

Abstract
Pancreatic neuroendocrine tumors (PNETs) are rare, indolent tumors that may occur sporadically or develop in association with well-recognized hereditary syndromes, particularly multiple endocrine neoplasia type 1 (MEN-1). We previously demonstrated that the hedgehog (HH) signaling pathway was aberrantly up-regulated in a mouse model that phenocopies the human MEN-1 syndrome, Men1l/l;RipCre, and that inhibition of this pathway suppresses MEN-1 tumor cell proliferation. We hypothesized that the HH signaling pathway is similarly upregulated in human PNETs. We performed immunohistochemical (IHC) staining for PTCH1 in human fresh and archival PNET specimens to examine whether human sporadic and MEN-1-associated PNETs revealed similar abnormalities as in our mouse model and correlated the results with clinical and demographic factors of the study cohort. PTCH1 staining was positive in 12 of 22 PNET patients (55%). Four of 5 MEN-1 patients stained for PTCH1 (p = 0.32 as compared with sporadic disease patients). Nine of 16 patients with metastatic disease stained for PTCH1 as compared with zero of 3 with localized disease only (p = 0.21). No demographic or clinical features appeared to be predictive of PTCH 1 positivity and PTCH 1 positivity per se was not predictive of clinical outcome. PTCH1, a marker of HH pathway up regulation, is detectable in both primary and metastatic tumors in more than 50% of PNET patients. Although no clinical or demographic factors predict PTCH1 positivity and PTCH1 positivity does not predict clinical outcome, the frequency of expression alone indicates that perturbation of this pathway with agents such as Vismodegib, an inhibitor of Smoothened (SMO), should be examined in future clinical trials.
Keywords: hedgehog, MEN-1, neuroendocrine, pancreas
Abbreviations
- ACTH
Adrenocorticotrophic hormone
- BCNS
basal cell nevus syndrome
- CgA
chromogranin A
- HH
hedgehog
- IHC
immunohistochemical
- MEN-1
multiple neuroendocrine tumor syndrome type 1
- NF-1
neurofibromatosis type 1
- PRRT
peptide radioreceptor therapy
- PNET
pancreatic neuroendocrine tumor
- PTCH 1
protein patched homolog 1
- SMO
smoothened
- VHL
von Hippel Lan- dau
- WHO
World Health Organization
Introduction
Neuroendocrine tumors (NETs) are rare, indolent tumors of various organs with variable presentations and responses to therapy.1 They are often subdivided clinically into pancreatic NETs (PNETs) and carcinoid tumors (of the alimentary tract, lungs and other sites) because of perceived differences in hereditary predisposition, syndromic presentation and response to therapy.1 PNETs may occur sporadically or develop in association with well-recognized hereditary syndromes (particularly multiple endocrine neoplasia type 1 [MEN-1] but also von Hippel Landau [VHL], Sturge Weber or neurofibromatosis type 1 [NF-1] syndromes).1-3
MEN-1 syndrome is an autosomal dominant condition that presents with 1) 4-gland parathyroid hyperplasia leading to hypercalcemia, 2) pituitary macroadenomas (primarily prolactinomas and ACTH producing lesions [Cushing's disease]), and 3) gastroenteropancreatic tumors (PNETs) that may be clinically silent or associated with various syndromes (especially Zollinger-Ellison syndrome and the insulinoma syndrome, among others).1-3 The MEN-1 gene resides on chromosome 11q13 and encodes for a 610 amino acid nuclear protein called menin that is mutated in patients with MEN-1.4 The recently characterized crystal structure of menin demonstrates that it acts as a scaffold protein in regulating gene transcription, cell proliferation, apoptosis and genome stability via its interaction with its various partners.5-8 However, MEN-1-affected individuals only develop clinically apparent phenotypic disease in early adulthood because they are protected from tumorigenesis earlier in life by virtue of having one normal tumor-suppressor MEN-1 allele inherited from their unaffected parent. For tumors to develop a second, somatic mutation must occur that inactivates the normal unaffected allele in at risk tissues leading to tumor development.9
Most sporadic PNETs develop in isolation, and are diagnosed approximately one decade later than those associated with MEN-1 syndrome.1 A landmark genomic study by Papadopolous et al identified 4 gene abnormalities in sporadic PNETs including MEN-1, Daxx, Atrx and Pten and demonstrated somatic MEN-1 abnormalities in greater than 40% of sporadic PNETs, indicating that tumorigenesis pathways may be similar for sporadic and hereditary PNET patients.10 For tumors to occur in sporadic patients one would expect 2 somatic mutations to occur inactivating both tumor-suppressing alleles.
