

Abstract
Epigenetics, or regulation of gene expression independent of DNA sequence, is the missing link between genotype and phenotype. Epigenetic memory, mediated by histone and DNA modifications, is controlled by a set of specialized enzymes, metabolite availability, and signaling pathways. A mostly unstudied subject is how sub-toxic exposure to several xenobiotics during specific developmental stages can alter the epigenome and contribute to the development of disease phenotypes later in life. Furthermore, it has been shown that exposure to low-dose xenobiotics can also result in further epigenetic remodeling in the germ line and contribute to increase disease risk in the next generation (multigenerational and transgenerational effects). We here offer a perspective on current but still incomplete knowledge of xenobiotic-induced epigenetic alterations, and their possible transgenerational transmission. We also propose several molecular mechanisms by which the epigenetic landscape may be altered by environmental xenobiotics and hypothesize how diet and physical activity may counteract epigenetic alterations.
Keywords: circadian clock, epigenetics, metabolism, transgenerational effects, toxicology
Epigenetics: A Signal Integrator among Genes, Metabolism, and Environment that Determines Phenotype
As multicellular organisms develop from a single cell, a complex biological program unfolds to create a multitude of cellular phenotypes. The developmental program consists of a genome, a gene regulatory network of transcription factors, and an epigenome directing genome accessibility and function. During the unfolding of the developmental program, the genomic information of an organism is constant, while the epigenome governing the utility of the genome is in a state of flux. In the past decade, epigenetic alterations associated with development and cell lineage commitment have garnered much attention. When Conrad Waddington first conceptualized Epigenetics in the late 1930's, he described it as “the science concerned with the causal analysis of development.” To illustrate his concept of epigenetics in development, Waddington depicted a fertilized egg rolling down canals to represent a cell entering lineage commitment.1 He suggested that the conditions surrounding the cell create an “epigenetic landscape” to dictate its fate. Today, it is known that Waddington's epigenetic landscape is fashioned through a collaboration of DNA methylation, histone modifications, non-coding RNA, and higher-order chromatin structure. The picture is apparently more complex, with histone modifications involving acetylation, methylation, phosphorylation, ADP-ribosylation, ubiquitylation, succinylation, sumoylation, and malonylation.2 At present, some of these modifications remain still poorly characterized in terms of their biological meaning.3 We have previously suggested that factors such as redox balance, oxygen, metabolism, and cofactor availability drive development by modifying the activity of enzymes that initiate and perpetuate epigenetic events.4 Lending credence to this principle is the observation that modifying these conditions in embryonic stem cells induces lineage commitment and the establishment of a new epigenotype consistent with that cell fate. While these observations strongly suggest that a linkage exists between the extracellular environment and epigenetics during development, they also portend developmental defects or later onset diseases of adulthood if this critical balance is altered.
Epigenetic events such as DNA methylation and histone acetylation and methylation are both heritable and exhibit plasticity.5 New epigenetic marks are created by the enzymatic addition of modifications to cytosine bases in DNA, and the posttranslational modification of histone tails. Other enzymes increase the dynamic nature of epigenetics by removing these modifications. A complete background on various epigenetic events, and their interactions with each other, exceed the scope of what will be discussed here (see refs.6-8). Epigenetic remodeling may result in pre-disposition to disease in later generations, as we will discuss in the next section.
We explore here the current knowledge regarding the mechanisms by which xenobiotic compounds can lead to persistent transgenerational remodeling of the epigenome. According to a classical definition, a xenobiotic is a chemical molecule found inside an organism that is not naturally produced by or expected to be present there. Xenobiotics can include clinically used drugs, environmental pollutants, cosmetics, and even dietary components.9
Pre-Conception and Maternal Exposure to Xenobiotics, Epigenetic Remodeling, and Disease Incidence in the Offspring
There is mounting evidence that environmental factors increase disease risk, in part by reprogramming the epigenome.10 It is well understood that the prenatal and neonatal developmental periods represent a time of enhanced susceptibility to reprogramming by environmental factors. Chief among the reasons is the rapid growth characterized by cell proliferation and differentiation that occurs during these life stages. Epigenetic mechanisms contribute to faithfully maintaining undifferentiated stem cells on one hand and organogenesis on the other.11,12 Early embryogenesis in mammals is consequently one of most critical periods for the appropriate establishment of the epigenome.13-16
The developmental origins of health and disease hypothesis
In 1989, David Barker refocused the evidence linking increased predisposition to disease in adulthood to suboptimal pregnancy environments when he associated birth weight and death rates from adult coronary heart disease.17 Most recently, this field has become known as the Developmental Origins of Health and Disease Hypothesis (DOHaD), proposing that environmental stimuli (including xenobiotics) during early life perturb normal development and increase risk for chronic diseases in later life. Epigenetic mechanisms have been suggested to be key mediators of this phenomenon.18
Environmental conditions experienced by progenitors (or founder individuals, F0) have been reported to additionally result in increased disease risk in the offspring (F1) or even the grand-offspring (F2). Strikingly, transmission of disease risk to the F1/F2 occurs in the absence of the environmental trigger.19 Several authors have proposed that transmission of phenotypes may be mediated by epigenetic reprogramming of germ cells and/or mature gametes;20 we refer to this phenomenon as transgenerational epigenetic inheritance. Many authors suggest that in order to be considered transgenerational inheritance, the epigenetic modifications and the inherited phenotypes should be detected through the third generation offspring (F3).21,22 The reason is that when an F0 gestating mother is exposed to an environmental challenge, her embryos/fetuses (F1) and the already developing germline (which will give rise to the F2) are also directly exposed. Therefore, F1 and F2 phenotypes could be the direct consequence to the developmental exposure event. The F3 would be the first generation where the phenotype is not due to the direct exposure to the external triggers (Fig. 1). If the exposure occurs after birth, then a true transgenerational effect occurs if the effect is observed in the F2 generation.23 The data available on true transgenerational effects of xenobiotic epigenetic toxicity are still scarce. Therefore, in the present review we will also present examples of xenobiotic-induced epigenetic remodeling found in F1 and F2 generations.
Figure 1.
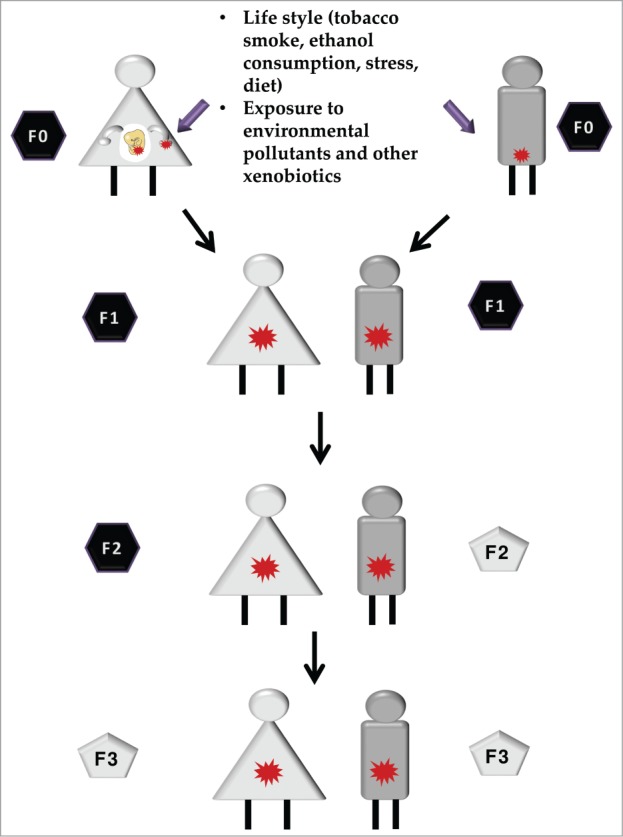
Intergenerational vs. transgenerational epigenetic inheritance. Epigenetic alterations can be induced by parental exposure to certain conditions such as lifestyle, including nutrition and exposure to different types of xenobiotics. Those changes, when present in germ cells and/or mature gametes (red stars) are transmitted to the offspring, in a phenomenon known as epigenetic inheritance. If the phenotype is observed further than the second or third generation (when the exposed progenitor is a gestating mother) a transgenerational epigenetic inheritance is observed (gray pentagons). If the phenotype is only observed in first generation (or second, for the exposure of a gestating mother), this is considered a parental effect and it is known as intergenerational epigenetic inheritance (black hexagons).
In this section, we discuss the evidence that exposure to xenobiotics during development interferes with normal epigenetic programming, permanently alters cell/tissue function, and, consequently, increases disease risk later in life. There are many gaps in our knowledge, not only because technological limitations exist but also because stability of epigenetic alterations is often an issue. This latter fact may explain why alterations in DNA methylation are more often investigated in the context of xenobiotic-induced epigenetic alterations. Published evidence is available for environmental tobacco exposure, air pollutants, endocrine disruptors, and various toxic metals (for a recent review see ref.24), as summarized below.
Lifestyle-derived epigenetic modulators: environmental tobacco smoke and ethanol consumption
Active tobacco smoking during pregnancy is associated with reduced fetal growth and increased fetal mortality.25,26 Likewise, maternal tobacco smoking has also been associated with altered pulmonary27 and immune function,28,29 as well as increased risk of cancer,30 obesity and type 2 diabetes in the offspring. Furthermore, children of either active or passive smokers showed reduced academic performance.31,32
Clinical studies support the thesis that maternal exposure to tobacco smoking may alter the fetal epigenome.33 Specifically, maternal exposure to environmental tobacco smoke (ETS) is associated with both global and locus-specific alterations in DNA methylation of cord blood and placenta of the offspring.33,34 One of the studies identified increased methylation in 26 CpGs, which mapped to 10 genes. Some of them are important for chemical detoxification (AHRR and CYP1A1) and tissue development (IGFI1). These epigenetic marks can be useful diagnostic markers to determine in utero exposure to smoking, although different promoters may be differently affected in terms of methylation (see29 for a review). Since different technologies have been used to detect DNA methylation changes associated with in utero ETS exposure, it is deemed necessary to confirm the same targets using a single, established technology. Prenatal ETS also induced lower global DNA methylation and increased methylation at specific loci in children35 and adult women.36 Genes exhibiting hypermethylation included AXL, coding for a receptor tyrosine kinase,33 which has several roles spanning from cancer to cell differentiation. The authors state correctly that the results should be interpreted with caution due to possible polymorphic imprinting. A recent study by Markunas et al. showed that maternal smoking during pregnancy resulted in altered methylation of 185 CpG islands in the offspring.37 Genes affected included pathways associated with nicotine dependence and smoking withdrawal (FRMD4A and ATP9A), as well as placental and embryonic development (MEG3).37 The former may contribute to problems experienced by smoking mothers in terms of maintaining pregnancy.38,39 Together, these studies suggest that prenatal ETS may induce epigenetic modifications in the offspring that can last for many years. Whether adult epigenetic marks are already established in utero or whether they appear secondarily as the individual ages remains to be determined. In addition, it remains unknown whether these epigenetic modifications are a consequence of disease or play a causal role. These difficulties suggest a central role for animal models with shorter generation times in unraveling the details of these events.
Prenatal alcohol exposure represents another social problem resulting in a wide range of phenotypic alterations, collectively known as fetal alcohol spectrum disorders (FASD). FASD is characterized by a cluster of neurodevelopmental disorders that include attention deficits, impaired learning and memory, increased anxiety, and behavioral disorders.40 Furthermore, it has been suggested that long-lasting effects of in utero exposure to alcohol consumption may be partly mediated by epigenetic mechanisms.41 In this regard, there is growing evidence that ethanol exposure influences DNA methylation, histone modifications, and regulation of non-coding RNAs in rodent models.41 Indeed, prenatal exposure to alcohol results in global DNA methylation changes in the pups.42,43 Liu et al.42 demonstrated that the expression of 84 genes was affected by differential promoter methylation of varying magnitude. These genes were determined to play a role in cancer, apoptosis, cell cycle, and olfaction. The authors reported increased methylation of genes related with metabolism (e.g., Cyp4f13) and decreased methylation of genes involved with developmental processes (Nlgn3, Elavl2, Sox21, and Sim1), imprinting (Igf2r) and chromatin (Hist1h3d). Fetal alcohol exposure also induced locus-specific alterations, including the insulin growth factor-II (IGF-II) locus.44 Many of these alterations are correlated to changes in expression of genes that may contribute, in part, to the reported FASD phenotypes.42 Although these studies suggest that alcohol exposure may induce the postnatal phenotypes through epigenetic remodeling, proof remains to be fully ascertained. In this regard, studies in the Agouti yellow viable mouse model (see section on Environmental pollutants: Endocrine-disrupting chemicals) constitute evidence that this may actually happen: fetal exposure to ethanol resulted in hypermethylation at the agouti locus and transcriptional silencing of the gene.45 These experiments therefore provide evidence that ethanol-induced alteration of DNA methylation may underlie phenotypic traits of FASD.
