

Abstract
Triple negative breast cancer cell lines have been reported to be resistant to the cyotoxic effects of temozolomide (TMZ). We have shown previously that a novel protein, human homolog of Xenopus gene which Prevents Mitotic Catastrophe (hPMC2) has a role in the repair of estrogen-induced abasic sites. Our present study provides evidence that downregulation of hPMC2 in MDA-MB-231 and MDA-MB-468 breast cancer cells treated with temozolomide (TMZ) decreases cell survival. This increased sensitivity to TMZ is associated with an increase in number of apurinic/apyrimidinic (AP) sites in the DNA. We also show that treatment with another alkylating agent, BCNU, results in an increase in AP sites and decrease in cell survival. Quantification of western blot analyses and immunofluorescence experiments reveal that treatment of hPMC2 downregulated cells with TMZ results in an increase in γ-H2AX levels, suggesting an increase in double strand DNA breaks. The enhancement of DNA double strand breaks in TMZ treated cells upon downregulation of hPCM2 is also revealed by the comet assay. Overall, we provide evidence that downregulation of hPMC2 in breast cancer cells increases cytotoxicity of alkylating agents, representing a novel mechanism of treatment for breast cancer. Our data thus has important clinical implications in the management of breast cancer and brings forth potentially new therapeutic strategies.
Keywords: abasic site, base excision repair, double strand breaks, temozolomide
Abbreviations
- AP
Apurinic/Apyrimidinic sites
- BCNU
1,3-bis-(2-chloroethyl)-1-nitrosourea
- BER
Base Excision Repair
- ERβ
Estrogen Receptor beta
- MX
Methoxyamine
- ODD
Oxidative DNA damage
- TMZ
Temozolomide
Introduction
Temozolomide (TMZ) is an oral, alkylating agent that efficiently crosses the blood-brain barrier and has demonstrated efficacy in the treatment of various tumors, including metastatic melanoma and glioblastoma multiforme.1,2 It has also been tested in patients with metastatic breast cancer and prostate cancer but inherent drug resistance has resulted in no clinical benefit.3,4 As a result, alkylating agents like TMZ are currently not as widely used in the management of breast cancer as they are in gliomas and melanomas; however, the possibility of overcoming resistance would make these agents useful additions to the chemotherapeutic management of breast cancer.
The antineoplastic efficacy of TMZ is dependent on its ability to methylate DNA, primarily at O6–guanine, N7-guanine and N3-adenine.5 The O6-methylguanine adduct is repaired by the DNA repair enzyme, O6-methylguanine methyltransferase (MGMT). As a result, MGMT is a well-known mechanism of resistance to alkylating agents. Attempts have been made to inactivate MGMT with the use of inherently nontoxic pseudosubstrates for the protein prior to administration of TMZ. In fact, work by Clemons et al. has shown that O6-(4-bromothenyl) guanine (PaTrin-2) is a potent inactivator of MGMT in the human breast cell line, MCF7, and in xenografts in vivo.6
However, repair by MGMT is not the sole mechanism responsible for resistance to alkylating agents. N7-methylguanine and N3-methyladenine comprising ~80% of TMZ-induced DNA lesions are recognized by DNA glycosylases and are substrates of the base excision repair (BER) pathway.5,7,8 BER is the predominant cellular mechanism that has evolved to repair small DNA base lesions. In BER, damaged bases are removed by a lesion-specific DNA glycosylase, generating abasic sites [apurinic/apyrimidinic (AP) sites].9 The abasic sites are recognized by the apurinic/apyrimidinic endonuclease, APE1, which incises the damaged strand, leaving 3'-OH and 5'-deoxyribose phosphate groups. DNA polymerase β (pol β) fills in the single nucleotide gap and the DNA ligase seals the final nick in the pathway.7,9 The cellular BER is rapid and efficient and it has been suggested that resistance to the therapeutic effects of TMZ is in part due to the robust repair by BER.10,11
The therapeutic efficacy of alkylating agents can be improved by blockage of BER. This includes modulating DNA glycosylase expression,12 blocking abasic site repair11,13 or by inhibition of Poly (ADP-ribose) polymerase 1 (PARP-1) by PARP inhibitors.14,15 PARP-1 is one of a family of enzymes that synthesize poly(ADP-ribose) and is known to function in BER by recruiting essential mediators to single-strand break intermediates.16 A variety of molecules have been developed to inhibit the action of PARP-1, thereby inhibiting efficient BER.17 These inhibitors are currently being tested alone or in combination with chemotherapeutic agents in the treatment of metastatic breast cancer and melanoma.18,19
Our present study focuses on the role of AP sites; the most common damage induced by alkylating therapeutic agents in TMZ cytoxicity. Previous work has shown that treatment with TMZ resulted in an increase in the number of AP sites in the DNA of colon and breast cancer cells.11,12 In addition, the cytotoxicity of TMZ was further enhanced by addition of methoxyamine (MX), a BER inhibitor that covalently binds AP sites to form methoxyamine-bound AP (MX-AP) sites.11,13 These MX-AP sites are resistant to recognition and repair by AP endonucleases, and persistence of these lesions generates single and double strand DNA breaks in the DNA eventually leading to cell death.
We have shown previously that a novel protein, human homolog of Xenopus gene which Prevents Mitotic Catastrophe (hPMC2), also known as RNA exonuclease 4 homolog (REXO4) has a role in the repair of estrogen-induced abasic sites.20 hPMC2 along with estrogen receptor β (ERβ) is required for tamoxifen-induced upregulation of the antioxidative gene Quinone Reductase (QR) at the Electrophile Response Element (EpRE).21 However, we also observed that downregulation of hPMC2 in MCF10A breast epithelial cells resulted in a significant increase in the number of AP sites.20
In this study we determined if TMZ-induced cytotoxicity was enhanced by downregulation of hPMC2 in triple negative breast cancer cells. MDA-MB-231 and MDA-MB-468 cells have been reported to be resistant to the cyotoxic effects of TMZ.22,23 Treatment with TMZ in hPMC2 downregulated cells resulted in an increase in the number of AP sites as well as a decrease in cell survival. We also observed a concomitant increase in DNA double strand breaks in hPMC2 downregulated cells treated with TMZ. Overall, downregulation of hPMC2 is an effective strategy to increase the sensitivity of TMZ-treated breast cancer cell lines. Our data thus has important clinical implications in the management of breast cancer and brings forth potentially new therapeutic strategies.
