

Abstract
Because STAT signaling is commonly activated in malignant gliomas as a result of constitutive EGFR activation, strategies for inhibiting the EGFR/JAK/STAT cascade are of significant interest. We, therefore, treated a panel of established glioma cell lines, including EGFR overexpressors, and primary cultures derived from patients diagnosed with glioblastoma with the JAK/STAT inhibitor cucurbitacin-I. Treatment with cucurbitacin-I depleted p-STAT3, p-STAT5, p-JAK1 and p-JAK2 levels, inhibited cell proliferation, and induced G2/M accumulation, DNA endoreduplication, and multipolar mitotic spindles. Longer exposure to cucurbitacin-I significantly reduced the number of viable cells and this decrease in viability was associated with cell death, as confirmed by an increase in the subG1 fraction. Our data also demonstrated that cucurbitacin-I strikingly downregulated Aurora kinase A, Aurora kinase B and survivin. We then searched for agents that exhibited a synergistic effect on cell death in combination with cucurbitacin-I. We found that cotreatment with cucurbitacin-I significantly increased Bcl-2/Bcl-xL family member antagonist ABT-737-induced cell death regardless of EGFR/PTEN/p53 status of malignant human glioma cell lines. Although >50% of the cucurbitacin-I plus ABT-737 treated cells were annexin V and propidium iodide positive, PARP cleavage or caspase activation was not observed. Pretreatment of z-VAD-fmk, a pan caspase inhibitor did not inhibit cell death, suggesting a caspase-independent mechanism of cell death. Genetic inhibition of Aurora kinase A or Aurora kinase B or survivin by RNA interference also sensitized glioma cells to ABT-737, suggesting a link between STAT activation and Aurora kinases in malignant gliomas.
Keywords: Aurora kinases, caspase-independent cell death, cell cycle arrest, Glioma
Abbreviations
- BSA
bovine serum albumin
- DMSO
dimethyl sulfoxide
- EGFR
epidermal growth factor receptor
- MTS
3-[4, 5-dimethylthiazol- 2yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H, tetrazolium
- FITC
fluorescein isothiocyanate
- NF-кB
nuclear factor кB
- PI3K
Phosphatidylinositol 3-Kinase
- PBS
phosphate-buffered saline
- PAGE
polyacrylamide gel electrophoresis
- PDGFR
platelet derived growth factor receptor
- TBS
Tris-buffered saline
- TRAIL
tumor necrosis factor–related apoptosis inducing ligand
- PI
propidium iodide
Introduction
Malignant gliomas are aggressive, highly invasive, morphologically heterogeneous tumors that have proven largely refractory to conventional treatment modalities such as surgery, irradiation, and cytotoxic chemotherapy. Despite recent research efforts in cancer therapy, the prognosis of patients with gliomas remains poor and their therapeutic resistance has not been fully understood.1-3 These tumors arise from a sequential accumulation of genetic aberrations and dysregulation of growth-factor signaling pathways, encompassing mutations and amplification of epidermal growth factor receptors (EGFR), deletion of the phosphatase and tensin homolog on chromosome 10 (PTEN), TP53 mutations, and p16 deletion and, less frequently, MDM2 amplification. These alterations activate critical downstream mediators that contribute to the neoplastic phenotype, and may constitute promising therapeutic targets.4,5
The epidermal growth factor receptor (EGFR) is frequently deregulated in malignant glioma, as a result of EGFR gene amplification, activating mutation, or both.6-11 EGFR variant III (EGFRviii), a common EGFR mutation, results from an in-frame deletion of exons 2 to 7, strongly and persistently stimulates several key signaling pathways, including phosphatidylinositol 3-kinase (PI3K)/Akt, extracellular regulated kinase (ERK), signal transducer and activator of transcription 3 (STAT3), BCL-xL, and nuclear factor ĸB (NF-ĸB).12-19 Although both EGFR and EGFRviii are associated with tumor growth and progression and are, thus, a logical target for anticancer therapy, the efficacy of EGFR-targeted small-molecule inhibitors or monoclonal antibodies in gliomas has been modest.
In studies using a large-scale short interfering RNA (siRNA) screening approach to identify critical “nodes” for cell death signaling, we identified several targets, including nuclear factor kappa B (NF-ĸB) and the proteasome, as well as Akt and Bcl-2 family members that, when inhibited, promoted apoptotic signaling in glioma cells.20-24 Bcl-2 antagonists can induce apoptosis as single agents only in cancer cells dependent on Bcl-2 and/or Bcl-xL for survival. As a single agent ABT-737, a novel Bcl-2/Bcl-xL inhibitor triggers apoptosis in various types of human cancers including multiple myeloma, leukemia, lymphoma and small cell lung cancer.25-29 However, we30-32 and others33 have demonstrated that ABT-737 minimally inhibits cell growth in glioma when used as a single agent. The complexity of the interactions between cell surface receptors and downstream signaling targets has called into question the clinical utility of blocking any target in isolation, and the results of single agent- based strategies have to date been disappointing in patients with glioma.2,4 Combination approaches can enhance ABT-737-induced glioma cell death, including use with the proteasomal inhibitor bortezomib,34 the survivin inhibitor YM-155,30 the PI3K/Akt inhibitor NVP-BKM120,31 the histone deacetylase inhibitor vorinostat,35 the alkylating agent temozolomide,36 or the death receptor ligand TRAIL.37 We performed a screen of ABT-737 in combination with other signaling inhibitors used at clinically achievable doses; combination of ABT-737 and cucurbitacin-I promoted apoptosis in malignant human glioma cell lines.
Signal-transducer-and-activator-of-transcription-3 (STAT3) regulates the transcription of several genes that are involved in cell cycle progression, anti-apoptosis, cell survival, and angiogenesis. STAT3 is activated in 60% of primary high-grade/malignant gliomas38 and closely linked with a variety of human malignancies, including leukemia,39 head and neck squamous cell carcinoma,40 and breast.41 Inhibition of JAK2/STAT3 signaling with cucurbitacin-I (JSI-124),42 a natural plant product, has been shown to have potent anti-cancer activities in hematological malignancies and solid tumors.43-47 Several lines of evidence have indicated that cucurbitacin-I promotes cell cycle arrest and/or apoptosis in glioma,48,49 lung,42,50 breast,51 and other malignancies.52-55 Previously we observed that blockade of the JAK/STAT pathway by cucurbitacin-I enhanced the efficacy of SRC family kinase (SFK) inhibition with dasatinib in glioma cells.48 Chumbalkar et al performed a tyrosine-directed search for signaling events downstream of ΔEGFR (EGFRviii) and identified STAT5 phosphorylation at Y699 as a key event.56 Later, Latha et al demonstrated that EGFRviii cooperated with STAT5 to regulate the Bcl-xL promoter.57 This prompted us to investigate the role of JAK/STAT inhibitor cucurbitacin-I and the Bcl-2/Bcl-xL antagonist ABT-737 in glioma cell lines. In this study, we report that cotreatment with cucurbitacin-I and ABT-737 induced G2/M arrest and apoptosis regardless of EGFR, PTEN, and TP53 mutation status in malignant human glioma cell lines.