In conclusion, the present section shows that 2 relevant components of human lifestyle (smoking and ethanol consumption) can result in epigenetic remodeling and impact the future offspring. Whether these epigenetic changes have transgenerational consequences remains to be determined.
Environmental pollutants: polycyclic aromatic hydrocarbons
Prenatal exposure to polycyclic aromatic hydrocarbons (PAHs) is associated with intrauterine growth restriction, reduced cognitive development, and behavioral disorders.10,24 A possible epigenetic mechanism is suggested by the observation that prenatal PAH exposure resulted in global hypomethylation in umbilical cord blood cells.46 This alteration persisted in offspring up to 3 years of age. Whether these epigenetic modifications contribute to disease risk later in life, or whether they are simply good markers of prenatal exposure, requires further investigation. In this regard, a follow-up study showed PAH-dependent DNA methylation in 30 specific loci, including the acyl-CoA synthetase long chain (ACSL) and, importantly, the interferon-gamma (IFNγ) genes.47,48 While the role of ACSL in lung disease remains to be elucidated, IFNγ has been previously reported to be involved in the development of asthma. Accordingly, in vitro studies conducted in lung cell lines confirmed that PAH exposure increased promoter DNA methylation and reduced IFNγ expression. These data constitute early support for a potential link between exposure to PAH and disease risk (asthma) mediated by epigenetic modifications.
Environmental pollutants: Endocrine-disrupting chemicals
Bisphenol A
Recent epidemiological studies show that prenatal exposure to Bisphenol A (BPA), which typically occurs through diet,49 is associated with intrauterine growth restriction, male reproductive abnormalities, altered behavior, asthma, and childhood obesity (reviewed in ref.50). Likewise, in utero BPA exposure in animal models resulted in phenotypes similar to those described in humans: dysfunction of the reproductive tract, altered brain development, and postnatal behavioral disorders.51-53
Classical experiments in the Agouti viable yellow (Avy) mouse model provided proof-of-principle that in utero exposure to BPA results in permanent epigenetic modifications that may lead to specific phenotypes.54 The agouti gene shows variable expression in genetically identical mice due to epigenetic regulation. The Avy allele results from the insertion of an Intracisternal A Particle (IAP) retrotransposon at the 5’ of the agouti gene.55 Importantly, the methyl groups of the IAP are established during development. Maternal exposure to BPA during gestation decreases DNA methylation at the Agouti locus of the offspring.54 This epigenetic shift also results in an increased prevalence of yellow, obese, diabetic mice in the offspring compared to lean black offspring of unexposed pregnancies. Another study involving BPA in utero toxicity shows that exposure to different BPA concentrations decreases expression of xenobiotic metabolic enzymes in the fetal liver, via epigenetic mechanisms.56 Although, as the authors state, other undetermined confounding factors may have played a role; this result implies a decreased capacity to metabolically deal with chemical entities, paving the way for the development of toxicity events later in life and possibly extending this effect to future generations.
In spite of the observations discussed above, it remains to be determined whether epigenetic reprogramming mediates the increase in postnatal disease risk associated with BPA. For example, prenatal BPA exposure in mice induces gender-specific changes in the expression of estrogen receptors (ER) in juvenile offspring (ERa and ERb).57 Importantly, altered ERa expression occurred in association with changes in DNA methylation. In another experimental setting, BPA administration during gestation and lactation in mice was shown to result in phenotypes reminiscent of metabolic syndrome in the offspring including obesity and cardiac dysfunction.58 Consistent with the sex-specificity noted above, global DNA methylation was increased in the hearts of male BPA-exposed offspring and decreased in the hearts of the females. In utero exposure to BPA was recently linked to the transgenerational inheritance of behavior, obesity, and reproductive disease.59,60 As defined above, transgenerational epigenetic inheritance implies that environmental cues induce epigenetic marks in the germ line. To date, not many studies have analyzed the epigenome of the gametes and, therefore, whether these transgenerational effects are mediated by epigenetic mechanisms remains unknown. However, it has been shown that exposure to plastic-derived endocrine disruptors, including BPA, bis(2-ehylexyl)phthalate (DEHP) and dibutylphathalate (DBP), cause transgenerational inheritance of obesity and testis and ovarian disease up to the F3 via the paternal line.59 Strikingly, these studies describe 197 regions with differential methylation in F3 sperm. Gene network analysis of the observed alterations involved the glial-derived neurotrophic factor (Gdnf) and neurotrophin 3 (Ntf3) pathways, as well as genes related to obesity processes. An elevated number of genes related to metabolism and metabolite transport was also found to be differentially methylated.59 Although there is no direct evidence that these sperm epimutations are also present in somatic tissues and contribute to the development of various diseases, male gametes may be carriers of xenobiotic-induced epigenomic information across generations.
Phthalates
Prenatal exposure to phthalates has been associated with shortened gestation, developmental abnormalities of reproductive tissues and increased adiposity in children.24 In rats, in utero exposure to di-(2-ethylhexyl)phthalate (DEHP) decreases androgen formation in testes of both fetal and adult animals.61 Reduced androgen production can be accounted for, in part, by the decreased expression of the mineralocorticoid receptor (MR) gene. Next, it was found that the MR gene promoter is hypomethylated in DEHP-exposed animals. It is then likely that in utero exposure to DEHP leads to epigenetically mediated MR dysfunction and concomitant reduced testosterone production. The evidence for epigenetic regulation of gene expression following phthalate exposure is further supported by studies in adult cancer cell lines exposed to DEHP, DBP, or butylbenzylphthalate (BBP).62 These authors showed that BBP induces ER transcription characterized by hypomethylation of ERα promoter CpG islands.
Vinclozolin
Fetal exposure to vinclozolin, a common fungicide with high affinity for the androgen receptor, influences sexual differentiation, gonadal development, and reproductive functions.63 From a DOHaD point of view, it is relevant to note that vinclozolin is the second most prevalent pesticide (43% of cases) detected in umbilical cord blood from growth-restricted newborns in a rural Chinese cohort.64 In rats, prenatal vinclozolin can induce epigenetic transgenerational inheritance of adult-onset disease.65 Transient exposure to vinclozolin during gestation causes spermatic cell defects and fertility problems to the offspring (F1). These phenotypic defects persist up to the fourth generation offspring (F4), even though the environmental factor is absent. These transgenerational effects appear to be mediated in part by epigenetic modifications in the male germ line.66 Exposure to this agent during gestation results in F0 to F3 transmission of low sperm counts and methylation patterns in critical genes for gender imprinted genes, as well as in kidney abnormalities and polycystic ovarian disease.67,68 Interestingly, the degree of transgenerational passage appears to be dependent on the rat strain used.68
Diethylstilbestrol
Prenatal and early neonatal exposure to diethylstilbestrol (DES), a non-steroidal estrogen, induces developmental anomalies of the female reproductive tract. Some of these effects are mediated by permanent promoter hypermethylation and reduced expression of the homeobox HOXA10 gene, which regulates uterine organogenesis of the offspring.69 Similarly, exposure of human endometrial cells to DES also altered expression of HOXA10.
DES-mediated harmful health effects are facilitated by its interaction with ER. Exposure to DES during gestation in mice induced long-term effects on cardiac function and structure in the offspring, in part through abnormal regulation of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) and calsequestrin 2 expression during physical cardiac challenge.70,71 These studies support the idea that in utero exposure to DES epigenetically alters DNA methylation, thus leading to altered gene expression that, in turn, may be responsible for the cardiac dysfunction.
In humans, in utero exposure to DES causes carcinomas in the uterus and other estrogen-responsive reproductive tissues in the daughters whose mothers took this potent synthetic estrogen.72,73 Interestingly, the cancers in these daughters exposed in utero emerged upon sexual maturation. This suggests that abnormal exposure to synthetic estrogens during development causes aberrant epigenetic programming in estrogen sensitive cell types that predisposes them to cancer in later life when exposed to additional endogenous estrogen.
In sum, exposure to endocrine-disrupting chemicals is clearly the best example so far of transmission of a modified epigenetic landscape to future generations, with effects observed as far as on the F3 generation, demonstrating a true transgenerational effect.
Environmental pollutants: heavy metals
Many studies have established an association between exposure to environmental heavy metals, such as arsenic, cadmium, lead, nickel, or mercury, and epigenetic modifications. Only a few studies, however, have addressed whether exposure to heavy metals in utero increases disease incidence in the offspring through epigenetic modifications.
Arsenic
In utero exposure to arsenic in humans has been linked to increased infant mortality, low birth weight, and developmental abnormalities.74,75 Arsenic exposure in utero has also been associated with both altered global methylation in cord blood76,77 and specific increased promoter methylation of genes involved in cancer risk, including p53,78 and p16.79 Follow-up studies are warranted to assess whether these reported epigenetic modifications are maintained later in life and contribute to disease risk.
Mercury
Methylmercury (MeHg) is a contaminant with potential neurotoxic effects, found in seafood, a source of polyunsaturated fatty acids cast as beneficial during pregnancy.80 Perinatal exposure to MeHg in mice results in persistent alterations on behavior and learning in the offspring.81 The behavioral alterations may partially be explained by reduced expression of the brain-derived neurotrophic factor (BDNF) in the hippocampus. Altered BDNF expression was associated with epigenetic marks typically linked to gene repression: DNA hypermethylation, increased H3K27me3, and reduced H3Ac. These studies suggest that early MeHg exposure induces long-lasting effects in behavior, in part through epigenetic modifications.82 This seminal observation deserves further validation in other model systems and global profiling will be required to understand whether the reported alteration is a more general event. Although evidence is lacking for generational passage of epigenetic modified marks, future studies will clarify this important issue.
Molecular Mechanisms of Epigenetic Remodeling by Xenobiotics
The driving force for the present review is the hypothesis that xenobiotic exposure during fetal development may alter gene expression patterns in a heritable manner through modifying the epigenome. Alterations in epigenetic control of gene expression that occur during fetal development disrupt normal unfolding of the program. This disruption can trigger developmental defects or more subtle changes that contribute mechanistically to late onset diseases of adulthood, as seen in the previous section. The stability of epigenetic marks during most of the lifespan limits the degree of alterations resulting from exposure to toxicants. In other words, the consequences of xenobiotic exposure in the context of epigenetic toxicity may be more severe when epigenetic reprogramming is in greatest flux, i.e., during gametogenesis and early embryogenesis.83 Moreover, epigenetic marks can also be remodeled after birth, although an acute and low dose exposure to a toxicant may have a stronger epigenetic effect when compared with a higher dose exposure in the adult if occurring in susceptibility time windows.84
It has been speculated that epigenetic remodeling may not only be transmitted to future generations by in utero exposure, but also through pre-conception mechanisms, not only involving paternal and maternal germline alterations, but also somatic epigenetic remodeling. In the latter case, the flux of epigenetic information is the opposite of what is expected (i.e., soma to germline) and may be mediated by hormones or circulating RNAs. This topic remains highly controversial at the moment and is discussed elsewhere.23
The enzymes responsible for initiating and perpetuating epigenetic events are reliant upon metabolic cofactors. In what manner could xenobiotics influence these cofactors and how could this affect epigenetic processes and transgenerational inheritance? Many xenobiotics generate free radicals as part of their chemistry, thus affecting a cell's redox state. Others draw upon metabolic cofactors to facilitate their removal, or utilize reducing equivalents for their detoxification. Some may interface with the circadian clock to disturb epigenetic regulation. We suggest that through each of these avenues, xenobiotics can perturb epigenetic regulation of gene expression during development by influencing the activities of enzymes responsible for epigenetic events (Fig. 2). In the next sub-sections, we will detail some of these potential mechanisms, which are often found in intricate connection.