We previously demonstrated that the hedgehog (HH) signaling pathway was aberrantly upregulated in a mouse model that phenocopies the human MEN-1 syndrome, Men1l/l;RipCre.11 Moreover, by pharmacologically inhibiting the HH signaling pathway with a HH antagonist, GDC-0449, we significantly suppressed MEN-1 tumor development.11 This pathway is potentially targetable in PNET patients as GDC-0449 (Vismodegib, Erivedge) is currently available clinically for the treatment of basal cell carcinoma.12 Vismodegib (Genentech, South San Francisco, California) acts by selectively binding to Smoothened (SMO), a 7-helix transmembrane receptor, thereby inhibiting activation of transcription factors of the glioma-associated oncogene family, to suppress tumor proliferation and growth.12,13 Activation of this pathway can be demonstrated by immunohistochemistry (IHC) staining for protein patched homolog 1 (PTCH1), the receptor for sonic hedgehog ligand and also a target gene upregulated by HH signaling.11-13 The aims of the current study were to 1) identify whether the same HH pathway abnormalities we previously demonstrated in mice were also present in human fresh and archival PNET specimens by IHC PTCH 1 staining, 2) to compare HH pathway IHC staining patterns in PNET specimens from MEN-1-associated and sporadic disease patients, and 3) to evaluate clinical features of patients that might predict the presence of HH pathway abnormalities in PNET patients. Our ultimate goal is to examine whether HH pathway inhibitors may play a role in mediating disease outcome for PNET patients.
Results
Table 1 demonstrates demographic and clinical data for the 22 patients enrolled in the current study. The mean age at biopsy was 50.8 ± 13.7 years and the mean duration of disease at biopsy was 2.0 ± 5.6 years. Eight patients were male (36%), 20 (91%) were Caucasian, one (4.5%) was Asian and one (4.5%) was African American. Thirteen patients (59%) had functional tumors and most of these were gastrinomas (7) or insulinomas (4). MEN-1 syndrome was present in 5 patients (23%) but only 4 were confirmed as such by genetic testing. At biopsy 3 patients (13%) were on somatostatin analog therapy (for a mean duration preceding biopsy of 5 months). Chromogranin A levels were available within 2 months of biopsy in 11 patients (50%) and averaged 36 times the upper limit of normal (including 2 of the 3 of the patients receiving somatostatin analog therapy; one was within normal limits and the other was 258 ng/ml with an upper limit of normal of 95 ng/ml). Four patients (18%) had previously received other antitumor therapies (chemotherapy in one, liver-directed therapy in 2 and peptide radioreceptor therapy [PRRT] in one). Three patients (14%) had previously undergone surgical resections (one had undergone multiple prior resections).
Table 1.
Demographic and clinical data for the study cohort
| Mean Age at Biopsy (years ±SD) | 50.8 ± 13.7 |
| Duration of Disease at Biopsy (years ± SD) | 2.0 ± 5.6 |
| Male (Number [%]) | 8 (36) |
| Ethnicity (Number [%]) | |
| White | 20 (91) |
| Asian | 1 (4.5) |
| African American | 1 (4.5) |
| Functional Syndrome (Number [%]) | |
| Absent | 9 (41) |
| Present | 13 (59) |
| Gastrinoma | 7 (32) |
| Insulinoma | 4 (18) |
| Other (1 VIPoma; 1 Somatostatinoma) | 2 (9) |
| Inherited Syndrome (Number [%]) | |
| Absent | 17 (77) |
| Present (all MEN-1) | 5 (23)* |
| Chromogranin A at Biopsy (% above upper level limit) | 3686** |
| Date range from CgA result to Biopsy (days) | 0-64 |
| Somatostatin Analog therapy at Biopsy (Number [%]) | 3 (13) |
| Duration of therapy at Biopsy (months ± SD) | 5 ± 1 |
| Prior Chemotherapy (Number [%]) | 1 (5) |
| Prior Liver directed therapy (Number [%]) | 2 (9) |
| Prior PRRT (number [%]) | 1 (5) |
| Prior surgical resections (Number [%]) | |
| None | 19 (86) |
| One | 2 (9) |
| Two | 0 (0) |
| Three or more | 1 (5) |
4 confirmed by genetic testing, 1 unconfirmed clinical diagnosis.