Results
Cucurbitacin inhibits growth in EGFR-overexpressing glioma cell lines and primary cultured glioma cells but not non-neoplastic human astrocytes
In our previous study we have demonstrated that cucurbitacin can induce cytotoxicity in a variety of glioblastoma derived cell lines.48 Because the epidermal growth factor receptor (EGFR) plays a prominent role in many tumors including glioblastoma and overexpression is observed in >50% of GBM patients,58-60 we used isogenic U87 cell lines expressing EGFR-WT and EGFRviii (Fig. S1A and S1B). U87, U87-EGFR-WT and U87-EGFRviii cultures were exposed to varying concentrations of cucurbitacin and MTS cell proliferation assay was performed after 24, 48 and 72 h of treatment. Vehicle-treated (DMSO) cells served as control. Cucurbitacin inhibited cell proliferation in a dose and time-dependent manner (Fig. 1A). The cucurbitacin IC50 (72 h) was 385.1 nmol/L for U87, and 295 and 266.4 nmol/L for U87-EGFR (WT) and U87-EGFRviii respectively.Figure 1B presents the effect of cucurbitacin in primary cultured glioma cells (GBM-1, GBM-8, GBM-9 and GBM-13) established from cells collected from 4 patients with histologically confirmed glioblastoma. The non-neoplastic human astrocytes (HA) showed no growth inhibition within the dose range (>2.0 μmol/L) tested, suggesting that cucurbitacin acts selectively against tumor cells (Fig. 1C).
Figure 1.

Anti-proliferative activity of cucurbitacin in glioma cell lines and non-neoplastic astrocytes. EGFR overexpressing human glioma cell lines U87-EGFR-WT, U87-EGFRviii and isogenic control, U87 (A), primary cultured glioma cells, GBM-1, GBM-8, GBM-9 and GBM-13 (B) and non-neoplastic human astrocytes (C) were grown on 96-well plates in growth medium and, after an overnight attachment period, were exposed to selected concentrations of inhibitor or vehicle (0, DMSO) for 24, 48, or 72 h (Day 1, Day 2 or Day 3). Cell proliferation inhibition was assessed semiquantitatively by spectrophotometric measurement of MTS bioreduction. Points represent the mean of 3 measurements carried out in triplicate.
Cucurbitacin inhibits Aurora kinase A, B and survivin
Previously we have shown that activated JAK1, JAK2, and STAT3 were inhibited with high-dose cucurbitacin in established glioma cell lines.48 In this study, we evaluated the protein expression of the phosphorylated forms of Akt, Bcl-2, XIAP as well as STAT target gene products such as Bcl-xL, Mcl-1, and cyclin D1 in the EGFR overexpressing U87-EGFRviii cell line. Modest or no inhibition of p-AKT, cyclin D1 and BCL-xL was observed after 24 h (Fig. 2A). No significant difference in the amount of proteins was noted (ERK, p38, JNK, nuclear forms of NF-ĸB, data not shown). However, levels of phospho-Stat-3 (at Tyr705), phospho-Stat-5 (at Tyr694), phospho-JAK-1 (at Tyr1022/1023), phospho-JAK-2 (at Tyr1007/1008) were reduced by cucurbitacin (Fig. 2B) in U87 and U87-EGFRviii cell lines. Interestingly, nanomolar concentrations of cucurbitacin led to a substantial reduction of Aurora kinase A, Aurora kinase B, and survivin in U87, U87-EGFR-WT, U87-EGFRviii (Fig. 2C, upper panel), LN229, T98G, and LNZ308 cell lines (Fig. 2C, lower panel). Then we carried out real time PCR on total RNA isolated from glioma cell lines. As shown in Figure 2C, a significant decrease in the mRNA expression of Aurora kinase A (2.5–4.5 fold), Aurora kinase B (20–25 fold), and survivin (25–30 fold), was observed in U87 cells treated with cucurbitacin (250 nmol/L) for 24 h. Similar results were observed in U87-EGFRviii, LNZ308, A172 and LN229 cell lines (data not shown).
Figure 2.

Cucurbitacin inhibits Aurora kinase A, Aurora kinase B and survivin. U87 and U87-EGFRviii (A and B) cells were treated with cucurbitacin (indicated concentration) for 24 h. (C), A panel of 4 glioma cell lines (U87, LN229, T98G and LNZ308) along with EGFR over expressing U87-EGFR-WT (U87-EGFR) and U87-EGFRviii cells were treated with cucurbitacin (indicated concentration) for 24 h. Cell extracts were subjected to Western blot analysis with indicated antibody. (D). U87 cells were treated with cucurbitacin (indicated concentration) for 24 h, after which total RNA was extracted and Aurora kinase A, Aurora kinase B and survivin mRNA levels were quantified by real time PCR as described in the Materials and Methods.
Cucurbitacin induces G2/M arrest in human glioma cell lines
Impaired regulation of Aurora kinase-A, Aurora kinase-B and survivin expression has been noted in proliferating cells to cause chromosomal abnormality and instability.61 Because preferential ablation of Aurora kinase A, Aurora kinase B or survivin by RNA interference62-64 produces a mitotic arrest, we investigated whether cucurbitacin influences the cell cycle profile in glioma cell lines. U87, U87-EGFRviii and other established cell lines such as T98G. LN18, LN229, A172, and LNZ308 were cultured with the indicated concentration of cucurbitacin for 24 h and analyzed by flow cytometry. A representative DNA profile (Fig. 3A) and a histogram obtained from multiple FACS analyses are shown in Figure 3B. Compared to the control (DMSO treated cells), cells accumulated in the G2/M phase with cucurbitacin treatment in a dose-dependent manner irrespective of EGFR or TP53 or PTEN status (see Table 1 for genetic features of the glioma cell lines). This was accompanied by the reduction of cells in the G1 and S phases of the cell cycle. After prolonged drug incubation (48 h, Fig. 3C upper panel; 72 h, Fig. 3C lower panel), there was an increase in the sub-G1 phase of cells, typical for late stages of apoptosis.
Figure 3.
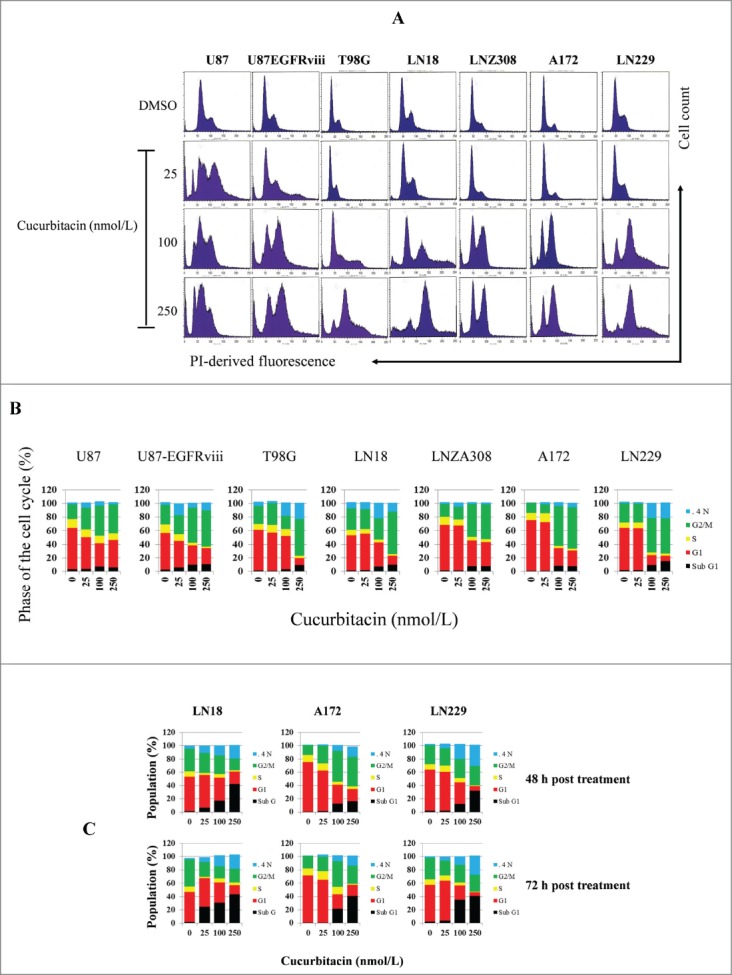
Cucurbitacin induces G2/M arrest regardless of EGFR/PTEN/p53 status in malignant human glioma cell lines. U87, U87-EGFRviii, T98G, LN18, LNZ308, A172 and LN229 cells were seeded at 60% confluence, allowed to attach overnight, and treated with cucurbitacin (indicated concentration) for 24 h. Control cells received DMSO (0). Cell cycle analysis using propidium iodide (PI) staining was performed as described in the Materials and Methods Section. Representative results of 3 independent experiments are presented here (A). Histogram represents the average of 3 experiments (B). LN18, A172 and LN229 cells were seeded at 60% confluence, allowed to attach overnight, and treated with cucurbitacin (indicated concentration) for 48 or 72 h. Control cells received DMSO (0). Cell cycle analysis was performed as described in the Materials and Methods Section (C).