Figure 2.
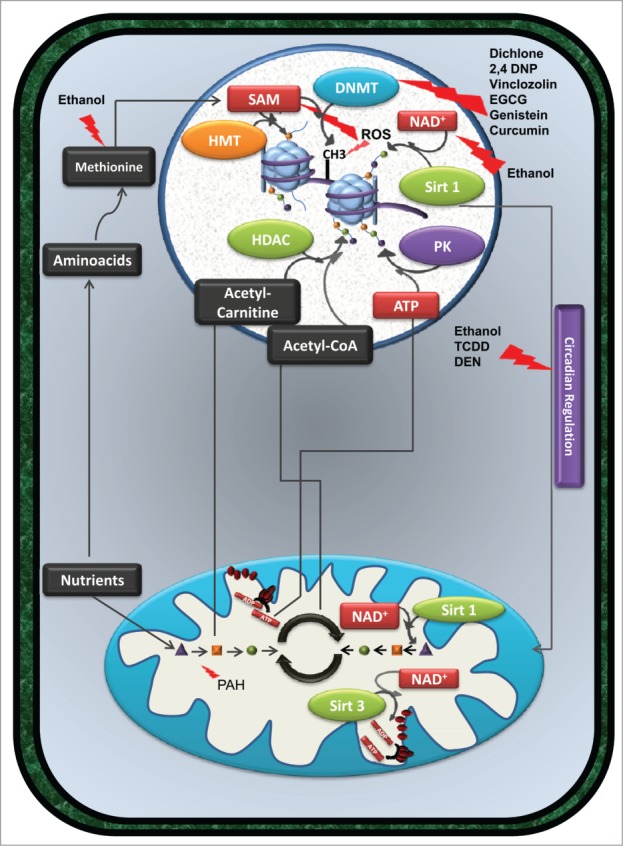
(See previous page). Exposure to xenobiotic compounds does not always cause cell death, but can induce persistent alterations in gene expression and/or metabolism. By interfering directly with the activity of specific enzymes or modulating the availability of specific metabolites, certain xenobiotics promote cellular epigenetic remodeling. Compounds such as dichlone, 2,4 DNP, vinclozolin, EGCG, genistein, and curcumin affect the activity of DNMT enzymes, altering DNA methylation patterns. Also, ethanol consumption, by interfering with methionine metabolism and consequently with SAM levels, results in a similar effect. Furthermore, the detoxification of ethanol by alcohol dehydrogenases decreases the availability of NAD+ and consequently influences the activity of HDAC such as sirtuin 1, inducing alterations in histone acetylation (green dots connected to histones in the figure). Alterations in the cellular redox balance also influence epigenetic regulation of gene expression. In fact, oxidizing conditions induced by certain xenobiotics inhibit the activity of SAM synthetase and consequently decrease the availability of SAM, necessary for DNMT and HMT activity. ROS produced by several compounds promotes the oxidation of DNA bases; a mutagenic event may interfere with DNA methylation. Interplay between epigenome, metabolism and circadian clocks can also be observed. Since the cellular response to xenobiotics can be regulated by the Clock gene system, compounds such as ethanol, TCDD, PCBs, and DEN alter the expression of clock gene expression and affect mitochondrial metabolism and consequently epigenetics. Metabolism altering agents, such as PAH, can limit the availability of certain metabolites, such as acetyl-carnitine, Acetyl-CoA or ATP, consequently affecting histone acetylation and phosphorylation (purple dots linked to histones in the figure) levels. HDAC enzymes such as Sirt1 and Sirt3 not only promote histone deacetylation but also promote deacetylation of several mitochondrial enzymes involved in mitochondrial metabolism and oxidative phosphorylation. Alterations in the acetylation status of those enzymes affect metabolism and metabolites availability, affecting epigenetic remodeling. These alterations, if occurring in developmental key points, can be passed to downstream generations, increasing their susceptibility to diseases. Abbreviations in figure: 2,4 DNP - 2,4 dichlorophenol; ADP - Adenosine diphosphate; ATP - Adenosine triphosphate; DEN – diethylnitrosamine; DMNT - DNA methyltransferases; EGCG - epigallocatechin-3-gallate; HDAC - histone deacetylases; HMT – histone methyltransferases; NAD+ - Nicotinamide adenine dinucleotide (oxidized form); PAH - polycyclic aromatic hydrocarbons; PK – protein kinase; SAM - s-adenosyl-L-methionine; Sirt1 - Sirtuin 1; Sirt3 – Sirtuin 3; TCDD - 2,3,7,8-tetrachlorodibenzo-p-dioxin, PCB – polychlorinated biphenyls.
Altering donors for epigenetic remodeling
Cellular and mitochondrial metabolism produce precursors that are used by the epigenetic machinery to acetylate, methylate, or phosphorylate targets in DNA and histones. Acetyl-coenzyme A (Acetyl-coA), acetylcarnitine, ATP, and s-adenosyl-L-methionine (SAM), among others, are used by the epigenetic machinery as acetyl, phosphate, or methyl donors. All of them are produced by cellular metabolism, with the mitochondrion being a fulcrum of metabolic processes, due to its role in nutrient and redox sensing.85 Xenobiotics that interfere with cell metabolism, namely with mitochondrial function, may alter the supply of substrates to the epigenetic machinery. Although the link between both observations has not been studied in detail, several xenobiotics that interfere with cell and mitochondrial metabolism also cause epigenetic alterations. As described in the section on Environmental pollutants: polycyclic aromatic hydrocarbons, maternal exposure to PAH results in hypermethylation of IFNγ47 and ACSL family member 3 genes48 in cord blood DNA. Interestingly, exposure to PAH present in house dust during winter but not summer results in decreased mitochondrial DNA content in the blood.86 The relationship between decreased mitochondrial DNA and epigenetic alterations and the potential to affect future generations were not explored, deserving further investigation in this and other cases.
S-adenosyl-methionine (SAM) is a critical metabolite required for epigenetic regulation of gene expression. As the carbon donor for methyltransferases, SAM exists at a critical intersection of metabolism and epigenetics. Both DNA and histone methyltransferases require SAM to establish the epigenotype of cells.87 There is good evidence to support the concept that xenobiotics directly influence SAM levels during development and, in kind, epigenetic regulation of gene expression. Several xenobiotics are detoxified via transmethylation reactions that require SAM as a cofactor. Although not performed in mammals, a study by Zhang et al.88 shows that 2,4 dichlorophenol (2,4 DNP), an environmental pollutant, increases DNA methylation in the livers of Carassius auratus (the common goldfish). These authors reported that this hypermethylation results from both increased DNA methyltransferases (DNMT) activity (see below) and also from increased SAM content. Whether this occurs in humans and the transgenerational consequences of such exposure is still to be determined.
Altering the cellular redox balance
Alterations in the cellular redox balance can also contribute to epigenetic remodeling caused by xenobiotics (Fig. 3). SAM is also a key component of the methionine cycle, a biochemical pathway linked to redox biology, epigenetics, and free radical production. Is it possible that the atypical redox state created by xenobiotics influences epigenetic regulation of gene expression in development? Development of an organism can be argued to represent a redox spectrum that becomes progressively more oxidizing.89–91 This spectrum is primarily the result of alterations in glutathione (GSH) production. As a general rule, undifferentiated cells have a higher GSH content compared to those that have differentiated.90,92 The effects of xenobiotic-induced oxidative stress in utero may be at different levels. Oxidizing conditions favor shunting sulfur from the methionine cycle into the production of GSH. Furthermore, oxidative stress inhibits SAM synthetase, the enzyme that synthesizes SAM from ATP and methionine (Fig. 3A).93,94 Combined, these observations suggest that toxic substances interfere with SAM levels at a point when they are required to establish the epigenotype of differentiating cells.
Figure 3.
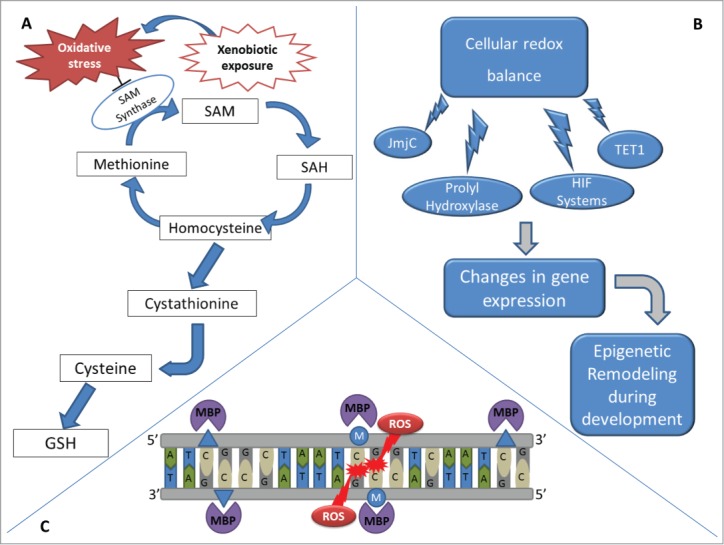
Effects of xenobiotic-induced redox alterations on epigenetics. (A) During embryonic development, xenobiotics may increase the production of ROS in the embryo, exposing proliferating cells to increased oxidative stress. Under oxidizing conditions, the shunting of sulfur from the methionine cycle to the production of GSH is observed. Additionally, oxidative stress also inhibits SAM synthetase. Thus, xenobiotics that interfere with cellular redox balance may change SAM availability when a delicate balance is needed to regulate the epigenotype of differentiating cells. (B) Cellular redox balance also interferes with demethylation enzymes, such as the JmjC superfamily and TET1, as well as with prolyl hydroxylases/hypoxia-inducible factors (HIF) systems, altering epigenetic settings during development and consequently altering gene expression. (C) Oxidation of methylated CpG sites due to the effect of xenobiotics can create either 5hmC or 8-OG and alters MBP binding kinetics, interfering with gene expression and re-methylation of the daughter DNA strands after cellular division.
Redox balance is also linked to the removal of epigenetic marks (Fig. 3B). The recent discovery of enzymes that remove methyl groups from DNA and histones has increased our understanding of concerted epigenetic alterations during development. Members of the JmjC superfamily of lysine demethylases and the Ten-11 translocation methylcytosine dioxygenase 1 (TET1) DNA demethylases are powerful oxidases capable of reversing histone and DNA methylation.95 These enzymes significantly impact the cell's epigenome (Fig. 3B). Histone lysines can be mono-, di-, or tri-methylated.96 Shifting a lysine's methylation status between these different degrees of methylation quickly influences chromatin structure. Likewise, TET1-mediated DNA demethylation can rapidly alter a cell's epigenetic landscape.97 Powering both enzyme families are 4 components: Fe(II), α-ketoglutarate, molecular oxygen, and ascorbate.97 These 4 components impact developing organisms by influencing the activities of these demethylases.
