

Abstract
Aberrant DNA methylation is a common epigenetic alteration found in colorectal adenomas and cancers and plays a role in cancer initiation and progression. Aberrantly methylated DNA loci can also be found infrequently present in normal colon tissue, where they seem to have potential to be used as colorectal cancer (CRC) risk biomarkers. However, detection and precise quantification of the infrequent methylation events seen in normal colon is likely beyond the capability of commonly used PCR technologies. To determine the potential for methylated DNA loci as CRC risk biomarkers, we developed MethyLight droplet digital PCR (ddPCR) assays and compared their performance to the widely used conventional MethyLight PCR. Our analyses demonstrated the capacity of MethyLight ddPCR to detect a single methylated NTRK3 allele from among more than 3125 unmethylated alleles, 25-fold more sensitive than conventional MethyLight PCR. The MethyLight ddPCR assay detected as little as 19 and 38 haploid genome equivalents of methylated EVL and methylated NTRK3, respectively, which far exceeded conventional MethyLight PCR (379 haploid genome equivalents for both genes). When assessing methylated EVL levels in CRC tissue samples, MethyLight ddPCR reduced coefficients of variation (CV) to 6–65% of CVs seen with conventional MethyLight PCR. Importantly, we showed the ability of MethyLight ddPCR to detect infrequently methylated EVL alleles in normal colon mucosa samples that could not be detected by conventional MethyLight PCR. This study suggests that the sensitivity and precision of methylation detection by MethyLight ddPCR enhances the potential of methylated alleles for use as CRC risk biomarkers.
Keywords: colorectal cancer, droplet digital PCR, DNA methylation, field cancerization effect, MethyLight, risk biomarkers
Abbreviations
- CRC
colorectal cancer
- CV
coefficients of variation
- ddPCR
droplet digital PCR
- LOD
limit of detection
- LOQ
limit of quantification
- NTC
no-template control
- PCR
polymerase chain reaction
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related death in the United States.1 The risk of CRC varies among people; those people with a personal or family history of colon polyps or CRC (excluding defined cancer family syndromes) can have a >10% lifetime risk of CRC.2-7 It appears that this increased risk is at least partly related to the effect of genetic variants and/or a field cancerization phenomenon, or ‘field effect’, in which the normal colon is primed to develop polyps or CRC.8-10 Biomarkers that can identify and accurately risk-stratify individuals have the potential to be used in personalized cancer prevention programs, which improve the effectiveness of CRC prevention and early detection.
Aberrantly methylated genes hold promise as risk biomarkers for colorectal cancer.11,12 For example, methylated mir342/EVL and other genes have been detected at higher frequency in the normal colon of people with CRC compared to average risk individuals, suggesting that they may indicate a field cancerization process.10,11,13 However, the potential of methylated genes as effective colon cancer risk biomarkers has not been fully realized and we postulate that this is because the methylated alleles present in normal colon mucosa are present at levels that are often below the detection limits of current PCR technologies. A more precise and sensitive method to detect low levels of methylated DNA would allow a better determination of whether methylated genes can be used as field effect markers. Droplet digital PCR (ddPCR) is a new technology that has the potential to precisely detect nucleic acid targets in various clinical specimens,14,15 but there are no published studies of its application to detecting methylated alleles.
In this study, we developed a MethyLight-based ddPCR assay to accurately quantify methylated NTRK3 and methylated EVL in tissue samples. We demonstrated that MethyLight ddPCR has a 25-fold lower limit of quantification (LOQ) and 20-fold lower limit of detection (LOD) than conventional MethyLight PCR. MethyLight ddPCR significantly improved precision and quantification to detect methylation in primary CRC tissues and normal colon mucosa biopsies. Our study shows the potential of MethyLight ddPCR-based assays to assess the use of methylated alleles as biomarkers for field cancerization and for the early detection of CRC.
Materials and Methods
Sample acquisition and preparation
Normal and matched cancer samples were collected from the Cooperative Human Tissue Network and ColoCare colon cancer cohort study (FHCRC). Normal colon mucosa biopsies were obtained from healthy subjects who underwent screening colonoscopy at the University of Pittsburgh Medical Center following protocols approved by the local IRB committee. Tissue samples were snap frozen in liquid nitrogen and transferred to a −80°C freezer for long-term storage. Stool samples were obtained from the EDRN GLNE CVC at the University of Michigan (PI Dean Brenner) following IRB approved protocols.