*11 with no CgA results at Biopsy.
Table 2 provides specific information regarding pathology, clinical features and PTCH 1 findings for individual patients. Primary tumor sizes ranged from 0.6 cm to 7.5 cm. Seventeen patients had pancreatic primary tumors and 7 had duodenal primary tumors (including 3 patients who had primary tumors in both the pancreas and duodenum). In one patient the primary tumor was not identified. Twelve of 22 tumors were NET grade 1, 9 were NET grade 2 and one was a neuroendocrine carcinoma (grade 3). Fifteen tumors were well differentiated, 6 were moderately differentiated and one was poorly differentiated. At the time of surgery, 3 patients had localized disease, 13 had regional lymph nodes only (including one patient with an isolated lymph node in the posterior pancreatic head) and 4 had widespread metastases. Information regarding local lymph node status at surgery was unavailable in 3 cases (one patient underwent a local pancreatic enucleation without lymph node dissection, one surgery was limited to an ovarian metastasis resection, and one patient underwent surgery at an outside hospital and we were unable to retrieve this information).
Table 2.
Pathology, clinical features and PTCH1 staining results
| Subject # | Specimen Size(s) (cm) | Primary Tumor site(s) | Tumor Grade | Differentiation | Lymph Node Metastases (Y/N) | Liver Metastases (Y/N) | Syndrome | CgA [pg/ml] (normal range) | MEN 1 (Y/N) | PTCH1 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.4 | Duodenum | 1 | Well | Y | N | Gastrinoma | 7663 (0–50 ) | N | +++ |
| 2 | 8 | Pancreas | 2 | Moderate | Y | Y | none | 258 (0–95) | N | ++ |
| 3 | 8 | Pancreas | 1 | Well | Y | N | none | 174 (0–95) | N | − |
| 4 | 5 | Pancreas | 2 | Moderate | Y | N | none | n/a | N | +++ |
| 5 | 0.8 | Duodenum | 1 | Well | Y | N | Gastrinoma | 432 (0-375) | N | ++ |
| 6 | 7.5 | Pancreas | 2 | Moderate | N | N | VIPoma | n/a | N | + |
| 7 | 1.3 | Pancreas | 2 | Well | N | N | Somato statinoma | n/a | N | + |
| 8 | 2 | Pancreas | 1 | Well | N | N | Insulinoma | n/a | N | − |
| 9 | 1.5 | Pancreas | 2 | Moderate | Y | N | none | n/a | N | ++ |
| 10 | 0.6/1.0 | Pancreas, Duodenum | 1 | Well | Y | Y | none | n/a | N | − |
| 11 | 4.5 | Pancreas | 2 | Well | Y | N* | none | n/a | N | +++ |
| 12 | 1.6 | Pancreas | 1 | Well | Y | N | none | <200 (0–95) | Y | + |
| 13 | 4.5/0.7 | Pancreas, Duodenum | 1 | Well | Y | N | Gastrinoma | 2725 (0–50) | N | − |
| 14 | 2.5 | Pancreas | 3 | Poor | Y | Y | Insulinoma | 15600 (0–95) | N | − |
| 15 | 1.5 | Duodenum | 2 | Moderate | Y | N | Insulinoma | 5.4 (1.9–15.0) | N | − |
| 16 | 4 | Pancreas | 2 | Moderate | Y | N | none | n/a | N | +++ |
| 17 | 3.1 | Unknown | 1 | Well | Y | N | Gastrinoma | 96.0 (n/a) | N | + |
| 18 | 1.1 | Duodenum | 1 | Well | Y | N | Gastrinoma | 1625 (0–115) | Y | ++ |
| 19 | 5.5/3.2 | Pancreas | 2 | Well | n/a** | Y | none | 48 (0–50) | N | +++ |
| 20 | n/a | Pancreas, Duodenum | 1 | Well | Y | N | Gastrinoma | n/a | Y | +++ |
| 21 | n/a | Pancreas | 1 | Well | n/a | N | none | n/a | Y | +++ |
| 22 | 1.1/2.2 | Pancreas | 1 | Well | n/a*** | N | Insulinoma | n/a | Y | +++ |
Abbreviations: CgA = chromogranin A; n/a = not available.
