

Abstract
Although age-associated gene expression and methylation changes have been reported throughout the literature, the unifying epigenomic principles of aging remain poorly understood. Recent explosion in availability and resolution of functional/regulatory genome annotation data (epigenomic data), such as that provided by the ENCODE and Roadmap Epigenomics projects, provides an opportunity for the identification of epigenomic mechanisms potentially altered by age-associated differentially methylated regions (aDMRs) and regulatory signatures in the promoters of age-associated genes (aGENs). In this study we found that aDMRs and aGENs identified in multiple independent studies share a common Polycomb Repressive Complex 2 signature marked by EZH2, SUZ12, CTCF binding sites, repressive H3K27me3, and activating H3K4me1 histone modification marks, and a “poised promoter” chromatin state. This signature is depleted in RNA Polymerase II-associated transcription factor binding sites, activating H3K79me2, H3K36me3, H3K27ac marks, and an “active promoter” chromatin state. The PRC2 signature was shown to be generally stable across cell types. When considering the directionality of methylation changes, we found the PRC2 signature to be associated with aDMRs hypermethylated with age, while hypomethylated aDMRs were associated with enhancers. In contrast, aGENs were associated with the PRC2 signature independently of the directionality of gene expression changes. In this study we demonstrate that the PRC2 signature is the common epigenomic context of genomic regions associated with hypermethylation and gene expression changes in aging.
Keywords: aging, epigenetics, epigenomics, polycomb, PRC2, ENCODE, GenomeRunner, methylation
Abbreviations
- aDMR
age-associated differentially methylated region
- aGEN
promoter of an age-associated gene
- ENCODE
Encyclopedia of DNA elements
- PRC2
Polycomb repressive complex 2
- TFBS
transcription factor binding site
Introduction
Aging affects all aspects of cellular regulation and maintenance, ultimately leading to detrimental physiological effects, impacting the well being of the whole organism. Several reports have identified age-associated genes (aGENs) whose gene expression patterns change with and predictive of age.1-4 In the last decade, age-associated epigenetic changes attracted significant attention,5,6 with DNA methylation being the best studied.1,7-17 In human subjects, age-related changes in DNA methylation have been detected mostly in whole blood,1,14,17 in purified subsets of blood,11,14 and in the brain.18,19 The relationship of age-related DNA methylation regions (aDMRs) with CpG islands,9 “bivalent domains,”14 and gene-centric regions, such as promoters, exonic, intronic and intergenic regions,11 Alu, LINE-1, and other repetitive elements11,20,21 has been extensively investigated. Yet, the unifying regulatory epigenomic mechanisms that may be targeted to neutralize and, potentially, reverse the detrimental effect of aging remain poorly understood.
Recent years have seen rapid growth of genomic organization and functional annotation data. The Encyclopedia of DNA Elements (ENCODE)22 and the NIH Roadmap Epigenomics23 projects have aimed to identify the functional elements in the human genome across the major cell types and tissues. We refer to these functional and regulatory regions as epigenomic elements, i.e., genomic properties other than the DNA sequence that describes functions, properties, or experimental values associated with specific genomic regions.24-27 Although the definition of “epigenomics” is still debated,28 we feel a more inclusive definition of epigenomic data is better suited to convey the concepts of this study.
Epigenomic marks are generally tissue- and cell-type specific,23,29,30 suggesting that age-dependent epigenomic changes may also be cell- or tissue-specific.31 However, several studies have shown that methylation changes can be defined independently of sex, tissue type, cellular composition, differentiation status, and array platform.7,10,12,14,17 In this study we used the ENCODE and Roadmap Epigenomics data to investigate potential cell- and tissue-type specificity of epigenomic signatures of aDMRs and aGENs.
It is generally thought that DNA methylation decreases with age.10,11,20 Yet, some promoter-specific CpG dinucleotides have been shown to become hypermethylated with age,9,10,14,17 which is a trend also associated with cancer progression.32 To investigate whether the directionality of age-associated changes is associated with similar epigenomic signature(s), we took advantage of published lists of aDMRs hyper- or hypo-methylated with age, as well as genes that increase or decrease their expression with age.
aDMRs have been linked to the promoters of key developmental genes, suggesting the former may directly affect gene expression.14,16-18 However, methylation and gene expression changes are not necessarily correlated.1,15,16 We investigated whether similar epigenomic signature(s) are shared by aDMRs that are associated vs. not associated with gene expression changes.
We found epigenomic marks from human embryonic stem cell lines most frequently and most significantly associated with aDMRs and aGENs, concordant with the hypothesis that age-associated methylation and gene expression changes are likely most relevant to developmental and differentiation processes. Both aDMRs and aGENs shared a similar combination of repressive (H3K27me3) and activating (H3K4me1) histone modification marks, as well as binding of Polycomb Repressive Complex 2 (PRC2) co-factors. Consequently, aDMRs and aGENs were enriched in chromatin regions labeled as “poised promoters.” On the contrary, binding sites of RNA Polymerase II co-transcription activators were absent in aDMRs and aGENs, concordant with the depletion of the “active promoter” chromatin state. This epigenomic signature was generally stable across cell lines, with the exception of monocyte-specific aDMRs15 and aGENs identified by de Magalhães et al.33 in a meta-analysis. Promoters of genes that increased/decreased their expression with age were associated with the same PRC2 signature. In contrast, aDMRs that were hyper- and hypo-methylated with age were associated with different epigenomic contexts, with hypomethylated aDMRs being enriched in enhancers. Prioritizing all genes by the presence of PRC2 signature identified that age-associated processes most often affect organ formation and molecular communications among cells.
Results
Age-associated methylated regions and genes are largely non-overlapping
Sets of age-associated methylated regions (aDMRs) and promoters of age-associated genes (referred hereafter as aGENs for brevity) were obtained from supplementary material of 8 studies (Table 1). The majority of the studies used the Illumina Infinium HumanMethylation450 BeadChip34 to identify aDMRs, with only one study using the Illumina HumanMethylation27 BeadChip.14 Genomic coordinates of aDMRs and aGENs were extracted from the platform-specific annotation files (Methods). Where information was available, aDMRs and aGENs were separated into sets positively and negatively correlated with age by methylation and expression, respectively. The aDMRs and aGENs from different studies were largely non-overlapping (Tables S1 and S2, Fig. S1, the fact also discussed in16), with only one aDMR being identified in 7 studies and 335 out of 16,854 aDMRs detected by 3 studies. These results suggest that different studies may point toward study-specific age-related epigenomic associations.
Table 1.