Results
Enhanced temozolomide-induced toxicity in hPMC2 downregulated cells
It has been shown previously that treatment with TMZ resulted in an increase in the number of AP sites in the DNA in colon cancer cell lines.11 An increase in the number of AP sites is a marker for DNA damage.24 The AP sites resulting from damage to the deoxyribose moiety of DNA will lead to aldehydic forms of DNA lesions which, if not repaired, are promutagenic.
We have shown previously that a novel protein, human homolog of Xenopus gene which Prevents Mitotic Catastrophe (hPMC2), also known as RNA exonuclease 4 homolog (REXO4) has a role in the repair of estrogen-induced AP sites.20 We had observed that downregulation of hPMC2 in non-tumorigenic MCF10A breast epithelial cells resulted in a significant increase in the number of AP sites. As a result, we determined the effect of TMZ-induced cytotoxicity in hPMC2 downregulated breast cancer cells.
MDA-MB-231 breast cancer cells were transfected with synthetic hPMC2 miRNAs that targets the hPMC2 3'-UTR and analyzed via Western blot for hPMC2 levels. Our results shown that transfection with miRNA resulted in downregulation of hPMC2 levels when compared to control miRNA transfected cells (Figs. 1A and B). The downregulation of hPMC2 appears specific because we did not observe downregulation of a related gene, Interferon-stimulated gene product of 20 kDa (ISG20), a protein that belongs to the same exonuclease family as hPMC239 Cells were then treated with TMZ for 24 hours and a biotinylated aldehyde-reactive probe (ARP) was used to detect the AP sites. Our results revealed that treatment with TMZ led to an increase in the number of AP sites. In addition, TMZ treatment of hPMC2 downregulated cells led to a 4-fold increase in the number of AP sites relative to the control cells (Fig. 1C).
Figure 1.
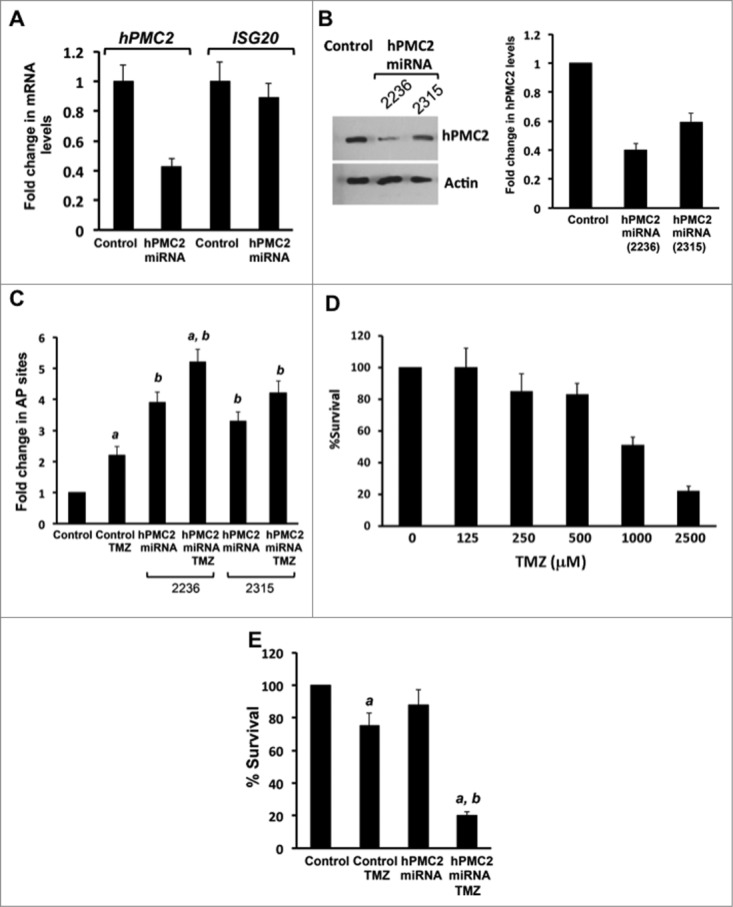
Enhanced temozolomide-induced toxicity in hPMC2 downregulated MDA-MB-231 cells. MDA-MB-231 cells were transfected with control or hPMC2 miRNAs (clones 2236 and/or 2315) and analyzed via (A) RT-PCR for hPMC2 and ISG20 levels. Cells were transfected with a mix of both miRNA clones. Transcript levels are expressed relative to GAPDH control and are based on 3 independent experiments. (B) Western blot for hPMC2 levels. Blots were probed with actin as a loading control. Image shown is a representative protein gel blot of 3 independent experiments. Quantification of hPMC2 expression in MDA-MB-231 cells. hPMC2 expression in control cells was given an arbitrary value of 1 and hPMC2 expression in downregulated cells was plotted relative to control. Error bars indicate SEM of 3 independent experiments. (C) Control and hPMC2 downregulated MDA-MB-231 cells were either untreated or treated with 250 μM TMZ for 24 hours. Genomic DNA was isolated from cells and a biotinylated Aldehyde-Reactive Probe (ARP) was used to detect the abasic sites. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment. (D) MDA-MB-231 cells were either untreated or treated with increasing concentrations of TMZ for 48 hours and maintained for a week. MTT assay was performed and the bars represent the fold change in % survival relative to untreated cells. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. (E) Control and hPMC2 downregulated MDA-MB-231 cells were either untreated or treated with 250 μM TMZ for 48 hours and maintained for a week. MTT assay was performed and the bars represent the fold change in % survival. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment.