Oxygen is potentially the most powerful morphogenic compound to influence an organism's development.4,89-91 Gradients of oxygen are responsible for directing the development of many organ systems in mammals. The discovery of demethylase enzymes, along with the prolyl hydroxylase/ hypoxia-inducible factors (HIF) system, links oxygen to the gene expression changes during development. Ascorbate is another critical cofactor that can possibly be involved in molding the epigenome of cells during development. Work from Ernst Wolvetang's group has established that ascorbate levels can influence the epigenotypes of human embryonic stem cells grown in tissue culture by increasing DNA demethylation.98,99
Oxidation of DNA bases by xenobiotics presents another way to induce epigenetic alterations. DNA oxidation is generally considered to be a mutagenic event.100 However, it is also capable of altering DNA methylation. Methylation of cytosine at the 5-position of cytosine's pyrimidine ring by DNMTs occurs almost exclusively at clusters of CpG dinucleotides, CpG islands in mammals, and is associated with gene silencing. Both members of the dinucleotide are subject to oxidation resulting in epigenetic alterations. The 5-methylcytosine is oxidized by the TET1 demethylases to create 5-hydroxymetylcytosine (5hmC). This oxidation also occurs by direct oxidation of 5-metylcytosine (5mC) via free radical attack. In both enzymatic and non-enzymatic direct oxidation mechanisms, 5hmC is removed from DNA by hmC glycosylase.97
The oxidation of guanine to 8-oxoguanine (8-OG) is an archetype for xenobiotic induced mutations. However, there are also epigenetic consequences associated with 8-OG. If 8-OG exists within a CpG it inhibits DNMT binding, resulting in the start of an epigenetic alteration. Methylated CpGs are also bound by several types of methyl DNA binding proteins (MBPs) that serve as a link between DNA methylation and histone modifications (Fig. 3C). MBPs play an important role in development to establish the epigenome of cells. Oxidation of a methylated CpG to create either 5hmC or 8-OG alters MBP binding kinetics and influences gene expression101 and remethylation of the daughter DNA strand after replication (Fig. 3C).
Altering DNA or chromatin remodeling enzyme activity and expression
The epigenetic machinery comprises a large set of primary enzymes and regulators, which are not only modulated by the available substrates and targets but also through several signaling pathways. Inhibition or stimulation of these players by epigenetic pharmacology is not uncommon and in fact is a proposed therapy against many cancers. Since this falls outside the scope of this review, the reader is directed to some excellent reviews.102–104
The activity of DNMT enzymes is often altered by xenobiotics either by effects at the protein or mRNA level. Examples include dichlone, a pesticide/fungicide, which mimics the DNMT inhibitor 5-azacytidine,105 or the aquatic pollutant 2,4-DNP, which increases DNMT expression and induces DNA hypermethylation in goldfish, as seen above.88
As described in the section on Lifestyle-derived epigenetic modulators: environmental tobacco smoke and ethanol consumption, cigarette smoking disturbs epigenetic regulation. Maternal tobacco smoking results in placental DNA methylation, which is directly associated with the gestational age.106 Several studies confirmed that alterations in DNA methylation result from in utero exposure to cigarette smoking.106,107 In fact, DNA methylation is suggested as a reliable marker of exposure to tobacco smoke,108 which has the potential to be screened in the offspring and even in following generations. Beyond damaging DNA directly,109 cigarette smoking or its components also modulate DNMT1 mRNA, protein and activity in multiple biological models including murine neurons,110 human bronchial epithelial cells111 and in different human tumors.112 Furthermore, cigarette smoke contains acrolein, which forms adducts with histone proteins and decreases histone acetylation.113
Another interesting example is vinclozolin, a fungicide presenting anti-androgenic effects in rats114 (see Environmental pollutants: Endocrine-disrupting chemicals). Vinclozolin treatment resulted in decreased DNMT1 and DNMT3L expression in offspring testis, which can explain the variable promoter methylation patterns found. A recent paper strengthened the transgenerational effect of vinclozolin exposure. The exposure of F0 gestating females to that fungicide resulted in close to 400 differently expressed genes, increased spermatogenic cell death, and male infertility in the F3 generation.115
Adult consumption of alcohol induces alterations in methionine metabolism and SAM levels that are accompanied by changes in DNA methylation or histone acetylation, contributing to the altered epigenome and the FAS phenotype, as described in the section on Lifestyle-derived epigenetic modulators: environmental tobacco smoke and ethanol consumption.116,117 Histone acetylation is removed through the activity of histone deacetylases (HDAC). Sirtuins, a class of NAD+ dependent HDACs linked to aging and cancer, have activities that are extremely sensitive to the cell's NAD+/NADH ratio.118 The detoxification of alcohol by alcohol dehydrogenase could influence these enzymes by modifying the availability of NAD+ and NAD+/NADH in cells. Also, since alcoholism results in folate deficiency, some of the epigenetic modifications described in offspring exposed in utero might be mediated by altered folate metabolism which affect enzyme-mediated DNA methylation and histone acetylation.119
Finally, DES-induced altered DNA methylation was also accompanied by increased expression of epigenetic regulators, such as DNMT1 and DNMT3a enzymes. DES-induced DNMT3a expression was observed in the male but not in the female offspring although DES increased DNA methylation of the calsequestrin 2 promoter in both male and female offspring. Neonatal exposure to DES in mice also altered the expression of DNMT1, DNMT3a, and DNMT3b.120
Xenobiotic epigenetic toxicity and the circadian clock
A clearly relevant interplay exists between the epigenome, metabolism, and circadian clocks.121 Response to xenobiotics is regulated by the circadian clock at multiple levels. One example is the hepatotoxicity of ethanol, which is apparently regulated by the clock gene Per1, since deletion of it decreases toxicity.122 Specifically, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the clock genes Bmal1 and Per2 in the ovary, disrupting in the process aryl hydrocarbon receptor (AhR) circadian expression.123 Other examples include diethylnitrosamine (DEN)-induced cytotoxicity on mouse primary hepatocytes, which is very dependent on the Clock gene system,124 and a role for hypoxia and HIF-1α in regulating AhR signaling via aryl hydrocarbon receptor nuclear translocator (ARNT) downstream of polychlorinated biphenyl (PCB) 126 exposure.125 Like Clock and other period-regulating proteins, HIF-1α, ARNT and AhR are all PAS (period circadian protein-ARNT- single-minded protein) domain-containing proteins. A link between metabolism and the circadian clock is evident in the demonstration that Clock-deficient (Clock-/-) mice have altered acetylation of liver mitochondrial proteins, resulting in a variation of metabolites over the circadian cycle.126 This variation of metabolites and gene expression with the circadian rhythm127 may impact epigenetics through variations in metabolites and enzyme expression.121,127 Moreover, it is hardly surprising that Clock, a key transcription factor for the circadian machinery, mediates histone acetyl transferase (HAT) activity which is counteracted by the circadian-controlled activity of Sirt1.128-130 From these examples, one can speculate that xenobiotics that interact with the Clock-controlled machinery may have a downstream persistent effect on metabolism and gene expression through epigenetic remodeling. This is more relevant if we consider that xenobiotic exposure during critical fetal epigenetic remodeling windows (see above) may be aggravated by the fact that the circadian clock is active from an early stage of development.131
miRNAs and epigenetic transgenerational transmission of xenobiotic-induced epigenetic remodeling
miRNAs are small, non-coding RNA molecules, usually smaller than 25 nucleotides, and which can negatively regulate gene expression.132 This type of conserved RNA is mostly originated from the nucleus, with recent data suggesting that miRNA may also exist in the mitochondrial matrix, where they are derived from mitochondrial DNA.133,134 While xenobiotics can acutely or chronically alter miRNA expression, differences in miRNA expression can alter the intra-cellular content in nuclear receptors and metabolizing enzymes, including CYP1B1 and CYP2E1 systems. The latter effects can alter the host response to xenobiotics; an excellent detailed review on this topic can be seen in Yokoi, 2013.135 Disturbances in miRNA patterns have been measured in vivo after exposure to several pollutants including 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD),136,137 tamoxifen,138 arsenic,139 and hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX).140 The link between miRNA pattern remodeling and physiological effects is far from being understood. Nonetheless, it appears that prenatal exposure to chemical agents can increase the susceptibility to disease of future generations in part by modulating miRNA expression profiles. For example, TCDD causes pre-natal alterations in miRNA expression patterns capable of affecting as many as 15 pathways downstream, including those related with inflammation, renal and urological diseases, cell death, and carcinogenesis.141 Still, it is not known whether the observed alterations in miRNA profiling contribute to acute toxicity.137 Although miRNA-mediated effects explain the possible increased incidence of chronic diseases in later adulthood, it does not immediately explain how the increased miRNA pattern alterations can translate into a transgenerational transmission. Very little if any work has been done in this regard, particularly in elucidating the role of miRNA alterations in xenobiotic toxicity. miRNA-mediated transgenerational transmission has been proposed in the context of circulating miRNA, which would link somatic alterations with epigenetic reprogramming in germ cells.142 Although the idea is clever, the theory is not supported by experimental data but rather by bioinformatics analysis. Cigarette smoking alters miRNA expression in human spermatozoa, thus aiding in understanding the possible transmission of garbled gene expression to the progeny, although this has never been demonstrated in future generations.143 With alternative in vivo systems, including zebrafish,144 transgenerational effects via miRNA should be better clarified in the future.
Can the Altered Epigenome be Reset?
The observations above suggest that the epigenome can be altered by xenobiotic effects, possibly resulting in persistent alterations in gene expression that span generations. If the link between xenobiotic transgenerational alterations of the epigenome and development of different pathologies is established, one relevant question is the potential to reset the epigenome to its original state, or even to potentiate xenobiotic effects. Although still under debate, it is possible that some dietary components or physical activity may modify the epigenome, possibly antagonizing previous xenobiotic-induced alterations.
Taking into account the knowledge regarding epigenetic remodeling by certain dietary components,145 the expression “you are what you eat” gains more relevance, especially in the context of pre-conception or in utero nutritional programming of chronic disease incidence in subsequent generations.25,146 The belief that food controls our health is not novel, but the control of gene expression by bioactive dietary compounds supplies an explanation for certain unanswered phenomena.145,147,148 On the positive side, certain dietary compounds such as genistein, a component of soy products, epigallocatechin-3-gallate (EGCG), found in green tea, sulforaphane, normally present in cruciferous vegetables, curcumin, present in curry and turmeric, caffeic acid, one of coffee components, and resveratrol, normally associated with grapes and wild berries, show beneficial properties against several pathologies, including cancer, cardiovascular, and neurodegenerative diseases.145 These diverse compounds interfere with DNA methylation and histone acetylation/deacetylation and methylation/demethylation, as well as with the expression of miRNAs and other non-coding RNAs, thereby altering cellular epigenetics and altering gene expression.145 Of particular importance, dietary constituents may be able to reverse abnormal gene activation or silencing observed during aging in distinct pathologies or induced by toxic compounds. Among pathologies, cancer is the most well studied example. The modulation of DNA methylation by curcumin in colorectal cancer cells, inducing both hyper- and hypo-methylation in partially methylated CpG loci, is a potential mechanism of colon cancer chemoprevention.149 EGCG inhibits DNMTs activity and reactivates tumor suppressor genes silenced by methylation in cancer cells.150 Genistein is also able to reverse DNA hypermethylation by a direct inhibition of DNMTs in esophageal cell carcinoma and prostate and breast cancer cells, reactivating tumor suppressor genes.151,152 An enhancement in histone acetylation and demethylation in renal and prostate cancer cells was also observed after genistein treatment.153,154
Although the literature is scarce regarding whether dietary components can modulate xenobiotic epigenetic toxicity, the evidence from cancer models strongly suggests that dietary manipulations may be effective in reverting epigenetic alterations.
Similar to diet, physical exercise can mitigate disturbances associated with several pathophysiological conditions at distinct levels of tissue and cellular organization.155-157 The beneficial effects related to “healthy” lifestyles, including those from exercise, are mediated by the modification of gene expression, which is orchestrated in part by epigenetic modifications to DNA.158,159 Through histone modifications, DNA methylation and expression of non-coding RNAs, physical exercise regulates the expression of genes involved in metabolic and inflammatory processes, aging, cancer, and in central nervous system degeneration. These genes include Peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1alpha), pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4), apoptosis-associated speck-like protein containing a CARD (PYCARD), and BDNF.160 It has been proposed that physical exercise regulates the epigenetic landscape indirectly through a metabolic remodeling, involving alterations in the production of tricarboxylic acid cycle intermediates that are involved in epigenetic engineering.161 The question remains whether exercise-induced epigenetic modifications can be transmitted to subsequent generations and, thus, cause a persistent modulation of gene expression. Since many genes epigenetically influenced by nutrition are also affected by exercise (or are involved in metabolic processes modified by exercise) it can be hypothesized that performing exercise during pregnancy could mediate adaptive conditions favoring positive health outcomes in the next generation through epigenetic modifications.162 Hopkins et al.163,164 showed that moderate-intensity exercise during the second half of pregnancy in healthy subjects induced a slight decrease in offspring weight and percentage of fat, with concomitant modulation effects in maternal and offspring blood cord metabolic-related outcomes, including IGF-I and -II, leptin and serum fatty acids. Whether this altered the epigenetic make-up in the fetus was not determined. Ronn et al. demonstrated that a 6-month exercise program in healthy men resulted in a genome-wide changes in DNA methylation in human adipose tissue, with a higher preponderance of increased promoter methylation.165 Furthermore, 6 weeks of voluntary wheel exercise prior to and during pregnancy prevented maternal high-fat diet-induced skeletal muscle PGC-1α promoter hypermethylation and increased its expression as well as its target genes. This effect correlated with an improvement of age-associated metabolic dysfunction observed at 9 months of age in the offspring.166 Whether different types, intensities and timings of exercise during pregnancy alter the potential epigenetic programming in different healthy and diseased cohorts is still worthy of investigation. To date, to the best of our knowledge, there are no data associating physical activity and reversion/prevention of xenobiotic-induced epigenetic toxicity.