This patient had a perihepatic lymph node identified at surgery but no liver metastases were identified.
*Away from operative site.
**Local enucleation.
Inspection of the PTCH1 stained sections revealed uptake primarily in pancreatic islets or tumor specimens. Figure 1 illustrates representative PTCH1 staining in a normal pancreatic islet (Fig. 1A) and a tumor expressing PTCH1 (Fig. 1C). In general, the number of positively staining cells was less homogeneous but denser in normal islets (Fig. 1A) as compared with primary or metastatic tumors (Fig. 1C). A representative tumor with undetectable PTCH1 staining is shown in Figure 1B. PTCH1 expression was undetectable in 6 of the 22 cases and 4 were only weakly positive (45%) (Table 2). Tissue specimens from 8 subjects stained robustly for PTCH1 and 4 exhibited intermediate staining (55%). Four of the 5 MEN-1 patients exhibited PTCH 1 positivity (3 robustly positive and one intermediately positive); one was only weakly positive.
Figure 1.
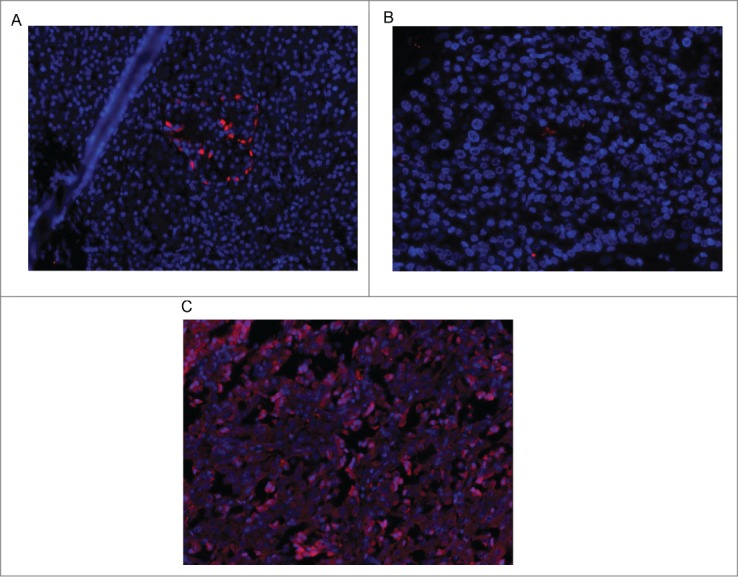
Immunofluorescence for PTCH 1 in a normal pancreatic islet (A), a tumor not expressing PTCH 1 (B), and a tumor expressing PTCH-1 (C).
Clinical and demographic features numerically (but not statistically) associated with positive PTCH 1 staining included MEN-1 status (4 of 5 [80%] with MEN-1 versus 8 of 17 [47%] without MEN-1; p = 0.32) and the presence of liver and/or lymph node metastases at the time of biopsy (9 of 16 [56%] with metastases vs. zero of 3 [0%] without; p = 0.21) (Table 2). The presence of liver metastases alone, age at biopsy, duration of disease at biopsy, presence of a neuroendocrine syndrome, size or site of the primary tumor, grade or differentiation of the primary tumor and chromogranin A level were not predictive of PTCH1 staining (Table 2, some data not shown).