Several studies have shown that treatment with increasing amounts of TMZ is cytotoxic, resulting in decrease in survival of colon, breast and ovarian cancer cells; however most of these studies used millimolar (mM) amounts of TMZ to observe cytotoxic effects.6,10,11,22,25 Studies using colon cancer cells have revealed that treatment with TMZ is cytotoxic, and the effect is potentiated by methoxyamine (MX) or PARP inhibitors.11,26 Work by Trivedi et al. has shown that MDA-MB-231 breast cancer cells are relatively insensitive to TMZ and sensitivity is increased by overexpression of N-methylpurine DNA glycosylase (MPG) or by loss of DNA polymerase β (pol β).22 In addition, Clemons et al. performed MTT assays with MCF7 breast cancer cells and showed that TMZ is cytotoxic, resulting in decrease in cell survival.6 In addition, the authors used micromolar (μM) concentrations of TMZ to observe cytotoxicity and the effect was potentiated with PaTrin-2, a guanine inhibitor that inactivates MGMT. Overall, these studies show that cytotoxic effects of TMZ in cancer cells is potentiated with the addition of agents that block BER.
We first examined the survival of MDA-MB-231 cells in response to increasing concentrations of TMZ. We observed at IC50 of about 1 mM. Our dose response is similar to the effect observed by Clemons et al. with MCF7 cells.6 In order to determine the effect of hPMC2 downregulation on TMZ-induced cytotoxicity, we performed the MTT assay in MDA-MB-231 cell lines with micromolar (μM) concentration of TMZ. Our results revealed that treatment with 250 μM TMZ resulted in a 20% decrease in survival of breast cancer cells relative to control (Fig. 1D). We observed a similar decrease in survival in hPMC2 downregulated cells. However, treatment of hPMC2 downregulated cells with TMZ revealed a dramatic decrease in survival of the cells by nearly 80% relative to control (Fig. 1E). Overall, our results indicated that TMZ induced cytotoxicity is effectively enhanced in hPMC2 downregulated cells.
In order to validate our findings in the MDA-MB-231 cell line, we used MDA-MB-468, another breast cancer cell line. We used miRNA to downregulate hPMC2 levels as confirmed by protein gel blot analyses (Fig. 2A). Our results with the AP site assay revealed that, as in MDA-MB-231 cells, the number of AP sites increased 3-fold in hPMC2 downregulated cells treated with TMZ relative to control cells (Fig. 2B). In addition, MTT assays also revealed that treatment of hPMC2 downregulated cells with TMZ resulted in a decrease in survival of cells by nearly 80% relative to control cells indicating that TMZ-induced cytotoxicity was enhanced in hPMC2 downregulated cells (Fig. 2C).
Figure 2.
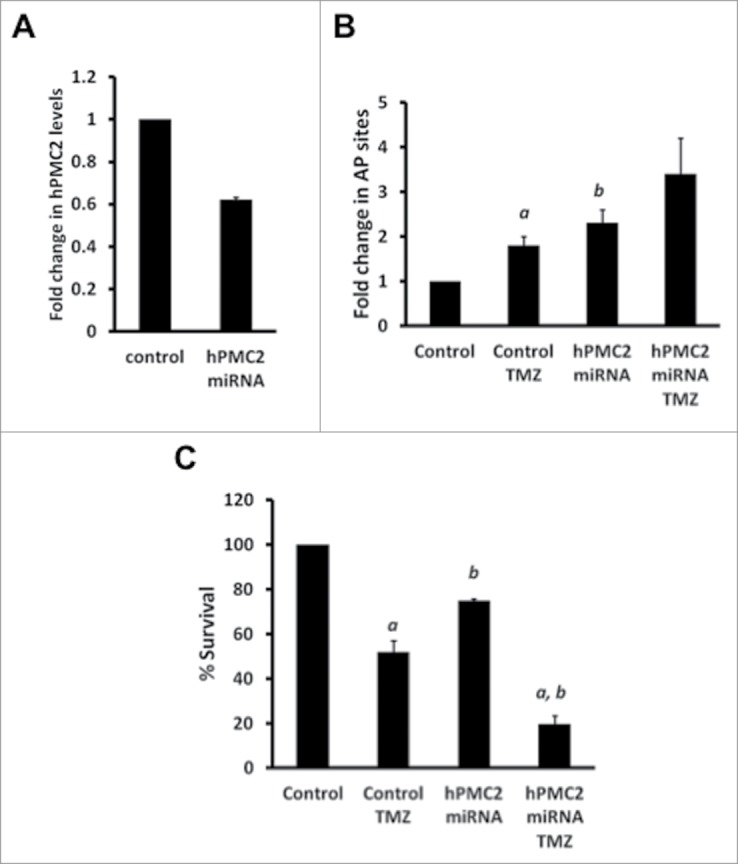
Enhanced temozolomide-induced toxicity in hPMC2 downregulated MDA-MB-468 cells. Control and hPMC2 downregulated MDA-MB-468 cells were analyzed via Western blot for hPMC2 levels. (A) Proteins were extracted from cells and processed for western blot analyses. Blots were probed with actin as a loading control. hPMC2 expression in control cells was given an arbitrary value of 1 and hPMC2 expression in downregulated cells was plotted relative to control. Error bars indicate SEM of 3 independent experiments. (B) Control and hPMC2 downregulated MDA-MB-468 cells were either untreated or treated with 125 μM TMZ for 24 hours. Genomic DNA was isolated from cells and a biotinylated ARP was used to detect the abasic sites. Error bars indicate SEM of 2 independent experiments, with triplicate values for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment. (C) Control and hPMC2 downregulated MDA-MB-468 cells were either untreated or treated with 125 μM TMZ for 48 hours and maintained for a week. MTT assay was performed and the bars represent the fold change in % survival. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment.