Datasets used in the current study
| Abbreviation | Brief description |
|---|---|
| Age-associated differentially methylated regions (aDMRs) | |
| Hannum_aDMRs_primary 1 | 71 methylation markers predictive of age, primary cohort, whole blood |
| Hannum_aDMRs_all 1 | 89 methylation markers predictive of age, primary and validation cohorts, whole blood |
| Alisch_aDMRs_all; 7 Alisch_aDMRs_all_pos; Alisch_aDMRs_all_neg | 2,078 regions correlated with age by methylation in peripheral blood; 479 of them showed positive correlation with age by methylation, 1,599 were found to be negatively correlated with age |
| Rakyan_aDMRs_all 14 | 131 regions correlated with age by methylation in whole blood, and show the same directional age-associated DNA methylation change in CD4+ T-cells and CD14+ monocytes. |
| Reynolds_aDMRs_tcells; 15 Reynolds_aDMRs_tcells_pos; Reynolds_aDMRs_tcells_neg | 2,595 regions correlated with age by methylation in CD4+ T-cells. Two,049 of them showed positive correlation with age by methylation, 546 were found to be negatively correlated with age |
| Reynolds_aDMRs_monocytes; 15 Reynolds_aDMRs_monocytes_pos; Reynolds_aDMRs_monocytes_neg | 2,259 regions correlated with age by methylation on CD14+ monocytes. 468 of them showed positive correlation with age by methylation, 1,791 were found to be negatively correlated with age |
| Steegenga_aDMRs_and_gene 16 | 726 methylation markers correlated with age- and gene expression changes in peripheral blood |
| Steegenga_aDMRs_not_gene 16 | 4,552 methylation markers correlated with age- but not with gene expression changes in peripheral blood |
| Steegenga_aDMRs_meta 16 | 7,477 age-associated methylation markers identified in multiple studies |
| Heyn_aDMRs 11 | 3,205 age-associated methylation markers differentially methylated in cord blood of newborns and CD4+ T cells of centenarian; 1,219 of them showed hypermethylation with age, 1,986 were found to be hypomethylated with age |
| Age-associated genes (aGENs) | |
| Hannum_genes_associated 1 | 326 genes correlated with age by expression, whole blood, data not related to methylation data |
| Hannum_genes_predictive; 1 Hannum_genes_predictive_pos; Hannum_genes_predictive_neg | 55 genes predictive of age based by their expression, whole blood, data not related to methylation data. Twenty-four of these genes showed positive correlation with age by expression, 31 were found to be negatively correlated with age |
| Genes_LongevityMap 47 | 752 manually curated list of age-associated genes |
| deMagalhaes_genes_all; 33 deMagalhaes_genes_pos; deMagalhaes_genes_neg | 73 age-associated genes. 56 genes overexpressed with age; 17 genes underexpressed with age. |
Epigenomic marks from human embryonic cell lines are most frequently and significantly associated with age-associated regions
The ENCODE and the Roadmap Epigenomic projects provide a wealth of cell type-specific genome annotation data. A total of 1,951 transcription factor binding site (TFBS) datasets (277 transcription factors, 91 cell lines), 722 histone mark data sets (41 marks, 69 cell lines), 136 chromatin state datasets (15 states, 9 cell lines) provided by the ENCODE project, and 979 histone marks data sets (127 cell- and tissue-types) provided by the Roadmap Epigenomics project were used in the analyses (Table S3).
To identify cell types with epigenomic marks most frequently and significantly enriched in aDMRs and aGENs, cell type-specific enrichment frequencies were compared with what can be observed by chance using Fisher's exact test (see Methods, Cell type enrichment analysis). Epigenomic marks from H7es cells, the undifferentiated human embryonic stem cell line, were most frequently and most significantly associated with aDMRs and aGENs (P = 3.94E-12). Other cell lines showing frequent epigenomic associations include K562 leukemia (P = 1.53E-06), HelaS3 cervical carcinoma (P = 3.10E-06), and H1hesc embryonic stem cell lines (P = 1.37E-05, Table S4A). These observations were confirmed by the cell type enrichment analysis of 127 cell- and tissue type-specific epigenomic datasets provided by the Roadmap Epigenomics project, with epigenomic marks from “ESC.H1,” H1 embryonic stem cell line, being most frequently and significantly associated with aDMRs and aGENs (P = 2.60E-12, Table S4B).
The epigenomic similarity analysis among cell types (see Methods, Epigenomic similarity among cell types) identified H1hesc and K562 cell lines as most epigenomically distinct from the other cell types (Fig. 1). The epigenomic similarity analysis of 127 cell- and tissue types used by the Roadmap Epigenomics project further highlighted a group of embryonic cell lines epigenomically distinct from the other cell types (Fig. S2). In summary, these results suggest preferential association of aDMRs and aGENs with epigenomic marks in cells with high developmental potential, such as embryonic stem cells.
Figure 1.
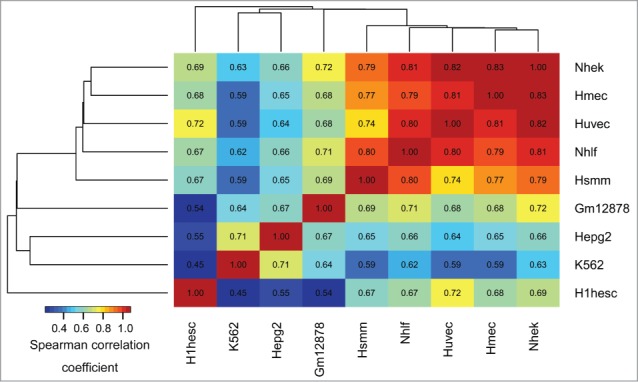
Epigenomic similarity among the ENCODE cell lines. We computed the Spearman correlation coefficient between cell type-specific epigenomic enrichment profiles (Methods). The resulting correlation matrix was clustered using Euclidean/average clustering metrics, and visualized with darker blue/red gradient representing weaker/stronger epigenomic similarity, respectively. Each cell shows the numerical value of the corresponding Spearman correlation coefficient.
aDMRs and aGENs share a similar repressive epigenomic signature
To identify whether aDMRs and aGENs from multiple studies were enriched in similar epigenomic signatures, their epigenomic similarity was assessed across all data sets (see Methods, Epigenomic similarity among aDMRs and aGENs, and35). Briefly, each set of aDMRs and aGENs was tested for enrichment in histone modification marks, TFBSs, and chromatin states obtained from H1hesc human embryonic cell line. The epigenomic data from H7es cell line was not used, as it provides insufficient data to evaluate epigenomic similarity, covering only 3 histone modification marks, H3K27ME3, H3K4ME3, and H3K36ME3. The aDMR- and aGEN set-specific enrichment P-values (epigenomic enrichment profiles) were collected, −log10-transformed, and correlated with each other using Spearman correlation coefficient (Fig. 2). Notably, the aGENs identified by a meta-analysis33 were the most epigenomically distinct. While these results suggest that the epigenomic signatures may be driven by study-specific conditions, the aDMRs and aGENs from different studies showed on average high epigenomic similarity (median Spearman correlation coefficient 0.45, median P -value < 1.00E-8). There results were confirmed by the epigenomic similarity analysis using the ENCODE and Roadmap Epigenomics data from all cell lines (Fig. S3A and S3B), showing even higher overall similarity among the aDMRs and aGENs from multiple studies (median Spearman correlation coefficient ranging from 0.51 to 0.55, median P -value < 1.00E-8). These results indicate that the largely non-overlapping sets of aDMRs and aGENs share similar epigenomic enrichments, suggesting the presence of a common epigenomic signature associated with them.
Figure 2.

Epigenomic similarity among aDMRs and aGENs using the ENCODE data, H1hesc cell line. We computed the Spearman correlation coefficient between aDMR-and aGEN-specific epigenomic enrichment profiles (Methods). The resulting correlation matrix was clustered using Euclidean/average clustering metrics, and visualized with darker blue/red gradient representing weaker/stronger epigenomic similarity, respectively. Each cell shows the numerical value of the corresponding Spearman correlation coefficient.
Age-associated regions are concurrently enriched in repressive (H3K27me3, EZH2) and activating (H3K4me1, H3K4me2) histone modification marks
The H3K27me3 repressive mark was the most frequent and most significantly enriched in the aDMRs and aGENs (Fig. 3, Fig. S4, Table S5A and S5B). It was accompanied by enrichment in histone methyltransferase EZH2, a subunit of Polycomb Repressive Complex 2 (PRC2), known to trimethylate H3K27me3 and repress transcription.17,36,37 Binding sites of another PRC2 subunit, SUZ12, and its interacting partner CTCF, along with the H2az histone modification mark known to co-localize with PRC2 at developmentally silenced genes36 were also noted. These results suggest potential association of PRC2 with age-related methylation and gene expression changes.