Table 1.
The genotypic features of the glioma cell lines used in this study
| Cell line | p53-gene status | PTEN-gene status |
|---|---|---|
| 1. LN18 | Heterozygous, WT/Mut238 | Wild type |
| 2. U87 | Homozygous, WT | Deleted |
| 3. T98G | Homozygous, Mut237 | Mutant |
| 4. LN229 | Heterozygous, WT/Mut164 | Wild type |
| 5. LNZ308 | Heterozygous, deleted/translocated | Deleted |
| 6. A172 | Heterozygous, WT/WT | Deleted |
Cotreatment with cucurbitacin and ABT-737 potentiates glioma cell death in a caspase-independent manner
Because survivin 30 signaling inhibited the clinical efficacy of ABT-737 and cucurbitacin affected survivin both at mRNA and protein levels (Fig. 2C and 2D), we hypothesized that cotreatment with cucurbitacin and ABT-737 may be a useful strategy for the treatment of glioma. To systematically compare the effect of ABT-737 and cucurbitacin, cells were treated with ABT-737 or cucurbitacin or the combination of both for 72 h and MTS assay was performed as described in the Materials and Methods. Cotreatment of ABT-737 and cucurbitacin was able to reduce proliferation of glioma cells significantly. Lower concentrations of ABT-737 and cucurbitacin sufficed to inhibit proliferation (significant reduction in U87, U87-EGFR-WT, U87-EGFRviii, T98G, LN18, and LNZ308 cell lines after 72 h, P < 0.005, t-test; Fig. S2). Cucurbitacin significantly increased the sensitivity of established and primary cultured glioma cells to ABT-737 treatment compared with cells treated with ABT-737 alone. Simultaneous treatment of ABT-737 plus cucurbitacin also had little or no effect on cell proliferation of non-neoplastic astrocytes, suggesting selectivity against glioma cells.
We next quantified the effects of the inhibitor combinations on apoptosis. U87, U87-EGFR-WT, U87-EGFRviii, A172, LN229 and LNZ308 cells treated with ABT-737 or cucurbitacin or the combination of both for 24 h were stained with annexin V and PI and analyzed by flow cytometry. Three experiments were performed in duplicate with similar results. A representative annexin V binding histogram (Fig. 4A and 4B) and a bar chart representing 3 independent experiments is shown in Figure 4C. Single agent ABT-737 or cucurbitacin resulted in only minimal or modest annexin V/PI staining. On the other hand, cotreatment with ABT-737 plus cucurbitacin enhanced annexin V/PI sensitivity (Fig. 4A–C). To understand whether the mechanism of cell death was caspase-dependent, an irreversible pan-caspase inhibitor (z-VAD-fmk) was used. U87, U87-EGFR-WT and U87-EGFRviii cells were treated with z-VAD-fmk, for 2 hours prior to treatment with cucurbitacin or ABT-737 or the combination of both for 20 h. Annexin V/PI analysis showed that cucurbitacin plus ABT-737-induced cell death was unaffected by the presence of z-VAD-fmk, suggesting a possible caspase-independent cell death pathway (Fig. 4D). We then used Western blot analysis to validate the results. LN18 cells treated with TRAIL (Tumor necrosis factor related apoptosis-inducing ligand) served as a positive control. In response to cucurbitacin or ABT-737 as a single agent or the combination of both, the 32- kDa procaspase-3 was not cleaved to a -p20, -p17 and –p12 kDa “active” form; nor were other caspase-processing events (Fig. 4E, F). Activation of caspase-3 (appearance of cleaved 19, 17, and 12 kDa fragments), caspase-7 (appearance of cleaved 35, 30, and 20 kDa fragments), caspase-8 (appearance of cleaved 43, 41, and 18 kDa fragments), caspase-9 (appearance of cleaved 37 and 17 kDa fragments) and PARP (appearance of 89 kDa fragment) was observed in LN18 cells treated with TRAIL (lane 11, Fig. 4E, F).
Figure 4.
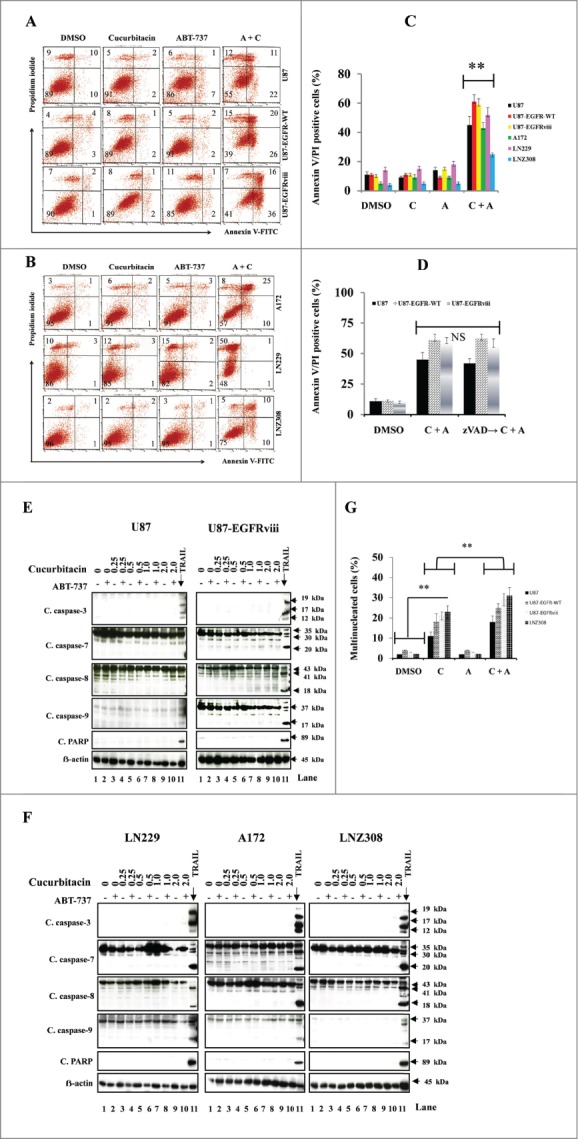
Cotreatment with cucurbitacin and ABT-737 potentiates glioma cell death in a caspase-independent manner. EGFR overexpressing human glioma cell lines U87-EGFR-WT, U87-EGFRviii and isogenic control, U87 (A), A172, LN229 and LNZ308 (B) were seeded at 60% confluence, allowed to attach overnight, and treated with cucurbitacin (C) or ABT-737 (A) or the combination of both (C + A) for 24 h. Control cells received an equivalent amount of DMSO. Apoptosis was analyzed by flow cytometry. The percentages of cells in each quadrant are indicated. (C) Bar chart represents data from 3 independent experiments (** P < 0.005 vs. untreated control or single agent). (D) U87, U87-EGFR-WT and U87-EGFRviii cells were pretreated with pan-caspase inhibitor zVAD-fmk (25 μmol/L) for 2 h followed by combination of cucurbitacin plus ABT-737 (C + A) for 24 h. Cell death was analyzed by flow cytometry. Data are representative of triplicate studies from 3 independent experiments (NS, not significant). U87 and U87-EGFRviii (E) LN229, A172 and LNZ308 (F) were seeded at 60% confluence, allowed to attach overnight, and treated with cucurbitacin (indicated concentration) with or without ABT-737 (500 nmol/L) for 24 h. Control cells received an equivalent amount of DMSO. Cell extracts were subjected to Western blot analysis with indicated antibody. LN18 treated with TRAIL (25 ng/ml for 3 h) served as positive control. G. U87, U87-EGFR-WT, U87-EGFRviii and LNZ308 cells were grown on chamber slides in growth medium, and, after an overnight attachment period, were exposed to cucurbitacin (C, 100 nmol/L) or ABT-737 (A, 1.0 μmol/L) or the combination of both (C + A) or vehicle (DMSO) for 72 h. Cells were washed once with PBS, fixed with 3.7% formaldehyde for 30 min and stained with DAPI for 2 h at room temperature. The slides were then washed in PBS, mounted, and examined under a fluorescent microscope. Cells with multi-lobed nuclei are presented in the bar graph. Points represent the mean of 3 measurements ± S.D (**, P < 0.005).
