylation of BCR-mediated signals, modulation of MAP kinases, and evidence of caspase-dependent apoptosis. (A) D1–1 cells were incubated with the Dried-I format of epratuzumab for the indicated times and cell lysates probed for phosphorylated Lyn, Syk, or PLCγ2. (B) The cell lysates of D1–1 cells obtained as described in (A) were probed for phosphorylated ERKs and JNK. (C) SP600125, a chemical inhibitor for JNK, protected D1–1 cells from apoptosis induced by plate-immobilized epratuzumab (hLL2*). (D) Western blot analysis of selective anti- and pro-apoptotic proteins following treatment of D1–1 cells with plate-immobilized epratuzumab (hLL2*) for 24, 48 and 72 h. The untreated sample at 72 h is shown. (E) Plate-immobilized epratuzumab (hLL2*; lanes 2–4) induced cleavages of caspase 3, caspase 9 and PARP, which were evident at 48 (lane 3) and 72 h (lane 4). The untreated sample at 72 h is shown (lane 1). (F) Z-VAD-fmk inhibited apoptosis in D1–1 cells induced by plate-immobilized epratuzumab (hLL2*).
Caspase-mediated apoptosis
The effect of the Dried-I format on the basal levels of selective pro-apoptotic and anti-apoptotic proteins, was evaluated in D1–1 cells following treatment for 24, 48 and 72 h. As shown in Figure 5D, the Dried-I format (sample: hLL2*) downregulated anti-apoptotic Bcl-xL and Mcl-1, while increasing the expression level of pro-apoptotic Bax; however, the results pertaining to Bcl-2 were less certain. The observed cleavage of caspase 3, caspase 9 and poly ADP ribose polymerase (PARP), as shown in Figure 5E, indicates the Dried-I format orchestrates a caspase-dependent apoptosis in D1–1 cells, which could be reduced from about 40% to a level similar to the untreated cells (about 15%) by the pan-caspase inhibitor, Z-VAD-fmk, at 10 or 25 μM (P < 0.02), as shown in Figure 5F. It is noted that untreated controls shown in Fig. 5D and E were taken at the 72-h time-point, and there was no change in untreated samples when examined at either 24 h or 48 h.
Decrease in Δψm and generation of ROS
In Figure 6A, the Dried-I format (subpanel: hLL2*) was shown to induce mitochondrial membrane depolarization, manifested as a decrease in Δψm, in about 45% of D1–1 cells, whereas no more than 20% of cells with comparable changes could be detected in the 5 controls. Similar results were observed for the Dried-I format in Ramos cells (data not shown) and in about 60% of Daudi cells treated with the Dried-I format (Fig. 6B, subpanel: hLL2*) or the Dried-II format (Fig. 6B, subpanel: HUV-EC/hLL2), which replaced plate-immobilized epratuzumab with soluble epratuzumab and plate-coated HEV-EC. To corroborate such findings, both the Dried-I (Fig. 6C, subpanel: hLL2*) and the Dried-II (Fig. 6C. subpanel: HUV-EC/hLL2) formats increased the generation of ROS in about 30% of the cells, as compared to about 6% in the untreated control, and about 9 to 10% in cells incubated with the Wet-I format (Fig. 6C, subpanel: hLL2), isotype control of the Dried-I format (Fig. 6C, subpanel: hMN-14*) or HEV-EC in the absence of epratuzumab (Fig. 6C, subpanel: HUV-EC).
Figure 6.

Effect on Δψm and ROS. (A) Treatment of D1–1 cells with the Dried-I format of epratuzumab (hLL2*) induced a decrease in Δψm. (B) Treatment of Daudi cells with the Dried-II format (HUV-EC/hLL2) also induced a decrease in Δψm, similar to that observed with the Dried-I format (hLL2*). (C) Both the Dried-I (hLL2*) and the Dried-II (HUV-EC/hLL2) formats of epratuzumab increased the generation of ROS in Daudi cells.
Effect on calcium mobilization and actin dynamics
In Daudi cells, pretreatment with either the Wet-I or the Dried-I format for 1 h notably reduced the amplitude of calcium ions released from intracellular stores following stimulation with anti-IgM, with a larger effect incurred by the plate-immobilized than the soluble epratuzumab; however, the subsequent entry of extracellular calcium was minimally affected (Fig. 7A). The ligation of CD22 by plate-immobilized epratuzumab appeared to stabilize F-actin from depolymerization by LatB, when analyzed at 5 min after the addition of LatB, as evidenced by the prominent staining of F-actin by rhodamin phalloidin, which was absent in the untreated Daudi cells, as shown in Figure 7B. Additional results shown in Figure 7C indicate that the addition of LatB did not affect the staining of F-actin in cells pretreated with hLL2*, but demolished the staining of F-actin in cells treated with soluble epratuzumab or the isotype control of the Dried-I format (hMN-14*).
Figure 7.

Decrease in intracellular calcium release and perturbation of actin dynamics. (A) Pretreatment of Daudi cells with the Wet-I (hLL2) or the Dried-I (hLL2*) format of epratuzumab for 1 h reduced the mobilization of intracellular calcium ions following stimulation with anti-IgM, but did not affect the subsequent entry of extracellular calcium. (B) The ligation of CD22 by plate-immobilized epratuzumab (hLL2*) stabilized the F-actin from depolymerization by LatB. (C) Before the addition of LatB (left panel), F-actin was visualized by staining with rhodamine phalloidin in untreated Daudi cells, as well as in cells pretreated with the Dried-I format of epratuzumab (hLL2*), the isotype control of the Dried-I format (hMN-14*), or the Wet-I format of epratuzumab (hLL2). The addition of LatB (right panel) did not affect the staining of F-actin in cells pretreated with hLL2*, but demolished the staining of F-actin in the other 3.