During subsequent follow up, one patient (#19, Table 2) died from multi-organ system failure 4 months after surgery with evidence of tumor progression (PTCH1 staining in this patient was robustly positive). The remaining 21 patients were all alive at follow up, which ranged from 11 to 123 months. Of these, 6 appeared to be cured (# 3, 10, 15, 17, 21 and 22, Table 2) with normal tumor markers and no recurrence on imaging after 13 to 74 months of follow up. One patient (#17, Table 2) required a second surgery to remove a peripancreatic lymph node, which was missed initially, and another (#15, Table 2) underwent a liver resection at the time of surgery in addition to removal of the primary tumor and regional nodes. PTCH1 staining was undetectable in 3 of these patients, negative (weakly positive only) in one and robustly positive in 2. Two additional patients (#5 and 6, Table 2) had no evidence of disease on imaging after 14 and 98 months of follow up but both of these patients underwent 2 subsequent debulking surgeries each and neither are likely to be cured of disease. One was negative on PTCH1 staining (stained only weakly positive) and one was (intermediately) positive. The 2 remaining patients with established widespread metastases at the index surgery exhibited a reduction in tumor bulk after somatostatin analog therapy alone after 11 months of follow up with weakly positive PTCH1 staining (#2, Table 2) and stable disease after somatostatin analog therapy, chemotherapy and liver directed therapy after 21 months of follow up with undetectable PTCH1 staining (#14, Table 2), respectively. Three patients (#1, 12 and 18, Table 2), 2 with MEN-1 syndrome, have remained free of new widespread metastases during follow up which ranged from 16 to 80 months without the need for any anti-tumor therapy. PTCH1 staining in these 3 patients was (robustly) positive, negative (stained weakly) and (intermediately) positive, respectively. One MEN-1 syndrome patient (#20, Table 2) with 102 months of follow up exhibited progressive lymph node disease for which somatostatin analog therapy was prescribed; this patient stained (robustly) positive for PTCH 1. Finally, 7 patients (# 4, 7, 8, 9, 11, 13 and 16, Table 2) developed newly recognized widespread metastases during follow up. Five of these patients (#7, 9, 11, 13 and 16, Table 2) with follow up ranging from 30 to 103 months have been managed with somatostatin analog therapy alone. Two were negative on PTCH 1 staining (one was weakly positive) and 3 were positive (one intermediately positive). The remaining 2 patients, both with follow of more than 120 months, required additional antitumor therapy; one stained (robustly) positive for PTCH1 (#4, Table 2) and one had undetectable PTCH1 staining (#8, Table 2). These data fail to reveal any correlation between the presence or degree of PTCH1 staining and clinical outcome.
Discussion
In the current study we have expanded on our prior work in Men1l/l;RipCre mice to demonstrate that indeed similar HH pathway abnormalities exist in human PNETs, both sporadic and hereditary. We identified PTCH1 positivity in more than half of the PNET patients studied and this was detectable in both primary and metastatic tumors.
It should be noted that we used a very stringent definition of positive PTCH1 staining in the current study. Specimens with weakly positive staining only were felt to be non-specific and were categorized as negative. Had we included weakly positive stains ("+" on our scale) as positive, the overall rate of PTCH1 positivity in the current study would have increased to 73% (16 of 22 cases).
PTCH1 staining could not be predicted based upon any clinical or demographic factors in the current study but there was a trend toward increased positivity in MEN-1 syndrome patients. However, this was a small study and it is quite possible that this finding was due to a type 1 error. Had we considered weakly positive PTCH1 staining as positive in the current study, all 5 MEN-1 syndrome patients would have been PTCH1 positive vs. 11 of 17 sporadic tumor patients (65%). We also failed to identify a difference in staining patterns between patients with localized and metastatic disease regardless of whether we considered weakly positive staining as positive or negative suggesting that HH pathway abnormalities occur early on in tumorigenesis. The regulation of HH signaling and thus PTCH1 expression in PNETs may be influenced by multiple factors. However, the high frequency of PTCH 1 positivity demonstrated in our study (> 50% of cases) suggests that Federal Drug Administration (FDA)-approved agents such as Vismodegib that target HH signaling may be suitable for treating patients with metastatic PNETs. In addition, we identified more intense PTCH1 staining in tumor tissue relative to normal pancreatic islet tissue, a factor that may be important to consider in any future trials of PTCH1 inhibition for patients with metastatic PNETs.
