

Abstract
Most monoclonal antibodies (mAbs) are administered to patients intravenously to ensure high bioavailability as rapidly as possible. The airways, however, are an attractive delivery route for mAbs for the treatment of lung diseases, making it possible to increase their concentration in the target organ while limiting their systemic passage. Several challenges must be overcome for translation into clinical practice. For example, the drug and device must be paired for the efficient and reliable deposition of a pharmacologically active and safe mAb in the lung region of interest. Mesh nebulizers appear to be the most effective aerosol-producing devices for delivering large amounts of biopharmaceutical while limiting protein instability during nebulization. We used metrological and analytic methods to analyze the effect of both antibody concentration and surfactant addition on aerosol performance and antibody integrity. These two factors had a limited effect on aerosol performance, but affected antibody aggregation. The addition of surfactants to antibody formulations at concentrations appropriate for lung administration markedly reduced the formation of medium or large aggregates, as shown by dynamic light scattering and fluorescence microscopy. Aggregation was also dependent on the type of mesh nebulizer, highlighting the need to optimize drug and device together.
Keywords: nebulization, formulation, aggregation, antibody, airways
Abbreviations
- B-35
Brij-35
- CMC
critical micellar concentration
- DLS
dynamic light scattering
- DPI
dry powder inhaler
- IgG
immunoglobulin G
- mAbs
monoclonal antibodies
- MALLS
multi-angle laser light scattering
- NaCl
sodium chloride
- PBS
phosphate-buffered saline
- pMDI
pressurized metered dose inhaler
- PS-20
Polysorbate 20
- PS-80
Polysorbate 80
- RI
refractive index
- SEC
size exclusion chromatography
- SD
standard deviation
- US FDA
United States Food and Drug Administration
- UV
ultraviolet
- VMD
volume mean diameter
Monoclonal antibodies (mAbs) and antibody-based therapies have proved successful for the treatment of cancers, inflammatory and autoimmune diseases, and numerous mAbs have “blockbuster” status (market worth > US$1 billion), placing them in a robust, dynamic position among biopharmaceuticals.1 Five mAbs were accorded breakthrough therapy status by the US FDA in 2013.2 Most mAbs are administered via the blood. The systemic route ensures that the highest bioavailability is achieved as rapidly as possible, but the passage of the mAb from the serum into the target organ may be limited.3,4
Less invasive routes of administration that do not require regular hospitalization are currently being explored for the treatment of long-term chronic diseases. For respiratory diseases, the airways are an obvious route for the local delivery of drugs. This route is routinely used in clinical practice for the delivery of small drug molecules, such as β2-adrenoreceptor agonists, muscarinic antagonists, and corticosteroids.5 The airways have recently been evaluated for the delivery of biopharmaceuticals, including mAbs. However, administration of proteins by inhalation is rare and only one protein drug, dornase alfa (Pulmozyme®), a recombinant human DNase used for the treatment of cystic fibrosis, is currently approved.6-14 Treatments based on mAb inhalation have yet to be validated.
We have shown that the airways constitute an effective administration route for the delivery of high concentrations of mAb to the lungs while limiting the passage of the drug into the bloodstream.9 The pulmonary delivery of mAbs is an attractive proposition for the treatment of pulmonary diseases, but it is challenging in terms of aerosol technology and the formulation of biological agents for inhalation. Further investigations of the behavior and fate of these complex molecules after their deposition in the lungs are also required.