Because Aurora kinase A, Aurora kinase B and survivin are essential for successful mitotic transition, particularly in cytokinesis,65-68 and mitotic catastrophe is characterized by the appearance of enlarged micro- and/or multinucleated cells, we examined the morphology of nuclei of cucurbitacin and ABT-737-treated cells. At least 100 cells were analyzed and the results are presented in Figure 4G. Staining of cucurbitacin and ABT-737-treated U87, U87-EGFR-WT, U87-EGFRviii and LNZ308 cells with DAPI revealed the presence of fragmented or lobulated nuclei in association with micronuclei, characteristics consistent with mitotic catastrophe. Cucurbitacin induced the accumulation of cells with multi-lobed nuclei, whereas multinucleated cells were rarely or minimally detected in the control or ABT-737 treated groups. Seventy-two hours after treatment with cucurbitacin, about 20% of the EGFR overexpressing U87 cells showed multi-nucleated cells. The fraction of multinucleated cells was significantly increased with the combination of cucurbitacin and ABT-737 (Fig. 4G).
Targeting Aurora kinase A, Aurora kinase B and survivin by RNA interference enhanced ABT-737-induced cytotoxicity
To determine the effects of Aurora kinase A, Aurora kinase B and survivin on ABT-737-induced cytotoxicity, U87 cells were transfected with the expression vector containing shRNA targeted against Aurora kinase A, Aurora kinase B or survivin. Western blot analysis was used to evaluate the levels of Aurora kinase A, Aurora kinase B and survivin protein expression. As shown in Figure 5 (left panel), abundant levels of Aurora kinase A, Aurora kinase B and survivin expression were seen in non-target shRNA transfected control U87 cells, whereas genetic interference with shRNA inhibited >50% of the respective proteins. To determine whether the inhibition of Aurora kinase A, Aurora kinase B or survivin expression results in enhanced sensitivity, cells were treated with varying doses of ABT-737. Annexin V/PI analysis clearly demonstrated that the suppression of Aurora kinase A, Aurora kinase B or survivin expression enhanced sensitivity to ABT-737 (Fig. 5 right panel).
Figure 5.
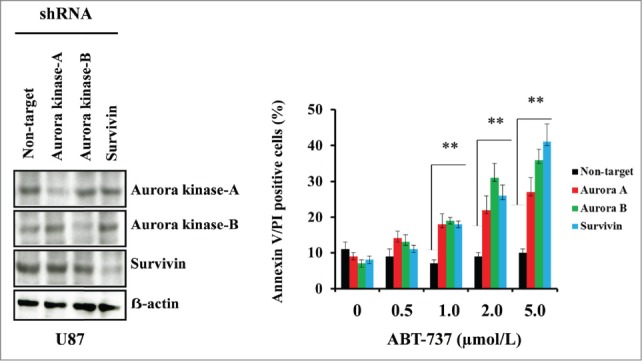
Targeting Aurora kinase A, Aurora kinase B and survivin by RNA interference enhanced ABT-737-induced cytotoxicity. U87 cells were transfected with non-target or Aurora kinase A, Aurora kinase B or survivin shRNA as described in the Materials and Methods. Forty-eight hours post-transfection, cells were treated with the indicated concentrations of ABT-737 for 24 h, and viability was assessed by annexin V/PI apoptosis assay (right panel; ** P < 0.005 versus non-target shRNA control). In parallel, cell lysates were collected and protein was subjected to Western blot analysis using indicated antibodies. Immunoblots were stripped and reprobed with β-actin (left panel). Data are representative of triplicate studies from 3 independent experiments (**, P <0.005).
Discussion
STAT signals downstream of EGFR are activated in 60% of glioblastoma patients and correlates inversely with survival. The EGFR/JAK/STAT pathway has generated significant interest as a key driver of tumor cell survival, proliferation, and invasion in GBM.38 Recently, using subcellular fractionation, Fan, et al 69 demonstrated that receptor phosphorylation of EGFRviii led to nuclear transport of EGFRviii and enhanced the formation of a complex between EGFRviii and STAT5. This complex serves as a transcriptional co-activator for a series of tumor-promoting genes such as cyclin D1, iNOS, B-myb, Aurora kinase and Cox-2.57,70 In this report we evaluated the effect of the JAK/STAT inhibitor, cucurbitacin on malignant human glioma cells in vitro. Application of cucurbitacin to glioma cell lines inhibited cell proliferation and induced cell-cycle arrest regardless of mutational status of p53, PTEN and EGFR. We observed that cucurbitacin diminished Aurora kinase A, Aurora kinase B and survivin expression. To the best of our knowledge, these observations have not been previously reported. In light of this observation, it is possible that cucurbitacin-induced STAT5 down regulation might reduce the transcription of Aurora kinases.