Discussion
In the present study, we confirmed that ligation of mIgM by a sufficient amount of anti-IgM (10 μg/mL) induced the phosphorylation of CD22, CD79a and CD79b, and the localization of all 3 phosphorylated proteins in lipid rafts, leading to cell death in D1–1, a subline of Daudi selected for a higher expression of mIgM. We further show that ligation of CD22 with plate-immobilized epratuzumab (the Dried-I format) induced a similar change in CD22, CD79a and CD79b, including phosphorylation, translocation into lipid rafts, and subsequent cell death. Thus, it appears that for a CD22-binding agent such as epratuzumab to kill Daudi cells in particular, and perhaps other CD22-expressing B-cell lymphomas, 2 critical events must occur in concert, (i) phosphorylation of CD22, CD79a and CD79b above a threshold level, and (ii) their movement to lipid rafts. This conclusion is supported by the finding that little or no cell death was observed for D1–1 cells with the Wet-II format comprising a secondary crosslinking GAH antibody at 10 μg/mL and either soluble epratuzumab at 7.5 μg/mL or a suboptimal amount of anti-IgM (1 μg/mL). The former treatment efficiently induced phosphorylation of CD22 (Fig. 3B, lane 5) and its localization to lipid rafts (Fig. 4B, lane 5), but was unable to phosphorylate CD79a and CD79b (Fig. 3B, lane 5), whereas the latter treatment failed to phosphorylate CD22, CD79a and CD79b to a detectable level (Fig. 3B, lane 6). On the other hand, combining these 2 treatments in the Wet-III format could result in phosphorylation of CD22, CD79a and CD79b (Fig. 3B, lane 7), their localization into lipid rafts (Fig. 4B, lane 4), and consequently, cell death, as observed for anti-IgM at 10 μg/mL or the Dried-I format of epratuzumab.
Binding of CD22 to beads coated with B3 antibody (a murine anti-hCD22 mAb) was reported to lower the threshold concentration of anti-IgM required for stimulating DNA synthesis in tonsillar B cells by 2 orders of magnitude, presumably due to sequestration of CD22 from mIgM by restricting the lateral movement of CD22 in the plane of the cell membrane.39 Our results show, however, that the ability of high-density epratuzumab, presented in the Dried-I or Particulate-I format, to engage CD22 along with co-clustering, rather than sequestration, of mIgM, constitutes a sufficient condition for cell killing in the total absence of anti-IgM, which may be further strengthened by the co-localization of both mIgM and CD22 in lipid rafts. Moreover, the binding of immobilized epratuzumab to CD22 is distinctive from that of a synthetic α2,6-linked sialic acid, which efficiently prevented CD22 from co-capping and co-localization with BCR in the lipid rafts after BCR ligation.40
Intriguingly, we did not observe any transient increase in intracellular calcium by immobilized epratuzumab in the Particulate-I format, but have noted a substantial decrease of anti-IgM-induced mobilization of intracellular calcium in Daudi cells pretreated with either the Dried-I or the Wet-I format of epratuzumab for 1 h. These results are consistent with 2 previous findings: one reporting that a copolymer comprising multiple copies of 2,4-dinitrophenyl (DNP) and a synthetic CD22 ligand (CD22L), which was capable of trans-binding to CD22 via colligation with BCR in a murine B cell line displaying a DNP-specific BCR, failed to induce any calcium flux;41 the other reporting that preincubating B cells with the IgG or F(ab′)2 of epratuzumab reduced the amplitude of calcium mobilization stimulated by anti-IgM/IgG.34 Thus, when both CD22 and BCR are co-clustered by immobilized epratuzumab or the DNP-CD22L copolymer, calcium signals resulting from BCR stimulation can be partially or completely suppressed, which is in contrast to the enhanced calcium flux found in B cells pretreated with certain anti-CD22 antibodies upon BCR activation, often attributed to sequestration of CD22 from BCR.42,43 Although certain intracellular events observed for the Dried-I format of epratuzumab and the DNP-CD22L copolymer were similar, such as a more sustained phosphorylation of CD22 and Lyn, differences in their opposing effect on pSyk and pPLCγ2 also should be noted.
Whereas the ability of an anti-mIg to induce calcium flux may or may not lead to cell death in B-cell lymphoma, as reported for B104,44 a human B-cell lymphoma line expressing BCR of both mIgM and mIgD, we show with the Dried-I format of immobilized epratuzumab that potent inhibition of cell proliferation with apoptosis also can be independent of calcium mobilization. Thus, besides the routine measurement of intracellular calcium as a marker for B-cell activation and cell surface binding to assess the affinity for CD22, the biological significance of CD22-targeting agents, particularly those derived from synthetic sialosides,40,41,45-47 should be substantiated with a suitable cytotoxicity assay, as exemplified by the capability of liposomal nanoparticles displaying both antigen and CD22L to induce antigen-specific B-cell apoptosis.48
Despite their difference in calcium mobilization, resemblances of anti-IgM and immobilized epratuzumab were revealed in several intracellular events, including caspase-dependent apoptosis, reduced Δψm, generation of ROS, and a similar profile of phosphorylated Lyn, Syk, PLCγ2, ERKs and JNKs. Moreover, the novel observation that some of the in vitro effects displayed by the Dried-I format of immobilized epratuzumab, including apoptosis, drop in Δψm, and generation of ROS, could be induced by co-cultivation of Daudi cells with HUV-EC in the presence of soluble epratuzumab (the Dried-II format) implies a physiological relevance of immobilized epratuzumab. Collectively, the present data provoke a hypothesis that the non-ligand-blocking epratuzumab may act in vivo by unmasking CD22 to facilitate the trans-interaction of B cells with vascular endothelium (Fig. 8A), thereby inducing the various in vitro effects of immobilized epratuzumab. Noting that cytokine-activated human endothelial cells (EC) express enhanced levels of CD22L49 and can adhere to B cells whose endogenous binding of CD22 to CD22L have been disrupted,50 one scenario, as depicted in Fig. 8B, would be the immobilization of the immune complexes comprising epratuzumab and B cells to the endothelium via the association of CD22 on B cells with the CD22L-containing glycoproteins on EC. Because human EC also express CD32A (FcγRIIA),51,52 an alternative explanation (Fig. 8C) would be the immobilization of the epratuzumab-B cell complex to the endothelium via the association of the Fc domain on epratuzumab with CD32A on EC. These 2 possibilities are not mutually exclusive, with both likely occurring in vivo, and provide a plausible mechanism mediated by epratuzumab that enables the strong interaction between B cells and EC due to concurrent engagement of multiple cell surface molecules present on both types of cells.