A prerequisite for successful inhalation therapy is the efficient and reliable deposition of sufficient numbers of particles in the pulmonary region of interest. This is dependent on aerosol technology, the performance of the device (e.g., aerosol output, particle size) and the physical characteristics of the drug formulation. Nebulizers are the most widely used inhalers for generating aerosols from protein solutions because the therapeutic dose is too large for delivery by either a pressurized metered dose inhaler (pMDI) or a dry powder inhaler (DPI). Three types of nebulizers are commercially available: (1) jet nebulizers, which use a source of air to spray the liquid into an aerosol and are the most commonly used devices for small molecules in clinical practice; (2) ultrasonic nebulizers, which use a piezoelectric system vibrating at high frequency to convert liquids into aerosols; and (3) mesh nebulizers, which use a vibrational element with a micropumping action to create aerosol particles. We and others have shown that it is feasible to generate aerosols containing large amounts of mAbs with mesh nebulizers, and that their aerodynamic properties are suitable for deposition in the lungs.7,8,15
The effect of aerosolization and drug formulation on the molecular integrity of the active mAb must be taken into account in the development of effective inhalation therapies. Like other therapeutic proteins, mAbs may undergo conformational changes, potentially decreasing their biological activity and causing them to become immunogenic. MAbs are susceptible to various stresses, such as high temperature, extreme pH, shear stress, surface adsorption, and freezing.2,16 Aerosol formation involves the dispersion/suspension of solid material or liquid droplets in a gaseous medium. This process is associated with physical stresses likely to induce changes in protein conformation. The development of inhaled antibody treatments is therefore a challenge for drug formulators. The use of MDIs for protein delivery is not recommended due to the instability associated with the propellants currently used in MDIs, but other aerosol technologies have been considered. Many proteins have been successfully formulated for DPIs, with various techniques, but DPI formulations are plagued by problems of hygroscopy and de-aggregation.15 Nebulizers have not yet proved completely successful for protein aerosolization. Moreover, several studies have shown that the use of jet and ultrasonic nebulizers results in lower levels of activity, a smaller proportion of protein monomers due to partial protein degradation, or higher levels of aggregation during nebulization with or without excipients. This may be due to the heat generated, ultrasonic waves in ultrasonic devices and shear force in jet nebulizers.10,15 Mesh nebulizers, however, seem to preserve protein integrity more effectively than other nebulizers because they do not usually generate large changes in temperature and recycling processes.8 Recently, Hertel et al. improved protein stability with a cooled device to reduce reservoir heating during mesh nebulization. Moreover, other options to stabilize proteins during mesh nebulization may be based on the optimization of formulations. 17,18
Given the recognized advantages of mesh nebulizers over other nebulizers for the pulmonary delivery of proteins, we tested three different mesh nebulizers. We evaluated the effect of antibody formulation on protein stability and aerosol performance. We studied a human polyclonal antibody containing various antibody isoforms, as well as one IgG1 and one IgG4 mAb. IgG1 is the most prevalent immunoglobulin isotype in the blood, and it is widely used for treatment purposes, together with IgG4.19 We monitored antibody instability by investigating the formation of different aggregate populations. We used size exclusion chromatography (SEC) to assess the proportion of monomers/dimers and small aggregates (<0.1 μm), dynamic light scattering (DLS) for medium-sized aggregates (<1 μm), and fluorescence microscopy for large aggregates (>2 μm), as aggregation is the most common consequence of changes in mAb conformation.
We investigated the influence of the device on antibody integrity. For that, we compared the aerosolization of a 10 mg/ml antibody solution in buffer without excipient by three different commercially-available mesh nebulizers using different technologies to generate droplets: two vibrating (PARI eFlow and Aerogen Solo) devices and one static (Omron) mesh nebulizer.20,21 The meshes selected had similar volume mean diameters (VMDs) in isotonic NaCl solution: 6.4 ± 0.1 μm (mean ± SD, n = 3), 5.1 ± 0.1 μm (mean ± SD, n = 4), and 4.1 ± 0.2 μm (mean ± SD, n = 3) for the Omron, Aerogen Solo and PARI eFlow nebulizers, respectively. We chose to use a concentration of 10 mg/ml, because this concentration is frequently encountered in formulations of injectable mAbs.19 As expected, an increase in the number of large aggregates after nebulization was demonstrated by fluorescence microscopy (Fig. 1). The amount of aggregate produced appeared to be device-dependent, with the PARI eFlow nebulizer forming fewer and larger aggregates. Following nebulization of this basic antibody formulation, it was not possible to analyze the DLS spectrum because there were too few (<70%) accurate acquisitions (Table 1). This problem may have resulted from presence of large aggregates (>2 μm) and other undetected forms (1–2 μm), increasing light scattering and interfering with monomer light scattering.