Enhanced BCNU-induced toxicity in hPMC2 downregulated cells
1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU) is a FDA approved chemotherapeutic agent. Like TMZ, BCNU has been used in the treatment of gliomas and in patients with brain metastasis from breast tumors.27 Although treatment with BCNU resulted in decreased survival of MCF7 cells, clinical trials for treatment of breast cancers indicate low patient response to BCNU.28 BCNU, like TMZ, produces a wide spectrum of modified bases that can be converted to AP sites by reaction with DNA glycosylases. The predominant DNA adducts involve alkylation at N7 of guanine and these lesions are substrates for BER.29,30 As a result, BCNU resistance is associated with enhanced capacity for repairing AP sites.29
Our results revealed that treatment of MDA-MB-231 cells with BCNU resulted in a significant increase in the number of abasic sites (Fig. 3A). Treatment of hPMC2 downregulated cells with BCNU led to a 2-fold increase in number of AP sites relative to control. Treatment with 50 μM BCNU resulted in a 50% decrease in survival of MDA-MB-231 cells (Fig. 3B). With hPMC2 downregulated cells, we observe a similar decrease in survival. However, treatment of hPMC2 downregulated cells with BCNU revealed a decrease in survival of the cells by nearly 80% relative to untreated control cells. Overall, our results indicate that BCNU induced cytotoxicity is effectively enhanced in hPMC2 downregulated cells.
Figure 3.
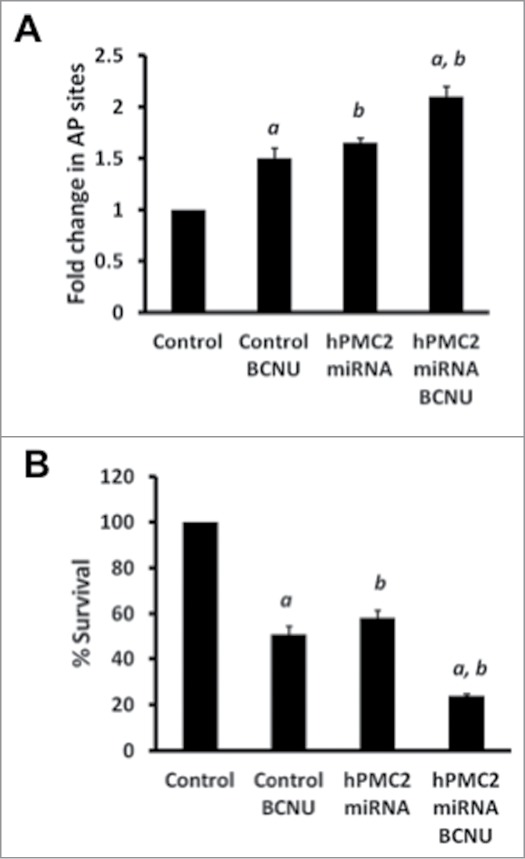
Enhanced BCNU-induced toxicity in hPMC2 downregulated MDA-MB-231 cells. (A) Control and hPMC2 downregulated MDA-MB-231 cells were either untreated or treated with 50 μM BCNU for 24 hours. Genomic DNA was isolated from cells and a biotinylated ARP was used to detect the abasic sites. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment. (B) Control and hPMC2 downregulated MDA-MB-231 cells were either untreated or treated with 50 μM BCNU for 48 hours and maintained for a week. MTT assay was performed and the bars represent the fold change in % survival. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment.
Treatment with MX results in a decrease in detectable AP sites
Methoxyamine (MX) is a BER inhibitor that covalently binds to AP sites to form methoxyamine-bound AP (MX-AP) sites.31,32 MX-AP sites are structurally modified AP sites resistant to cleavage by AP endonucleases. The persistence of MX-AP sites leads to cell death and as a result, MX has been used to improve the therapeutic efficacy of alkylating agents.11,13 We used MX in order to confirm that TMZ-induced cytotoxicity was due to an increase in number of AP sites. ARP and MX have a similar reactivity with the aldehyde group in AP sites, and as a result, they competitively bind to the AP sites in the DNA.33 Using the ARP assay, we measured the detectable AP sites produced as a result of treatment with TMZ and/or transfection with hPMC2 miRNA. Because ARP detects only free AP sites and not MX-AP sites, treatment with MX would result in a decrease in detectable AP sites. Our results with MDA-MB-231 cells revealed that co-treatment with MX reduced detectable AP sites in each of the experimental condition (Fig. 4). This is not due to the decrease of AP sites but to the occupancy of AP sites by MX, rendering them unavailable for ARP. This further supports that the observed TMZ-induced cytotoxicity in hPMC2 downregulated cells can be attributed to an increase in AP sites.
Figure 4.
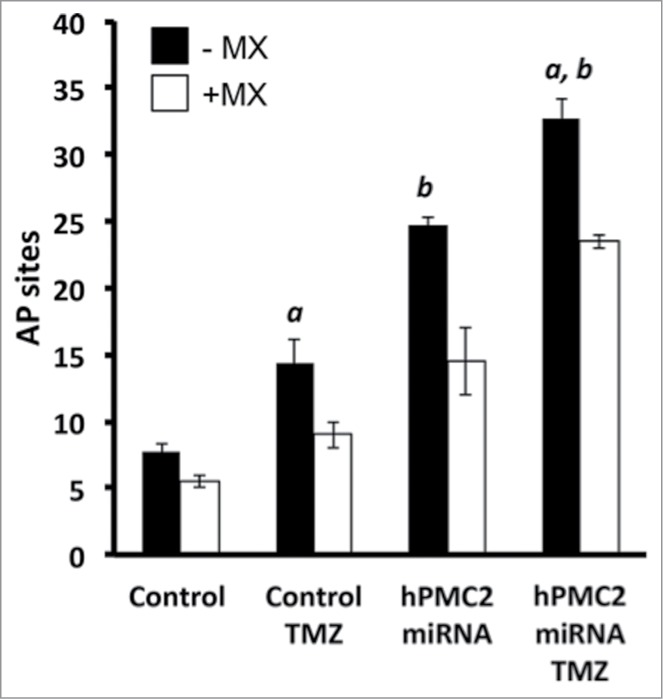
Treatment with MX results in a decrease in detectable AP sites. Control and hPMC2 downregulated MDA-MB-231 cells were either untreated or treated with 250 μM TMZ for 24 h. A parallel set of control and hPMC2 downregulated cells with and without TMZ was treated with 12.5 mM MX. Genomic DNA was isolated from cells and a biotinylated ARP was used to detect the abasic sites. Black columns in the bar graph represent number of AP sites in cells with or without TMZ treatment while the white columns represent the number of AP sites due to MX treatment with or without TMZ. The bracket represents the decrease in the number of detectable AP sites due to MX treatment. Error bars indicate SEM of 2 independent experiments with triplicates for each treatment. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment.