DNA extraction
DNA was extracted from tissue samples using the NucleoSpin Tissue kit (Macherey-Nagel Cat #740952) and treated with RNase A (Life Technologies Cat #EN0531) following the manufacturer's instructions, after extraction, DNA samples were loaded on a 1.5% agarose gel to check the quality of the DNA and make sure there was no contamination with RNA. DNA concentration was measured using NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE). Stool DNA was extracted from stool samples as previously published with the exception that DNA capture probes were not used.16
Bisulfite conversion and recovery
DNA samples (500 ng) were bisulfite converted using the EZ DNA Methylation Kit (ZymoResearch Cat #D5002) following the manufacturer's instructions. The bisulfite-converted samples were eluted in a 20-µL volume and stored at −80°C until needed.
MethyLight ddPCR
The MethyLight ddPCR reaction mixture consisted of the 2X ddPCR Supermix for Probes (BioRad Cat #186–3010), and locus specific primers and probes. The primer and probe sequences for methylated EVL (NP_057421) and methylated NTRK3 (NM_002530) were designed using ABI PrimerExpress software Version 5.0.17 (Applied Biosystems, Life Technologies), and synthesized with a FAM reporter. We determined the relative amounts of each sample through a C-LESS-C1 assay,18 which amplifies the total amount of DNA in a PCR reaction. The C-LESS probe was synthesized with a VIC reporter. A complete list of all MethyLight primer and probe sequences is provided in Table 1. The location of CpGs in the MethyLight primers and probes and the amplicon for methylated NTRK3, methylated EVL and C-less-C1 assays is provided in Figure S1. The primer and probes were used at final concentrations of 900 and 250 nmol/L, respectively. Various amounts of bisulfite-converted DNA were used in a final volume of 20 μL. Each 20-μL PCR reaction was loaded into the Bio-Rad DG8 disposable droplet generation cartridge (BioRad, Hercules, CA). A volume of 70 μL of droplet generation oil was loaded into adjacent oil wells. The microfluidic chip was loaded into a droplet generator (BioRad, Hercules, CA). The resulting water-in-oil droplets were pipette-transferred from the outlet well to a 96-well polypropylene plate. The plate was heat-sealed with PX1™ PCR Plate Sealer (BioRad, Hercules, CA), placed on a T100TM Thermal Cycler (BioRad, Hercules, CA) and amplified to the endpoint. The thermal cycling conditions were 95°C for 10 min then 40 cycles of 95°C for 15 s and 60°C for 1 min (2.5°C/s ramp rate) with a final 10 min hold at 98°C. After PCR amplification, the 96-well PCR plate containing droplets was loaded into a QX200 droplet reader (Bio-Rad). The ddPCR system partitions the 20 μL PCR reaction into an average of 15,000 nanoliter droplets and each droplet from each well of the plate was read with a 2-color fluorescence reader to determine how many droplets were positive for the methylated NTRK3 or EVL(in FAM), as well as for the control reaction C-LESS-C1(in VIC). All methylation quantification experiments included no-template-controls (NTC) wells, which contained all the components of the reaction except for the DNA template, control wells containing 2,000 pg of 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655) and 2,000 pg of 100% unmethylated EpiTect Unmethyl control DNA (QIAGEN Cat #59665). No amplification signal was detected in NTC wells. Data was analyzed using the QuantaSoft software version 1.4.0.99 (BioRad, Hercules, CA). The number of methylated DNA molecules was measured as concentrations, i.e., the methylated NTRK3 or EVL copies per μL of PCR mixture.
Table 1.
Primer and probe sequences used in MethyLight assays
| Assay | Primer/probe | Primer/probe sequence (5′ to 3′) | References |
|---|---|---|---|
| NTRK3 | Forward | CGGCGTTCGCGATGGT | Ref.16 |
| Reverse | ACCTTTAAAACGCCGAACGAT | ||
| Probe | FAM-TTAGACGTTGAAGGATTTTGTA | ||
| EVL | Forward | AACGACTCCGAATCCTCGAA | |
| Reverse | GCGAATAGTAACGCGCGTATT | ||
| Probe | FAM-CGCGAACTAATCTCAACA | ||
| C-LESS-C1 | Forward | TTGTATGTATGTGAGTGTGGGAGAGAGA | Ref.17 |
| Reverse | TTTCTTCCACCCCTTCTCTTCC | ||
| Probe | VIC-CCTCCCCCTCTAACTCTAT-MGBNFQa |
MGBNFQ refers to a Minor Groove Binder non-fluorescent quencher in the 3′ terminus of the probe.