Figure 8.
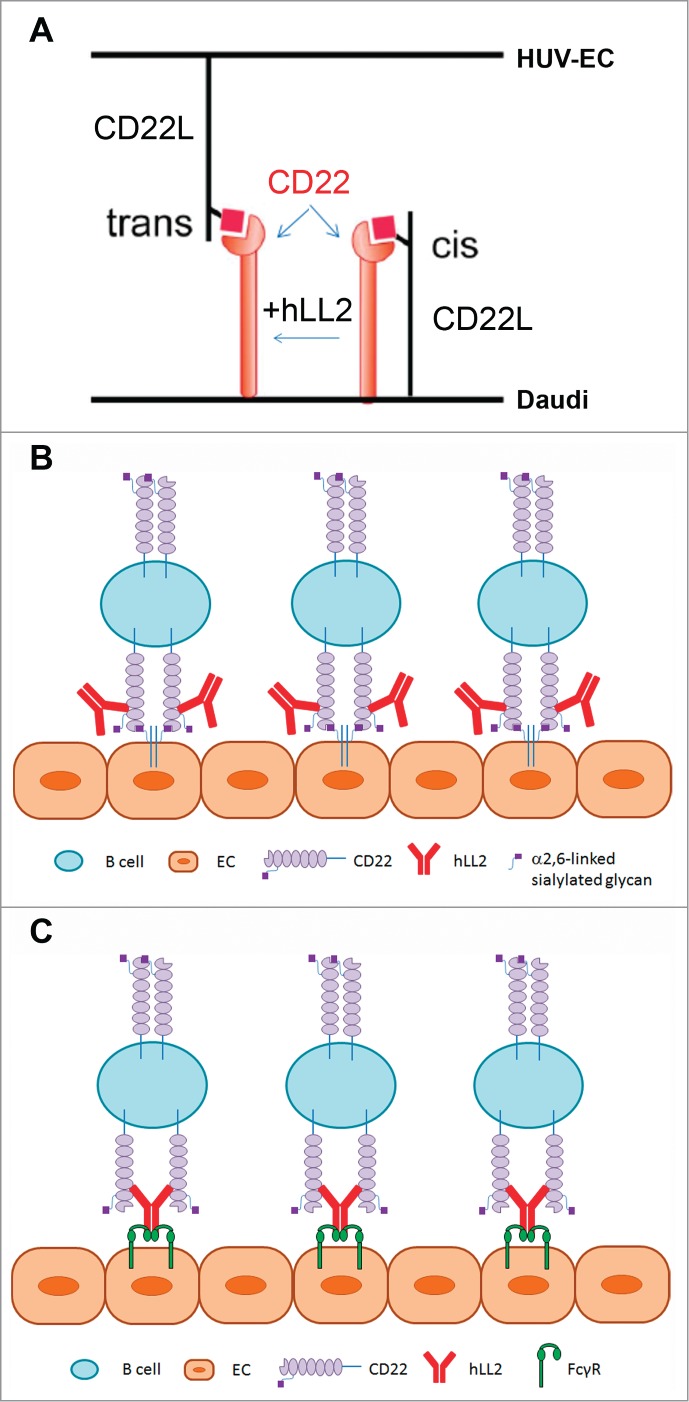
Schematics of proposed mechanism of epratuzumab-mediated interaction of endothelial cells with CD22-expressing B cells. (A) CD22 can interact with CD22L (sialylated glycoproteins) on the same (cis) or different cells (trans). To induce the trans interaction, it is necessary to overcome the cis interaction, which may be provided by non-ligand-blocking epratuzumab. Because Daudi cells have a high levels of CD22L, the binding of CD22 to (activated) endothelial cells are inhibited by cis-binding. The ligation of epratuzumab to CD22 is likely to break up the cis-interaction, and because it is not a blocking antibody, epratuzumab may not interfere with the further binding of CD22 to the CD22L expressed on activated endothelial cells. Thus, epratuzumab plays an indirect role to facilitate an efficient binding of B cells to endothelial cells, which mimics the direct binding of B cells to immobilized epratuzumab. (B) Epratuzumab enables the attachment of CD22-expressing B cells to EC in the endothelium via the trans-interaction of CD22 with CD22L. (C) Epratuzumab links CD22-expressing B cells to EC in the endothelium via the Fc-FcγR binding.
Knowing that binding of CD22 by soluble epratuzumab leads to internalization raises the question whether internalization of CD22 plays a role in the mechanism of cell killing. Taking a cue from CD20, which also interacts with BCR and affects calcium mobilization and its own degradation,53 the expression levels of CD22 as well as BCR on the cell surface may be critical for the activity of anti-CD22 mAbs, and needs further investigation. On the other hand, we speculate that immobilized epratuzumab may delay or prevent the internalization of BCR or CD22, or both, by changing actin dynamics and stabilizing the co-localized CD22 and BCR in the lipid rafts, leading to functional inactivation of BCR.