Downregulation of hPMC2 is associated with increases in DNA double-strand breaks
Enzymes such as the human AP endonuclease (APE1), the major AP endonuclease are known to catalyze the removal of abasic sites and avert potential mutagenic effects of oxidative damage.34 In order to ensure that the levels of APE1 were not affected by hPMC2 downregulation, we tested the level of expression of APE1. Western blot analyses in MDA-MB-231 cells reveal that the level of expression of APE1 is not affected either by downregulation of hPMC2 or treatment with TMZ (Fig. 5A).
Figure 5.

TMZ-induced increase in DNA damage in hPMC2 downregulated MDA-MB-231 cells. MDA-MB-231 cells were transfected with hPMC2 miRNA and were either untreated or treated with 250 μM TMZ for 24 hours and analyzed via Western blot for APE1, γ-H2AX, and actin levels. (A) Representative protein gel blot of APE1 and actin levels of 2 independent experiments. (B) Representative western blot of γ-H2AX and actin levels of 2 independent experiments. (C) Quantification of γ-H2AX expression in MDA-MB-231 cells. γ-H2AX expression in control cells was given an arbitrary value of 1 and γ-H2AX expression in downregulated cells was plotted relative to control. Error bars indicate SEM of 2 independent experiments. a, significance (P < 0.05) vs. untreated cells; b, significance (P < 0.05) vs. control transfected cells with the same treatment. (D) Cells were analyzed using comet assay. Results are expressed in terms of the Olive Moment, which is the summation of each tail intensity integral value, multiplied by its relative distance from the center of the head and divided by the total comet intensity.
H2AX, a histone protein is rapidly phosphorylated in Ser139 when DNA breaks are introduced in mammalian cells.35 As a result, detection of γ-H2AX is useful in monitoring the induction of DNA damage response pathways. It has been previously shown that accumulation of TMZ-induced BER intermediates triggers the γ-H2AX-mediated DNA damage response.10,11 As a result, we tested the levels of γ-H2AX by western blotting in control or hPMC2 downregulated cells treated with TMZ. Our results revealed that as expected, treatment of control MDA-MB-231 cells with TMZ resulted in an increase in γ-H2AX levels (Figs. 5B and C). We also observed that TMZ treatment in hPMC2 downregulated cells resulted in a 3-fold increase in γ-H2AX levels compared to control untreated cells (Figs. 5B and C). Because γ-H2AX is well known as a marker of DNA double-strand breaks, increased levels of γ-H2AX represents the augmentation of DNA double strand breaks in hPMC2 downregulated cells treated with TMZ. The enhancement of DNA double strand breaks in TMZ treated cells upon downregulation of hPCM2 is also revealed by the comet assay (Fig. 5D).
Expression of hPMC2 in human breast tissue
We then determined if hPMC2 is a viable target for breast cancer therapy by assessing hPMC2 protein expression in human breast cancers. Tissue microarrays containing normal breast and breast cancer tissue were stained for hPMC2 (Fig. 6). The measured outcomes were hPMC2 intensity and percent. We also considered the product of the 2 (specifically, intensity x percent / 10). We find (as shown in Table 1) a marginally significant mean difference in hPMC2 intensity between tumor and normal tissue (p = 0.06). No significant mean differences were found for hPMC2 percent or the product of percent and intensity. Regardless, our data showing expression of hPMC2 in human breast tumors makes it a good target for the development of agents that can enhance the TMZ-induced cytotoxicity.
Figure 6.
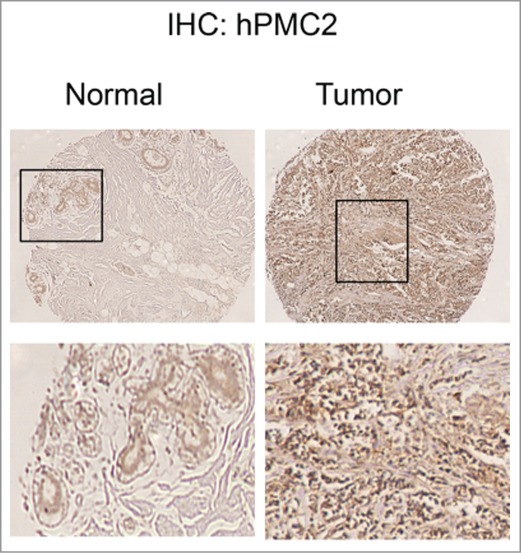
hPMC2 expression in human breast tissue samples. Sections obtained from normal breast and breast tumors were stained for endogenous hPMC2 protein.
Table 1.
Means and paired t-test p-values for hPMC2 expression in 48 matched pairs
| Mean |
|||||
|---|---|---|---|---|---|
| Measure | Tumor | Normal | Difference(T-N) | StdDev of Difference | P-Value |
| Intensity | 2.2 | 1.9 | 0.3 | 1.1 | 0.06 |
| Percent | 33.2 | 34.8 | −1.5 | 24.9 | 0.71 |
| Product | 7.9 | 8.0 | −0.5 | 8.4 | 0.97 |
Discussion
Our studies provide evidence that downregulation of hPMC2 increases TMZ- and BCNU-induced cytotoxicity in breast cancer cells. This increased sensitivity to TMZ and BCNU in hPMC2 downregulated cells is associated with an increase in the number of AP sites resulting in double strand DNA breaks in the DNA. This is confirmed by protein gel blot and immunofluorescence experiments that reveal an increase in γ-H2AX, a marker of DNA double strand damage.
Alkylating agents like TMZ and BCNU are a major class of chemotherapeutic drugs; however, intrinsic drug resistance to these agents is the most common cause of treatment failure.2,13,36 Efforts to overcome drug resistance by targeting specific DNA repair pathways have yielded promising results. Inactivation of MGMT with O6-benzylguanine (BG) potentiated the sensitivity to alkylating agents such as TMZ and BCNU.37,38 However, this treatment is limited by a narrow therapeutic index due to severe myelosuppression.37 As with other targeted therapies, agents that block a single pathway are not completely effective as tumors maintain a complex machinery to repair DNA lesions induced by therapeutic agents. As a result, there is a constant need to develop new drugs to improve therapeutic efficacy.