Figure 1.
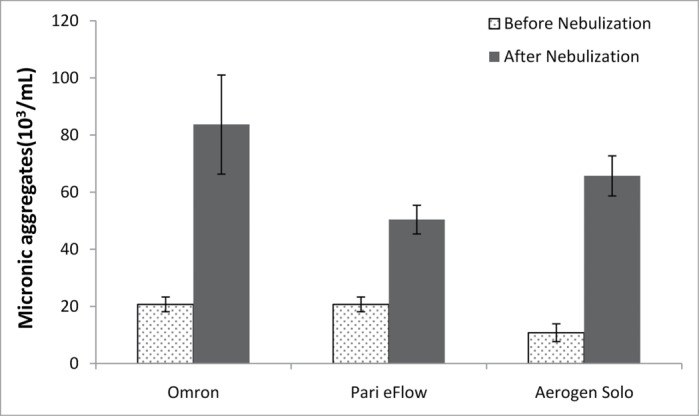
Large antibody aggregates were analyzed by fluorescence microscopy before and after nebulization with the three nebulizers (Omron, PARI eFlow, Aerogen Solo), for a 10 mg/ml antibody solution.
Table 1.
Characteristics of a 10 mg/ml solution of antibody in PBS pH 7.2 after nebulization with three nebulizers. Each result is the mean (± SD) of three nebulizations
| Nebulizer | Hydrodynamic diameter (nm ± SD) | Percentage monomers (% mass) | Size exclusion chromatography (% monomers/% other aggregates) | VMD (μm) (mean ± SD) | Flow rate (ml/min ± SD) |
|---|---|---|---|---|---|
| Omron | NA | NA | 91.1 ± 0.6 / 8.9 ± 0.5 | 5.6 ± 0.1 | 0.23 ± 0.04 |
| Aerogen Solo | NA | NA | 91.3 ± 2.5 / 8.7 ± 1.9 | 4.1 ± 0.1 | 0.50 ± 0.01 |
| PARI eFlow | NA | NA | 90.6 ± 0.5 / 9.5 ± 0.6 | 3.8 ± 0.3 | 0.45 ± 0.05 |
An analysis of dimers and small aggregates by SEC (Fig. 2) revealed no significant difference in the size-exclusion chromatogram of the antibody before and after nebulization. This suggests that nebulization with mesh devices produced more large aggregates than antibody oligomers. Mesh nebulization generated mostly medium-sized and large aggregates when the antibody was formulated in buffer only. Aggregate size depended on the device, but not on the type of aerosol technology. Light microscopy showed that the Omron and Aerogen Solo mesh nebulizers generated larger aggregates than the PARI nebulizer, which generated smaller structures undetectable by this method.
Figure 2.

A and 2B. Size exclusion chromatography of a 10 mg/ml antibody solution in PBS before (solid line) and after (dotted line) nebulization with the Aerogen Solo. (A) UV signal; (B) MALLS signal. The left is Molecular Weight determined by MALLS and the right axis is light scatter in arbitrary units.
The physical characteristics of the aerosol and device performances with the antibody (10 mg/ml in phosphate-buffered saline (PBS) pH 7.2) were similar for the various mesh devices tested (Table 1). The flow rate was <0.5 ml/minute for all nebulizers tested. The highest flow rate was that for the Aerogen Solo (0.5 ml/minute) and the lowest was that for the Omron (0.2 ml/minute). All nebulizers generated aerosol droplets with diameters of less than 5 μm (Table 1): the lowest VMD was obtained with the PARI eFlow (3.8 μm), followed by the Aerogen Solo (4.1 μm), and the Omron had a markedly higher VMD (5.6 μm), as expected from the characteristics of this device.