Figure 3.
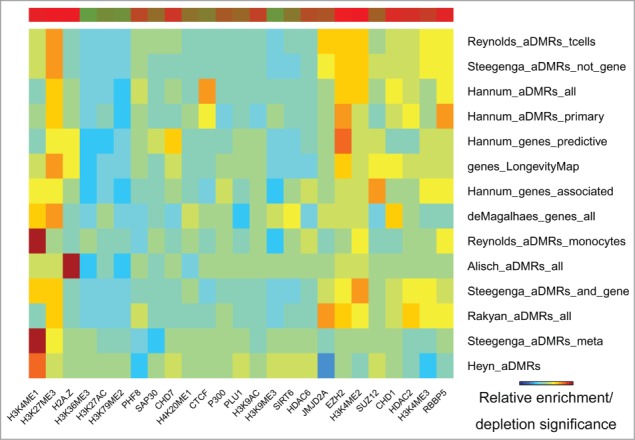
Histone modification marks (ENCODE) enriched/depleted in aDMRs and aGENs. Darker blue/red gradient highlights depleted/enriched associations, respectively. Red/green bar gradient defines frequency of epigenomic marks enriched/depleted in aDMRs and aGENs, respectively.
Despite being enriched in repressive histone modification marks and TFBSs, the aDMRs and aGENs were also enriched in activating H3K4me1, H3K4me2, and H3K4me3 histone modification marks, known to surround transcription start sites.38 These activating marks have been shown to co-localize with the H3K27me3 repressive mark in “bivalent domains” in embryonic stem cells,39 confirming the functional role of aDMRs in orchestrating developmental regulation through such domains.14 In summary, these results suggest that binding of the PRC2 complex may be involved in regulation of aGENs poised for active transcription, and that its regulatory effect may be altered by the presence of aDMRs.
H3K27ac, H3K79me3, and H3K36me3 marks of active transcription are depleted in age-associated regions
H3K27ac, a mark of active promoters and enhancers, H3K36me3, a mark elevated predominantly in transcribed regions of active genes and peaked at the 3′ end of genes,40 and H3K79me3, a promoter mark, were all depleted in the aDMRs and aGENs (Fig. 3, Fig. S4, Table S5A and S5B). Depletion of these marks in aGENs was the most pronounced, as compared with aDMRs. These findings are consistent with the observation that H3K27ac is enriched in enhancers poised for active transcription and depleted in active enhancers,41 further supporting the association of aDMRs and aGENs with “bivalent domains.”
Repressive transcription factor binding sites are enriched in age-associated regions
EZH2 and SUZ12 transcriptional repressors and histone modifiers were identified among the most frequent binding sites enriched in aDMRs and aGENs (Fig. 4, Table S5C). Two other transcriptional repressors, SIN3A and CtBP2, were also highly enriched. Another enriched transcription factor, E2F6, which plays a crucial role in controlling cell cycle and tumor suppressor proteins, also possesses repressive activity upon E2F-dependent transcription. Enrichment of aDMRs and aGENs in Rad21, a double-strand-break repair protein and a part of cohesin complex interacting with PRC2 complex and participating in controlling transcription of silenced and active genes42 was also noted. These results emphasize enrichment of aDMRs and aGENs in binding sites of other repressive regulators known to interact with PRC2.
Figure 4.
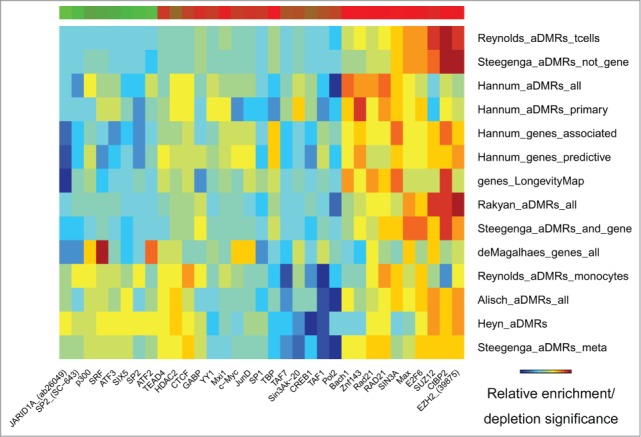
Transcription factor binding sites (ENCODE) enriched/depleted in aDMRs and aGENs. Darker blue/red gradient highlights depleted/enriched associations, respectively. Red/green bar gradient defines frequency of epigenomic marks enriched/depleted in aDMRs and aGENs, respectively.
RNA Polymerase II and other transcription initiation factors are depleted in age-associated regions
Concordant with the presence of the PRC2 signature, aDMRs and aGENs were depleted in RNA Polymerase II binding sites, and sites of other proteins necessary for RNA Polymerase II-dependent transcription, such as TAF1, TAF7, TBP, CREB1 (Fig. 4, Table S5C), and a group of multi-functional transcription factors, such as the activators/repressors SP1 and SP2 and development and oncogenesis regulators SRF, SIX5, JunD, c-Myc. JARID1A, an H3K4 demethylase required for haematopoietic stem cell self-renewal,43,44 showed marginal enrichment in some, but an overall strong depletion, in the majority of aDMRs and aGENs. These results further strengthen the connection of aDMRs and aGENs with the binding of Polycomb repressive complex 2 in the absence of activating transcription factors.
“Poised promoters” but not “active promoters” are enriched, and “heterochromatin” chromatin states are depleted in age-associated regions
Epigenomic enrichment analysis of chromatin states identified the “poised promoter” state as the most frequently enriched in aDMRs and aGENs, followed by the “strong enhancer” state (Fig. 5, Table S5D). Consistent with the depletion of RNA Polymerase II and activating TFBSs, the “active promoter” chromatin state was among the most depleted. A group of monocyte-specific,15 pediatric-teenager,7 pediatric-centenarian,11 and meta aDMRs16 showed a weaker enrichment in “poised promoter” but a stronger depletion in “active promoter” chromatin states. The “transcriptional elongation” state was uniformly depleted in all sets of aDMRs and aGENs. However, the “heterochromatin” state was also depleted, suggesting the presence of open chromatin around aDMRs and aGENs. These results further confirm the association of aDMRs and aGENs with poised promoters and enhancer regions controlled by repressive transcription factors and the PRC2 complex.
Figure 5.
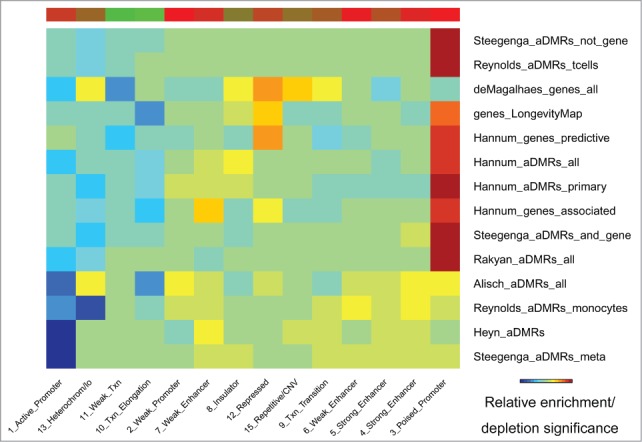
Chromatin states (ENCODE) enriched/depleted in aDMRs and aGENs. Darker blue/red gradient highlights depleted/enriched associations, respectively. Red/green bar gradient defines frequency of epigenomic marks enriched/depleted in aDMRs and aGENs, respectively.