In conclusion, we provide evidence for the mechanism of action by which immobilized epratuzumab induces cytotoxic and cytostatic effects in CD22-expressing B-lymphoma lines with BCR of the IgM isotype. Our findings add to the existing knowledge that immobilized antibodies, such as those directed at CD3,54 CD47,55 or CD40,56 display different biological ability on target cells from their soluble counterparts, and establish that ligation of CD22 by immobilized epratuzumab perturbs BCR-mediated signals in malignant B cells without the involvement of anti-BCR antibodies. We also uncover, for the first time, a role of immobilized epratuzumab to stabilize F-actin and the potential of soluble epratuzumab to promote the adhesion of B cells to endothelial cells, which may occur in vivo to manifest the various biological activities observed for the immobilized epratuzumab in vitro. Future studies will assess if similar effects are involved in the depletion and modulation of B cells in healthy individuals, as well as SLE patients. Since plate- or bead-immobilized epratuzumab may represent a surrogate in-vitro mechanism of antibody crosslinking in vivo, the current study suggests that other agents comprising multiple epratuzumab molecules are worth investigating, such as CD22-targeting immunoliposomes. These can be generated to provide a high number of epratuzumab molecules on the surface of each liposome, as has been shown for immunoliposomes comprising the anti-HER2 trastuzumab,57 anti-CD74 milatuzumab,58 or an anti-transferrin receptor antibody.59
Materials and Methods
Cell lines, antibodies, and reagents
All cell lines, except D1–1 (a subline of Daudi generated in-house), were obtained from the American Type Culture Collection and have been authenticated by Promega using Short Tandem Repeat (STR) analysis. Phospho-specific antibodies for ERK, JNK, PLCγ2, and Lyn were obtained from Cell Signaling, as were antibodies specific for β-actin, Bcl-xL, Mcl-1, Caspase-3, Caspase-9, and PARP. The sources of other antibodies were as follows: Santa Cruz Biotech for CD22, CD79a, CD79b, Bcl-2, and Bax; Millipore for anti-tyrosine (4G10); and Jackson ImmunoResearch for F(ab′)2 fragment of affinity purified goat anti-human IgM, Fc5μ fragment-specific (anti-IgM), F(ab′)2 fragment of affinity purified goat anti-human IgG Fcγ fragment-specific (GAH), and rhodamine-conjugated F(ab′)2 fragment of affinity-purified goat anti-human IgG, F(ab′)2 fragment-specific (rhodamine-anti-IgG). The anti-CEACAM5 antibody, labetuzumab (hMN-14), was supplied by Immunomedics and served as an isotype control. Cell culture media and supplements, fluorescein isothiocyanate-conjugated DNase I (FITC-DNase I), Alexa Fluor 488 conjugated annexin V (488-annexin V), tetramethylrhodamine/ethyl ester (TMRE), and 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCF-DA), were supplied by Invitrogen. Rhodamine phalloidin was obtained from Cytoskeleton. One Solution Cell Proliferation assay reagent was obtained from Promega. Phosphosafe and RIPA buffers were procured from EMD chemicals. Latrunculin B (LatB), Jasplakinolide (JP), and all other chemicals were obtained from Sigma-Aldrich.
Immobilization of epratuzumab
To prepare the Dried-I format, epratuzumab in the form of IgG or F(ab′)2 at the indicated concentrations in bicarbonate buffer (50 mM; pH 9.6) was added to non-tissue-culture flat-bottom or U-bottom plates as specified, and incubated at 4°C overnight, followed by washing with 2X RPMI-1640 medium containing 5% fetal bovine serum (FBS) on the next day before use. Control antibodies were immobilized in the same fashion. To prepare the Dried-II format, 2 mL of HUV-EC-C cells (1.5 × 104 /mL) were added to 6-well, tissue-culture-treated flat-bottom plates, incubated overnight, and washed before use. To prepare the Particulate-I format, epratuzumab (50 μg) was conjugated to 200 μL of carboxyl polystyrene particles (3.0 to 3.4 μm, 5% w/v; Spherotech) in 1 mL of 2-(N-morpholino)ethanesulfonic acid buffer containing 20 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide for 30 min according to the manufacturer's protocol. Conjugated particles were washed 3X with phosphate-buffered saline (PBS) and reconstituted with 200 μL of PBS containing 0.05% bovine serum albumin (BSA) for use as the stock solution. To prepare the Particulate-II format, FAST FLOW Immobilized rProtein A (40 to 165 μm; Repligen) was incubated with 100 μL of epratuzumab (1 mg/mL) and supernatants were analyzed to estimate the amounts of epratuzumab noncovalently linked to the sepharose beads. The resulting epratuzumab-bound beads were washed 3X with PBS and reconstituted in 100 μL of the RPMI-1640 medium.
Cell culture and cytotoxicity assay
Cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, L-glutamine (2 mM), penicillin (200 U/mL), and streptomycin (100 μg/mL) in a humidified incubator at 37oC with 5% CO2. To evaluate the functional activity of epratuzumab in the Dried-I format, different amounts of IgG (starting from 20 μg/mL with 5-fold serial dilution to 0.01 ng/mL) were immobilized in non-tissue-culture-treated, U-bottom, 96-well plates overnight. Daudi cells (1 × 104 cells per well) were seeded and incubated for 3 days. For D1–1 and Ramos cells, IgG or F(ab’)2 of epratuzumab at 5, 10, and 20 μg/mL was immobilized in 48-well plates overnight. After washing, cells were seeded (1 × 104 cells per well) and incubated for 4 days. The number of viable cells was then determined using the MTS assay per the manufacturer's protocol, and plotted as percent of the untreated. Activity of soluble epratuzumab and anti-IgM also was evaluated in parallel.
Annexin V binding assay
Cells in 6-well plates (2 × 105 cells/well) were treated for 24 or 48 h with epratuzumab immobilized to polystyrene beads (Particulate-I) or plates (Dried-I), washed, resuspended in 100 μL of annexin-binding buffer, stained with 5 μL of 488-annexin V and 0.5 μL of 7-aminoactinomycin D (7-AAD) for 20 min, added 400 μL of annexin-binding buffer, and analyzed by flow cytometry (FACS Calibur; Becton Dickinson). Alternatively, cells were resuspended in 100 μL of annexin-binding buffer, stained with 5 μL of 488-annexin V for 20 min, added 400 μL of annexin-binding buffer containing 7-AAD, and analyzed. When required, cells were pretreated with the indicated inhibitors for 2 h before adding the test article.