Surfactants are often used to prevent the formation of aggregates in biological formulations. We hypothesized that they would protect the antibody during aerosolization. Surfactants are encountered in various formulations of commercially nebulized or injectable products, at concentrations ranging from 0.001 to 0.1% (w/v).19 Polysorbates are the most widely used surfactants in the pharmaceutical industry. They are added during the formulation of inhaled corticoids (fluticasone and budesonide) to facilitate the dispersion or dissolution of the drugs during nebulization.22 They are also used in formulations of antibodies for intravenous (i.v.) administration to prevent aggregation.19 As previously shown, the ability of the surfactants such as polysorbates to stabilize a protein and protect against aggregation depends on the protein–to-surfactant ratio.23
We therefore used the three mesh nebulizers to nebulize 10 mg/ml solutions of antibody in PBS pH 7.2 supplemented with various concentrations of Polysorbate 20 (PS-20), ranging from 0.00001 to 0.01% (w/v). We chose to use PS-20 because this compound is frequently present in injectable antibody formulations and other inhalable drug formulations. In this case, for large aggregates, we took the initial levels in the samples (number of aggregates per ml) before nebulization as the baseline values, and the amounts of aggregate following nebulization are expressed as a percentage increase (n = 3). Except for the PARI eFlow nebulizer, which induced only a slight increase in the number of large aggregates, PS-20 limited the formation of the large aggregates (>2 μm) after nebulization, as demonstrated by fluorescence microscopy. A minimum concentration of 10−4% (w/v) PS-20 was required to decrease the amount of large aggregates for both the Omron and Aerogen Solo nebulizers (Fig. 3). The protective effect was therefore not correlated with the PS-20 critical micellar concentration (CMC) (0.007% in water), as previously shown for other proteins.24
Figure 3.
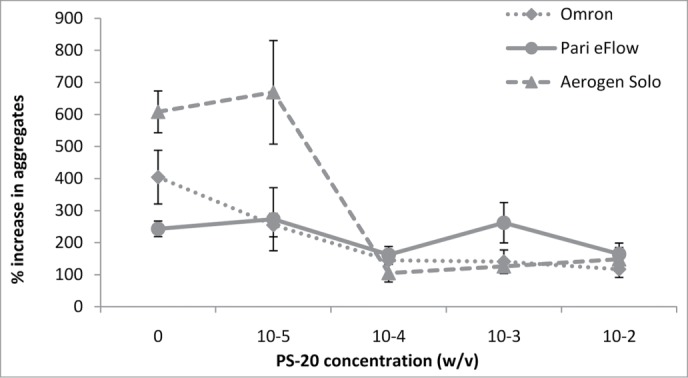
The percentage increase in the number of large antibody aggregates was analyzed by fluorescence microscopy following nebulization, for the three nebulizers (Omron, PARI eFlow, Aerogen Solo).
For all devices, we obtained DLS measurements indicating that PS-20 protected against the formation of medium-sized aggregates. A marked decrease in the amount of aggregate was obtained with PS-20: 10−5% (w/v), 10−4% (w/v), and 10−3% (w/v) for the Omron, Aerogen Solo and PARI eFlow, respectively (Table 2). By contrast, no difference before and after nebulization was observed for monomers/dimers and other aggregates (<0.2 μm) by SEC analysis (n = 3; Table 1). We also studied formulations with other nonionic polyoxyethylene surfactants: Polysorbate 80 (PS-80), which is commonly used in commercially inhalable or injectable products, and Brij 35 (B-35), which is used in various protein methods but not in drug formulations.19,22,25 They also stabilized the antibody during nebulization, limiting the formation of medium-sized and large aggregates (data not shown). However, the concentration required to stabilize the antibody during nebulization depended on the surfactant molecule (0.01% [w/v] for PS-80 vs. 0.0001% [w/v] for B-35), but was not correlated with CMC (0.0017% and 0.011% in water, respectively).