Conventional MethyLight PCR
The conventional MethyLight PCR was performed in one 20-μL PCR reaction well of a 96-well plate. The reaction mixture consisted of the 2X iTAQ Universal Probes Supermix (BioRad Cat # 172–5130), and the same locus-specific primers and probes used in MethyLight ddPCR. The primer and probes were used at final concentrations of 900 and 250 nmol/L, respectively. Bisulfite-converted DNA was used as a template for MethyLight PCR assay in a final reaction volume of 20 μL.Each MethyLight PCR reaction was performed using the CFX96 Touch™ Real-Time PCR Detection System (BioRad). The thermal cycling conditions were: 95°C for 3 minutes followed by 49 cycles of 95°C for 5 seconds and 60°C for 1 minute. To assess the specificity of each MethyLight PCR to only detect methylated alleles, an initial experiment was conducted using CpGenome Universal Unmethylated DNA (EMD Millipore Cat # S7822) and 100% unmethylated bisulfite-converted EpiTect Unmethyl control DNA (QIAGEN Cat #59665). We did not detect amplification signal in those samples. Non-template-control (NTC) wells gave no amplification signal. We routinely evaluated each MethyLight reaction by including positive control wells containing 4,500 pg of 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655), negative control wells containing 4,500 pg of 100% unmethylated EpiTect Unmethyl control DNA (QIAGEN Cat #59665), and NTC wells. A five-point standard curve was determined using serial dilutions of the 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655) with water at 45,000 pg, 4,500 pg, 450 pg, 45 pg, and 4.5 pg per 20-μL reaction. Data was analyzed using the Bio-Rad CFX manager software version 3.1 (BioRad, Hercules, CA) and Cq was determined with the Single Threshold method. Intra-assay variation in concentration measured by %CV was less than 15%.
MethyLight ddPCR evaluation experiments
To analyze the intra-assay variation, each sample was replicated at least 4 times in the same assay. The operational definition of limits of quantification (LOQ) is the lowest concentration of methylated alleles in a sample that can be measured with %CV less than 15%. LOQ was determined by serial dilutions of the 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655) with the 100% unmethylated EpiTect Unmethyl control DNA (QIAGEN Cat #59665). The same methylated and unmethylated control DNAs have been used extensively in other DNA methylation studies.19-21 Samples with methylation percentages of 100%, 20%, 4%, 0.8%, 0.16%, 0.032%, 0.006%, and 0% with 20 ng of total DNA were tested at least 4 times in a run in at least 3 independent runs. The operational definition of limits of detection (LOD) is the lowest nonzero concentration in a sample at which replicate measurements give positive qualitative results. LOD was determined by serial dilutions of the 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655) with water. A set of samples containing 40,000 pg, 20,000 pg, 10,000 pg, 5000 pg, 2500 pg, 1250 pg, 250 pg, 125 pg, 62.5 pg, 31.25 pg, and 0 pg were tested at least 4 times in a run in at least 3 independent runs.
EVL methylation analysis in clinical samples
The bisulfite-converted DNA from clinical samples was further diluted by 10-fold for EVL methylation analysis. The conventional MethyLight PCR assays were performed by mixing 4 μL of diluted bisulfite-converted samples with 2X iTAQ Universal Probes Supermix (BioRad Cat # 172–5130), 250 nmol/L of EVL-specific forward and reverse primers and 900 nmol/L probe in a 20-μL reaction volume per reaction, as described above. The quantity of methylated EVL (in pictograms) in clinical samples is determined from a 5-point standard curve derived from the 100% methylated EpiTect Methyl control DNA (QIAGEN Cat #59655) as described above. The clinical samples were assayed with the standards in the same experimental run. For comparing EVL methylation measurements in the clinical samples by the two different PCR based methods, the conventional MethyLight PCR measurements (picograms of methylated EVL per reaction) was converted to the number of methylated EVL copies per μL using the linear regression equation shown in Figure 2B: Y = 0.1435X−0.442, in which X represents the amount of input DNA (copies per μL) and Y represents the concentration of methylated EVL in copies per μL reaction mixture. For the MethyLight ddPCR, the 4 μL of diluted bisulfite-converted samples were mixed with 2X ddPCR Supermix for Probes (BioRad Cat #186–3010), 250 nmol/L of EVL-specific forward and reverse primers and 900 nmol/L probe in a 20 μL reaction volume per reaction. Each 20-μL reaction mixture was partitioned into an average of 15,000 nanoliter droplets, thermocycled, and read with a 2-color reader as described above. CRC samples with high levels of methylated EVL were run in 4 replicate wells and colon mucosa samples with low levels of methylated EVL were run in 8 replicate wells. We combined the droplet counts (positive and negative) from all replicated wells to yield a ‘merged’ well. The concentration and Poisson confidence intervals for each ‘merged’ well were computed using the QuantaSoft software version 1.4.0.99 (BioRad, Hercules, CA). Each sample was measured in at least 2 independent runs.