Immunoblot analysis
Daudi, D1–1 or Ramos cells (2 × 107 cells) were added to plates coated with epratuzumab (10 μg/mL) and incubated for the indicated times. Cells were washed with PBS, lysed in ice-cold Phosphosafe buffer, and the lysates clarified by centrifugation at 13,000 × g. Protein samples (25 μg/lane) were resolved by SDS-PAGE on 4–20% gradient tris-glycine gels, followed by transfer onto nitrocellulose membranes, and probed with appropriate antibodies.
Immunoprecipitation
D1–1 cells (5 × 106 cell/well) in 6-well plates were incubated with test articles for the indicated times. After lysing the cells in ice-cold RIPA buffer, immunoprecipitation was performed using phospho-tyrosine antibody (4G10; 1:200 dilution). Samples (20 μL) were separated by SDS-PAGE and transferred onto a nitro-cellulose membrane, followed by probing with the indicated antibodies.
Isolation of lipid rafts
Lipid rafts were prepared as described previously34 and briefly below. Cells (5 × 107) were untreated or treated for 2 h with various test articles and lysed in 1 mL of cell lysis buffer (Cell Signaling) containing 1% Triton and 1% protease inhibitor cocktail on ice for 30 min. The lysates were transferred to ultracentrifuge tubes (Sorvall Ultracrimp Tube, 11.5-mL; Fisher Scientific), mixed with 1 mL of 80% sucrose in lysis buffer, overlaid with 5 mL of 35% sucrose and 4.5 mL of 5% sucrose, then centrifuged using Beckman SW55Ti rotor at 50,000 rpm (200,000 × g) for 3 h at 4 °C. Fractions of 1 mL were collected from the top, the protein concentration of each fraction determined with the Bio-Rad protein assay kit, and 20 μg sample of each fraction was resolved by 12–20%SDS-PAGE, followed by immunoblots with appropriate antibodies.
Measurement of Δψm and ROS
Daudi or D1–1 cells (2 × 105 cells/well) were incubated for 48 h with test articles as indicated, washed, stained with either TMRE (50 nM) or CM-H2DCF-DA (1 μM) for 30 min in the dark at 37°C, washed 3X with PBS, and analyzed by flow cytometry for Δψm or ROS, respectively, as described previously.60
Immunofluorescence microscopy
Daudi cells (2 × 106 per sample) were pretreated with test articles as indicated at 37°C for 1 h, incubated with or without LatB for 5 min, washed, fixed, permeabilized with 4% formalin and 0.1% Triton-100, and stained with Rhodamin-phalloidin and FITC-DNase I for 30 min in the dark at room temperature. Cells were then washed, resuspended in mounting solution containing DAPI, and examined by fluorescent microscopy.
Calcium mobilization assay
Daudi cells were loaded with Fluo-3 AM and Fura Red AM dye for 30 min at room temperature in the dark. For measurement of intracellular calcium flux, cells were washed 2x with an assay buffer comprising HBSS (1.25 mM CaCl2, 10 mM HEPES, 1% BSA), and resuspended in the same buffer, from which 1 mL (2 × 106 cells) was dispensed into each vial and incubated with a test antibody (20 μg/mL) or the Dried-I format of immobilized epratuzumab (20 μg/mL) as indicated, for 1 h at 37°C. All samples were kept on ice until analysis. Baseline fluorescence from each sample was monitored for 1 min before stimulating with anti-IgM (25 μg/mL), and the signal collected for the next 8 min. To monitor calcium influx, cells were washed with HBSS buffer (no Ca2+, 10 mM HEPES, 1% BSA, 1.5 mM EGTA), baseline was recorded for 1 min before stimulating with anti-IgM (25 μg/mL). After 4 min, 5 mM CaCl2 was added to the sample and the signal continuously monitored for another 6 min. The ratio of the geometric mean fluorescence intensity of fluo-3 (Em 530/30 nm) to Fura Red (Em 610/20 nm) was plotted against time and analyzed by Flowjo software.
Statistical analysis
Data obtained from in vitro studies were plotted using Prism software (version 4.03). Comparisons of mean values between 2 treatments were determined by Student's t-test, assuming a normal distribution for the data. A 2-tailed t-test was used when comparing different samples. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
CHC, YW and DMG are current employees of Immunomedics, Inc. PG declares no conflict of interest.
Acknowledgment
We thank Rongxiu Li for excellent technical assistance.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Author Contributions
CHC directed the project, analyzed the data, and wrote the manuscript. YW and PG performed the experiments, analyzed the data, and wrote the manuscript. DMG oversaw the research and wrote the manuscript.