Patients with PNETS may present with early stage disease that is surgically curable especially if they have syndromic features.1,2 However, few patients are actually cured with surgery and those with MEN-1 syndrome are generally not subjected to surgery with a curative intent because hereditary PNET patients have multifocal disease.1,2 Other patients present with regional or more-widespread disease and, although they are not generally curable with surgery, debulking is often performed in the hopes of slowing down subsequent disease progression.14 Nevertheless, most patients with widespread metastatic PNET disease ultimately succumb to the disease despite the availability of multiple palliative interventions.1,2,15-22
Somatostatin analog therapy is usually the first non-surgical therapy used in patients with slowly progressive disease because it is generally well tolerated. Octreotide has not been studied in PNET patients specifically but the PROMID trial in widely metastatic small bowel carcinoid tumors was the first to show tumor stabilization in a randomized clinical setting and this has become the de facto first line treatment for many patients.15 The CLARINET trial, which evaluated the impact of another somatostatin analog, lanreotide, in a double-blind, randomized trial of metastatic patients with both alimentary tract carcinoids as well as PNETs, was just recently published with a positive result, further cementing the value of using somatostatin analog therapy in these patients. 16 However, somatostatin analogs only produce tumor stabilization and this only persists for a finite period of time.
The next level of palliative therapy, which includes various liver-directed approaches (such as chemoembolization, bland embolization, radiofrequency ablation, radioactive eluting beads and even liver transplantation) or other systemic therapies (such as chemotherapy, PRRT or targeted therapies) all have limited durations of efficacy and side effects which limit their use.1,2,17-22 There clearly is an unmet need for more effective, well-tolerated anti-tumor therapy in patients with metastatic PNET tumors.
On the basis of our data, PTCH1 positivity does not appear to predict clinical outcome but this is in the absence of any therapy directed against the HH pathway specifically. Vismodegib, an FDA-approved antitumor agent that selectively inhibits SMO to suppress growth of basal cell carcinomas, directly inhibits the HH pathway.12,13 This agent appears to be well tolerated and effective in the treatment of basal cell skin cancers.12,13,23-25 In patients with a hereditary PTCH1 mutation leading to the basal cell nevus syndrome (BCNS), a condition associated with hundreds of basal cell carcinomas, the response rate to therapy with Vismodegib was 100%.24 However, patients with sporadic basal cell cancers respond less frequently (approximately 30–50%) possibly because of acquired resistance to therapy.23,25 Moreover, patients with faster growing solid tumors such as pancreatic or lung cancers do not appear to benefit from Vismodegib despite the established presence of HH pathway abnormalities.25,26 In the context of our current study confirming perturbations of the HH pathway in the majority of PNET patients, we believe that HH pathway inhibitor agents, such as Vismodegib, given alone or in combination with somatostatin analogs, should be studied in MEN-1 syndrome-associated and sporadic, locally advanced or early metastatic PNET patients.
Methods
The University of Pennsylvania NET center enrolls patients into an IRB-approved clinical database and biobank study entitled "Clinicopathologic correlates of NETs" funded by the Commonwealth of Pennsylvania and administered through the Abramson Cancer Center (Grant # 040-0427-4-561074-XXXX-2433-8341). Prospective patients are recruited during routine clinical visits to the NET Center comprising their normal course of care. Once informed consent has been obtained, participating individuals are entered into our longitudinal Redcap (Vanderbilt University, Nashville, USA) database that houses demographic data, pathology reports, imaging and biomarker results, therapeutic interventions and clinical outcome information. Study subjects also provide blood for biobanking and allow access to fresh tumor tissue in the event that they undergo surgery after enrollment, or archival paraffin imbedded tissue in the event that surgery had already been performed previous to their enrollment. Tissue samples are processed through the Cooperative Human Tissue Network (CHTN; grant # U01 CA 44974).