Figure 2.
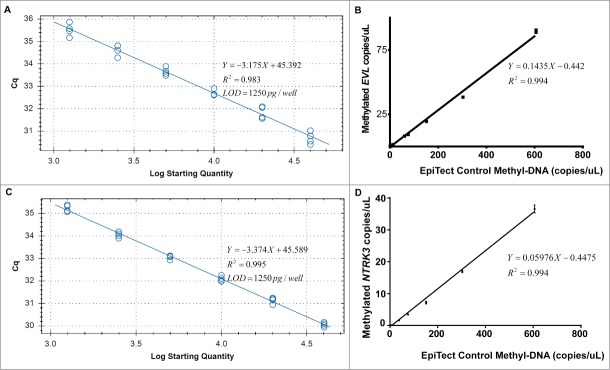
Comparative analysis of limit of detection (LOD) for conventional MethyLight PCR and MethyLight ddPCR. We prepared a two-fold dilution series of EpiTect 100% methylated DNA with water. Standard curve of quantification in conventional MethyLight PCR shows LOD = 1250 pg or 379 haploid genome equivalents per well for (A) EVL and (C) NTRK3. Standard curve of quantification in MethyLight ddPCR shows LOD = 62.5 pg or 19 haploid genome equivalents per well for (B) EVL and LOD = 125 pg or 38 haploid genome equivalents per well for (D) NTRK3.
Statistical analysis
For MethyLight ddPCR, each sample was partitioned into an average of 15,000 droplets per well and replicated in multiple wells. Each droplet serves as an independent unit for PCR reaction. After PCR, each droplet was read with a 2-color fluorescence detector to determine the number of positive and negative events. An event with fluorescence amplitude value more than the set detection threshold was considered a methylation-positive event. The droplet counts (positive and negative) from all replicated wells were combined to yield a ‘merged’ well. The concentration and 95% Poisson confidence intervals for each ‘merged’ well were computed using the QuantaSoft software version 1.4.0.99 (BioRad, Hercules, CA). For LOQ and LOD studies, the linear regression between the ddPCR measurements in copies per μL and the amount of input DNA was performed with GraphPad Prism version 6.02 (GraphPad Software Inc., La Jolla, CA). For the conventional MethyLight PCR, the Cq values of a range of standard DNA with known quantity were linear regressed against the log transformation of starting quantity using the Bio-Rad CFX manager software version 3.1 (BioRad, Hercules, CA).
Results
MethyLight ddPCR has a 25-fold lower limit of quantification (LOQ) and 20-fold lower limit of detection (LOD) than conventional MethyLight PCR
We evaluated the performance of MethyLight ddPCR assays for two representative methylated genes, EVL and NTRK3, and compared the results to those of conventional MethyLight PCR using two metrics: limit of quantification (LOQ) and limit of detection (LOD, defined in the Materials and Methods section). To assess the limit of quantification, we prepared a five-fold dilution series by serially diluting 100% methylated EpiTect Methyl control DNA into 100% unmethylated EpiTect UnMethyl control DNA (Each well contained 20 ng or 6060 haploid human genome equivalents of total input DNA, assuming one haploid human genome is 3.3 pg of DNA.). Conventional MethyLight PCR could detect down to one methylated NTRK3 allele in a background of 125 unmethylated alleles (LOQ = 0.8%, R2 = 0.908) (Fig. 1A). In MethyLight ddPCR, each of the serial diluted samples was partitioned into an average of 15,000 droplets per well and replicated in 4 wells. 1-D ddPCR analysis plot showed the number of positive droplets decreased as methylated DNA was serial diluted with unmethylated DNA (Fig. 1B). We were able to detect levels of methylated NTRK3 in mixed samples as low as 0.032%, equivalent to 1 methylated NTRK3 allele among 3125 unmethylated alleles, with excellent quantitative linearity (LOQ = 0.032%, R2 = 0.996). This is a 25-fold lower LOQ than conventional MethyLight PCR (Fig. 1C). As expected, no amplification signals were detected from no-template control (NTC) wells. We observed that increasing the amount of total DNA loaded into each reaction mix enhanced the LOQ of the MethyLight ddPCR assay (i.e., LOQ with 8 ng of total DNA was 1:312 compared to 1:3125 with 20 ng of total DNA). These results demonstrate the superior sensitivity of MethyLight ddPCR for absolute quantification of methylated NTRK3 compared to conventional MethyLight PCR.