References
- 1. Leung SO, Goldenberg DM, Dion AS, Pellegrini MC, Shevitz J, Shih LB, Hansen HJ. Construction and characterization of a humanized, internalizing, B-cell (CD22)-specific, leukemia/lymphoma antibody, LL2. Mol Immunol 1995; 32:1413-27; PMID:8643111; http://dx.doi.org/ 10.1016/0161-5890(95)00080-1 [DOI] [PubMed] [Google Scholar]
- 2. Leonard JP, Coleman M, Ketas JC, Chadburn A, Furman R, Schuster MW, Ashe M, Schuster SJ, Wegener WA, Hansen HJ, et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin's lymphoma:phase I/II clinical trial results. Clin Cancer Res 2004; 10:5327-34; PMID:15328168; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0294 [DOI] [PubMed] [Google Scholar]
- 3. Leonard JP, Schuster SJ, Emmanouilides C, Couture F, Teoh N, Wegener WA, Coleman M, Goldenberg DM. Durable complete responses from therapy with epratuzumab and rituximab:final results from an international multicenter, phase 2 study in recurrent, indolent, non-Hodgkin lymphoma. Cancer 2009; 113:2714-23; http://dx.doi.org/ 10.1002/cncr.23890 [DOI] [PubMed] [Google Scholar]
- 4. Raetz EA, Cairo MS, Borowitz MJ, Blaney SM, Krailo MD, Leil TA, Reid JM, Goldenberg DM, Wegener WA, Carroll WL, et al. Chemotherapy reintroduction with epratuzumab in children with acute lymphoblastic leukemia in marrow relapse:a Children's Oncology Group pilot study. J Clin Oncol 2008; 26:3756-62; PMID:18669463; http://dx.doi.org/ 10.1200/JCO.2007.15.3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Advani AS, McDonough S, Coutre S, Wood B, Radich J, Mims M, O’Donnell M, Elkins S, Becker M, Othus M, et al. SWOG S0910:a phase 2 trial of clofarabine/cytarabine/epratuzumab for relapsed/refractory acute lymphocytic leukaemia. Br J Haematol 2014; 165:504-9; PMID:24579885; http://dx.doi.org/ 10.1111/bjh.12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace DJ, Goldenberg DM. Epratuzumab for systemic lupus erythematosus. Lupus 2013; 22:400-5; PMID:23553783; http://dx.doi.org/ 10.1177/0961203312469692 [DOI] [PubMed] [Google Scholar]
- 7. Wallace DJ, Gordon C, Strand V, Hobbs K, Petri M, Kalunian K, Houssiau F, Tak PP, Isenberg DA, Kelley L, et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic erythematosus:results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology (Oxford) 2013; 52:1313-22; PMID:23542611; http://dx.doi.org/ 10.1093/rheumatology/ket129 [DOI] [PubMed] [Google Scholar]
- 8. Strand V, Petri M, Kalunian K, Gordon C, Wallace DJ, Hobbs K, Kelley L, Kilgallen B, Wegener WA, Goldenberg DM. Epratuzumab for patients with moderate to severe flaring SLE:health-related quality of life outcomes and corticosteroid use in the randomized controlled ALLEVIATE trials and extension study SL0006. Rheumatology (Oxford) 2014; 53:502-11; PMID:24273022; http://dx.doi.org/ 10.1093/rheumatology/ket378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steinfeld SD, Tant L, Burmester GR, Teoh NKW, Wegener WA, Goldenberg DM, Prodier O. Epratuzumab (humanised anti-CD22 antibody) in primary Sjögren's syndrome:an open-label phase I/II study. Arthritis Res Ther 2006; 8:R129; PMID:16859536; http://dx.doi.org/ 10.1186/ar2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O’Reilly MK, Paulson JC. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol Sci 2009; 30:240-8; http://dx.doi.org/ 10.1016/j.tips.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tedder TF, Tuscano J, Sato S, Kehrl JH. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev Immunol 1997; 15:481-504; PMID:9143697; http://dx.doi.org/ 10.1146/annurev.immunol.15.1.481 [DOI] [PubMed] [Google Scholar]
- 12. Law CL, Aruffo A, Chandran KA, Doty RT, Clark EA. Ig domains 1 and 2 of murine CD22 constitute the ligand-binding domain and bind multiple sialylated ligands expressed on B and T cells. J Immunol 1995; 155:3368-76; PMID:7561031 [PubMed] [Google Scholar]
- 13. Wilson GL, Fox CH, Fauci AS, Kehrl JH. cDNA cloning of the B cell membrane protein CD22:a mediator of B-B cell interactions. J Exp Med 1991; 173:137-46; PMID:1985119; http://dx.doi.org/ 10.1084/jem.173.1.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ravetch JV, Lanier LL. Immune inhibitory receptors. Science 2000; 290:84-9; PMID:11021804; http://dx.doi.org/ 10.1126/science.290.5489.84 [DOI] [PubMed] [Google Scholar]
- 15. Sato S, Tuscano JM, Inoaki M, Tedder TF. CD22 negatively and positively regulates signal transduction through the B lymphocyte antigen receptor. Semin Immunol 1998; 10:287-97; PMID:9695185; http://dx.doi.org/ 10.1006/smim.1998.0121 [DOI] [PubMed] [Google Scholar]
- 16. Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003; 72:395-447; PMID:12651740; http://dx.doi.org/ 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- 17. John B, Herrin BR, Raman C, Wang YN, Bobbitt KR, Brody BA, Justement LB. The B cell coreceptor CD22 associates with AP50, a clathrin-coated pit adaptor protein, via tyrosine-dependent interaction. J Immunol 2003; 170:3534-43; PMID:12646615; http://dx.doi.org/ 10.4049/jimmunol.170.7.3534 [DOI] [PubMed] [Google Scholar]
- 18. Shan D, Press OW. Constitutive endocytosis and degradation of CD22 by human B cells. J Immunol 1995; 154:4466-75; PMID:7722303 [PubMed] [Google Scholar]