Increasing evidence indicates that aberrant expression of Aurora kinase A, Aurora kinase B and survivin results in chromosomal instability, aneuploidy and malignant transformation. Aurora kinase A encodes a protein that localizes to the spindle poles, contributes to separation and maturation of the centrosomes during mitosis, plays a pivotal role in G2/M cell cycle progression and is frequently amplified in several types of cancer.71,72 Aurora kinase B encodes a chromosomal passenger protein that is required for proper mitosis and cytokinesis.73 Here, we have demonstrated that glioma cells treated with cucurbitacin develop spindle abnormalities and multinucleated cells at a high frequency, consistent with the results of previous reports on Aurora kinase A, Aurora kinase B and survivin inhibition in a variety of experimental systems. Thus, it is plausible that the mechanism by which Aurora kinase A, Aurora kinase B and survivin inhibition leads to cell death is related to its ability to induce polyploidy.72,74
In our initial screen of cucurbitacin with other signaling inhibitors; simultaneous treatment of cucurbitacin with ABT-737 at clinically achievable doses in vitro resulted in a synergistic interaction. However, we were unable to detect the active forms of caspases-3, -7, -8 -9 and PARP in cucurbitacin and ABT-737 treated cell lines. Pan-caspase inhibitor zVAD-fmk did not block cucurbitacin and ABT-737-induced cell death. This could be due to the involvement of a number of non-caspase proteases (such as lysosomal cathepsins) that are capable of executing cell death. It has been suggested that mitotic catastrophe is not a genuine cell death executioner mechanism, but a pathway that precedes cell death, in which caspase activation and other markers of classical apoptosis are completely absent.75 Our study clearly demonstrates that the combination of ABT-737 and cucurbitacin promoted mitotic spindle failure, G2/M arrest which was accompanied by an increase in the sub-G1 fraction of cells, and eventual cell death. Genetic knockdown of Aurora kinase A, aurora kinase B or survivin dramatically increased ABT-737 induced glioma cell death. This is in agreement with our previous report in which we demonstrated that cotreatment with the survivin inhibitor YM-155 potentiated ABT-737-induced cytotoxicity in a synergistic manner.30 In summary, our data reveal that the treatment of glioma cells with cucurbitacin potentiated ABT-737-induced cytotoxicity by down-regulating Aurora kinase A, Aurora kinase B and survivin regardless of EGFR/PTEN/p53 status, suggesting the utilization of these agents for GBM.
Materials and Methods
Cell lines
Primary cultures were obtained from freshly resected tissues (histologically confirmed glioblastoma) after surgical removal under an Institutional Review Board-approved protocol for acquisition and the use of tumor tissue collected at the time of tumor resection. We also obtained primary glioma cells from Conversant Biologics (Huntsville, AL). Established malignant human glioma cell lines such as T98G, A172 and LN229 were obtained from the American Type Culture Collection (Manassas, VA). LN18 and LNZ308 were provided by Dr. Nicolas de Tribolet (Lausanne, Switzerland). The establishment of the parental human glioblastoma cell line, U87 and its derivatives, which overexpress exogenous wild-type EGFR (U87-EGFR-WT), or constitutively active EGFR with a genomic deletion of exons 2–7 (U87-EGFRviii) has been described elsewhere.76 The cell lines were kindly provided by Dr. Shi-Yuan Cheng (Northwestern University Feinberg School of Medicine, Chicago, IL). PTEN and TP53 status (Table 1) of these glioma cell lines have been characterized elsewhere.77,78 Human astrocytes (HA) and growth media were obtained from ScienCell Research Laboratories (Carlsbad, CA). Cell culture conditions of these cell lines were as previously described.7,30,31,79
Reagents and antibodies
ABT-737 and cucurbitacin-I (referred as cucurbitacin in this manuscript) were purchased from Chemie Tek (Indianapolis, IN) and Sigma (St. Louis, MO) respectively. All antibodies were purchased from Cell Signaling Technology (Beverly, MA).
Cell proliferation analysis
Cells (5 × 103/well) were plated in 96-well microtiter plates (Costar, Cambridge, MA) in 100 μl of growth medium, and after overnight attachment, exposed for the indicated days to inhibitors or vehicle (DMSO). After the treatment interval (24, 48, and 72 h) at 37°C, cells were washed in medium, and the number of viable cells was determined using a colorimetric cell proliferation assay (CellTiter96 Aqueous NonRadioactive Cell Proliferation Assay; Promega, Madison, WI)80 essentially by incubating cells in the MTS solution for 2 h and by determining the absorbance at 490 nm wavelength, as reported previously.31 Dose response curves and IC50 values were obtained using GraphPad Prism (GraphPad Software, Inc.., La Jolla, CA) by taking the absorption values from an MTS assay.
Annexin V apoptosis assay
Apoptosis induction in vehicle- or inhibitor-treated cells was assayed by the detection of membrane externalization of phosphatidylserine using an Annexin V assay kit (Molecular Probes, Invitrogen) as described previously.31 2 × 105 cells were harvested at various intervals after treatment, washed with ice-cold phosphate-buffered saline (PBS) and resuspended in 200 μl of binding buffer. Annexin V-FITC and 1 μg/ml propidium iodide were added and cells were incubated for 15 min in a dark environment. Labeling was analyzed by flow cytometry with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
Cell cycle analysis
The effect of varying concentrations of inhibitors on cell cycle distribution was determined by flow cytometric analysis of the nuclear DNA content as previously described.31 Briefly, cells grown exponentially to 50–60% confluency were exposed to the inhibitors or DMSO for a range of intervals, harvested, washed in ice-cold PBS, and fixed in 70% ethanol. DNA was stained by incubating the cells in PBS containing propidium iodide (50 μg/ml) and RNase A (1 mg/ml) for 60 min at room temperature, and fluorescence was measured and analyzed using a Becton Dickinson FACScan and Cell Quest software (Becton Dickinson Immunocytometry Systems, San Jose, CA).
Western blotting analysis
Treated and untreated cells were washed in cold PBS and lysed in buffer containing 30 mM HEPES, 10% glycerol, 1% Triton X-100, 100 mM NaCl, 10 mM MgCl2, 5 mM EDTA, 2 mM Na3VO4, 2 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 0.8 μM aprotinin, 50 μM bestatin, 15 μM E-64, 20 μM leupeptin, and 10 μM pepstatin A for 15 min on ice. Samples were centrifuged at 12,000 g for 15 min, supernatants were isolated, and protein was quantified using Protein Assay Reagent (Pierce Chemical, Rockford, IL). Equal amounts of protein were separated by SDS PAGE and electrotransferred onto a nylon membrane (Invitrogen). Nonspecific antibody binding was blocked by incubation of the blots with 4% bovine serum albumin in Tris-buffered saline (TBS)/Tween 20 (0.1%) for 1 h at room temperature. The blots were then probed with appropriate dilutions of primary antibody overnight at 4°C. The antibody-labeled blots were washed 3 times in TBS/Tween 20 for 15 min and then incubated with a 1:2000 dilution of horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Inc..) in TBS/Tween 20 at room temperature for 1 h. After additional washing in TBS/Tween 20, the proteins were visualized by Western Blot Chemiluminescence Reagent (Cell Signaling Technology Inc., Beverly, MA). Where indicated, the blots were reprobed with antibodies against β-actin to ensure equal loading and transfer of proteins.
Quantitative real-time PCR array
Changes in the gene expression were determined using real-time PCR (RTPCR) assay. Cells (5 × 105/well) were plated in 6-well plates (Costar, Cambridge, MA) in 2.0 ml of growth medium, and after overnight attachment, exposed to inhibitors or vehicle (DMSO) for 24 h. After the treatment, cells were washed in PBS and total RNA was isolated using RNeasy mini kit (Qiagen) as per the manufacturer's protocol. RNA concentration and purity were checked by UV spectrophotometry, and RNA integrity was determined by electrophoresis. Primers were purchased from Qiagen that enable gene expression analysis using SYBR Green-based quantitative real-time PCR. cDNA synthesis was performed using QuantiTect Reverse Transcription Kit (Qiagen # 205311). The RT-PCR reactions were run on the MyIQ (Bio-Rad) and the results, normalized to GAPDH, were determined using iQ5 software (Bio-Rad).