One of the most common damage induced by alkylating agents is the formation of abasic sites. Previous work has shown that treatment with either TMZ or BCNU resulted in an increase in the number of AP sites in colon and breast cancer cells.11,12 In addition, sensitivity was enhanced by the addition of MX, a BER inhibitor that forms MX-AP sites. MX has been shown to potentiate the cytotoxic effects of TMZ and BCNU in colon cancer cells and xenografts11,13 and sensitivity to TMZ in breast cancer cells.12
In this present work, we have focused on hPMC2 and its role in the repair of AP sites. We have shown that downregulation of hPMC2 in 2 breast cancer cell lines, MDA-MB-231 and MDA-MB-468, increases the cytotoxicity of TMZ and BCNU. Increased sensitivity was associated with a significant increase in the number of AP sites suggesting an accumulation of toxic BER intermediates. The removal of AP sites is accomplished by APE1, which cleaves DNA adjacent to the AP sites. In general, AP sites are repaired rapidly and efficiently in mammalian cells by BER.39 However, a substantial increase in number of AP sites in hPMC2 downregulated cells treated with TMZ/BCNU could result in an “imbalance” that could outrun the downstream steps that handle toxic BER intermediates and repair DNA. It has been shown previously that TMZ-induced AP sites can lead to single-strand and double strand breaks in the DNA that are cytotoxic.10,11 Quantification of our western blot analyses revealed that there was an increase in γ-H2AX levels when hPMC2 downregulated cells were treated with TMZ. H2AX is phosphorylated in response to double-strand breaks; as a result, the increase in γ-H2AX is likely due to an increase in unprocessed AP sites that in turn caused DNA strand breaks. Overall, the increased sensitivity observed in hPMC2 downregulated cells treated with either TMZ or BCNU was associated with a significant increase in AP sites and double strand breaks suggesting an accumulation of toxic BER intermediates. This high-level production of toxic intermediates could eventually lead to cell death.
TMZ has demonstrated efficacy in the treatment of various tumors, including gliomas and melanomas; however, it has been ineffective in treatment of breast cancer in the clinic. Breast cancer is the most commonly diagnosed cancer in women, accounting for 23% of cases.40 However, despite several advances, 30–40% of women will be diagnosed with metastatic cancer and eventually succumb to the disease. Recent retrospective studies suggest that triple-negative breast cancer (TNBC), lacking estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) are particularly sensitive to DNA-damaging chemotherapy with alkylating agents.41 Our studies reveal that downregulation of hPMC2 potentiates the sensitivity of alkylating agents by increasing AP sites and DNA double strand breaks. These findings implicate hPMC2 as a target in the development of therapeutic approaches for increasing cellular sensitivity to alkylating agents.
Materials and Methods
Tissue culture
MDA-MB-231 and MDA-MB-468 breast cancer cells were obtained from American Type Culture Collection and maintained as previously described.42 The cells were transfected with control or hPMC2 miRNAs using FuGene HD transfection reagent as described previously.20 Control miRNA encodes miRNA for LacZ (β-galactosidase). After 48 hours, cells were treated with TMZ for 24 hours. Cells were harvested and the whole cell lysates were analyzed by protein gel blotting.
Drugs
Temozolomide and BCNU (Sigma, MO, USA) were prepared fresh before use. Methoxyamine (Sigma, MO, USA) was stored as 2.5 M stock at −20°C for a maximum of 1 month.
Western blot analysis
Total protein from cells was extracted using M-PER Mammalian Protein Extraction reagent (Thermo Fisher Scientific, Rockford, IL) and analyzed via western blotting as previously described.20 hPMC2 was detected using an antibody generated in the Montano laboratory. The γ-H2AX antibody was obtained from Millipore (MA, USA) and APE1 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The signal in each case was detected with either Super Femto reagents (Thermo Fisher Scientific, Rockford, IL) or ECL (GE Healthcare, Piscataway, NJ) and signal intensities were normalized to their respective actin loading controls. The chemiluminescence in each case was quantified by using AlphaImager software (Proteinsimple, Santa Clara, CA).
Reverse transcription (RT) PCR analyses
Cells were subjected to reverse transcription-PCR (RT-PCR) analyses as previously described.43 The primers sequences used were.
hPMC2
(forward)5′-AGGGCAGAATTCTAGTGGGG-3′
(reverse)5′- TTCACCATGACGTACAGCCT-3′
ISG20
(forward)5′- GTACGACAAGTTCATCCGGC -3′
(reverse)5′- TCGTAGATTGTGTAGCCGCT -3′
Detection of AP sites
Control or hPMC2 downregulated MDA-MB-231 or MDA-MB-468 cells were treated with 250 μM TMZ or 50 μM BCNU for 24 h. In case of MX treatment, 12.5 mM of MX was also added to the cells for 24 h. DNA extraction was performed with a DNA isolation kit produced by Dojindo Molecular Technologies (Rockville, MD, USA). Aldehyde-reactive probe labeling and quantification of abasic sites were performed with an AP sites assay kit (Dojindo Molecular Technologies) as previously described.20
MTT assay
Control or hPMC2 downregulated MDA-MB-231 or MDA-MB-468 cells were plated in 96-well plates and treated with TMZ or BCNU for 48 hours. The cells were then maintained for a week and MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed as instructed by the manufacturer (Sigma, MO, USA).
Comet assay
Control or hPMC2 downregulated MDA-MB-231 cells were treated with either vehicle or TMZ for 24 hours. Cells were analyzed using CometAssay (Trevigen, Gaithersburg, MD, USA) according to the manufacturer's instructions. Comets were analyzed using CometScore software (TriTek, Sumerduck, VA, USA).