In this review we have primarily focused on the evidences to support that diet and exercise can contribute to maintain a “healthy” epigenome. Yet, there are additional lifestyle factors, such as psychosocial events, which can contribute to modulate the epigenome as well. This modulation may work in both ways, either exacerbating or mitigating xenobiotic-derived effects. Although this topic is out of the scope of the present review, there are good examples that this may occur in humans. For example, psychosocial stress can alter the regulation of the glucocorticoid receptor by modifying its promoter methylation.167 Furthermore, lower methylation of the glucocorticoid receptor gene was measured in blood samples from humans suffering from posttraumatic stress disorder.168 Of interest and not surprisingly, the methylation state of the glucocorticoid receptor gene promoter in genomic DNA from cord blood mononuclear cells was significantly associated with maternal anxiety.169 How stress-initiated epigenetic events in the womb cross-talk with xenobiotic effects at the same level remain to be elucidated.
Epigenome Toxicology: Perpetuating the phenotype? – A perspective
The previous notion that a toxicological response was based in the loss of cell or organism viability has changed and now acknowledges that a toxic response can occur for much lower dosages than previously thought. Xenobiotics may cause a persistent up- or down-regulation of signaling pathways or gene expression patterns, thereby resulting in non-lethal cellular changes that will impact the long term cell physiology under basal or stress conditions.
Exposure of the maternal and paternal lineages to xenobiotics can not only cause immediate toxicity, but can also result in alterations of the epigenome in germ cells, the fetus, and in germ cells of the fetus that may lead to persistent gene expression alterations in later generations. This type of toxicity is currently impossible to foresee when predicting molecule safety and only multi-generational animal studies can be used to conclude a true transgenerational effect. Still, some of the observed effects may depend on the strain of animal used68 and may not be reproducible. The same may occur in humans. Although a study spanning 3 human generations is obviously difficult to attain, it is possible that individual differences in the epigenome or in its regulatory machinery may impact the transgenerational effects of a certain agent. This possible idiosyncratic nature of transgenerational transmission of toxicity responses further complicates the picture. Since multiple mechanisms and targets can be involved, and since there is not real knowledge of the exposure times and dosages (and in which time-frame) necessary for the effect to be observed in future generations, this implies that novel assessment methods and models are necessary. It is not simply necessary to understand the different up and downstream mediators and effectors of each toxicity pathway (for example, ref.170); the focus of modern toxicology should be how each molecule or mixture of molecules disturbs each of those partners.171
Disturbance of epigenetic markers by foreign agents is clearly a novel challenge for new toxicology. Studies conducted so far and presented here are only the tip of the iceberg, since the list of xenobiotics with relevance for human health is immense. A deeper knowledge of how these agents interact with the epigenetic landscape in cells and how those changes are passaged to future generations should be incorporated in future toxicological studies. We believe that this review, based on experimental data and personal perspectives from the authors, will pave the way for obtaining critical knowledge in this field.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are extremely thankfully to Alexandra Holy, MBA/MPP, Mills College, Oakland, CA, USA for proofreading English language in this manuscript.
Funding
Supported by the Foundation for Science and Technology (FCT Portugal) and co-funded by FEDER/Compete and National Budget, grants PTDC/DTP-FTO/1180/2012, SFRH/BDP/4225/2007, PTDC/DTP-DES/1071/2012 - FCOMP-01-0124-FEDER-028617, PTDC/DTP-DES/1246/2012 - FCOMP-01-0124-FEDER-028618, PEst-OE/SAU/UI0617/2011 and PEst-C/SAU/LA0001/2013-2014; by NIEHS grant P42 ES013663-5099; by NIH grants P30 CA086862-8604, R01 CA115438, PO1 HD21350 R24 RR025866, R24 RR031459, R21 HD071306 and R21 HD072518; by Fundação Montepio Geral, Portugal; by Ministerio de Economia y Competitividad, Spain grant BFU2011-2973; Instituto de Salud Carlos III, Spain grant CP11/00312 and by The European Foundation for the Study of Diabetes (EFS/Novo Nordisk Programme, 2010 grant 80800). VAS is supported by QREN project #4832, ref. CENTRO-07-ST24-FEDER-002008.
References
- 1.Waddington CH. Principles of embryology. New York: Macmillan; 1956. [Google Scholar]
- 2.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381–95; PMID:21321607; http://dx.doi.org/ 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci 2010; 35:618–26; PMID:20685123; http://dx.doi.org/ 10.1016/j.tibs.2010.05.006 [DOI] [PubMed] [Google Scholar]
- 4.Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med 2007; 43:1023–36; PMID:17761298; http://dx.doi.org/ 10.1016/j.freeradbiomed.2007.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 2014; 157:95–109; PMID:24679529; http://dx.doi.org/ 10.1016/j.cell.2014.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzio EA, Soliman KF. Basic concepts of epigenetics: impact of environmental signals on gene expression. Epigenetics 2012; 7:119–30; PMID:22395460; http://dx.doi.org/ 10.4161/epi.7.2.18764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science 2010; 330:612–6; PMID:21030644; http://dx.doi.org/ 10.1126/science.1191078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 2007; 128:635–8; PMID:17320500; http://dx.doi.org/ 10.1016/j.cell.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 9.Johnson CH, Patterson AD, Idle JR, Gonzalez FJ. Xenobiotic Metabolomics: Major Impact on the Metabolome. Annu Rev Pharmacol 2012; 52:37–56; PMID:21819238; http://dx.doi.org/ 10.1146/annurev-pharmtox-010611-134748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollati V, Baccarelli A. Environmental epigenetics. Heredity (Edinb) 2010; 105:105–12; PMID:20179736; http://dx.doi.org/ 10.1038/hdy.2010.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corry GN, Tanasijevic B, Barry ER, Krueger W, Rasmussen TP. Epigenetic regulatory mechanisms during preimplantation development. Birth Defects Res C Embryo Today 2009; 87:297–313; PMID:19960551; http://dx.doi.org/ 10.1002/bdrc.20165 [DOI] [PubMed] [Google Scholar]
- 12.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447:425–32; PMID:17522676; http://dx.doi.org/ 10.1038/nature05918 [DOI] [PubMed] [Google Scholar]
- 13.Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics 2011; 6:791–7; PMID:21636976; http://dx.doi.org/ 10.4161/epi.6.7.16209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lees-Murdock DJ, Walsh CP. DNA methylation reprogramming in the germ line. Adv Exp Med Biol 2008; 626:1–15; PMID:18372787; http://dx.doi.org/ 10.1007/978-0-387-77576-0_1 [DOI] [PubMed] [Google Scholar]
- 15.Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet 2005; 14(Spec No 1):R47–58; PMID:15809273; http://dx.doi.org/ 10.1093/hmg/ddi114 [DOI] [PubMed] [Google Scholar]
- 16.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 2002; 241:172–82; PMID:11784103; http://dx.doi.org/ 10.1006/dbio.2001.0501 [DOI] [PubMed] [Google Scholar]
- 17.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989; 2:577–80; PMID:2570282; http://dx.doi.org/ 10.1016/S0140-6736(89)90710-1 [DOI] [PubMed] [Google Scholar]
- 18.Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res 2007; 61:5R–10R; PMID:17413851; http://dx.doi.org/ 10.1203/pdr.0b013e318045bedb [DOI] [PubMed] [Google Scholar]
- 19.Zambrano E, Martinez-Samayoa PM, Bautista CJ, Deas M, Guillen L, Rodriguez-Gonzalez GL, Guzman C, Larrea F, Nathanielsz PW. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J Physiol 2005; 566:225–36; PMID:15860532; http://dx.doi.org/ 10.1113/jphysiol.2005.086462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet 2012; 13:153–62; PMID:22290458; http://dx.doi.org/ 10.1038/nrm3288 [DOI] [PubMed] [Google Scholar]
- 21.Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol 2008; 25:2–6; PMID:17949945; http://dx.doi.org/ 10.1016/j.reprotox.2007.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8:253–62; PMID:17363974; http://dx.doi.org/ 10.1038/nrg2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma A. Transgenerational epigenetic inheritance: focus on soma to germline information transfer. Prog Biophys Mol Biol 2013; 113:439–46; PMID:23257323; http://dx.doi.org/ 10.1016/j.pbiomolbio.2012.12.003 [DOI] [PubMed] [Google Scholar]
- 24.Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol 2011; 31:363–73; PMID:21256208; http://dx.doi.org/ 10.1016/j.reprotox.2010.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abel EL. Smoking during pregnancy: a review of effects on growth and development of offspring. Hum Biol 1980; 52:593–625; PMID:7009384 [PubMed] [Google Scholar]
- 26.Rantakallio P. The effect of maternal smoking on birth weight and the subsequent health of the child. Early Hum Dev 1978; 2:371–82; PMID:750195; http://dx.doi.org/ 10.1016/0378-3782(78)90064-6 [DOI] [PubMed] [Google Scholar]
- 27.Grabenhenrich LB, Gough H, Reich A, Eckers N, Zepp F, Nitsche O, Forster J, Schuster A, Schramm D, Bauer CP, et al.. Early-life determinants of asthma from birth to age 20 years: A German birth cohort study. J Allergy Clin Immunol 2014; 133(4):979–88; PMID:24461583; http://dx.doi.org/ 10.1016/j.jaci.2013.11.035 [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Joad JP, Zhong C, Pinkerton KE. Effects of environmental tobacco smoke exposure on pulmonary immune response in infant monkeys. J Allergy Clin Immunol 2008; 122:400–6, 6 e1-5; PMID:18502491; http://dx.doi.org/ 10.1016/j.jaci.2008.04.011 [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Joad JP, Abel K, Spinner A, Smiley-Jewell S, Liu H, Pinkerton KE. Effects of environmental tobacco smoke on the developing immune system of infant monkeys. J Allergy Clin Immunol 2007; 120:445–51; PMID:17482667; http://dx.doi.org/ 10.1016/j.jaci.2007.03.028 [DOI] [PubMed] [Google Scholar]
- 30.Wilhelm-Benartzi CS, Koestler DC, Houseman EA, Christensen BC, Wiencke JK, Schned AR, Karagas MR, Kelsey KT, Marsit CJ. DNA methylation profiles delineate etiologic heterogeneity and clinically important subgroups of bladder cancer. Carcinogenesis 2010; 31:1972–6; PMID:20802236; http://dx.doi.org/ 10.1093/carcin/bgq178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makin J, Fried PA, Watkinson B. A comparison of active and passive smoking during pregnancy: long-term effects. Neurotoxicol Teratol 1991; 13:5–12; PMID:2046627; http://dx.doi.org/ 10.1016/0892-0362(91)90021-N [DOI] [PubMed] [Google Scholar]
- 32.Rantakallio P. A follow-up study up to the age of 14 of children whose mothers smoked during pregnancy. Acta Paediatr Scand 1983; 72:747–53; PMID:6637474; http://dx.doi.org/ 10.1111/j.1651-2227.1983.tb09805.x [DOI] [PubMed] [Google Scholar]