Transient transfection
Optimal 29mer-pRS-shRNA constructs were obtained from Origene (Rockville, MD). Sequences specific for human Aurora kinase A, Aurora kinase B or survivin and non-target control shRNA sequences were used for this study. Cells were seeded in 6-well plates and allowed to reach 70% to 80% confluence. Transfection of targeting or control (non-targeting) shRNA was performed by using FuGene 6 according to the manufacturer's recommendations (Roche Applied Science, Indianapolis, IN). One μg of shRNA in 100 μL Opti-MEM medium was mixed with 2 μL of FuGene 6. After the mixture was incubated at room temperature for 20 min, complete medium was added to make the total volume up to 2 mL. After 48 h, medium was changed and cells were incubated with inhibitors for 24 h. Cell viability (annexin V/propidium iodide binding) or Western blot analysis were carried out as described above.
Fluorescence microscopy
Cells were grown on chamber slides (Nalge Nunc, Naperville, IL) in growth medium, and, after an overnight attachment period, were exposed to selected concentrations of inhibitor or vehicle (DMSO) for various intervals. Cells were washed once with PBS, fixed with 3.7% formaldehyde for 30 min and stained with DAPI for 2 h at room temperature. The slides were then washed in PBS, mounted, and examined under a fluorescent microscope. Morphological changes in response to inhibitor treatment were evaluated by microscopic inspection.
Statistical analysis
Unless otherwise stated, data are expressed as mean ± SD. The significance of differences between experimental conditions was determined using a 2-tailed Student's t test. Differences were considered significant at p values <0.05.
Authorship contributions:
Participated in research design: Daniel R. Premkumar and Ian F. Pollack. Conducted experiments: Esther P. Jane and Daniel R. Premkumar. Performed data analysis: Esther P. Jane, Daniel R. Premkumar, and Ian F. Pollack. Wrote or contributed to the writing of the manuscript: Daniel R. Premkumar and Ian F. Pollack
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Jay Venkatachari, Robert Lacomy and Alexis Styche for technical assistance.
Funding
This work was supported by National Institutes of Health [Grant P01NS40923] (to I.F.P); by the Walter L. Copeland fund of The Pittsburgh Foundation (to D.R.P); by a grant from Ian's Friends Foundation and Connor's Cure Foundation (to I.F.P).
References
- 1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008; 359:492-507; PMID:18669428; http://dx.doi.org/ 10.1056/NEJMra0708126 [DOI] [PubMed] [Google Scholar]
- 2. DeAngelis LM. Brain tumors. N Engl J Med 2001; 344:114-23; PMID:11150363 [DOI] [PubMed] [Google Scholar]
- 3. Pollack IF. Brain tumors in children. N Engl J Med 1994; 331:1500-7; PMID:7969301 [DOI] [PubMed] [Google Scholar]
- 4. Ziegler DS, Kung AL, Kieran MW. Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol 2008; 26:493-500; PMID:18202424; http://dx.doi.org/ 10.1200/JCO.2007.13.9717 [DOI] [PubMed] [Google Scholar]
- 5. Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther 2007; 6:1909-19; PMID:17620423; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-0047 [DOI] [PubMed] [Google Scholar]
- 6. Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 353:2012-24; PMID:16282176; http://dx.doi.org/ 10.1056/NEJMoa051918 [DOI] [PubMed] [Google Scholar]
- 7. Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res 1996; 56:5079-86; PMID:8895767 [PubMed] [Google Scholar]
- 8. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C et al. ,, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007; 21:2683-710; PMID:17974913; http://dx.doi.org/ 10.1101/gad.1596707 [DOI] [PubMed] [Google Scholar]
- 9. Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol 2001; 12:745-60; PMID:11484948; http://dx.doi.org/ 10.1023/A:1011177318162 [DOI] [PubMed] [Google Scholar]
- 10. Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S,Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res 2006; 12:5064-73; PMID:16951222; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0913 [DOI] [PubMed] [Google Scholar]
- 11. Ge H, Gong X, Tang CK. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J Cancer 2002; 98:357-61; PMID:11920586; http://dx.doi.org/ 10.1002/ijc.10224 [DOI] [PubMed] [Google Scholar]
- 12. Li B, Yuan M, Kim IA, Chang CM, Bernhard EJ, Shu HK. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004; 23:4594-602; PMID:15077177; http://dx.doi.org/ 10.1038/sj.onc.1207602 [DOI] [PubMed] [Google Scholar]
- 13. Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3'-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res 2003; 63:2742-6; PMID:12782577 [PubMed] [Google Scholar]
- 14. Lorimer IA, Lavictoire SJ. Activation of extracellular-regulated kinases by normal and mutant EGF receptors. Biochim Biophys Acta 2001; 1538:1-9; PMID:11341977; http://dx.doi.org/ 10.1016/S0167-4889(00)00129-4 [DOI] [PubMed] [Google Scholar]
- 15. Prigent SA, Nagane M, Lin H, Huvar I,Boss GR, Feramisco JR, Cavenee WK, Huang HS. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J Biol Chem 1996; 271:25639-45; PMID:8810340; http://dx.doi.org/ 10.1074/jbc.271.41.25639 [DOI] [PubMed] [Google Scholar]
- 16. de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM,You MJ, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev 2008; 22:449-62; PMID:18258752; http://dx.doi.org/ 10.1101/gad.1606508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000; 19:2474-88; PMID:10851046; http://dx.doi.org/ 10.1038/sj.onc.1203527 [DOI] [PubMed] [Google Scholar]
- 18. Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci U S A 1998; 95:5724-9; PMID:9576951; http://dx.doi.org/ 10.1073/pnas.95.10.5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu JL, Abe T, Inoue R, Fujiki M, Kobayashi H. IkappaBalphaM suppresses angiogenesis and tumorigenesis promoted by a constitutively active mutant EGFR in human glioma cells. Neurol Res 2004; 26:785-91; PMID:15494123; http://dx.doi.org/ 10.1179/016164104225014139 [DOI] [PubMed] [Google Scholar]
- 20. Thaker NG, Zhang F, McDonald PR, Shun TY, Lazo JS, Pollack IF. Functional genomic analysis of glioblastoma multiforme through short interfering RNA screening: a paradigm for therapeutic development. Neurosurg Focus 2010; 28:E4; PMID:20043719 [DOI] [PubMed] [Google Scholar]
- 21. Thaker NG, Pollack IF. Molecularly targeted therapies for malignant glioma: rationale for combinatorial strategies. Expert Rev Neurother 2009; 9:1815-36; PMID:19951140; http://dx.doi.org/ 10.1586/ern.09.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thaker NG, Zhang F, McDonald PR, Shun TY,Lewen MD, Pollack IF, Lazo JS. Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol Pharmacol 2009; 76:1246-55; PMID:19783622; http://dx.doi.org/ 10.1124/mol.109.058024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thaker NG, McDonald PR, Zhang F, Kitchens CA, Shun TY, Pollack, Lazo JS. Designing, optimizing, and implementing high-throughput siRNA genomic screening with glioma cells for the discovery of survival genes and novel drug targets. J Neurosci Methods 2010; 185:204-12; PMID:19782703; http://dx.doi.org/ 10.1016/j.jneumeth.2009.09.023 [DOI] [PubMed] [Google Scholar]