For the current study, we recruited 22 patients with MEN-1 or sporadic PNETs. Our Redcap database was queried for demographic and clinical information including tumor extent at the time of tissue acquisition and all pathology slides were re-reviewed and reclassified according to the minimal dataset for NET diagnosis. In cases in which formalin-fixed, paraffin embedded blocks were available, all available slides were reviewed; representative blocks of tumor (primary and/or metastasis) were identified, and 5 micron sections were cut on charged slides for subsequent IHC staining. In patients undergoing surgery, fresh tumor tissue was procured, snap frozen in liquid nitrogen, and subsequently stored at −80 degrees for subsequent analysis. The study pathologist reviewed all available pathology slides and the tumors were classified according to the World health organization (WHO) recommendations regarding diagnostic terminology for NETs. This included grading of the tumors according to their pattern and cytologic features and assessment of mitotic rate and proliferative activity as determined by immunostaining for Ki67.27,28 The tumors were diagnosed as Neuroendocrine Tumor grade 1 or 2 or Neuroendocrine carcinoma (NEC or Tumor grade 3).
Tissue specimens containing tumor and normal adjacent pancreas or metastatic tissue underwent PTCH 1 staining as follows. Tissue sections were de-paraffinized in xylene substitute solution and serially rehydrated with decreasing concentrations of alcohol. The tissues were permeabilized in 0.2 % Triton X-100 prior to addition of primary antibody. Images were captured using a Nikon Eclipse E800 fluorescence microscope equipped with a CCD digital camera. Anti-PTCH1 antibody used for immunostaining was purchased from Proteintech Group (17520-1-AP). Based on the intensity of the immunofluorescence staining, PTCH1 levels were graded on an intensity scale of ‘+’ (weakly positive) to ‘++’ (intermediately positive) to ‘+++’ (robustly positive). In all cases we used slides prepared from the adjacent normal tissue as negative controls. We considered specimens with ‘++’ and ‘+++’ staining to be positive for PTCH 1.
Contributions of Authors
Study concept and design: BG, XH, VL, DM. Specimen Procurement: RR, DF, MR, BB. Pathology review and specimen preparation: VL. PTCH1 staining and quantification: BG, XH. Data collection: MR, BB, DM. Data analysis: BG, VL, DM. Manuscript preparation: All authors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Commonwealth of PA grant: 040-0427-4-561074-XXXX-2433-8341 to DM. CHTN grant: UO1 CA 44974. NIH grant: 1-RO1-CA-178856-01 to XH. Caring for Carcinoid Foundation – AACR grant: 11-60-33 to XH.
References
- 1. Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008;135:1469-1492; PMID:18703061; http://dx.doi.org/ 10.1053/j.gastro.2008.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jensen RT, Berna MJ, Bingham MD, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management and controversies. Cancer 2008;113(7 Suppl):1807-43; http://dx.doi.org/ 10.1002/cncr.23648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997; 18:276(5311):404-7; http://dx.doi.org/ 10.1126/science.276.5311.404 [DOI] [PubMed] [Google Scholar]
- 4. Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer 2005; 5:367-75; PMID:15864278; http://dx.doi.org/ 10.1038/nrc1610 [DOI] [PubMed] [Google Scholar]
- 5. Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999; 96, 143-152; PMID:9989505; http://dx.doi.org/ 10.1016/S0092-8674(00)80967-8 [DOI] [PubMed] [Google Scholar]
- 6. Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 2004;13, 587-597; PMID:14992727; http://dx.doi.org/ 10.1016/S1097-2765(04)00081-4 [DOI] [PubMed] [Google Scholar]