Immunohistochemistry
Tissue array containing sections of human breast tumors and matching normal breast tissues were obtained from US. Biomax (Rockville, MD) and Cooperative Breast Cancer Tissue Resource (CBCTR). We carried out immunohistochemical staining to detect hPMC2 levels as previously described.44 The staining score was the product of the intensity of HEXIM1 nuclear staining and percentage of hPMC2 positive cells. For statistical analyses we use a one-sample (paired) t-test to test the null hypothesis of a mean difference (tumor minus normal for each individual) equal to zero.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grant (CA092240) to M.M.M and Department of Defense Breast Cancer Postdoctoral award (BC087610) to N.K.
References
- 1.Biasco G, Pantaleo MA, Casadei S. Treatment of brain metastases of malignant melanoma with temozolomide. N Engl J Med 2001; 345: 621–2; PMID:11529230; http://dx.doi.org/ 10.1056/NEJM200108233450817 [DOI] [PubMed] [Google Scholar]
- 2.Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates A, et al.. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 2000; 18: 158–66; PMID:10623706 [DOI] [PubMed] [Google Scholar]
- 3.Trudeau ME, Crump M, Charpentier D, Yelle L, Bordeleau L, Matthews S, Eisenhauer E. Temozolomide in metastatic breast cancer (MBC): a phase II trial of the National Cancer Institute of Canada - Clinical Trials Group (NCIC-CTG). Ann Oncol 2006; 17: 952–6; PMID:16565212; http://dx.doi.org/ 10.1093/annonc/mdl056 [DOI] [PubMed] [Google Scholar]
- 4.van Brussel JP, Busstra MB, Lang MS, Catsburg T, Schroder FH, Mickisch GH. A phase II study of temozolomide in hormone-refractory prostate cancer. Cancer Chemother Pharmacol 2000; 45: 509–12; PMID:10854140; http://dx.doi.org/ 10.1007/s002800051027 [DOI] [PubMed] [Google Scholar]
- 5.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science 2001; 291: 1284–9; PMID:11181991; http://dx.doi.org/ 10.1126/science.1056154 [DOI] [PubMed] [Google Scholar]
- 6.Clemons M, Kelly J, Watson AJ, Howell A, McElhinney RS, McMurry TB, Margison GP. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br J Cancer 2005; 93: 1152–6; PMID:16278661; http://dx.doi.org/ 10.1038/sj.bjc.6602833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobol RW, Wilson SH. Mammalian DNA beta-polymerase in base excision repair of alkylation damage. Prog Nucleic Acid Res Mol Biol 2001; 68: 57–74; PMID:11554313; http://dx.doi.org/ 10.1016/S0079-6603(01)68090-5 [DOI] [PubMed] [Google Scholar]
- 8.Tentori L, Graziani G. Pharmacological strategies to increase the antitumor activity of methylating agents. Curr Med Chem 2002; 9: 1285–301; PMID:12052167; http://dx.doi.org/ 10.2174/0929867023369916 [DOI] [PubMed] [Google Scholar]
- 9.Lindahl T, Wood RD. Quality control by DNA repair. Science 1999; 286: 1897–905; PMID:10583946; http://dx.doi.org/ 10.1126/science.286.5446.1897 [DOI] [PubMed] [Google Scholar]
- 10.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res 2005; 65: 6394–400; PMID:16024643; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0715 [DOI] [PubMed] [Google Scholar]
- 11.Yan L, Bulgar A, Miao Y, Mahajan V, Donze JR, Gerson SL, Liu L. Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase IIalpha. Clin Cancer Res 2007; 13: 1532–9; PMID:17332299; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1595 [DOI] [PubMed] [Google Scholar]
- 12.Rinne M, Caldwell D, Kelley MR. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol Cancer Ther 2004; 3: 955–67; PMID:15299078 [PubMed] [Google Scholar]
- 13.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther 2003; 2: 1061–6; PMID:14578471 [PubMed] [Google Scholar]
- 14.Stachelek GC, Dalal S, Donigan KA, Campisi Hegan D, Sweasy JB, Glazer PM. Potentiation of temozolomide cytotoxicity by inhibition of DNA polymerase beta is accentuated by BRCA2 mutation. Cancer Res; 70: 409–17; PMID:20028873; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tentori L, Leonetti C, Scarsella M, D'Amati G, Vergati M, Portarena I, Xu W, Kalish V, Zupi G, Zhang J, et al.. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res 2003; 9: 5370–9; PMID:14614022 [PubMed] [Google Scholar]
- 16.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 2007; 35: 7665–75; PMID:17982172; http://dx.doi.org/ 10.1093/nar/gkm933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res 2007; 13: 1383–8; PMID:17332279; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2260 [DOI] [PubMed] [Google Scholar]
- 18.Cheng CL, Johnson SP, Keir ST, Quinn JA, Ali-Osman F, Szabo C, Li H, Salzman AL, Dolan ME, Modrich P, et al.. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair-deficient malignant glioma xenograft. Mol Cancer Ther 2005; 4: 1364–8; PMID:16170028; http://dx.doi.org/ 10.1158/1535-7163.MCT-05-0128 [DOI] [PubMed] [Google Scholar]
- 19.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al.. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–34; PMID:19553641; http://dx.doi.org/ 10.1056/NEJMoa0900212 [DOI] [PubMed] [Google Scholar]