- 24. Kitchens CA, McDonald PR, Shun TY, Pollack IF, Lazo JS. Identification of chemosensitivity nodes for vinblastine through small interfering RNA high-throughput screens. J Pharmacol Exp Ther 2011; 339:851-8; PMID:21880871; http://dx.doi.org/ 10.1124/jpet.111.184879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L, Lin X, Hierman JS, Wilburn DL, Watkins DN; et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res 2008; 68:2321-8; PMID:18381439; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10:375-88; PMID:17097560; http://dx.doi.org/ 10.1016/j.ccr.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 27. Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J; Warner RB, Ng SC, Fesik SW, Elmore SW, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res 2007; 67:1176-83; PMID:17283153; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2203 [DOI] [PubMed] [Google Scholar]
- 28. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar; Willis SN, Scott CL, Day CL, Cory S, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10:389-99; PMID:17097561; http://dx.doi.org/ 10.1016/j.ccr.2006.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S; Avet-Loiseau H, Bataille R, Moreau P, Le Gouill S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood 2011; 118:3901-10; PMID:21835956; http://dx.doi.org/ 10.1182/blood-2010-11-317438 [DOI] [PubMed] [Google Scholar]
- 30. Jane EP, Premkumar DR, DiDomenico JD, Hu B, Cheng SY, Pollack IF. YM-155 potentiates the effect of ABT-737 in malignant human glioma cells via survivin and Mcl-1 downregulation in an EGFR-dependent context. Mol Cancer Ther 2013; 12:326-38; PMID:23325792; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jane EP, Premkumar DR, Morales A, Foster KA, Pollack IF. Inhibition of phosphatidylinositol 3-kinase/AKT signaling by NVP-BKM120 promotes ABT-737-induced toxicity in a caspase-dependent manner through mitochondrial dysfunction and DNA damage response in established and primary cultured glioblastoma cells. J Pharmacol Exp Ther 2014; 350:22-35; PMID:24741074; http://dx.doi.org/ 10.1124/jpet.114.212910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Premkumar DR, Jane EP, DiDomenico JD, Vukmer NA, Agostino NR, Pollack IF. ABT-737 synergizes with bortezomib to induce apoptosis, mediated by Bid cleavage, Bax activation, and mitochondrial dysfunction in an Akt-dependent context in malignant human glioma cell lines. J Pharmacol Exp Ther 2012; 341:859-72; PMID:22393246; http://dx.doi.org/ 10.1124/jpet.112.191536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, Macher-Goeppinger S, Radlwimmer B, Wiestler OD, Herold-Mende C; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008; 27:6646-56; PMID:18663354; http://dx.doi.org/ 10.1038/onc.2008.259 [DOI] [PubMed] [Google Scholar]
- 34. Premkumar DR, Jane EP, Agostino NR, DiDomenico JD, Pollack IF. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol Carcinog 2013; 52:118-33; PMID:22086447; http://dx.doi.org/ 10.1002/mc.21835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foster KA, Jane EP, Premkumar DR, Morales A, Pollack IF. Co-administration of ABT-737 and SAHA induces apoptosis, mediated by Noxa upregulation, Bax activation and mitochondrial dysfunction in PTEN-intact malignant human glioma cell lines. J Neurooncol 2014; PMID:25139025 [DOI] [PubMed] [Google Scholar]
- 36. Voss V, Senft C, Lang V, Ronellenfitsch MW, Steinbach JP, Seifert V, Kögel D. The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res 2010; 8:1002-16; PMID:20587533; http://dx.doi.org/ 10.1158/1541-7786.MCR-09-0562 [DOI] [PubMed] [Google Scholar]
- 37. Cristofanon S, Fulda S. ABT-737 promotes tBid mitochondrial accumulation to enhance TRAIL-induced apoptosis in glioblastoma cells. Cell Death Dis 2012; 3:e432; PMID:23190604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res 2008; 14:6042-54; PMID:18829483; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benekli M, Baumann H, Wetzler M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J Clin Oncol 2009; 27:4422-32; PMID:19667270; http://dx.doi.org/ 10.1200/JCO.2008.21.3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agulnik M, da Cunha Santos G, Hedle D, Nicklee T, Dos Reis PP, Ho J, et al. Predictive and pharmacodynamic biomarker studies in tumor and skin tissue samples of patients with recurrent or metastatic squamous cell carcinoma of the head and neck treated with erlotinib. J Clin Oncol 2007; 25:2184-90; PMID:17538163; http://dx.doi.org/ 10.1200/JCO.2006.07.6554 [DOI] [PubMed] [Google Scholar]
- 41. Tweardy D, Chang JC. Stat5: from breast development to cancer prognosis, prediction, and progression. J Clin Oncol 2011; 29:2443-4; PMID:21576641; http://dx.doi.org/ 10.1200/JCO.2010.34.2014 [DOI] [PubMed] [Google Scholar]
- 42. Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res 2003; 63:1270-9; PMID:12649187 [PubMed] [Google Scholar]
- 43. Shi X, Franko B, Frantz C, Amin HM, Lai R. JSI-124 (cucurbitacin I) inhibits Janus kinase-3/signal transducer and activator of transcription-3 signalling, downregulates nucleophosmin-anaplastic lymphoma kinase (ALK), and induces apoptosis in ALK-positive anaplastic large cell lymphoma cells. Br J Haematol 2006; 135:26-32; PMID:16939498; http://dx.doi.org/ 10.1111/j.1365-2141.2006.06259.x [DOI] [PubMed] [Google Scholar]
- 44. Jung SH, Lee YK, Lee HJ, Choi NR, Vo MC, Hoang MD; Lim MS, Nguyen-Pham TN, Kim HJ, et al. Dendritic cells loaded with myeloma cells pretreated with a combination of JSI-124 and bortezomib generate potent myeloma-specific cytotoxic T lymphocytes in vitro. Exp Hematol 2014; 42:274-81; PMID:24407159; http://dx.doi.org/ 10.1016/j.exphem.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 45. Gheeya JS, Chen QR, Benjamin CD, Cheuk AT,Tsang P, Chung JY, et al. Screening a panel of drugs with diverse mechanisms of action yields potential therapeutic agents against neuroblastoma. Cancer Biol Ther 2009; 8:2386-95; PMID:19946221; http://dx.doi.org/ 10.4161/cbt.8.24.10184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Molavi O, Ma Z, Mahmud A, Alshamsan A, Samuel J, Lai R, Kwon GS, Lavasanifar A. Polymeric micelles for the solubilization and delivery of STAT3 inhibitor cucurbitacins in solid tumors. Int J Pharm 2008; 347:118-27; PMID:17681440; http://dx.doi.org/ 10.1016/j.ijpharm.2007.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tseng LM, Huang PI, Chen YR, Chen YC, Chou YC, Chen YW; Chang YL, Hsu HS, Lan YT, Chen KH, et al. Targeting signal transducer and activator of transcription 3 pathway by cucurbitacin I diminishes self-renewing and radiochemoresistant abilities in thyroid cancer-derived CD133+ cells. J Pharmacol Exp Ther 2012; 341:410-23; PMID:22328572; http://dx.doi.org/ 10.1124/jpet.111.188730 [DOI] [PubMed] [Google Scholar]
- 48. Premkumar DR, Jane EP, Agostino NR, Scialabba JL, Pollack IF. Dasatinib synergizes with JSI-124 to inhibit growth and migration and induce apoptosis of malignant human glioma cells. J Carcinog 2010; 9; PMID:20808823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z, Zhang J. JSI-124 inhibits glioblastoma multiforme cell proliferation through G(2)/M cell cycle arrest and apoptosis augment. Cancer Biol Ther 2008; 7:1243-9; PMID:18487947; http://dx.doi.org/ 10.4161/cbt.7.8.6263 [DOI] [PubMed] [Google Scholar]
- 50. Hsu HS, Huang PI, Chang YL, Tzao C, Chen YW, Shih HC, Hung SC, Chen YC, Tseng LM, Chiou SH. Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in nonsmall cell lung cancer-derived CD133-positive cells. Cancer 2011; 117:2970-85; PMID:21225866; http://dx.doi.org/ 10.1002/cncr.25869 [DOI] [PubMed] [Google Scholar]