Figure 1.
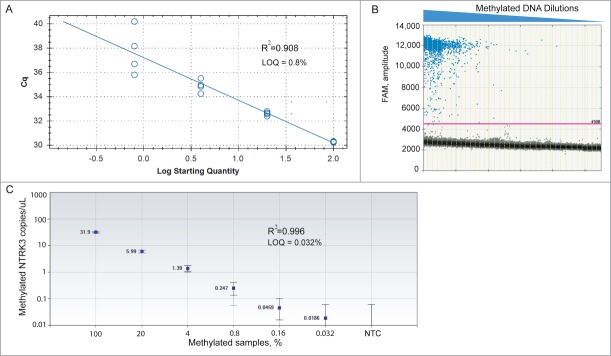
Comparative analysis of limit of quantification (LOQ) for NTRK3 MethyLight ddPCR and conventional MethyLight PCR. We prepared a 5-fold dilution series of EpiTect 100% methylated control DNA into EpiTect 100% Unmethylated control DNA (total 20 ng of DNA per well). (A) Standard curve of quantification in conventional MethyLight PCR shows relationship between Cq value and log transformation of percentage of methylation. LOQ = 0.8%. (B) 1-D ddPCR analysis plot shows detection of methylated NTRK3 in serial dilutions of mixed samples. Each sample was partitioned into an average of 15,000 droplets per well and replicated in 4 wells. The droplet counts (positive and negative) from all replicated wells were combined to yield a ‘merged’ well. An event with fluorescence amplitude value > 4500 was considered a methylation-positive event (threshold line was set at 4500). The number of methylation-positive events decreased as the methylated DNA was further diluted with unmethylated DNA. (C) ddPCR can detect levels of methylated NTRK3 in mixed samples as low as 0.032%. The concentration and Poisson confidence intervals for each ‘merged’ well were computed using the QuantaSoft software version 1.4.0.99 (BioRad, Hercules, CA). Concentrations represent the average measurement of methylated NTRK3 copies per μL of PCR mixture in merged wells for each sample. Error bars indicate the Poisson 95% confidence intervals for each measurement. R2= 0.996 shows good linear correlation between measured concentration of methylated NTRK3 and expected percentage of methylation. NTC: no-template control.
To assess the absolute LOD, we prepared serial dilutions of 100% methylated EpiTect Methyl control DNA using water. With the optimal experiment conditions, the LOD of the conventional MethyLight PCR was 1250 pg of DNA per well for both the EVL and NTRK3 assays, which is 379 haploid genome equivalents of methylated EVL and methylated NTRK3 (Fig. 2A, C). In contrast, the LOD of the MethyLight ddPCR assay for EVL was 62.5 pg of DNA, which is 19 haploid genome equivalents of methylated EVL and 20-fold lower than the conventional MethyLight PCR (Fig. 2B). The LOD of the MethyLight ddPCR assay for NTRK3 was 125 pg of DNA, which is 38 haploid genome equivalents of methylated NTRK3 and 10-fold lower than conventional MethyLight PCR (Fig. 2D).
MethyLight ddPCR improved precision to detect methylated DNA in primary CRC tissues
We evaluated the performance of MethyLight ddPCR in the quantification of methylated EVL in clinical samples. We assessed two groups of tissue samples whose EVL methylation status had been determined by pyrosequencing: primary colon cancer tissues with a high proportion of methylated EVL DNA; and normal colon mucosa biopsies with a very low proportion of methylated EVL DNA. For CRC tissue samples with high levels of methylated EVL (N = 4), we compared the mean methylation and %CV generated by MethyLight ddPCR to conventional Methylight PCR for each sample (Fig. 3). MethyLight ddPCR reduced coefficients of variance (CV) to 6–65% of CVs seen with conventional MethyLight PCR.
Figure 3.
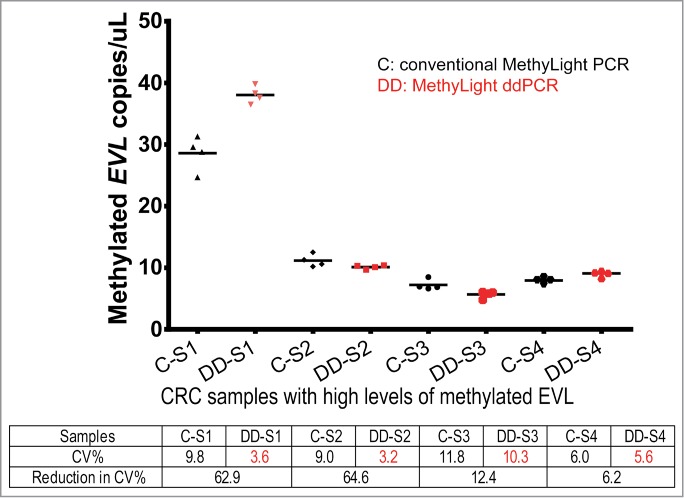
Quantification of methylated EVL in colorectal cancer tissue samples by conventional MethyLight PCR (black) and MethyLight ddPCR (red). CV% and reduction in CV, measured as (CVS—CVd)/(CVS), are listed for each sample.