Table 2.
Minimum concentration of PS-20 required to prevent the production of medium-sized aggregates during the nebulization of an antibody solution (10 mg/ml in PBS pH 7.2) with three nebulizers
| Nebulizer | Minimum PS-20 concentration (% w/v) |
|---|---|
| Omron | 10−5 |
| Aerogen Solo | 10−4 |
| PARI eFlow | 10−3 |
The nature of the surfactant had no effect on the aerodynamic properties of the aerosol (data not shown). Most parameters remained stable in the presence of increasing concentrations of surfactant (Table 3). For confirmation of these results with mAbs, we evaluated the impact of surfactants on the stability of two mAbs, a human IgG1 and a human IgG4, formulated in buffer without PS during mesh nebulization. Higher percentages of surfactants markedly reduced the formation of medium-sized or large aggregates, as shown by DLS and fluorescence microscopy (Table 4). This effect was dependent on PS concentration and independent of the device used.
Table 3.
Characteristics of a 10 mg/ml antibody solution in PBS pH 7.2 supplemented with various concentrations of PS-20, after nebulization with the Aerogen Solo nebulizer. Each result is the mean (± SD) of three nebulizations
| PS-20 concentration% (w/v) | Hydrodynamic diameter (nm ± SD) | Percentage monomers (% mass ± SD) | VMD (μm) (mean ± SD) | Flow rate (ml/min ± SD) | Viscosity (mm2/s ± SD) |
|---|---|---|---|---|---|
| 0% | NA | NA | 4.1 ± 0.1 | 0.50 ± 0.01 | 1.07 ± 1.81E-02 |
| 0.00001% | NA | NA | 3.8 ± 0.5 | 0.54 ± 0.04 | 1.05 ± 7.34E-03 |
| 0.0001% | NA | NA | 3.8 ± 0.2 | 0.49 ± 0.01 | 1.03 ± 5.23E-03 |
| 0.001% | 11.4 ± 0.5 | 99.9 ± 0.2 | 3.6 ± 0.6 | 0.49 ± 0.01 | 1.03 ± 2.00E-03 |
| 0.01% | 11.2 ± 0.3 | 99.9 ± 0.2 | 3.8 ± 0.4 | 0.48 ± 0.01 | 1.02 ± 5.46E-03 |
Table 4.
Characteristics of solutions of two monoclonal antibodies (IgG1 and IgG4) in different buffers with various concentrations of polysorbate, before and after nebulization. Each result is the mean (± SD) of three nebulizations
| DLS (% of monomer ± SD) |
Large antibody aggregates (103/ml ± SD) |
||||||
|---|---|---|---|---|---|---|---|
| Nebulizer | Type of surfactant | Surfactant concentration (% w/v) | Before nebulization | After nebulization | Before nebulization | After nebulization | |
| Mab 1 – Buffer 1* | Aerogen solo | PS-20 | 0 | 99.6 | 99.1 | 85.6 | 128.8 |
| 0.01 | 99.2 | 98.6 | 27.9 | 45.9 | |||
| 0.1 | 99.9 | 100 | 33.3 | 29.7 | |||
| Mab 2 – Buffer 2** | PARI eFlow | None | 0 | 100 | NA | 918.0 | NA |
| PS-20 | 0.01 | 100 | 97.3 ± 3.8 | 607.5 | 1024.7 ± 356.4 | ||
| 0.1 | 100 | 99.6 ± 0.3 | 783.0 | 625.5 ± 232.4 | |||
| PS-80 | 0.01 | 100 | 99.4 ± 0.4 | 388.8 | 954.0 ± 623.5 | ||
| 0.1 | 100 | 99.9 ± 0.0 | 702.0 | 688.5 ± 269.0 | |||
n = 1 nebulization; **n = 3 nebulizations; NA, not analyzable.