- 19. O’Reilly MK, Tian H, Paulson JC. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J Immunol 2011; 186:1554-63; http://dx.doi.org/ 10.4049/jimmunol.1003005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Razi N, Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci USA 1998; 95:7469-74; PMID:9636173; http://dx.doi.org/ 10.1073/pnas.95.13.7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engels P, Nojima Y, Rothstein D, Zhou LJ, Wilson GL, Kehrl JH, Tedder TF. The same epitope on CD22 of B lymphocytes mediates the adhesion of erythrocytes, T and B lymphocytes, neutrophils, and monocytes. J Immunol 1993; 150:4719-32; PMID:7684411 [PubMed] [Google Scholar]
- 22. Leprince C, Draves KE, Geahlen RL, Ledbetter JA, Clark EA. CD22 associates with the human surface IgM-B-cell antigen receptor complex. Proc Natl Acad Sci USA 1993; 90:3236-40; PMID:8475064; http://dx.doi.org/ 10.1073/pnas.90.8.3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petri RJ, Schnetkamp PPM, Patel KD, Awasthi-Kalia M, Deans JP. Transient translocation of the B cell receptor and Src homology 2 domain-containing inositol phosphatase to lipid rafts:evidence towards a role in calcium regulation. J Immunol 2000; 165:1220-7; PMID:10903719; http://dx.doi.org/ 10.4049/jimmunol.165.3.1220 [DOI] [PubMed] [Google Scholar]
- 24. Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22:a requirement for Lyn. J Exp Med 1998; 187:807-11; PMID:9480991; http://dx.doi.org/ 10.1084/jem.187.5.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2002; 2:945-56; PMID:12461567; http://dx.doi.org/ 10.1038/nri955 [DOI] [PubMed] [Google Scholar]
- 26. Tuscano J, Engel P, Tedder TF, Kehr JH. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22L interactions impairs T-cell proliferation. Blood 1996; 87:4723-30; PMID:8639842 [PubMed] [Google Scholar]
- 27. Pezzutto A, Dörken B, Moldenhauer G, Clark EA. Amplification of human B cell activation by a monoclonal antibody to the B cell-specific antigen CD22, Bp 130/140. J Immunol 1987; 138:98-103; PMID:3097151 [PubMed] [Google Scholar]
- 28. Pezzutto A, Rabinovitch PS, Dörken B, Moldenhauer G, Clark EA. Role of the CD22 human B cell antigen in B cell triggering by anti-immunoglobulin. J Immunol 1988; 140:1791-5; PMID:3257985 [PubMed] [Google Scholar]
- 29. Nitschke L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr Opin Immunol 2005; 17:290-7; PMID:15886119; http://dx.doi.org/ 10.1016/j.coi.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 30. Dörner T, Kaufmann J, Wegener WA, Teoh N, Goldenberg DM, Burmester GR. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther 2006; 8:R74; PMID:16630358; http://dx.doi.org/ 10.1186/ar1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carnahan J, Stein R, Qu Z, Hess K, Cesano A, Hansen HJ, Goldenberg DM. Epratuzumab, a CD22-targeting recombinant humanized antibody with a different mode of action from rituximab. Mol Immunol 2007; 44:1331-41; PMID:16814387; http://dx.doi.org/ 10.1016/j.molimm.2006.05.007 [DOI] [PubMed] [Google Scholar]
- 32. Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dörner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis 2008; 67:450-7; PMID:17673490; http://dx.doi.org/ 10.1136/ard.2007.075762 [DOI] [PubMed] [Google Scholar]
- 33. Daridon C, Blassfeld D, Reiter K, Mei HE, Giesecke C, Goldenberg DM, Hansen A, Hostmann A, Frolich D, Dӧrner T. Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res Ther 2010; 12:R204; PMID:21050432; http://dx.doi.org/ 10.1186/ar3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sieger N, Fleischer SJ, Mei HE, Reiter K, Shock A, Burmester GR, Daridon C, Dӧrner T. CD22 ligation inhibits downstream B-cell receptor signaling and Ca2+ flux upon activation. Arthritis Rheum 2013; 65:770-9; PMID:23233360; http://dx.doi.org/ 10.1002/art.37818 [DOI] [PubMed] [Google Scholar]
- 35. Rossi EA, Goldenberg DM, Michel R, Rossi DL, Wallace DJ, Chang CH. Trogocytosis of multiple B-cell surface markers by CD22-targeting epratuzumab. Blood 2013; 122:3020-9; PMID:23821660; http://dx.doi.org/ 10.1182/blood-2012-12-473744 [DOI] [PubMed] [Google Scholar]
- 36. Rossi EA, Chang CH, Goldenberg DM. Anti-CD22/CD20 bispecific antibody with enhanced trogocytosis for treatment of lupus. PLOS ONE, 2014; 9:e98315; http://dx.doi.org/ 10.1371/journal.pone.0098315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carnahan J, Wang P, Kendall R, Chen C, Hu S, Boone T, Juan T, Talvenheimo J, Montestruque S, Sun J, et al. Epratuzumab, a humanized monoclonal antibody targeting CD22:characterization of in vitro properties. Clin Cancer Res 2003; 9:3982S-90S; PMID:14506197 [PubMed] [Google Scholar]
- 38. Qu Z, Goldenberg DM, Cardillo TM, Shi V, Hansen HJ, Chang CH. Bispecific anti-CD20/22 antibodies inhibit B-cell lymphoma proliferation by a unique mechanism of action. Blood 2008; 111:2211-9; PMID:18025153; http://dx.doi.org/ 10.1182/blood-2007-08-110072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doody GM, Justement LB, Delibrias CC, Matthews RJ, Lin J, Thomas ML, Fearon DT. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science 1995; 269:242-4; PMID:7618087; http://dx.doi.org/ 10.1126/science.7618087 [DOI] [PubMed] [Google Scholar]
- 40. Yu J, Sawada T, Adachi T, Gao X, Takematsu H, Kozutsumi Y, Ishida H, Kiso M, Tsubata T. Synthetic glycan ligand excludes CD22 from antigen receptor-containing lipid rafts. Biochem Biophys Res Commun 2007; 360:759-64; PMID:17631277; http://dx.doi.org/ 10.1016/j.bbrc.2007.06.110 [DOI] [PubMed] [Google Scholar]