- 7. Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol 2007; 265-266, 34-41; PMID:17222504; http://dx.doi.org/ 10.1016/j.mce.2006.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunag J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUN D but has opposite effects on transcription. Nature 2012;482:542-6; PMID:22327296; http://dx.doi.org/ 10.1038/nature10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knudson A. Mutation and cancer: statistical study of retinoblastoma. PNAS 1971; 68: 820-3; PMID:5279523; http://dx.doi.org/ 10.1073/pnas.68.4.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011. Mar 4; 331(6021):1199-203; http://dx.doi.org/ 10.1126/science.1200609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, Ng JM, Curran T, Hua X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res 2013; 73(8):2650-8; PMID:23580576; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meiss F, Zeiser R. Vismodegib. Recent Results Cancer Res 2014; 201:405-17; http://dx.doi.org/ 10.1007/978-3-642-54490-3_25 [DOI] [PubMed] [Google Scholar]
- 13. Gould SE, Low JA, Marsters JC, Jr, Robarge K, Rubin LL, de Sauvage FJ, Sutherlin DP, Wong H, Yauch RL. Discovery and preclinical development of vismodegib. Expert Opin Drug Discov. 2014 Aug;9(8):969-84. [DOI] [PubMed] [Google Scholar]
- 14. Norton JA, Warren RS, Kelly MG, Zuraek MB, Jensen RT. Aggressive surgery for metastatic liver neuroendocrine tumors. Surgery 2003; 134;1057-1063; PMID:14668741; http://dx.doi.org/ 10.1016/j.surg.2003.07.025 [DOI] [PubMed] [Google Scholar]
- 15. Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Bläker M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009; 27: 4656-4663; PMID:19704057; http://dx.doi.org/ 10.1200/JCO.2009.22.8510 [DOI] [PubMed] [Google Scholar]
- 16. Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, Cadiot G, Wolin EM, Capdevila J, Wall L, et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. NEJM 2014; 371: 224-233; PMID:25014687; http://dx.doi.org/ 10.1056/NEJMoa1316158 [DOI] [PubMed] [Google Scholar]
- 17. O'Toole D, Ruszniewski P. Chemoembolization and other ablative therapies for liver metastases of gastrointestinal endocrine tumours. Best Prac Res Clin Gastroenterol 2005; 19: 585-594; PMID:16183529; http://dx.doi.org/ 10.1016/j.bpg.2005.02.011 [DOI] [PubMed] [Google Scholar]
- 18. Ruutiainen AT, Soulen MC, Tuite CM, et al. Chemoembolization and bland embolization of neuroendocrine tumor metastases to the liver. J Vasc and Intervent Radiol 2007; 18: 847-855; http://dx.doi.org/ 10.1016/j.jvir.2007.04.018 [DOI] [PubMed] [Google Scholar]
- 19. Strosberg JR, Fine RL, Choi J, Nasir A, Coppola D, Chen DT, Helm J, Kvols L. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 2011; 117: 268-275; PMID:20824724; http://dx.doi.org/ 10.1002/cncr.25425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, Feelders RA, van Aken MO, Krenning EP. Treatment with the radiolabeled somatostatin analog [177Lu-DOTA0, Tyr3] octreotate: toxicity, efficacy, and survival. J Clin Oncol 2008; 26: 2124-2130; PMID:18445841; http://dx.doi.org/ 10.1200/JCO.2007.15.2553 [DOI] [PubMed] [Google Scholar]
- 21. Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. NEJM 2011; 364: 501-513; PMID:21306237; http://dx.doi.org/ 10.1056/NEJMoa1003825 [DOI] [PubMed] [Google Scholar]
- 22. Yao JC, Phan AT, Jehl V, Shah G, Meric-Bernstam F. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res 2013; 73:1449-1453; PMID:23436795; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3923 [DOI] [PubMed] [Google Scholar]
- 23. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. NEJM 2012; 366:2171-2179; PMID:22670903; http://dx.doi.org/ 10.1056/NEJMoa1113713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, Coppola C, Chanana AM, Marji J, Bickers DR, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. NEJM 2012; 366:2180-2188; PMID:22670904; http://dx.doi.org/ 10.1056/NEJMoa1113538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med 2014; 4:a013581; PMID:24985127; http://dx.doi.org/ 10.1101/cshperspect.a013581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res 2011; 17:2502-2511; PMID:21300762; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scarpa A, Mantovani W, Capelli P, Beghelli S, Boninsegna L, Bettini R, Panzuto F, Pederzoli P, delle Fave G, Falconi M. Pancreatic endocrine tumors: improved TNM staging and histopathological grading permit a clinically efficient prognostic stratification of patients. Mod Pathol 2010; 23:824-833; PMID:20305616; http://dx.doi.org/ 10.1038/modpathol.2010.58 [DOI] [PubMed] [Google Scholar]
- 28. Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND. (eds). WHO classification of tumours of the digestive system. (2010) IARC Press, Lyon, pp. 10-12. [Google Scholar]