MethyLight ddPCR enabled detection and absolute quantification of low-frequency methylation events in normal colon mucosa biopsies
We applied MethyLight ddPCR to DNA isolated from normal colon mucosa biopsy samples (N = 9). These samples contain low levels of EVL methylation, which could not be detected by conventional MethyLight PCR. With input DNA of 8 ng in each well and 8 wells of replications, we detected a substantial number of DNA molecules present in each sample based on an assessment of DNA quantities using a duplexed C-LESS-C1 assay (Fig. 4). Using MethyLight ddPCR, we were able to selectively detect and absolutely quantify methylated EVL in all 9 samples (Fig. 4).
Figure 4.
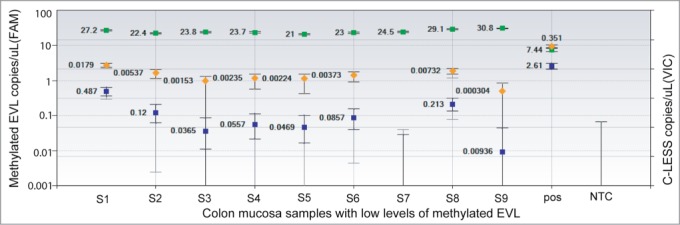
Quantification of methylated EVL in colon mucosa biopsy samples by MethyLight ddPCR. Genomic DNA samples were bisulfite converted and analyzed by MethyLight ddPCR. Each sample (8 ng or 2424 haploid genome equivalents of total input DNA) was partitioned into an average of 15,000 droplets per well and replicated in 8 wells. The droplet counts (positive and negative) from all replicated wells were combined to yield a ‘merged’ well. The concentration and Poisson confidence intervals for each ‘merged’ well were computed using the QuantaSoft software version 1.4.0.99 (BioRad, Hercules, CA). For each sample, the results are presented as the concentration of methylated EVL (copies per μL, blue). The DNA concentration in each sample was estimated based on a C-LESS-C1 assay (copies per μL, green). The fractional abundance of methylated EVL to C-LESS in each sample is also shown (orange). Error bars indicate the Poisson 95% confidence intervals for each measurement. Results with conventional MethyLight PCR are not shown because no amplicon was generated using this method. NTC: no template control. Pos: EpiTect 100% methylated control DNA 2 ng or 606 haploid genome equivalents per well.
Discussion
In this study, we have demonstrated the superiority of our MethyLight ddPCR assay over the widely used conventional MethyLight PCR assay to detect and quantify the methylated alleles EVL and NTRK3. The most prominent advantages are improvement in the lower limits of detection, the absolute quantification of methylated alleles, and in precision, especially for detecting infrequently methylated alleles. Our MethyLight ddPCR assay achieved an LOQ of 0.032% for measuring methylated NTRK3, which is 25-fold lower than the conventional MethyLight PCR. We were able to quantify 19 haploid genome equivalents of methylated EVL and 38 haploid genome equivalents of methylated NTRK3 by MethyLight ddPCR: a 20-fold and 10-fold decrease in LOD respectively, when compared to 379 haploid genome equivalents for both genes by conventional MethyLight PCR. Another advantage of MethyLight ddPCR over conventional MethyLight PCR is the improvement in precision when measuring colon cancer tissue samples containing high levels of methylated EVL. Our method achieved a 6–65% reduction in %CV compared to the conventional MethyLight PCR. The most important advantage of MethyLight ddPCR is that it enables quantification of infrequent methylation events, as demonstrated by our study using methylated EVL in normal colon mucosa biopsy samples. While conventional MethyLight PCR was unable to detect methylated EVL in any of the normal colon mucosa biopsies that were tested, MethyLight ddPCR was able to reliably detect and quantitate the methylated EVL in all of these samples. This is especially important for the identification of field cancerization effects and for the early detection of CRC, which rely on the detection of rare methylation events in colon tissues, body fluids, or stool samples. A stool-based molecular assay to detect CRC would provide a simple, inexpensive, and non-invasive method of screening. However, the technical challenges of developing stool-based molecular assays include the presence of contaminating DNA, the poor quality of DNA extracted from stool samples, and the presence of PCR inhibitors in the DNA, which all likely will attenuate the performance of assays like MethyLight. Recent technical advances, such as digital PCR MethyLight, are well suited to address these technical challenges and to improve the sensitivity and precision of the biomarker assays used on clinical samples. With this in mind, we have done preliminary experiments and shown ddPCR MethyLight improved the