- 41. Courtney AH, Puffer EB, Pontrello JK, Yang ZQ, Kiessling LL. Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation. Proc Natl Acad Sci USA 2009; 106:2500-5; PMID:19202057; http://dx.doi.org/ 10.1073/pnas.0807207106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rudge EU, Cutler AJ, Pritchard NR, Smith KGC. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and FcgRII-mediated B cell suppression. J Exp Med 2002; 195:1079-85; PMID:11956299; http://dx.doi.org/ 10.1084/jem.20011435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan VWF, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol 1998; 8:545-53; PMID:9601638; http://dx.doi.org/ 10.1016/S0960-9822(98)70223-4 [DOI] [PubMed] [Google Scholar]
- 44. Ishigami T, Kim K-M, Horiguchi Y, Higaki Y, Hata D, Heike T, Katamura K, Mayumi M, Mikawa H. Anti-IgM antibody-induced cell death in a human B lymphoma cell line, B104, represents a novel programmed cell death. J Immunol 1992; 148:360-8; PMID:1729359 [PubMed] [Google Scholar]
- 45. O’Reilly MK, Collins BE, Han S, Liao L, Rillahan C, Kitov PI, Bundle DR, Paulson JC. Bifunctional CD22 ligands use multimeric immunoglobulins as protein scaffolds in assembly of immune complexes on B cells. J Am Chem Soc 2008; 130:7736-46; http://dx.doi.org/ 10.1021/ja802008q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abdu-Allah HHM, Tamanaka T, Yu J, Zhuoyuan L, Sadagopan M, Adachi T, Tsubata T, Kelm S, Ishida H, Kiso M. Design, synthesis, and structure-affinity relationships of novel series of sialosides as CD22-specific inhibitors. J Med Chem 2008; 51:6665-81; PMID:18841881; http://dx.doi.org/ 10.1021/jm8000696 [DOI] [PubMed] [Google Scholar]
- 47. Abdu-Allah HHM, Watanabe K, Completo GC, Sadagopan M, Hayashizaki K, Takaku C, Tamanaka T, Takematsu H, Kozutsumi Y, Paulson JC. CD22-antagonists with nanomolar potency:the synergistic effect of hydrophobic group at C-2 and C-9 of sialic acid scaffold. Bioorg Med Chem 2011; 19:1966-71; PMID:21349726; http://dx.doi.org/ 10.1016/j.bmc.2011.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest 2013; 123:3074-83; PMID:23722906; http://dx.doi.org/ 10.1172/JCI69187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hanasaki K, Varki A, Stamenkovic I, Bevilacqua MP. Cytokine-induced β-galactoside α-2,6-sialyltransferase in human endothelial cells mediates α2,6-sialylation of adhesion molecules and CD22 ligands. J Biol Chem 1994; 269:10637-43; PMID:8144653 [PubMed] [Google Scholar]
- 50. Hanasaki K, Varki A, Powell LD. CD22-mediated cell adhesion to cytokine-activated human endothelial cells. Positive and negative regulation by α2-6-sialylation of cellular glycoproteins. J Biol Chem 1995; 270; 7533-42; PMID:7706300; http://dx.doi.org/ 10.1074/jbc.270.13.7533 [DOI] [PubMed] [Google Scholar]
- 51. Groger M, Sarmay G, Fiebiger E, Wolfe K, Petzelbauer P. Dermal microvascular endothelial cells express CD32 receptors in vivo and in vitro. J Immunol 1996;156:1549-56; PMID:8568259 [PubMed] [Google Scholar]
- 52. Pan LF, Kreisle RA, Shi YD. Detection of Fcg receptors on human endothelial cells stimulated with cytokines tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-g). Clin Exp Immunol 1998; 112:533-8; PMID:9649226; http://dx.doi.org/ 10.1046/j.1365-2249.1998.00597.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walshe CA, Beers SA, French RR, Chan CH, Johnson PW, Packham GK, Glennie MJ, Cragg MS. Induction of cytosolic calcium flux by CD20 is dependent upon B cell antigen receptor signaling. J Biol Chem 2008; 83:16971-84; http://dx.doi.org/ 10.1074/jbc.M708459200 [DOI] [PubMed] [Google Scholar]
- 54. Geppert TD, Lipsky PE. Accessory cell independent proliferation of human T4 cells stimulated by immobilized monoclonal antibodies to CD3. J Biol Chem 1987; 138:1660-6. [PubMed] [Google Scholar]
- 55. Mateo V, Lagneaux L, Bron D, Brion G, Armant M, Delespesse G, Sarfati M. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat Med 1999; 5:1277-84; PMID:10545994; http://dx.doi.org/ 10.1038/15233 [DOI] [PubMed] [Google Scholar]
- 56. Watanabe S, Kagamu H, Yoshizawa H, Fujita N, Tanaka H, Tanaka J, Gejyo F. The duration of CD40 signaling directs biological ability of dendritic cells to induce antitumor immunity. J Immunol 2003; 171:5828-36; PMID:14634092; http://dx.doi.org/ 10.4049/jimmunol.171.11.5828 [DOI] [PubMed] [Google Scholar]
- 57. Chiu GNC, Edwards LA, Kapanen AI, Malinen MM, Dragowska WH, Warburton C, Chikh GG, Fang KY, Tans S, Sy J, et al. Modulation of cancer cell survival pathways using multivalent liposomal therapeutic antibody constructs. Mol Cancer Ther 2007; 6:844-55; PMID:17339368; http://dx.doi.org/ 10.1158/1535-7163.MCT-06-0761 [DOI] [PubMed] [Google Scholar]
- 58. Hertlein E, Triantafilou G, Sass EJ, Hessler JD, Zhang X, Jarjoura D, Lucas DM, Muthusamy N, Goldenberg DM, Lee RJ, et al. Milatuzumab immunoliposomes induce cell death in CLL by promoting accumulation of CD74 on the surface of B cells. Blood 2010; 116:2554-8; PMID:20574049; http://dx.doi.org/ 10.1182/blood-2009-11-253203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J, Tian S, Petros RA, Napier ME, DeSimone JM. The complex role of multivalency in nanoparticles targeting the transferrin receptor for cancer therapies. J Am Chem Soc 2010; 132:11306-13; PMID:20698697; http://dx.doi.org/ 10.1021/ja1043177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gupta P, Goldenberg DM, Rossi EA, Chang CH. Multiple signaling pathways induced by hexavalent, monospecific, anti-CD20 and hexavalent, bispecific, anti-CD20/CD22 humanized antibodies correlate with enhanced toxicity to B-cell lymphomas and leukemias. Blood 2010; 116:3258-67; PMID:20628151; http://dx.doi.org/ 10.1182/blood-2010-03-276857 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.