Repressive signature is associated with aDMRs and aGENs positively correlated with age, while enhancers are preferentially associated with aDMRs hypomethylated with age
Some studies provide the directionality of age-association changes, that is, whether aDMRs are hyper/hypomethylated with age, and whether aGENs are positively/negatively correlated with age by expression. This directionality of changes is referred to hereafter as “positive/negative.” Sets of positive and negative aDMRs and aGENs were clustered by their epigenomic similarity to identify epigenomically distinct groups. This clustering, as expected, separated positive and negative aDMRs into two distinct groups, with the exception of monocyte-specific positive aDMRs15 (Fig. 6, Fig. S5). Notably, positive and negative aGENs did not show epigenomic differences, sharing the same epigenomic signature with aDMRs hypermethylated with age. These results suggest that aDMRs hyper- or hypo-methylated with age may be associated with differential epigenomic signatures, whereas the same epigenomic mechanisms may drive increasing or decreasing gene expression changes of aGENs.
Figure 6.
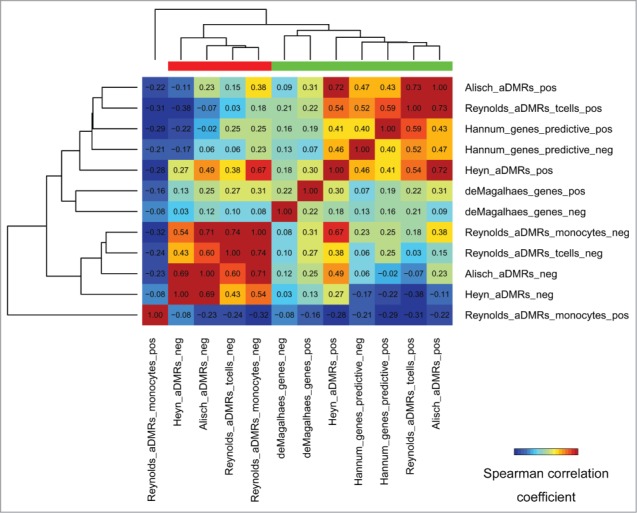
Epigenomic similarity among aDMRs hyper/hypomethylated with age, and aGENs showing increasing/decreasing gene expression with age (“pos/neg” postfixes). Pos/Neg aDMR-and aGEN epigenomic enrichment profiles obtained using H1hesc epigenomic data from the ENCODE project (see Methods) were correlated to each other using the Spearman correlation coefficient metric. The resulting correlation matrix was clustered using Euclidean/average clustering metrics, and visualized with darker blue/red gradient representing weaker/stronger epigenomic similarity, respectively. Each cell shows the numerical value of the corresponding Spearman correlation coefficient. Red/green bars define groups of aDMRs and aGENs showing high inter-group similarity.
In order to identify differential epigenomic signatures between the groups of aDMRs hyper- or hypomethylated with age, we compared them using differential epigenomic analysis (see Methods). The repressive H3K27me3 mark was not significantly associated with negative aDMRs, while being highly enriched in positive aDMRs. On the contrary, H3K36me3, a mark of transcribed regions, was more significantly depleted in positive aDMRs (Table S6). Several epigenomic marks were identified as differentially enriched, that is, significantly enriched in positive aDMRs and aGENs while depleted in negative aDMRs and aGENs, or vice versa. Among them were repressor EZH2, histone deacetylase HDAC2, lysine-specific demethylase JMJD2A (aka KDM4A),45 and a component of methyltransferase complex, RBBP546 (Table S6). These results highlight clear differences in histone code and histone modifiers associated with positive and negative aDMRs.
Twenty-three transcription factors also showed differential directionality and significance of enrichment. The repressive EZH2, SUZ12, CtBP2, E2F6, Znf143, SIN3A, YY1, and Max TFBSs were significantly enriched in positive aDMRs, and depleted in negative aDMRs. The transcription-promoting TAF1, TBP and RNA Polymerase II factors, on the other hand, were significantly depleted in the negative aDMRs. Comparison of chromatin states identified “strong enhancer” and “weak enhancer” as enriched in negative aDMRs. Positive aDMRs, on the other hand, were significantly depleted in “weak transcription” chromatin states (Supplementary Table S6). These results emphasize the PRC2 epigenomic signature as associated with aDMRs hypermethylated with age, while linking hypomethylated aDMRs with enhancers.
Study-specific epigenomic associations are generally stable across cell lines
In order to identify potential cell type-specific enrichments in each study, sets of aDMRs and aGENs were analyzed individually (Supplementary Results S1). Different sets of aDMRs and aGENs emphasized the aforementioned histone modification marks differentially (Supplementary Results S1A, D), but consistently enriched/depleted across cell types. For example, aDMRs predictive of age1 did not show depletions, while being enriched in EZH2, CTCF, H3K27me3 and other repressive and activating histone marks. Other aDMRs and aGENs showed consistent enrichment and depletion patterns, with enrichments in H1hesc epigenomic data among the most significant. The pediatric-teenager aDMRs7 showed the strongest association with the H2az histone mark. The monocyte-specific aDMRs,15 meta-aDMRs16 and pediatric-centenarian aDMRs11 were the most prominently enriched in H3K4me1 across the majority of cell types. These results suggest that the age-associated histone code may be similarly regulated in any cell type.
Similarity of the enrichment patterns across cell types was also supported by the analysis of TFBSs (Supplementary Results S1B). As described in the previous section, a group of aDMRs, such as pediatric-teenager aDMRs,7 and T-cell-specific aDMRs,15 showed depletion in RNA Polymerase II related TFBSs, while the others14-16 also showed enrichment in EZH2, SUZ12, CtBP2, RAD21, Znf143, SIN3A, and E2F6. Again, enrichments in H1hesc epigenomic data were most pronounced. In contrast with other sets of aDMRs and aGENs, meta-aGENs from33 showed enrichment in RNA Polymerase II, TAF1, TBP and other transcription promoting transcription factors. This observation placed TFBSs in a category of regulatory marks most diversely associated with aDMRs and aGENs.
Cell type-specific chromatin states analysis showed consistent enrichment of aDMRs and aGENs in “poised promoter,” “strong enhancer” and “repressed” chromatin states, and depletion in “heterochromatin,” “transcription elongation” and “active promoter” states (Supplementary Results S1C). Several sets of aDMRs and aGENs,1,14,47 including T-cell-specific aDMRs,15 were strongly enriched in the “poised promoter” state, while the others,7,11,16,33 including monocyte-specific aDMRs,15 were predominantly enriched in “strong enhancer” state. Although meta-aGENs33 were enriched in “strong enhancer” and depleted in “heterochromatin” chromatin state, comparable with other aGENs and aDMRs, they were also enriched in the “active promoter” states. In summary, each set of aDMRs and aGENs emphasized different aspects of the repressive PRC2 signature that was generally similar across multiple cell types.