- 33.Suter MA, Anders AM, Aagaard KM. Maternal smoking as a model for environmental epigenetic changes affecting birthweight and fetal programming. Mol Hum Reprod 2013; 19:1–6; PMID:23139402; http://dx.doi.org/ 10.1093/molehr/gas050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, et al.. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425–31; PMID:22851337; http://dx.doi.org/ 10.1289/ehp.1205412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breton CV, Salam MT, Gilliland FD. Heritability and role for the environment in DNA methylation in AXL receptor tyrosine kinase. Epigenetics 2011; 6:895–8; PMID:21555911; http://dx.doi.org/ 10.4161/epi.6.7.15768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flom JD, Ferris JS, Liao Y, Tehranifar P, Richards CB, Cho YH, Gonzalez K, Santella RM, Terry MB. Prenatal smoke exposure and genomic DNA methylation in a multiethnic birth cohort. Cancer Epidemiol Biomarkers Prev 2011; 20:2518–23; PMID:21994404; http://dx.doi.org/ 10.1158/1055-9965.EPI-11-0553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markunas CA, Xu Z, Harlid S, Wade PA, Lie RT, Taylor JA, Wilcox AJ. Identification of DNA Methylation Changes in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect 2014; 122:1147–53; PMID:24906187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Talbot P, Lin S. The effect of cigarette smoke on fertilization and pre-implantation development: assessment using animal models, clinical data, and stem cells. Biol Res 2011; 44:189–94; PMID:22513422; http://dx.doi.org/ 10.4067/S0716-97602011000200-011 [DOI] [PubMed] [Google Scholar]
- 39.Pineles BL, Park E, Samet JM. Systematic review and meta-analysis of miscarriage and maternal exposure to tobacco smoke during pregnancy. AmJ Epidemiol 2014; 179:807–23; PMID:24518810; http://dx.doi.org/ 10.1093/aje/kwt334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pruett D, Waterman EH, Caughey AB. Fetal alcohol exposure: consequences, diagnosis, and treatment. Obstet Gynecol Surv 2013; 68:62–9; PMID:23322082; http://dx.doi.org/ 10.1097/OGX.0b013e31827f238f [DOI] [PubMed] [Google Scholar]
- 41.Ungerer M, Knezovich J, Ramsay M. In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res 2013; 35:37–46; PMID:24313163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009; 4:500–11; PMID:20009564; http://dx.doi.org/ 10.4161/epi.4.7.9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol Clin Exp Res 1991; 15:395–8; PMID:1877725; http://dx.doi.org/ 10.1111/j.1530-0277.1991.tb00536.x [DOI] [PubMed] [Google Scholar]
- 44.Downing C, Johnson TE, Larson C, Leakey TI, Siegfried RN, Rafferty TM, Cooney CA. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: effects of a methyl-supplemented diet. Alcohol 2011; 45:65–71; PMID:20705422; http://dx.doi.org/ 10.1016/j.alcohol.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 2010; 6:e1000811; PMID:20084100; http://dx.doi.org/ 10.1371/journal.pgen.1000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbstman JB, Tang D, Zhu D, Qu L, Sjodin A, Li Z, Camann D, Perera FP. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[apyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect 2012; 120:733–8; PMID:22256332; http://dx.doi.org/ 10.1289/ehp.1104056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang WY, Levin L, Talaska G, Cheung YY, Herbstman J, Tang D, Miller RL, Perera F, Ho SM. Maternal exposure to polycyclic aromatic hydrocarbons and 5'-CpG methylation of interferon-gamma in cord white blood cells. Environ Health Perspect 2012; 120:1195–200; PMID:22562770; http://dx.doi.org/ 10.1289/ehp.1103744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, Ho SM. Relation of DNA methylation of 5'-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One 2009; 4:e4488; PMID:19221603; http://dx.doi.org/ 10.1371/journal.pone.0004488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson NK, Chuang JC, Morgan MK, Lordo RA, Sheldon LS. An observational study of the potential exposures of preschool children to pentachlorophenol, bisphenol-A, and nonylphenol at home and daycare. Environ Res 2007; 103:9–20; PMID:16750524; http://dx.doi.org/ 10.1016/j.envres.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 50.Rochester JR. Bisphenol A and human health: a review of the literature. Reprod Toxicol 2013; 42:132–55; PMID:23994667; http://dx.doi.org/ 10.1016/j.reprotox.2013.08.008 [DOI] [PubMed] [Google Scholar]
- 51.Calhoun KC, Padilla-Banks E, Jefferson WN, Liu L, Gerrish KE, Young SL, Wood CE, Hunt PA, Vandevoort CA, Williams CJ. Bisphenol a exposure alters developmental gene expression in the fetal rhesus macaque uterus. PLoS One 2014; 9:e85894; PMID:24465770; http://dx.doi.org/ 10.1371/journal.pone.0085894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hunt PA, Lawson C, Gieske M, Murdoch B, Smith H, Marre A, Hassold T, VandeVoort CA. Bisphenol A alters early oogenesis and follicle formation in the fetal ovary of the rhesus monkey. Proc Natl Acad Sci U S A 2012; 109:17525–30; PMID:23012422; http://dx.doi.org/ 10.1073/pnas.1207854109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolstenholme JT, Rissman EF, Connelly JJ. The role of Bisphenol A in shaping the brain, epigenome and behavior. Horm Behav 2011; 59:296–305; PMID:21029734; http://dx.doi.org/ 10.1016/j.yhbeh.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 2007; 104:13056–61; PMID:17670942; http://dx.doi.org/ 10.1073/pnas.0703739104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 1999; 23:314–8; PMID:10545949; http://dx.doi.org/ 10.1038/15490 [DOI] [PubMed] [Google Scholar]
- 56.Nahar MS, Kim JH, Sartor MA, Dolinoy DC. Bisphenol A-associated alterations in the expression and epigenetic regulation of genes encoding xenobiotic metabolizing enzymes in human fetal liver. Environ Mol Mutagen 2014; 55:184–95; PMID:24214726; http://dx.doi.org/ 10.1002/em.21823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kundakovic M, Gudsnuk K, Franks B, Madrid J, Miller RL, Perera FP, Champagne FA. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci U S A 2013; 110:9956–61; PMID:23716699; http://dx.doi.org/ 10.1073/pnas.1214056110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel BB, Raad M, Sebag IA, Chalifour LE. Lifelong exposure to bisphenol a alters cardiac structure/function, protein expression, and DNA methylation in adult mice. Toxicol Sci 2013; 133:174–85; PMID:23418087; http://dx.doi.org/ 10.1093/toxsci/kft026 [DOI] [PubMed] [Google Scholar]
- 59.Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS One 2013; 8:e55387; PMID:23359474; http://dx.doi.org/ 10.1371/journal.pone.0055387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolstenholme JT, Edwards M, Shetty SR, Gatewood JD, Taylor JA, Rissman EF, Connelly JJ. Gestational exposure to bisphenol a produces transgenerational changes in behaviors and gene expression. Endocrinology 2012; 153:3828–38; PMID:22707478; http://dx.doi.org/ 10.1210/en.2012-1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinez-Arguelles DB, Culty M, Zirkin BR, Papadopoulos V. In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology 2009; 150:5575–85; PMID:19819939; http://dx.doi.org/ 10.1210/en.2009-0847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang SC, Lee BM. DNA methylation of estrogen receptor alpha gene by phthalates. J Toxicol Environ Health A 2005; 68:1995–2003; PMID:16326419; http://dx.doi.org/ 10.1080/15287390491008913 [DOI] [PubMed] [Google Scholar]
- 63.Uzumcu M, Suzuki H, Skinner MK. Effect of the anti-androgenic endocrine disruptor vinclozolin on embryonic testis cord formation and postnatal testis development and function. Reprod Toxicol 2004; 18:765–74; PMID:15279874; http://dx.doi.org/ 10.1016/j.reprotox.2004.05.008 [DOI] [PubMed] [Google Scholar]
- 64.Wickerham EL, Lozoff B, Shao J, Kaciroti N, Xia Y, Meeker JD. Reduced birth weight in relation to pesticide mixtures detected in cord blood of full-term infants. Environ Int 2012; 47:80–5; PMID:22796478; http://dx.doi.org/ 10.1016/j.envint.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anway MD, Leathers C, Skinner MK. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 2006; 147:5515–23; PMID:16973726; http://dx.doi.org/ 10.1210/en.2006-0640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308:1466–9; PMID:15933200; http://dx.doi.org/ 10.1126/science.1108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paoloni-Giacobino A. Epigenetic effects of methoxychlor and vinclozolin on male gametes. Vitam Horm 2014; 94:211–27; PMID:24388192; http://dx.doi.org/ 10.1016/B978-0-12-800095-3.00008-0 [DOI] [PubMed] [Google Scholar]
- 68.Guerrero-Bosagna C, Covert TR, Haque MM, Settles M, Nilsson EE, Anway MD, Skinner MK. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod Toxicol 2012; 34:694–707; PMID:23041264; http://dx.doi.org/ 10.1016/j.reprotox.2012.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bromer JG, Wu J, Zhou Y, Taylor HS. Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming. Endocrinology 2009; 150:3376–82; PMID:19299448; http://dx.doi.org/ 10.1210/en.2009-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haddad R, Kasneci A, Sebag IA, Chalifour LE. Cardiac structure/function, protein expression, and DNA methylation are changed in adult female mice exposed to diethylstilbestrol in utero. Can J Physiol Pharmacol 2013; 91:741–9; PMID:23984849; http://dx.doi.org/ 10.1139/cjpp-2013-0014 [DOI] [PubMed] [Google Scholar]
- 71.Haddad R, Kasneci A, Mepham K, Sebag IA, Chalifour LE. Gestational exposure to diethylstilbestrol alters cardiac structure/function, protein expression and DNA methylation in adult male mice progeny. Toxicol Appl Pharmacol 2013; 266:27–37; PMID:23142472; http://dx.doi.org/ 10.1016/j.taap.2012.10.018 [DOI] [PubMed] [Google Scholar]
- 72.Herbst AL. Summary of the changes in the human female genital tract as a consequence of maternal diethylstilbestrol therapy. J Toxicol Environ Health Suppl 1976; 1:13–20; PMID:994230 [PubMed] [Google Scholar]
- 73.Bibbo M, Al-Naqeeb M, Baccarini I, Gill W, Newton M, Sleeper KM, Sonek RN, Wied GL. Follow-up study of male and female offspring of DES-treated mothers a preliminary report. J Reprod Med 1975; 15:29–32; PMID:1171234 [PubMed] [Google Scholar]
- 74.Rahman A, Persson LA, Nermell B, El Arifeen S, Ekstrom EC, Smith AH, Vahter M. Arsenic exposure and risk of spontaneous abortion, stillbirth, and infant mortality. Epidemiology 2010; 21:797–804; PMID:20864889; http://dx.doi.org/ 10.1097/EDE.0b013e3181f56a0d [DOI] [PubMed] [Google Scholar]
- 75.Rahman A, Vahter M, Smith AH, Nermell B, Yunus M, El Arifeen S, Persson LA, Ekstrom EC. Arsenic exposure during pregnancy and size at birth: a prospective cohort study in Bangladesh. AmJ Epidemiol 2009; 169:304–12; PMID:19037006; http://dx.doi.org/ 10.1093/aje/kwn332 [DOI] [PubMed] [Google Scholar]
- 76.Koestler DC, Avissar-Whiting M, Houseman EA, Karagas MR, Marsit CJ. Differential DNA methylation in umbilical cord blood of infants exposed to low levels of arsenic in utero. Environ Health Perspect 2013; 121:971–7; PMID:23757598; http://dx.doi.org/ 10.1289/ehp.1205925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pilsner JR, Hall MN, Liu X, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Yunus M, Rahman M, Graziano JH, et al.. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One 2012; 7:e37147; PMID:22662134; http://dx.doi.org/ 10.1371/journal.pone.0037147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Intarasunanont P, Navasumrit P, Waraprasit S, Chaisatra K, Suk WA, Mahidol C, Ruchirawat M. Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ Health 2012; 11:31; PMID:22551203; http://dx.doi.org/ 10.1186/1476-069X-11-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kile ML, Baccarelli A, Hoffman E, Tarantini L, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostofa G, Hsueh YM, Wright RO, et al.. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ Health Perspect 2012; 120:1061–6; PMID:22466225; http://dx.doi.org/ 10.1289/ehp.1104173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jones ML, Mark PJ, Waddell BJ. Maternal dietary omega-3 fatty acids and placental function. Reproduction 2014; 147(5):R143–52; PMID:24451224; http://dx.doi.org/ 10.1530/REP-13-0376 [DOI] [PubMed] [Google Scholar]