detection and quantification of methylated NTRK3 in DNA extracted from stool samples (Figure S2). Other approaches for improving the detection of methylated DNA sequences in clinical specimens include DNA capture methods to enrich for the target of interest. For example, a recent study used methyl-binding protein to capture and enrich methylated Vimentin from stool samples.22 However, the efficiency of currently used capture methods requires improvement in order to obtain optimal capture of all the targets in a sample. Furthermore, advances in whole genome amplification and next-generation sequencing from single-cells have enabled detection of rare molecular events in clinical specimens and are also promising methods for biomarker assays.23-25
Unlike conventional MethyLight PCR, the droplets in MethyLight ddPCR are cycled to endpoint and each droplet is read as positive or negative.15 Thus, amplification efficiency is less of a concern in MethyLight ddPCR compared to conventional MethyLight PCR. Poisson statistics are used to accurately determine the absolute number of starting copies of methylated alleles. These statistics also point to the detection of a single, rare methylation event (i.e., methylated allele) among a large background of unmethylated alleles by incorporating more replications per sample. The sensitivity of our MethyLight ddPCR assay is primarily governed by the number of individual droplet PCR reactions being run per sample, and the amount of input DNA. Given the absence of background signal in the NTC wells and our experience with different amount of input DNA per reaction, we predict that higher amounts of total input DNA per reaction than used in our studies would further decrease the LOQ.
As a proof of principle study for applying ddPCR to DNA methylation analysis and biomarker discovery, our conclusion is based on the comparison of only two methylated genes, EVL and NTRK3. Ongoing work by our group is evaluating the use of MethyLight ddPCR for the assessment of other risk biomarker candidates. We anticipate that the improvements in sensitivity, precision and quantification by MethyLight ddPCR will have a direct impact on risk biomarker assessment by precisely detecting rare methylation events that have been beyond the limit of detection of conventional MethyLight PCR. Our findings support further assessment of the value of MethyLight ddPCR in methylated gene based biomarker research.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Colin Prichard, our lab colleague Dr. Vivek Nandakumar and Dr. Hector Macias from BioRad for their critical comments. We also thank Missy Tuck for her assistance in providing the stool samples.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This research was supported by National Cancer Institute (NCI) RO1CA194663, P30CA15704, UO1CA152756, U54CA143862, and P01CA077852 (WMG); Burroughs Wellcome Fund Translational Research Award for Clinician Scientist (WMG); NIH 2T32DK007742-16 (MY); the Lattner Foundation (WMG), R.A.C.E. Charities (WMG).
References
- 1.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin 2014; 64:104-17; PMID:24639052 [DOI] [PubMed] [Google Scholar]
- 2.Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 2001; 96:2992-3003; PMID:11693338; http://dx.doi.org/ 10.1111/j.1572-0241.2001.04677.x [DOI] [PubMed] [Google Scholar]
- 3.Andrieu N, Launoy G, Guillois R, Ory-Paoletti C, Gignoux M. Familial relative risk of colorectal cancer: a population-based study. Eur J Cancer 2003; 39:1904-11; http://dx.doi.org/ 10.1016/S0959-8049(03)00420-9 [DOI] [PubMed] [Google Scholar]
- 4.Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer 2006; 42:216-27; PMID:16338133; http://dx.doi.org/ 10.1016/j.ejca.2005.09.023 [DOI] [PubMed] [Google Scholar]
- 5.Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC. A prospective study of family history and the risk of colorectal cancer. N Engl J Med 1994; 331:1669-74; PMID:7969357; http://dx.doi.org/ 10.1056/NEJM199412223312501 [DOI] [PubMed] [Google Scholar]
- 6.Hemminki K, Li X. Familial colorectal adenocarcinoma from the Swedish family-cancer database. Int J Cancer 2001; 94:743-8; PMID:11745471; http://dx.doi.org/ 10.1002/ijc.1533 [DOI] [PubMed] [Google Scholar]
- 7.Slattery ML, Kerber RA. Family history of cancer and colon cancer risk: the Utah population database. J Natl Cancer Inst 1994; 86:1618-26; PMID:7932826; http://dx.doi.org/ 10.1093/jnci/86.21.1618 [DOI] [PubMed] [Google Scholar]