Genes with an age-associated epigenomic signature are enriched in organ morphogenesis, cell signaling, extracellular matrix and brain-related functions
In order to identify genes showing the identified age-associated epigenomic signature in their promoters, independent of cell- or tissue specificity, we performed reverse epigenomic analysis of the promoters of all genes (Table S7). Briefly, for each gene, a promoter-specific score was calculated using a set of promoter-specific epigenomic marks weighted by their average significance identified from the enrichment analysis of aDMRs and aGENs (Methods). This analysis was designed to prioritize the promoters of genes having, i.e., H3K27me3 and other repressive marks, TFBSs, and the “poised promoter” chromatin state, and under-emphasize the promoters with activating H3K36me3, H3K27ac, RNA Polymerase II-related TFBSs and the “active promoter” chromatin state. Therefore, it was not surprising to identify “H3K27me3 bound,” “Suz12 targets” and similar gene signatures from MSigDB (P adj. = 5.52E-64/9.86E-48, respectively, Table S8) via functional enrichment analysis on the top 500 genes, prioritized by the age-associated epigenomic signature. 71 of these genes were identified as targets of the hsa-miR-1275 miRNA (P adj. = 9.260E-3), which participates in PRC2-mediated silencing and is regulated by the level of the H3K27me3 repressive mark,48 supporting the notion these genes share the age-associated repressive epigenomic signature.
Transmembrane protein 61 (TMEM61) and solute carrier family 10, member 4 (SLC10A4) were among the top genes potentially regulated by the age-associated epigenomic signature (Table S7), suggesting involvement of cell signaling processes. Functional enrichment analysis further identified “gated channel activity” (P adj. = 4.36E-5) molecular function and “intrinsic component of plasma membrane” (P adj. = 3.61E-11) cellular components as the most significant gene ontologies (Table S9). Suggestive of age-related developmental processes, “organ morphogenesis” and “cell-cell signaling” were the most significant biological processes (P adj. = 1.16E-10/1.73E-10, respectively). Of note were enrichments in “extracellular matrix” cellular component functional category (P adj. = 1.41E-8), and “extracellular matrix organization” Reactome canonical pathway (P adj. = 3.97E-5) – processes increasingly recognized in the context of aging.49 A separate group of functional enrichments included the “synaptic transmission” biological process (P adj. = 3.56E-8) and the “neuroactive ligand-receptor interaction” KEGG canonical pathway (P adj. = 1.79E-4, Table S10), which suggest involvement of genes regulated by the age-associated epigenomic signature in brain-related processes. This observation was further supported by enrichment in a gene set involved in neurotransmission and neurodevelopment from50 (P adj. = 3.968E-5). There results outline core biological processes that may be affected by the PRC2 epigenomic signature in aging.
Discussion
This study identified the epigenomic signature of Polycomb Repressive Complex 2 binding (EZH2, SUZ12, CTCF) to be associated with aDMRs and aGENs across multiple studies. This finding is in line with the notion that PRC2 plays a direct role in modulating longevity and stress resistance.37 The association of “poised promoters,” marked by the repressive H3K27me3, H3K9me3 and activating H3K4me1, H3K4me2, H3K4me3 histone modification marks, is consistent with previous findings of “bivalent domains,”11,14,15,23,51 and further establishes PRC2 as a repressor of regulatory regions poised for active transcription. We found clear differences between hyper- and hypo-methylated aDMRs, but not between aGENs positively and negatively correlated with age, linking hypermethylated aDMRs with the PRC2 signature and hypomethylated aDMRs with enhancers. Although epigenomic marks from the human embryonic stem cell line were most strongly associated with aDMRs and aGENs, the PRC2 epigenomic signature was generally consistent across the cell lines. These findings highlight PRC2 as a key regulator of age-related processes and strengthen the notion that it may be used as a target for increasing longevity37 and, perhaps, reversing detrimental effects of aging.
Our selection of normal human embryonic stem cells was guided by the following:1) epigenomic data derived from these cell lines showed the most frequent and most significant enrichments in the age-associated regions, confirmed by the ENCODE and Roadmap Epigenomics data; 2) these cells are pluripotent, i.e., able to differentiate into multiple lineages, thus being relevant to investigation of the epigenomic context of development and aging; 3) epigenomic enrichments across multiple cell lines were generally similar; and 4) DNA methylation changes in genes required for stem cell differentiation have been demonstrated by others.17,52 Notably, the other cell lines showing frequent enrichments in age-associated regions resembled embryonic cell lines in their developmental properties. For example, the multipotent K562, a highly undifferentiated lymphoblastoid cell line, is able to develop into different blood cell progenitors,53 resembling the developmental potential of haematopoietic stem cells. Furthermore, monoclonal antibodies against K562 cells also recognize human haematopoietic pluripotent stem cells.54 Our findings of concordant age-related epigenomic associations across multiple cell types confirm previous studies that reported a high correlation of the age-associated loci in multiple tissues and cell types.7,14,17
The consistency of the epigenomic signature shared by different sets of aDMRs and aGENs cannot be attributed solely to their overlapping genomic locations. In fact, aDMRs from different studies were largely non-overlapping (Tables S1 and S2), and the number of aDMRs in each study differed. It is important to note that some sets of aGENs were defined independently of aDMRs,1,33,47 yet, they showed the same epigenomic enrichments as the aDMRs. Note that age-associated regions do not have to be overlapping in order to show similar enrichment patterns, due to the fact that histone modification regions and chromatin states span large stretches of the genome and can overlap with multiple aDMRs and aGENs. If some aDMRs were missed in one study, due to tissue-specific, technical or data processing differences, the enrichment analysis would still identify similar enrichment patterns if they were driven by the same biological effect. Notably, both aDMRs associated and not associated with proximal gene expression changes16 possessed a similar PRC2 epigenomic signature, suggesting that some aDMRs may exert their regulatory effect via long range interactions, or indirectly affect gene expression by enhancing the effect of other aDMRs.
In human embryonic cell lines, EZH2 and PRC2 have been shown to repress key developmental genes poised for activation during ES cells differentiation,55,56 regulating pluripotency and differentiation through accumulation of H3K27me3 on chromatin.57 However, Polycomb proteins can also facilitate productive gene expression required for establishing the identity and for differentiation of stem cells,42,58 reviewed in59. This activation is accompanied by replacement of H327me3 by H3K27ac41 and increasing levels of H2az, H3K4me1, and RNA Polymerase II transcription.51 The predominant enrichment of the H2az histone modification mark in pediatric-teenager aDMRs7 suggests that some tissues and developmental stages utilize additional mechanisms together with PRC2 to facilitate active developmental processes. Our study cautions against labeling the PRC2 epigenomic signature as repressive only in context of age-associated changes and warrants further investigation of the role of PRC2 in aging on cell-, tissue- and developmental stage-specific level.
The directionality of age-associated changes deserves further attention. While some studies report predominant DNA hypermethylation14 or hypomethylation21,52 with age, little is known about epigenomic mechanisms regulating the increase or decrease of gene expression. Our observation that the directionality of gene expression is not associated with differential epigenomic signatures highlights the dual role of PRC2 as both a repressor and a facilitator of age-associated gene expression changes. Our findings that PRC2 is strongly associated with hypermethylated aDMRs further suggest that PRC2-driven regulation may be affected by the increase of methylation with age.
Although we observed enrichment of PRC2 binding co-factors, and depletion in transcription activation mark H3K36me3 in aDMRs hypermethylated with age, we also noted the enrichment of demethylase JMJD2A and methyltransferase RBBP5. The role of Jumonji domain-containing protein 2A (JMJD2A), which targets methylated H3K36me3 and prevents senescence by modulating p53 and Rb pathways,60 has been well studied in cancer.45 Its potential role in aging has also been described.61 The presence of JMJD2A and RBBP5 in the absence of H3K36me3 in aDMRs hypermethylated with age suggests activity of p53 and Rb pathways at early developmental stages and their silencing followed by increased methylation over time.