- 81.Onishchenko N, Karpova N, Sabri F, Castren E, Ceccatelli S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J Neurochem 2008; 106:1378–87; PMID:18485098; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05484.x [DOI] [PubMed] [Google Scholar]
- 82.Ceccatelli S, Bose R, Edoff K, Onishchenko N, Spulber S. Long-lasting neurotoxic effects of exposure to methylmercury during development. J Intern Med 2013; 273:490–7; PMID:23600401; http://dx.doi.org/ 10.1111/joim.12045 [DOI] [PubMed] [Google Scholar]
- 83.Migicovsky Z, Kovalchuk I. Epigenetic memory in mammals. Front Genet 2011; 2:28; PMID:22303324; http://dx.doi.org/ 10.3389/fgene.2011.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J 2012; 53:289–305; PMID:23744968; http://dx.doi.org/ 10.1093/ilar.53.3-4.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010; 10:12–31; PMID:19796712; http://dx.doi.org/ 10.1016/j.mito.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pieters N, Koppen G, Smeets K, Napierska D, Plusquin M, De Prins S, Van De Weghe H, Nelen V, Cox B, Cuypers A, et al.. Decreased mitochondrial DNA content in association with exposure to polycyclic aromatic hydrocarbons in house dust during wintertime: from a population enquiry to cell culture. PLoS One 2013; 8:e63208; PMID:23658810; http://dx.doi.org/ 10.1371/journal.pone.0063208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem 2012; 23:853–9; PMID:22749138; http://dx.doi.org/ 10.1016/j.jnutbio.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Li H, Qiu Q, Qi Y, Huang D, Zhang Y. 2,4-Dichlorophenol induces global DNA hypermethylation through the increase of S-adenosylmethionine and the upregulation of DNMTs mRNA in the liver of goldfish Carassius auratus. Comp Biochem Physiol C Toxicol Pharmacol 2014; 160:54–9; PMID:24316013; http://dx.doi.org/ 10.1016/j.cbpc.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 89.Sohal RS, Allen RG. Relationship between metabolic rate, free radicals, differentiation and aging: a unified theory In: Molecular biology of aging, Basic Life Sciences. Woodhead AD, Blackett AD, Hollander A, eds. New York: Plenum Press; 1985:75–104. [DOI] [PubMed] [Google Scholar]
- 90.Sohal RS, Allen RG, Nations C. Oxygen free radicals play a role in cellular differentiation: an hypothesis. J Free Radic Biol Med 1986; 2:175–81; PMID:3553300; http://dx.doi.org/ 10.1016/S0748-5514(86)80067-8 [DOI] [PubMed] [Google Scholar]
- 91.Allen RG, Balin AK. Oxidative influence on development and differentiation: an overview of a free radical theory of development. Free Radic Biol Med 1989; 6:631–61; PMID:2666278; http://dx.doi.org/ 10.1016/0891-5849(89)90071-3 [DOI] [PubMed] [Google Scholar]
- 92.Warshaw JB, Wilson CW 3rd, Saito K, Prough RA. The responses of glutathione and antioxidant enzymes to hyperoxia in developing lung. Pediatr Res 1985; 19:819–23; PMID:4034284; http://dx.doi.org/ 10.1203/00006450-198508000-00008 [DOI] [PubMed] [Google Scholar]
- 93.Corrales F, Ochoa P, Rivas C, Martin-Lomas M, Mato JM, Pajares MA. Inhibition of glutathione synthesis in the liver leads to S-adenosyl-L-methionine synthetase reduction. Hepatology 1991; 14:528–33; PMID:1874498 [PubMed] [Google Scholar]
- 94.Corrales F, Gimenez A, Alvarez L, Caballeria J, Pajares MA, Andreu H, Pares A, Mato JM, Rodes J. S-adenosylmethionine treatment prevents carbon tetrachloride-induced S-adenosylmethionine synthetase inactivation and attenuates liver injury. Hepatology 1992; 16:1022–7; PMID:1398482; http://dx.doi.org/ 10.1002/hep.1840160427 [DOI] [PubMed] [Google Scholar]
- 95.Hitchler MJ, Domann FE. Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome. Free Radic Biol Med 2012; 53:2178–87; PMID:23022407; http://dx.doi.org/ 10.1016/j.freeradbiomed.2012.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 2006; 7:715–27; PMID:16983801; http://dx.doi.org/ 10.1038/nrg1945 [DOI] [PubMed] [Google Scholar]
- 97.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev 2011; 25:2436–52; PMID:22156206; http://dx.doi.org/ 10.1101/gad.179184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chung TL, Turner JP, Thaker NY, Kolle G, Cooper-White JJ, Grimmond SM, Pera MF, Wolvetang EJ. Ascorbate promotes epigenetic activation of CD30 in human embryonic stem cells. Stem cells 2010; 28:1782–93; PMID:20715184; http://dx.doi.org/ 10.1002/stem.500 [DOI] [PubMed] [Google Scholar]
- 99.Prowse AB, Doran MR, Cooper-White JJ, Chong F, Munro TP, Fitzpatrick J, Chung TL, Haylock DN, Gray PP, Wolvetang EJ. Long term culture of human embryonic stem cells on recombinant vitronectin in ascorbate free media. Biomaterials 2010; 31:8281–8; PMID:20674971; http://dx.doi.org/ 10.1016/j.biomaterials.2010.07.037 [DOI] [PubMed] [Google Scholar]
- 100.D'Errico M, Parlanti E, Dogliotti E. Mechanism of oxidative DNA damage repair and relevance to human pathology. Mutat Res 2008; 659:4–14; PMID:18083609; http://dx.doi.org/ 10.1016/j.mrrev.2007.10.003 [DOI] [PubMed] [Google Scholar]
- 101.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 2004; 32:4100–8; PMID:15302911; http://dx.doi.org/ 10.1093/nar/gkh739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Campbell RM, Tummino PJ. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest 2014; 124:64–9; PMID:24382391; http://dx.doi.org/ 10.1172/JCI71605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest 2014; 124:56–63; PMID:24382390; http://dx.doi.org/ 10.1172/JCI69736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature 2013; 502:480–8; PMID:24153301; http://dx.doi.org/ 10.1038/nature12751 [DOI] [PubMed] [Google Scholar]
- 105.Ceccaldi A, Rajavelu A, Ragozin S, Senamaud-Beaufort C, Bashtrykov P, Testa N, Dali-Ali H, Maulay-Bailly C, Amand S, Guianvarc'h D, et al.. Identification of novel inhibitors of DNA methylation by screening of a chemical library. ACS Chem Biol 2013; 8:543–8; PMID:23294304; http://dx.doi.org/ 10.1021/cb300565z [DOI] [PubMed] [Google Scholar]
- 106.Maccani JZ, Koestler DC, Houseman EA, Marsit CJ, Kelsey KT. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics 2013; 5:619–30; PMID:24283877; http://dx.doi.org/ 10.2217/epi.13.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet 2013; 4:132; PMID:23882278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shenker NS, Ueland PM, Polidoro S, van Veldhoven K, Ricceri F, Brown R, Flanagan JM, Vineis P. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology 2013; 24:712–6; PMID:23867811; http://dx.doi.org/ 10.1097/EDE.0b013e31829d5cb3 [DOI] [PubMed] [Google Scholar]
- 109.Huang J, Okuka M, Lu W, Tsibris JC, McLean MP, Keefe DL, Liu L. Telomere shortening and DNA damage of embryonic stem cells induced by cigarette smoke. Reprod Toxicol 2013; 35:89–95; PMID:22824788; http://dx.doi.org/ 10.1016/j.reprotox.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 110.Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, Guidotti A. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci U S A 2008; 105:16356–61; PMID:18852456; http://dx.doi.org/ 10.1073/pnas.0808699105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, et al.. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 2010; 29:3650–64; PMID:20440268; http://dx.doi.org/ 10.1038/onc.2010.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kwon YM, Park JH, Kim H, Shim YM, Kim J, Han J, Park J, Kim DH. Different susceptibility of increased DNMT1 expression by exposure to tobacco smoke according to histology in primary non-small cell lung cancer. J Cancer Res Clin Oncol 2007; 133:219–26; PMID:17053888; http://dx.doi.org/ 10.1007/s00432-006-0160-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen D, Fang L, Li H, Tang MS, Jin C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J Biol Chem 2013; 288:21678–87; PMID:23770671; http://dx.doi.org/ 10.1074/jbc.M113.476630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buckley J, Willingham E, Agras K, Baskin LS. Embryonic exposure to the fungicide vinclozolin causes virilization of females and alteration of progesterone receptor expression in vivo: an experimental study in mice. Environ Health 2006; 5:4; PMID:16504050; http://dx.doi.org/ 10.1186/1476-069X-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guerrero-Bosagna C, Savenkova M, Haque MM, Nilsson E, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of altered Sertoli cell transcriptome and epigenome: molecular etiology of male infertility. PLoS One 2013; 8:e59922; PMID:23555832; http://dx.doi.org/ 10.1371/journal.pone.0059922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zakhari S. Alcohol metabolism and epigenetics changes. Alcohol Res 2013; 35:6–16; PMID:24313160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal 2011; 15:551–89; PMID:20919933; http://dx.doi.org/ 10.1089/ars.2010.3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pereira CV, Lebiedzinska M, Wieckowski MR, Oliveira PJ. Regulation and protection of mitochondrial physiology by sirtuins. Mitochondrion 2012; 12:66–76; PMID:21787885; http://dx.doi.org/ 10.1016/j.mito.2011.07.003 [DOI] [PubMed] [Google Scholar]
- 119.Halsted CH, Medici V. Aberrant hepatic methionine metabolism and gene methylation in the pathogenesis and treatment of alcoholic steatohepatitis. Int J Hepatol 2012; 2012:959746; PMID:22007317; http://dx.doi.org/ 10.1155/2012/959746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sato K, Fukata H, Kogo Y, Ohgane J, Shiota K, Mori C. Neonatal exposure to diethylstilbestrol alters expression of DNA methyltransferases and methylation of genomic DNA in the mouse uterus. Endocr J 2009; 56:131–9; PMID:18997445; http://dx.doi.org/ 10.1507/endocrj.K08E-239 [DOI] [PubMed] [Google Scholar]
- 121.Aguilar-Arnal L, Sassone-Corsi P. The circadian epigenome: how metabolism talks to chromatin remodeling. Curr Opin Cell Biol 2013; 25:170–6; PMID:23385084; http://dx.doi.org/ 10.1016/j.ceb.2013.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang T, Yang P, Zhan Y, Xia L, Hua Z, Zhang J. Deletion of circadian gene Per1 alleviates acute ethanol-induced hepatotoxicity in mice. Toxicology 2013; 314:193–201; PMID:24144995; http://dx.doi.org/ 10.1016/j.tox.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 123.Tischkau SA, Jaeger CD, Krager SL. Circadian clock disruption in the mouse ovary in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Lett 2011; 201:116–22; PMID:21182907; http://dx.doi.org/ 10.1016/j.toxlet.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matsunaga N, Kohno Y, Kakimoto K, Hayashi A, Koyanagi S, Ohdo S. Influence of CLOCK on cytotoxicity induced by diethylnitrosamine in mouse primary hepatocytes. Toxicology 2011; 280:144–51; PMID:21167249; http://dx.doi.org/ 10.1016/j.tox.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 125.Vorrink SU, Severson PL, Kulak MV, Futscher BW, Domann FE. Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines. Toxicol Appl Pharmacol 2014; 274:408–16; PMID:24355420; http://dx.doi.org/ 10.1016/j.taap.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Masri S, Patel VR, Eckel-Mahan KL, Peleg S, Forne I, Ladurner AG, Baldi P, Imhof A, Sassone-Corsi P. Circadian acetylome reveals regulation of mitochondrial metabolic pathways. Proc Natl Acad Sci U S A 2013; 110:3339–44; PMID:23341599; http://dx.doi.org/ 10.1073/pnas.1217632110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci U S A 2012; 109:5541–6; PMID:22431615; http://dx.doi.org/ 10.1073/pnas.1118726109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006; 125:497–508; PMID:16678094; http://dx.doi.org/ 10.1016/j.cell.2006.03.033 [DOI] [PubMed] [Google Scholar]