- 51. Lieblein JC, Ball S, Hutzen B, Sasser AK, Lin HJ, Huang TH, Hall BM, Lin J. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 2008; 8:302; PMID:18939993; http://dx.doi.org/ 10.1186/1471-2407-8-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brady MT, Miller A, Sait SN, Ford LA, Minderman H, Wang ES, Lee KP, Baumann H, Wetzler M. Down-regulation of signal transducer and activator of transcription 3 improves human acute myeloid leukemia-derived dendritic cell function. Leuk Res 2013; 37:822-8; PMID:23628554; http://dx.doi.org/ 10.1016/j.leukres.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishdorj G, Johnston JB, Gibson SB. Cucurbitacin-I (JSI-124) activates the JNK/c-Jun signaling pathway independent of apoptosis and cell cycle arrest in B leukemic cells. BMC Cancer 2011; 11:268; PMID:21702955; http://dx.doi.org/ 10.1186/1471-2407-11-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ishdorj G, Johnston JB, Gibson SB. Inhibition of constitutive activation of STAT3 by curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Mol Cancer Ther 2010; 9:3302-14; PMID:21159613; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0550 [DOI] [PubMed] [Google Scholar]
- 55. Chen CL, Hsieh FC, Lieblein JC, Brown J, Chan C, Wallace JA, Cheng G, Hall BM, Lin J. Stat3 activation in human endometrial and cervical cancers. Br J Cancer 2007; 96:591-9; PMID:17311011; http://dx.doi.org/ 10.1038/sj.bjc.6603597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chumbalkar V, Latha K, Hwang Y, Maywald R, Hawley L, Sawaya R; Diao L, Baggerly K, Cavenee WK, Furnari FB, et al. Analysis of phosphotyrosine signaling in glioblastoma identifies STAT5 as a novel downstream target of DeltaEGFR. J Proteome Res 2011; 10:1343-52; PMID:21214269; http://dx.doi.org/ 10.1021/pr101075e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Latha K, Li M, Chumbalkar V, Gururaj A, Hwang Y, Dakeng S, et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int J Cancer 2013; 132:509-20; PMID:22729867; http://dx.doi.org/ 10.1002/ijc.27690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shinojima N, Tada K, Shiraishi S, Kamiryo T,Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 2003; 63:6962-70; PMID:14583498 [PubMed] [Google Scholar]
- 59. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res 1991; 51:2164-72; PMID:2009534 [PubMed] [Google Scholar]
- 60. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60:1383-7; PMID:10728703 [PubMed] [Google Scholar]
- 61. Wang Q, Hirohashi Y, Furuuchi K, Zhao H, Liu Q,Zhang H, Murali R, Berezov A, Du X, Li B; et al. The centrosome in normal and transformed cells. DNA Cell Biol 2004; 23:475-89; PMID:15307950; http://dx.doi.org/ 10.1089/1044549041562276 [DOI] [PubMed] [Google Scholar]
- 62. Beltrami E, Plescia J, Wilkinson JC, Duckett CS, Altieri DC. Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J Biol Chem 2004; 279:2077-84; PMID:14581472; http://dx.doi.org/ 10.1074/jbc.M309479200 [DOI] [PubMed] [Google Scholar]
- 63. Kimura M, Yoshioka T, Saio M, Banno Y, Nagaoka H, Okano Y. Mitotic catastrophe and cell death induced by depletion of centrosomal proteins. Cell Death Dis 2013; 4:e603; PMID:23598415; http://dx.doi.org/ 10.1038/cddis.2013.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nabzdyk CS, Lancero H, Nguyen KP, Salek S, Conte MS. RNA interference-mediated survivin gene knockdown induces growth arrest and reduced migration of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 2011; 301:H1841-9; PMID:21856925; http://dx.doi.org/ 10.1152/ajpheart.00089.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hoar K, Chakravarty A, Rabino C, Wysong D, Bowman D, Roy N, Ecsedy JA. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol 2007; 27:4513-25; PMID:17438137; http://dx.doi.org/ 10.1128/MCB.02364-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li J, Anderson MG, Tucker LA, Shen Y, Glaser KB, Shah OJ. Inhibition of Aurora B kinase sensitizes a subset of human glioma cells to TRAIL concomitant with induction of TRAIL-R2. Cell Death Differ 2009; 16:498-511; PMID:19079141; http://dx.doi.org/ 10.1038/cdd.2008.174 [DOI] [PubMed] [Google Scholar]
- 67. Lee PY, Chen CL, Lin ZZ, Cheng AL, Chen EI, Whang-Peng J, Huang CY. The Aurora kinases inhibitor VE-465 is a novel treatment for glioblastoma multiforme. Oncology 2013; 84:326-35; PMID:23636005; http://dx.doi.org/ 10.1159/000347021 [DOI] [PubMed] [Google Scholar]
- 68. Lens SM, Wolthuis RM, Klompmaker R, Kauw J, Agami R, Brummelkamp T, Kops G, Medema RH. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J 2003; 22:2934-47; PMID:12805209; http://dx.doi.org/ 10.1093/emboj/cdg307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, Chen J, Lau J, Knobbe-Thomsen C, Weller M, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013; 24:438-49; PMID:24135280; http://dx.doi.org/ 10.1016/j.ccr.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010; 29:3997-4006; PMID:20473332; http://dx.doi.org/ 10.1038/onc.2010.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hochegger H, Hegarat N, Pereira-Leal JB. Aurora at the pole and equator: overlapping functions of Aurora kinases in the mitotic spindle. Open Biol 2013; 3:120185; PMID:23516109; http://dx.doi.org/ 10.1098/rsob.120185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang XH, Rao M, Loprieato JA, Hong JA, Zhao M, Chen GZ, Humphries AE, Nguyen DM, Trepel JB, Yu X, Schrump DS. Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer Biol Ther 2008; 7:1388-97; PMID:18708766; http://dx.doi.org/ 10.4161/cbt.7.9.6415 [DOI] [PubMed] [Google Scholar]
- 73. Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol 2007; 8:798-812; PMID:17848966; http://dx.doi.org/ 10.1038/nrm2257 [DOI] [PubMed] [Google Scholar]
- 74. Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res 2006; 12:6869-75; PMID:17145803; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1405 [DOI] [PubMed] [Google Scholar]
- 75. Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res 2005; 11:3155-62; PMID:15867207; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-2223 [DOI] [PubMed] [Google Scholar]
- 76. Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, Huang CM, Gill GN, Wiley HS, Cavenee WK. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997; 272:2927-35; PMID:9006938; http://dx.doi.org/ 10.1074/jbc.272.5.2927 [DOI] [PubMed] [Google Scholar]
- 77. Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 1997; 94:12479-84; PMID:9356475; http://dx.doi.org/ 10.1073/pnas.94.23.12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weller M, Rieger J, Grimmel C, Van Meir EG, De Tribolet N,Krajewski S, Reed JC, von Deimling A, Dichgans J. Predicting chemoresistance in human malignant glioma cells: the role of molecular genetic analyses. Int J Cancer 1998; 79:640-4; PMID:9842975; http://dx.doi.org/ 10.1002/(SICI)1097-0215(19981218)79:6%3c640::AID-IJC15%3e3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 79. Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther 2011; 10:198-208; PMID:21220502; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Riss TL MR. Comparison of MTT, XTT and a novel tetrazolium compound MTS for in vitro proliferation and chemosensitivity assays. Mol Biol Cell 1992; 3:184 [Google Scholar]