A set of aGENs identified by a meta-analysis of gene expression profiles from mice, rats and humans33 was markedly different in its enrichment in RNA Polymerase II and other transcription promoting factors from the overall repressive signature of PRC2 binding. This may be due to pre-selection of genes that consistently change their expression across organisms, hence, emphasizing their RNA Polymerase II-driven transcription. This observation suggests that meta-analysis may emphasize actively transcribed genes while eliminating some important age-related associations.
To extend our finding on gene-centric level, while considering potential limitations of gene expression meta-analysis, we reverse engineered the promoters of all genes in the human genome for presence of the PRC2 signature. In addition to “cell-cell signaling” and “extracellular matrix” processes, increasingly recognized in the context of aging,49 we identified several development-related biological processes, ranging from general “organ morphogenesis” and “tissue development” to more specific “sensory organ development” and “neurogenesis” (Table S8). It is important to link these observations with our findings that the PRC2 signature is associated with aDMRs hypermethylated with age. Increase in methylation has traditionally been viewed as leading to decrease in gene expression,29,32 suggesting these biological processes being downregulated with age. Indeed, prenatal hypomethylation following increased hypermethylation with age has been observed in normal brain development,19 further suggesting that genes enriched in “neurogenesis,” “synaptic transmission” and other brain-related processes identified in our study as being regulated by the PRC2 signature also undergo repression with age through hypermethylation.
The breadth of cell- and tissue specificity of the ENCODE and the Roadmap Epigenomics data allows unprecedented insights into the epigenomics of aging. More work is required in order to build a comprehensive map of common and cell- and tissue-specific epigenomic signatures enriched in aDMRs and aGENs across the whole human body, such as the Illumina Human BodyMap 2.062 and the Digital Aging Atlas63 projects covering gene expression changes in different tissues. As more age-associated genomic regions are identified, such as the age-associated miRNAs, long non-coding RNAs, epigenome quantitative trait loci from age-oriented epigenome-wide association studies, the methods described in this study will allow an even more comprehensive understanding of the genomics and epigenomics of aging. Last, but not least, identification of hypomethylated aDMRs associated with enhancers, generally regulating gene expression via long-range interactions,64 underscores the importance of long-distance interactions of the age-associated genomic regions in the 3D structure of the genome. This understanding will help find a systemic approach to amending the detrimental effects of age in each cell in the human body.
Methods
Data sources and pre-processing
Genomic coordinates of all probes on the Illumina Infinium HumanMethylation27 and HumanMethylation450 BeadChips were extracted from the corresponding probe annotation files (GEO accession numbers GPL8490 and GPL13534, respectively). Unless otherwise specified, hg19 human genome assembly coordinate system was used. The coordinates of the HumanMethylation27 chip were converted from hg18 to hg19 genome assembly using the liftOver tool through the UCSC genome browser.65 See https://github.com/mdozmorov/Aging/tree/master/Illumina for details.
Genomic coordinates of aDMRs were obtained from the lists of probes from supplementary material of each study by cross-matching them to the corresponding probe annotation file. Genomic coordinates of aGENs were obtained from the lists of gene names from supplementary material of each study, and genomic coordinates of their promoters were extracted using the refGene table via the UCSC genome browser MySQL database (accessed on January 10, 2015). A promoter was defined as the 2,000 bp region upstream of a gene's transcription start sites. See https://github.com/mdozmorov/Aging/tree/master/data for details.
The human hg19 genome annotation data from the ENCODE project was obtained through the UCSC genome database65 (accessed on 10/12/2014), converted to the BED format and stored in a local SQLite database. Three groups of epigenomic data were used: histone modifications, transcription factor binding sites experimentally obtained by ChIP-seq, and chromatin state segmentation by Hidden Markov Models (Table S3). The additional histone modification data from the Roadmap Epigenomics covering 127 cell- and tissue-types66 was downloaded from https://www.broadinstitute.org/˜anshul/projects/roadmap/peaks/consolidated/broadPeak/ (accessed on 03/16/2015).
Cell type enrichment analysis
Cell type enrichment analysis was performed to identify cell type specificity of the epigenomic marks showing most frequent and most significant enrichments in the aDMRs and aGENs. Cell type-specific and a total number of epigenomic datasets were compared with cell type-specific and a total number of epigenomic data sets identified as significantly enriched (P-values < 1.00E-10) in aDMRs and aGENs. The P-values of cell types showing most frequent and most significant enrichments that could happen by chance were calculated using Fisher's exact test. See https://github.com/mdozmorov/Aging/blob/master/R.Aging/cell_type_enrichment.R for more details.
Epigenomic enrichment analysis
The epigenomic enrichment analysis was performed using GenomeRunner.35,67,68 Briefly, the enrichment analysis evaluates whether a set of aDMRs or aGENs is statistically significantly co-localizes with epigenomic datasets. The two-tailed Fisher's exact test was used to calculate enrichments, or depletions, of such co-localization. A ‘background’, or ‘universe’ of all chip-specific CpGs, or the promoters of all genes, was used to evaluate epigenomic enrichments that occur by chance. When testing a set of aDMRs or aGENs for enrichment in multiple epigenomic data sets, the enrichment P-values were corrected for multiple testing using a False Discovery Rate (FDR) calculation. The P-values are then −log10-transformed, and a “−“ sign is added to denote depleted associations. This transformation converts the P-values into a linear scale, where larger/smaller numbers denote more statistically significant enriched/depleted associations, respectively. The following analyses use such transformed P-values, unless explicitly stated otherwise.
Epigenomic similarity analysis
Epigenomic similarity among aDMRs and aGENs
When several sets of aDMRs and aGENs are analyzed, a natural question may be asked – how similar is the epigenomic context associated with these aDMRs and aGENs? To answer this, the epigenomic similarity analysis35 that compares the epigenomic enrichment profiles among the sets of aDMRs and aGENs was designed. An epigenomic enrichment profile is defined as a vector of P-values obtained by testing a set of aDMRs or aGENs for enrichment in several epigenomic datasets. If two sets of aDMRs are co-localized in similar epigenomic data sets, their epigenomic enrichment profiles will be similar. On the other hand, if the 2 sets of aDMRs are enriched in different epigenomic datasets, their epigenomic enrichment profiles will be different.
Epigenomic similarity among cell types
The same approach is used to evaluate epigenomic similarity among the cell types. Instead of collecting aDMR-or aGEN-specific epigenomic enrichment profiles, the epigenomic enrichment profiles for each cell type were assembled and compared using Spearman correlation coefficients.
Differential epigenomic enrichment analysis
If a group of aDMRs and aGENs is epigenomically different from another group, e.g., Figure 6, epigenomic data sets differentially enriched in aDMRs and aGENs between those groups can be identified. For each epigenomic mark, the distributions of −log10-transformed P-values are compared between the groups using a moderated t-statistics, as implemented in the limma R package.69 See https://github.com/mdozmorov/Aging/blob/master/R.Aging/GR_ALL.Rmd for more details.
Clustering and Visualization
The –log10-transformed P-values are also used for visualization purposes. For the epigenomic enrichment analysis, the transformed P-values are assembled into an n × m matrix, where n (rows) is the number of aDMRs and aGENs and m (columns) is the number of epigenomic marks. To allow the user to focus on the most significant enrichments, this matrix is filtered to remove epigenomic marks not showing any significant enrichment with at least one set of aDMRs or aGENs. The resulting matrix is then clustered using “Euclidean” distance to measure dissimilarity between rows and columns, and the “average” agglomeration method. The clustered matrix is then visualized using Matlab-like color gradient, with darker blue/red gradient indicating under/overrepresented enrichments, respectively.