- 8.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953; 6:963-8; PMID:13094644; http://dx.doi.org/ 10.1002/1097-0142(195309)6:5%3c963::AID-CNCR2820060515%3e3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- 9.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, et al.. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005; 97:1330-8; PMID:16174854; http://dx.doi.org/ 10.1093/jnci/dji275 [DOI] [PubMed] [Google Scholar]
- 10.Belshaw NJ, Pal N, Tapp HS, Dainty JR, Lewis MP, Williams MR, Lund EK, Johnson IT. Patterns of DNA methylation in individual colonic crypts reveal aging and cancer-related field defects in the morphologically normal mucosa. Carcinogenesis 2010; 31:1158-63; PMID:20395289; http://dx.doi.org/ 10.1093/carcin/bgq077 [DOI] [PubMed] [Google Scholar]
- 11.Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, et al.. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene 2008; 27:3880-8; PMID:18264139; http://dx.doi.org/ 10.1038/onc.2008.10 [DOI] [PubMed] [Google Scholar]
- 12.Woodson K, Weisenberger DJ, Campan M, Laird PW, Tangrea J, Johnson LL, Schatzkin A, Lanza E. Gene-specific methylation and subsequent risk of colorectal adenomas among participants of the polyp prevention trial. Cancer Epidemiol, Biomarkers Prev 2005; 14:1219-23; PMID:15894675; http://dx.doi.org/ 10.1158/1055-9965.EPI-04-0726 [DOI] [PubMed] [Google Scholar]
- 13.Ahuja N, Li Q, Mohan M, Baylin S, Issa J-P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58:5489-94; PMID:9850084 [PubMed] [Google Scholar]
- 14.Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, Vessella RL, Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 2013; 10:1003-5; PMID:23995387; http://dx.doi.org/ 10.1038/nmeth.2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al.. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011; 83:8604-10; PMID:22035192; http://dx.doi.org/ 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagore KS, Lawson MJ, Yucaitis JA, Gage R, Orr T, Shuber AP, Ross ME. Sensitivity and specificity of a stool DNA multitarget assay panel for the detection of advanced colorectal neoplasia. Clin Colorectal Cancer 2003; 3:47-53; PMID:12777192; http://dx.doi.org/ 10.3816/CCC.2003.n.011 [DOI] [PubMed] [Google Scholar]
- 17.Luo Y, Kaz AM, Kanngurn S, Welsch P, Morris SM, Wang J, Lutterbaugh JD, Markowitz SD, Grady WM. NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet 2013; 9:e1003552; PMID:23874207; http://dx.doi.org/ 10.1371/journal.pgen.1003552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisenberger DJ, Trinh BN, Campan M, Sharma S, Long TI, Ananthnarayan S, Liang G, Esteva FJ, Hortobagyi GN, McCormick F, et al.. DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. Nucleic Acids Res 2008; 36:4689-98; PMID:18628296; http://dx.doi.org/ 10.1093/nar/gkn455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics 2010; 283:341-9; PMID:20165882; http://dx.doi.org/ 10.1007/s00438-010-0522-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wojdacz TK, Dobrovic A, Hansen LL. Methylation-sensitive high-resolution melting. Nat Protocols 2008; 3:1903-8; PMID:19180074; http://dx.doi.org/ 10.1038/nprot.2008.191 [DOI] [PubMed] [Google Scholar]
- 21.Wiesmann F, Veeck J, Galm O, Hartmann A, Esteller M, Knuchel R, Dahl E. Frequent loss of endothelin-3 (EDN3) expression due to epigenetic inactivation in human breast cancer. Breast Cancer Res 2009; 11:R34; PMID:19527488; http://dx.doi.org/ 10.1186/bcr2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou H, Harrington J, Rego RL, Ahlquist DA. A novel method to capture methylated human DNA from stool: implications for colorectal cancer screening. Clin Chem 2007; 53:1646-51; PMID:17712002; http://dx.doi.org/ 10.1373/clinchem.2007.086223 [DOI] [PubMed] [Google Scholar]
- 23.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Science U S A 2011; 108:9530-5; PMID:21586637; http://dx.doi.org/ 10.1073/pnas.1105422108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L, Ma F, Chapman A, Lu S, Xie XS. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annual review of genomics and human genetics 2015; 16:1-24; PMID:26077818 [DOI] [PubMed] [Google Scholar]
- 25.Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, Tacey M, Wong R, Singh M, Karapetis CS, et al.. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2015; 00:1-8; PMID:25851626 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.