We found that surfactants limited the formation of large aggregates during mesh nebulization at a range of concentrations commonly used to stabilize mAbs in injectable solutions. This may be because surfactants preferentially accumulate at the air-water interface, coating hydrophobic surfaces and antibody partitioning at the interface.24,26 They may also interact directly with antibody molecules, protecting them against interfacial stress.5 However, other factors may also cause antibody instability and aggregation during nebulization. The three nebulizers had similar membrane pore sizes, but generated different aggregate populations (Fig. 3; Tables 1 and 2). Thus, the device used, the piezoelectric frequency or the interaction between solution and membrane during nebulization may affect the formation of antibody aggregates.
A crucial parameter in drug development is the amount of the biodrug that must be delivered to achieve pharmacological activity in vivo. One inherent limitation of aerosol delivery is the nebulization time that patients can tolerate, which is directly correlated with the volume administered. In such conditions, delivery of the correct dose may require an increase in the amount of protein in a constant volume. Antibody concentration, however, is known to affect aggregation.5 Extremely high or low concentrations have been reported to favor aggregate formation for many proteins.8 We therefore evaluated the effect of antibody concentration, within the range of concentrations used in clinical practice for i.v. formulations, on antibody stability during nebulization. We nebulized antibody solutions with concentrations of 2 to 40 mg/ml, with the Aerogen Solo mesh nebulizer. As shown in Figure 4 and Table 5, antibody concentrations of up to 10 mg/ml resulted in the formation of numerous large (>2 μm) aggregate populations, as observed by fluorescence microscopy, and other undetectable smaller species (probably 1 to 2 μm in diameter) interfering with light scattering by the monomer after nebulization. Higher concentrations of antibody (≥15 mg/ml; Figure 4) were more stable during nebulization, forming less aggregate. This may be because protein molecules are surface-active and tend to partition at the interface.17 If the numbers of molecules accumulating at the surface are constant for each aerosol particle generated by a device, then higher concentrations result in larger numbers of antibodies within the aerosol particles being protected against interfacial stress (Fig. 5). Increasing antibody concentration further may not enhance protection further because very high concentrations of protein are prone to aggregation due to protein-protein interaction.16 Additional experiments are required to evaluate the maximal concentration of antibody that can be used in aerosol formulations with minimal levels of aggregate formation.
Figure 4.
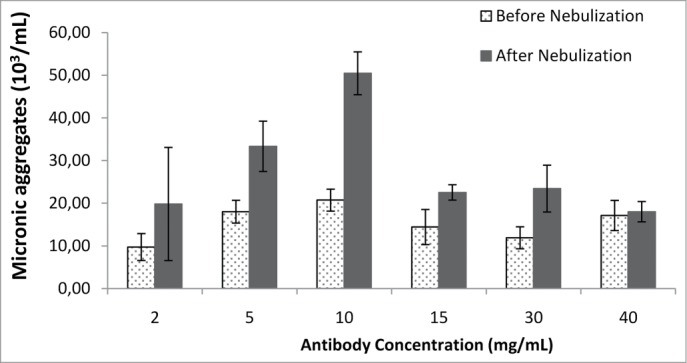
Large antibody aggregates were analyzed by fluorescence microscopy before and after nebulization with the Aerogen Solo nebulizer, for various antibody concentrations.
Table 5.