A similar visualization strategy is used to visualize the results of the epigenomic similarity analysis. The n × n matrix of Spearman correlation coefficients is clustered and visualized using Matlab-like color gradient. See https://github.com/mdozmorov/Aging/blob/master/R.Aging/GR_ALL.Rmd for more details.
Reverse epigenomic analysis
Genomic coordinates of the promoters of all genes defined in RefSeq database were obtained from the UCSC genome browser database65 (accessed on 10/12/2014). The promoters of each gene were annotated for the presence of 26 histone modification marks, 76 TFBSs and 15 chromatin states. Each annotation was up-weighted by the significance of an average −log10-transformed enrichment P-value of a given epigenomic category. Note that if an epigenomic category was identified as depleted, the absence of it was up-weighted. A summary score of such up-weighted annotations was calculated for each gene (see https://github.com/mdozmorov/Aging/blob/master/R.Aging/annotation_enrichment.R for more details).
Functional enrichment analysis
Gene-centric functional and canonical pathway enrichment analysis was performed using ToppGene Suite.70
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The author thanks the members of the Epigenome Data Analysis and Coordination Center and the NIH Roadmap Epigenomic project who have processed and made publicly available the epigenomic data used in this study. The author is very grateful to Yana Bromberg, Jonathan Wren, Cory Giles, Amy Olex for their review and critique of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J-B, Gao Y, et al.. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49:359–67; PMID:23177740; http://dx.doi.org/ 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin C-S, Jan YN, Kenyon C, Bargmann CI, Li H. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet 2004; 36:197–204; PMID:14730301; http://dx.doi.org/ 10.1038/ng1291 [DOI] [PubMed] [Google Scholar]
- 3.Park S-K, Prolla TA. Lessons learned from gene expression profile studies of aging and caloric restriction. Ageing Res Rev 2005; 4:55–65; PMID:15619470; http://dx.doi.org/ 10.1016/j.arr.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 4.Pletcher SD, Stumpf MPH. Population genomics: ageing by association. Curr Biol 2002; 12:R328–30; PMID:12007435; http://dx.doi.org/ 10.1016/S0960-9822(02)00832-1 [DOI] [PubMed] [Google Scholar]
- 5.D'Aquila P, Rose G, Bellizzi D, Passarino G. Epigenetics and aging. Maturitas 2013; 74:130–6; PMID:23245587; http://dx.doi.org/ 10.1016/j.maturitas.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 6.Huidobro C, Fernandez AF, Fraga MF. Aging epigenetics: causes and consequences. Mol Aspects Med 2013; 34:765–81; PMID:22771540; http://dx.doi.org/ 10.1016/j.mam.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 7.Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res 2012; 22:623–32; PMID:22300631; http://dx.doi.org/ 10.1101/gr.125187.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, Kahn RS, Ophoff RA. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One 2009; 4:e6767; PMID:19774229; http://dx.doi.org/ 10.1371/journal.pone.0006767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al.. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602; PMID:19680444; http://dx.doi.org/ 10.1371/journal.pgen.1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernández AF, Bayón GF, Urdinguio RG, Toraño EG, García MG, Carella A, Petrus-Reurer S, Ferrero C, Martinez-Camblor P, Cubillo I, et al.. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res 2015; 25:27–40; PMID:Can't; http://dx.doi.org/ 10.1101/gr.169011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al.. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A 2012; 109:10522–7; PMID:22689993; http://dx.doi.org/ 10.1073/pnas.1120658109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013; 14:R115; PMID:24138928; http://dx.doi.org/ 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AA, Akman K, Calimport SRG, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res 2012; 15:483–94; PMID:23098078; http://dx.doi.org/ 10.1089/rej.2012.1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rakyan VK, Down TA, Maslau S, Andrew T, Yang T-P, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al.. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 2010; 20:434–9; PMID:20219945; http://dx.doi.org/ 10.1101/gr.103101.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, Burke G, Post W, Shea S, Jacobs DR Jr, et al.. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun 2014; 5:5366; PMID:25404168; http://dx.doi.org/ 10.1038/ncomms6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steegenga WT, Boekschoten MV, Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, Teschendorff AE, Butcher LM, Beck S, Müller M. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr) 2014; 36:9648; PMID:24789080; http://dx.doi.org/ 10.1007/s11357-014-9648-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al.. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 2010; 20:440–6; PMID:20219944; http://dx.doi.org/ 10.1101/gr.103606.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ, et al.. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet 2011; 20:1164–72; PMID:21216877; http://dx.doi.org/ 10.1093/hmg/ddq561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, Colantuoni C, Weinberger DR, Kleinman JE, Lipska BK. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet 2012; 90:260–72; PMID:22305529; http://dx.doi.org/ 10.1016/j.ajhg.2011.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, Sparrow D, Vokonas P, Baccarelli A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234–9; PMID:19150625; http://dx.doi.org/ 10.1016/j.mad.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jintaridth P, Mutirangura A. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 2010; 41:194–200; PMID:20145203; http://dx.doi.org/ 10.1152/physiolgenomics.00146.2009 [DOI] [PubMed] [Google Scholar]
- 22.ENCODE Project Consortium . The ENCODE (ENCyclopedia Of DNA Elements) project. Science 2004; 306:636–40; PMID:15499007; http://dx.doi.org/ 10.1126/science.1105136 [DOI] [PubMed] [Google Scholar]
- 23.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al.. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 2010; 28:1045–8; PMID:20944595; http://dx.doi.org/ 10.1038/nbt1010-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev 2009; 23:781–3; PMID:19339683; http://dx.doi.org/ 10.1101/gad.1787609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardison RC. Genome-wide epigenetic data facilitate understanding of disease susceptibility association studies. J Biol Chem 2012; 287:30932–40; PMID:22952232; http://dx.doi.org/ 10.1074/jbc.R112.352427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inbar-Feigenberg M, Choufani S, Butcher DT, Roifman M, Weksberg R. Basic concepts of epigenetics. Fertil Steril 2013; 99:607–15; PMID:23357459; http://dx.doi.org/ 10.1016/j.fertnstert.2013.01.117 [DOI] [PubMed] [Google Scholar]
- 27.Leung A, Schones DE, Natarajan R. Using epigenetic mechanisms to understand the impact of common disease causing alleles. Curr Opin Immunol 2012; 24:558–63; PMID:22857822; http://dx.doi.org/ 10.1016/j.coi.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ptashne M. Epigenetics: core misconcept. Proc Natl Acad Sci U S A 2013; 110:7101–3; PMID:23584020; http://dx.doi.org/ 10.1073/pnas.1305399110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan A-C, Galm O, et al.. A DNA methylation fingerprint of 1628 human samples. Genome Res 2012; 22:407–19; PMID:21613409; http://dx.doi.org/ 10.1101/gr.119867.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genom 2011; 12:231; PMID:21569359; http://dx.doi.org/ 10.1186/1471-2164-12-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson RF, Atzmon G, Gheorghe C, Liang HQ, Lowes C, Greally JM, Barzilai N. Tissue-specific dysregulation of DNA methylation in aging. Aging Cell 2010; 9:506–18; PMID:20497131; http://dx.doi.org/ 10.1111/j.1474-9726.2010.00577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahuja N, Issa JP. Aging, methylation and cancer. Histol Histopathol 2000; 15:835–42; PMID:10963127 [DOI] [PubMed] [Google Scholar]