- 20.Krishnamurthy N, Ngam CR, Berdis AJ, Montano MM. The exonuclease activity of hPMC2 is required for transcriptional regulation of the QR gene and repair of estrogen-induced abasic sites. Oncogene 2011; 30: 4731–9; PMID:21602889; http://dx.doi.org/ 10.1038/onc.2011.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sripathy SP, Chaplin LJ, Gaikwad NW, Rogan EG, Montano MM. hPMC2 is required for recruiting an ERbeta coactivator complex to mediate transcriptional upregulation of NQO1 and protection against oxidative DNA damage by tamoxifen. Oncogene 2008; 27: 6376–84; PMID:18663360; http://dx.doi.org/ 10.1038/onc.2008.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trivedi RN, Wang XH, Jelezcova E, Goellner EM, Tang JB, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Mol Pharmacol 2008; 74: 505–16; PMID:18477668; http://dx.doi.org/ 10.1124/mol.108.045112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patil R, Portilla-Arias J, Ding H, Inoue S, Konda B, Hu J, Wawrowsky KA, Shin PK, Black KL, Holler E, et al.. Temozolomide delivery to tumor cells by a multifunctional nano vehicle based on poly(beta-L-malic acid). Pharm Res 2010; 27: 2317–29; PMID:20387095; http://dx.doi.org/ 10.1007/s11095-010-0091-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res 1998; 58: 222–5; PMID:9443396 [PubMed] [Google Scholar]
- 25.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res 2007; 13: 260–7; PMID:17200364; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1920 [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin Cancer Res 1999; 5: 2908–17; PMID:10537360 [PubMed] [Google Scholar]
- 27.Jacot W, Gerlotto-Borne MC, Thezenas S, Pouderoux S, Poujol S, About M, Romieu G. Carmustine and methotrexate in combination after whole brain radiation therapy in breast cancer patients presenting with brain metastases: a retrospective study. BMC Cancer 2010; 10: 257; PMID:20525352; http://dx.doi.org/ 10.1186/1471-2407-10-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weydert CJ, Zhang Y, Sun W, Waugh TA, Teoh ML, Andringa KK, Aykin-Burns N, Spitz DR, Smith BJ, Oberley LW. Increased oxidative stress created by adenoviral MnSOD or CuZnSOD plus BCNU (1,3-bis(2-chloroethyl)-1-nitrosourea) inhibits breast cancer cell growth. Free Radic Biol Med 2008; 44: 856–67; PMID:18155673; http://dx.doi.org/ 10.1016/j.freeradbiomed.2007.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alarcon K, Demeunynck M, Lhomme J, Carrez D, Croisy A. Potentiation of BCNU cytotoxicity by molecules targeting abasic lesions in DNA. Bioorg Med Chem 2001; 9: 1901–10; PMID:11425593; http://dx.doi.org/ 10.1016/S0968-0896(01)00097-9 [DOI] [PubMed] [Google Scholar]
- 30.Chaney SG, Sancar A. DNA repair: enzymatic mechanisms and relevance to drug response. J Natl Cancer Inst 1996; 88: 1346–60; PMID:8827012; http://dx.doi.org/ 10.1093/jnci/88.19.1346 [DOI] [PubMed] [Google Scholar]
- 31.Liuzzi M, Talpaert-Borle M. A new approach to the study of the base-excision repair pathway using methoxyamine. J Biol Chem 1985; 260: 5252–8; PMID:2580833 [PubMed] [Google Scholar]
- 32.Rosa S, Fortini P, Karran P, Bignami M, Dogliotti E. Processing in vitro of an abasic site reacted with methoxyamine: a new assay for the detection of abasic sites formed in vivo. Nucleic Acids Res 1991; 19: 5569–74; PMID:1719478; http://dx.doi.org/ 10.1093/nar/19.20.5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc Natl Acad Sci U S A 2000; 97: 686–91; PMID:10639140; http://dx.doi.org/ 10.1073/pnas.97.2.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson DM 3rd, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem 1995; 270: 16002–7; PMID:7608159; http://dx.doi.org/ 10.1074/jbc.270.27.16002 [DOI] [PubMed] [Google Scholar]
- 35.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273: 5858–68; PMID:9488723; http://dx.doi.org/ 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 36.Dinnes J, Cave C, Huang S, Milne R. A rapid and systematic review of the effectiveness of temozolomide for the treatment of recurrent malignant glioma. Br J Cancer 2002; 86: 501–5; PMID:11870527; http://dx.doi.org/ 10.1038/sj.bjc.6600135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L, Nakatsuru Y, Gerson SL. Base excision repair as a therapeutic target in colon cancer. Clin Cancer Res 2002; 8: 2985–91; PMID:12231545 [PubMed] [Google Scholar]
- 38.Yan L, Donze JR, Liu L. Inactivated MGMT by O6-benzylguanine is associated with prolonged G2/M arrest in cancer cells treated with BCNU. Oncogene 2005; 24: 2175–83; PMID:15735757; http://dx.doi.org/ 10.1038/sj.onc.1208250 [DOI] [PubMed] [Google Scholar]
- 39.Wood RD . DNA repair in eukaryotes. Annu Rev Biochem 1996; 65: 135–67; PMID:8811177; http://dx.doi.org/ 10.1146/annurev.bi.65.070196.001031 [DOI] [PubMed] [Google Scholar]
- 40.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108; PMID:15761078; http://dx.doi.org/ 10.3322/canjclin.55.2.74 [DOI] [PubMed] [Google Scholar]
- 41.Colleoni M, Cole BF, Viale G, Regan MM, Price KN, Maiorano E, Mastropasqua MG, Crivellari D, Gelber RD, Goldhirsch A, et al.. Classical cyclophosphamide, methotrexate, and fluorouracil chemotherapy is more effective in triple-negative, node-negative breast cancer: results from two randomized trials of adjuvant chemoendocrine therapy for node-negative breast cancer. J Clin Oncol 2010; 28: 2966–73; PMID:20458051; http://dx.doi.org/ 10.1200/JCO.2009.25.9549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montano MM, Katzenellenbogen BS. The quinone reductase gene: a unique estrogen receptor-regulated gene that is activated by antiestrogens. Proc Natl Acad Sci U S A 1997; 94: 2581–6; PMID:9122238; http://dx.doi.org/ 10.1073/pnas.94.6.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogba N, Chaplin L, Doughman YQ, Fujinaga K, Montano MM. HEXIM1 regulates E2/ERα-mediated expression of Cyclin D1 in mammary cells via modulation of P-TEFb. Cancer Research 2008; 68: 7015–24; PMID:18757415; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ketchart W, Ogba N, Kresak A, Albert JM, Pink JJ, Montano MM. HEXIM1 is a critical determinant of the response to tamoxifen. Oncogene 2011; 30: 3563–9; PMID:21423213; http://dx.doi.org/ 10.1038/onc.2011.76 [DOI] [PMC free article] [PubMed] [Google Scholar]