- 129.Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci 2010; 123:3837–48; PMID:21048160; http://dx.doi.org/ 10.1242/jcs.051649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Masri S, Sassone-Corsi P. The circadian clock: a framework linking metabolism, epigenetics and neuronal function. Nat Rev Neurosci 2013; 14:69–75; PMID:23187814; http://dx.doi.org/ 10.1038/nrn3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kowalska E, Moriggi E, Bauer C, Dibner C, Brown SA. The circadian clock starts ticking at a developmentally early stage. J Biol Rhythms 2010; 25:442–9; PMID:21135160; http://dx.doi.org/ 10.1177/0748730410385281 [DOI] [PubMed] [Google Scholar]
- 132.Ranganathan K, Sivasankar V. MicroRNAs - Biology and clinical applications. J Oral Maxillofac Pathol 2014; 18:229–34; PMID:25328304; http://dx.doi.org/ 10.4103/0973-029X.140762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Duarte FV, Palmeira CM, Rolo AP. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014; 5:865–86; PMID:25264560; http://dx.doi.org/ 10.3390/genes5040865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bandiera S, Mategot R, Girard M, Demongeot J, Henrion-Caude A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic Biol Med 2013; 64:12–9; PMID:23792138; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.06.013 [DOI] [PubMed] [Google Scholar]
- 135.Yokoi T, Nakajima M. microRNAs as mediators of drug toxicity. Ann Rev Pharmacol Toxicol 2013; 53:377–400; PMID:23189953; http://dx.doi.org/ 10.1146/annurev-pharmtox-011112-140250 [DOI] [PubMed] [Google Scholar]
- 136.Yoshioka W, Higashiyama W, Tohyama C. Involvement of microRNAs in dioxin-induced liver damage in the mouse. Toxicol Sci 2011; 122:457–65; PMID:21602190; http://dx.doi.org/ 10.1093/toxsci/kfr130 [DOI] [PubMed] [Google Scholar]
- 137.Moffat ID, Boutros PC, Celius T, Linden J, Pohjanvirta R, Okey AB. microRNAs in adult rodent liver are refractory to dioxin treatment. Toxicol Sci 2007; 99:470–87; PMID:17698510; http://dx.doi.org/ 10.1093/toxsci/kfm189 [DOI] [PubMed] [Google Scholar]
- 138.Pogribny IP, Tryndyak VP, Boyko A, Rodriguez-Juarez R, Beland FA, Kovalchuk O. Induction of microRNAome deregulation in rat liver by long-term tamoxifen exposure. Mutat Res 2007; 619:30–7; PMID:17343880; http://dx.doi.org/ 10.1016/j.mrfmmm.2006.12.006 [DOI] [PubMed] [Google Scholar]
- 139.Ray PD, Yosim A, Fry RC. Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead, and mercury: strategies and challenges. Front Genet 2014; 5:201; PMID:25076963; http://dx.doi.org/ 10.3389/fgene.2014.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang B, Pan X. RDX induces aberrant expression of microRNAs in mouse brain and liver. Environ Health Perspect 2009; 117:231–40; PMID:19270793; http://dx.doi.org/ 10.1289/ehp.11841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singh NP, Singh UP, Guan H, Nagarkatti P, Nagarkatti M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS One 2012; 7:e45054; PMID:23024791; http://dx.doi.org/ 10.1371/journal.pone.0045054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sharma A. Novel transcriptome data analysis implicates circulating microRNAs in epigenetic inheritance in mammals. Gene 2014; 538:366–72; PMID:24487054; http://dx.doi.org/ 10.1016/j.gene.2014.01.051 [DOI] [PubMed] [Google Scholar]
- 143.Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics 2012; 7:432–9; PMID:22441141; http://dx.doi.org/ 10.4161/epi.19794 [DOI] [PubMed] [Google Scholar]
- 144.Baker TR, Peterson RE, Heideman W. Using zebrafish as a model system for studying the transgenerational effects of dioxin. Toxicol Sci 2014; 138:403–11; PMID:24470537; http://dx.doi.org/ 10.1093/toxsci/kfu006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hardy TM, Tollefsbol TO. Epigenetic diet: impact on the epigenome and cancer. Epigenomics 2011; 3:503–18; PMID:22022340; http://dx.doi.org/ 10.2217/epi.11.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Roseboom TJ, van der Meulen JHP, Ravelli ACJ, Osmond C, Barker DJP, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol 2001; 185:93–8; PMID:11738798; http://dx.doi.org/ 10.1016/S0303-7207(01)00721-3 [DOI] [PubMed] [Google Scholar]
- 147.Tollefsbol TO. Dietary epigenetics in cancer and aging. Cancer Treat Res 2014; 159:257–67; PMID:24114485; http://dx.doi.org/ 10.1007/978-3-642-38007-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chouliaras L, van den Hove DL, Kenis G, Keitel S, Hof PR, van Os J, Steinbusch HW, Schmitz C, Rutten BP. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr Alzheimer Res 2012; 9:536–44; PMID:22272625; http://dx.doi.org/ 10.2174/156720512800618035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, Goel A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS One 2013; 8:e57709; PMID:23460897; http://dx.doi.org/ 10.1371/journal.pone.0057709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003; 63:7563–70; PMID:14633667 [PubMed] [Google Scholar]
- 151.Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res 2005; 11:7033–41; PMID:16203797; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-0406 [DOI] [PubMed] [Google Scholar]
- 152.King-Batoon A, Leszczynska JM, Klein CB. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen 2008; 49:36–45; PMID:18181168; http://dx.doi.org/ 10.1002/em.20363 [DOI] [PubMed] [Google Scholar]
- 153.Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, Saini S, Tanaka Y, Dahiya AV, Khatri G, et al.. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 2009; 30:662–70; PMID:19221000; http://dx.doi.org/ 10.1093/carcin/bgp042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, Majid S, Igawa M, Dahiya R. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer 2008; 123:552–60; PMID:18431742; http://dx.doi.org/ 10.1002/ijc.23590 [DOI] [PubMed] [Google Scholar]
- 155.Ascensao A, Lumini-Oliveira J, Oliveira PJ, Magalhaes J. Mitochondria as a target for exercise-induced cardioprotection. Current Drug Targets 2011; 12:860–71; PMID:21269265; http://dx.doi.org/ 10.2174/138945011795529001 [DOI] [PubMed] [Google Scholar]
- 156.Marques-Aleixo I, Oliveira PJ, Moreira PI, Magalhaes J, Ascensao A. Physical exercise as a possible strategy for brain protection: Evidence from mitochondrial-mediated mechanisms. Prog Neurobiol 2012; 99:149–62; PMID:22940590; http://dx.doi.org/ 10.1016/j.pneurobio.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 157.Ascensao A, Martins MJ, Santos-Alves E, Goncalves IO, Portincasa P, Oliveira PJ, Magalhaes J. Modulation of hepatic redox status and mitochondrial metabolism by exercise: Therapeutic strategy for liver diseases. Mitochondrion 2013; 13:862–70; PMID:23880173; http://dx.doi.org/ 10.1016/j.mito.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 158.Denham J, Marques FZ, O'Brien BJ, Charchar FJ. Exercise: Putting Action into Our Epigenome. Sports Med 2014; 44:189-209; PMID: 24163284 [DOI] [PubMed] [Google Scholar]
- 159.McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol 2009; 587:5951–8; PMID:19884317; http://dx.doi.org/ 10.1113/jphysiol.2009.181065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ntanasis-Stathopoulos J, Tzanninis JG, Philippou A, Koutsilieris M. Epigenetic regulation on gene expression induced by physical exercise. J Musculoskelet Neuronal Interact 2013; 13:133–46; PMID:23728100 [PubMed] [Google Scholar]
- 161.Pareja-Galeano H, Sanchis-Gomar F, Garcia-Gimenez JL. Physical exercise and epigenetic modulation: elucidating intricate mechanisms. Sports Med 2014; 44:429–36; PMID:24399634; http://dx.doi.org/ 10.1007/s40279-013-0138-6 [DOI] [PubMed] [Google Scholar]
- 162.Donovan EL, Miller BF. Exercise during pregnancy: developmental origins of disease prevention? Exerc Sport Sci Rev 2011; 39:111; PMID:21701279; http://dx.doi.org/ 10.1097/JES.0b013e31821f7d78 [DOI] [PubMed] [Google Scholar]
- 163.Hopkins SA, Baldi JC, Cutfield WS, McCowan L, Hofman PL. Effects of exercise training on maternal hormonal changes in pregnancy. Clin Endocrinol 2011; 74:495–500; PMID:21198740; http://dx.doi.org/ 10.1111/j.1365-2265.2010.03964.x [DOI] [PubMed] [Google Scholar]
- 164.Hopkins SA, Baldi JC, Cutfield WS, McCowan L, Hofman PL. Exercise training in pregnancy reduces offspring size without changes in maternal insulin sensitivity. J Clin Endocrinol Metab 2010; 95:2080–8; PMID:20335449; http://dx.doi.org/ 10.1210/jc.2009-2255 [DOI] [PubMed] [Google Scholar]
- 165.Ronn T, Volkov P, Davegardh C, Dayeh T, Hall E, Olsson AH, Nilsson E, Tornberg A, Nitert MD, Eriksson KF, et al.. A Six Months Exercise Intervention Influences the Genome-wide DNA Methylation Pattern in Human Adipose Tissue. Plos Genet 2013; 9; e1003572; PMID:23825961; http://dx.doi.org/ 10.1371/journal.pgen.1003572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ, Yan Z. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1alpha gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014; 63:1605–11; PMID:24430439; http://dx.doi.org/ 10.2337/db13-1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Martin-Blanco A, Ferrer M, Soler J, Salazar J, Vega D, Andion O, Sanchez-Mora C, Arranz MJ, Ribases M, Feliu-Soler A, et al.. Association between methylation of the glucocorticoid receptor gene, childhood maltreatment, and clinical severity in borderline personality disorder. J Psychiatr Res 2014; 57:34–40; PMID:25048180; http://dx.doi.org/ 10.1016/j.jpsychires.2014.06.011 [DOI] [PubMed] [Google Scholar]
- 168.Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, Makotkine I, Daskalakis NP, Marmar CR, Meaney MJ. Lower Methylation of Glucocorticoid Receptor Gene Promoter 1F in Peripheral Blood of Veterans with Posttraumatic Stress Disorder. Biol Psychiatry 2015; 77:356–64; PMID:24661442; http://dx.doi.org/ 10.1016/j.biopsych.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 169.Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A, Schops G, Dom M, Van Bree R, Freson K, et al.. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiat Res 2013; 47:880–91; PMID:23566423; http://dx.doi.org/ 10.1016/j.jpsychires.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 170.McMullen PD, Bhattacharya S, Woods CG, Sun B, Yarborough K, Ross SM, Miller ME, McBride MT, Lecluyse EL, Clewell RA, et al.. A map of the PPARalpha transcription regulatory network for primary human hepatocytes. Chem Biol Interact 2013; 209C:14–24 [DOI] [PubMed] [Google Scholar]
- 171.Stokes SE, Winn LM. NF-kappaB Signaling Is Increased in HD3 Cells Following Exposure to 1,4-Benzoquinone: Role of Reactive Oxygen Species and p38-MAPK. Toxicol Sci 2013; 137(2):303–10; PMID:24213144 [DOI] [PubMed] [Google Scholar]