- 33.de Magalhães JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009; 25:875–81; PMID:Can't; http://dx.doi.org/ 10.1093/bioinformatics/btp073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al.. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288–95; PMID:21839163; http://dx.doi.org/ 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 35.Dozmorov MG, Giles CB, Koelsch KA, Wren JD. Systematic classification of non-coding RNAs by epigenomic similarity. BMC Bioinformat 2013; 14 Suppl 14:S2; PMID:24267974; http://dx.doi.org/ 10.1186/1471-2105-14-S14-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 2008; 135:649–61; PMID:18992931; http://dx.doi.org/ 10.1016/j.cell.2008.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ. Polycomb repressive complex 2 and trithorax modulate drosophila longevity and stress resistance. Proc Natl Acad Sci U S A 2010; 107:169–74; PMID:20018689; http://dx.doi.org/ 10.1073/pnas.0907739107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823–37; PMID:17512414; http://dx.doi.org/ 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 39.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al.. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315–26; PMID:16630819; http://dx.doi.org/ 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- 40.Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem 2005; 280:17732–6; PMID:15760899; http://dx.doi.org/ 10.1074/jbc.M500796200 [DOI] [PubMed] [Google Scholar]
- 41.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931–6; PMID:21106759; http://dx.doi.org/ 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaaf CA, Misulovin Z, Gause M, Koenig A, Gohara DW, Watson A, Dorsett D. Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet 2013; 9:e1003560; PMID:23818863; http://dx.doi.org/ 10.1371/journal.pgen.1003560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, Garcia BA, Bernstein E, Lowe SW. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A 2012; 109:8971–6; PMID:22615382; http://dx.doi.org/ 10.1073/pnas.1119836109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart MH, Albert M, Sroczynska P, Cruickshank VA, Guo Y, Rossi DJ, Helin K, Enver T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal. Blood 2015; 125(13):2075–8; PMID:25655602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 2013; 73:2936–42; PMID:23644528; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang P, Chaturvedi C-P, Tremblay V, Cramet M, Brunzelle JS, Skiniotis G, Brand M, Shilatifard A, Couture J-Fcc. A phosphorylation switch on RbBP5 regulates histone H3 Lys4 methylation. Genes Dev 2015; 29:123–8; PMID:25593305; http://dx.doi.org/ 10.1101/gad.254870.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Budovsky A, Craig T, Wang J, Tacutu R, Csordas A, Lourenço J, Fraifeld VE, de Magalhães JP. LongevityMap: a database of human genetic variants associated with longevity. Trends Genet 2013; 29:559–60; PMID:23998809; http://dx.doi.org/ 10.1016/j.tig.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 48.Katsushima K, Shinjo K, Natsume A, Ohka F, Fujii M, Osada H, Sekido Y, Kondo Y. Contribution of microRNA-1275 to Claudin11 protein suppression via a polycomb-mediated silencing mechanism in human glioma stem-like cells. J Biol Chem 2012; 287:27396–406; PMID:22736761; http://dx.doi.org/ 10.1074/jbc.M112.359109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robert L, Labat-Robert J. Longevity and aging: role of genes and of the extracellular matrix. Biogerontology 2015; 16:125–9; PMID:25502365; http://dx.doi.org/ 10.1007/s10522-014-9544-x [DOI] [PubMed] [Google Scholar]
- 50.Gratac∫s M, Costas J, de Cid R, Bayés M, González JR, Baca-García E, de Diego Y, Fernández-Aranda F, Fernández-Piqueras J, Guitart M, et al.. Identification of new putative susceptibility genes for several psychiatric disorders by association analysis of regulatory and non-synonymous SNPs of 306 genes involved in neurotransmission and neurodevelopment. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:808–16; PMID:Can't; http://dx.doi.org/ 10.1002/ajmg.b.30902 [DOI] [PubMed] [Google Scholar]
- 51.Cui K, Zang C, Roh T-Y, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009; 4:80–93; PMID:19128795; http://dx.doi.org/ 10.1016/j.stem.2008.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 2011; 117:e182-9; PMID:21427290; http://dx.doi.org/ 10.1182/blood-2011-01-331926 [DOI] [PubMed] [Google Scholar]
- 53.Lozzio BB, Lozzio CB, Bamberger EG, Feliu AS. A multipotential leukemia cell line (K-562) of human origin. Proc Soc Exp Biol Med 1981; 166:546–50; PMID:7194480; http://dx.doi.org/ 10.3181/00379727-166-41106 [DOI] [PubMed] [Google Scholar]
- 54.Kubota Y, Tanaka T, Irino S. Monoclonal antibodies raised against K562 cells reacted with human haematopoietic pluripotent stem cells. Leuk Res 1991; 15:195–204; PMID:2030600; http://dx.doi.org/ 10.1016/0145-2126(91)90121-9 [DOI] [PubMed] [Google Scholar]
- 55.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K-i, et al.. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301–13; PMID:16630818; http://dx.doi.org/ 10.1016/j.cell.2006.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci 2010; 35:323–32; PMID:20346678; http://dx.doi.org/ 10.1016/j.tibs.2010.02.009 [DOI] [PubMed] [Google Scholar]
- 57.Cai L, Rothbart SB, Lu R, Xu B, Chen W-Y, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, et al.. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell 2013; 49:571–82; PMID:23273982; http://dx.doi.org/ 10.1016/j.molcel.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, Fischle W, Bonaldi T, Pasini D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 2014; 53:49–62; PMID:24289921; http://dx.doi.org/ 10.1016/j.molcel.2013.10.030 [DOI] [PubMed] [Google Scholar]
- 59.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 2013; 20:1147–55; PMID:24096405; http://dx.doi.org/ 10.1038/nsmb.2669 [DOI] [PubMed] [Google Scholar]
- 60.He J, Kallin EM, Tsukada Y-I, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Biol 2008; 15:1169–75; PMID:18836456; http://dx.doi.org/ 10.1038/nsmb.1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lorbeck MT, Singh N, Zervos A, Dhatta M, Lapchenko M, Yang C, Elefant F. The histone demethylase Dmel\Kdm4A controls genes required for life span and male-specific sex determination in Drosophila. Gene 2010; 450:8–17; PMID:19786080; http://dx.doi.org/ 10.1016/j.gene.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Asmann YW, Necela BM, Kalari KR, Hossain A, Baker TR, Carr JM, Davis C, Getz JE, Hostetter G, Li X, et al.. Detection of redundant fusion transcripts as biomarkers or disease-specific therapeutic targets in breast cancer. Cancer Res 2012; 72:1921–8; PMID:22496456; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3142 [DOI] [PubMed] [Google Scholar]
- 63.Craig T, Smelick C, Tacutu R, Wuttke D, Wood SH, Stanley H, Janssens G, Savitskaya E, Moskalev A, Arking R, et al.. The Digital Ageing Atlas: integrating the diversity of age-related changes into a unified resource. Nucleic Acids Res 2015; 43:D873–8; PMID:25232097; http://dx.doi.org/ 10.1093/nar/gku843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature 2012; 489:109–13; PMID:22955621; http://dx.doi.org/ 10.1038/nature11279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996–1006; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol 2015; PMID:25690853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dozmorov MG, Cara LR, Giles CB, Wren JD. GenomeRunner: automating genome exploration. Bioinformatics 2012; 28:419–20; PMID:22155868; http://dx.doi.org/ 10.1093/bioinformatics/btr666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dozmorov MG, Wren JD, Alarcón-Riquelme ME. Epigenomic elements enriched in the promoters of autoimmunity susceptibility genes. Epigenetics 2014; 9:276–85; PMID:24213554; http://dx.doi.org/ 10.4161/epi.27021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:Article3; PMID:16646809 [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009; 37:W305–11; PMID:19465376; http://dx.doi.org/ 10.1093/nar/gkp427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.