Characteristics of solutions of various concentrations of antibody in PBS pH 7.2, after nebulization with the Aerogen Solo nebulizer. Each result is the mean (± SD) of three nebulizations
| Antibody concentration (mg/ml) | Hydrodynamic diameter (nm ± SD) | Percentage monomers (mean% mass) | VMD (μm) (mean ± SD) | Flow rate (ml/min ± SD) | Viscosity (mm2/s ± SD) |
|---|---|---|---|---|---|
| 2 | NA | NA | 4.4 ± 0.3 | 0.55 ± 0.01 | 0.97 ± 1.02E-03 |
| 5 | NA | NA | 3.6 ± 0.2 | 0.48 ± 0.01 | 1.02 ± 4.40E-03 |
| 10 | NA | NA | 4.1 ± 0.1 | 0.50 ± 0.01 | 1.07 ± 1.81E-02 |
| 15 | 12.6 ± 1.42 | 94.4 ± 9.0 | 4.0 ± 0.1 | 0.47 ± 0.01 | 1.09 ± 2.00E-02 |
| 30 | 11.9 ± 0.06 | 99.6 ± 0.2 | 4.1 ± 0.1 | 0.42 ± 0.01 | 1.17 ± 7.38E-03 |
| 40/45 | 12.6 ± 0.35 | 99.6 ± 0.5 | 4.0 ± 0.2 | 0.40 ± 0.01 | 1.28 ± 9.97E-03 |
Figure 5.
Model of the spatial distribution of antibodies in aerosol droplets as a function of antibody concentration.
Formulations including high concentrations of antibody were associated with a lower nebulizer output rate (0.55 ml/min for low concentrations and 0.40 ml/min for high concentrations). The duration of aerosol inhalation required was, therefore, significantly longer (Table 5). Despite the physical stability of high-concentration antibody formulations, patient compliance may therefore limit the delivery of very high concentrations of antibody to the lung in aerosol form by nebulization.
Finally, we evaluated the possible additive protective effects of antibody concentration and surfactant for nebulizer formulations. We prepared solutions of antibodies at different concentrations (2, 5, 10, 15, 30, and 40 mg/ml) in PBS and with various concentrations of PS-20 (0.00001, 0.0001, and 0.001% w/v) and nebulized them with the Aerogen Solo mesh nebulizer. At high concentrations of antibody (30 and 40 mg/ml), PS-20 was not required to prevent aggregation. At the lowest concentration of antibody, the maximum PS-20 concentration required for antibody stability was 0.001% (w/v) (Table 6). These results are consistent with the findings of Mahler et al.,23 who showed that the ability of surfactants, such as polysorbates, to stabilize a protein and protect it against aggregation depends on the protein-to-surfactant ratio.
Table 6.
Minimum concentration of PS-20 required to limit the formation of medium-sized and large aggregates
| Antibody concentration (mg/ml) | ||||||
|---|---|---|---|---|---|---|
| 2 | 5 | 10 | 15 | 30 | 40 | |
| Minimum PS-20 concentration (% w/v) | 0.001 | 0.0001 | 0.0001 | 0.0001 | 0 | 0 |
Our results indicate that mesh nebulization does not affect the generation of oligomeric structures, regardless of the formulation (surfactant, protein concentration) or device used. However, the formation of medium-sized and large aggregates in antibody formulations for nebulization was limited by high concentrations of surfactants or protein, and the sizes of the different aggregate populations generated depended on the device. Higher antibody or surfactant concentrations did not significantly modify the VMD of the aerosol cloud, which was suitable for pulmonary delivery. These findings are consistent with previous observations for solutions of various molecules with different viscosities.
The success of inhaled antibody treatments depends on the performance of the aerosol generators their ability to deliver effective, reproducible pulmonary deposition, and the stability of the molecules during aerosolization. Both these factors must be optimized to achieve the desired pharmacological effect in the lungs. Mesh nebulizers clearly have several technical advantages over jet/ultrasonic nebulizers, MDI and DPI for the delivery of inhaled antibodies. They can deliver large therapeutic doses and better preserve the molecular integrity of the proteins. Our findings confirm the utility of adding surfactants and increasing protein concentration to stabilize antibody formulations during vibrating-membrane nebulization. However, formulations should be optimized for each “drug and device” pairing, taking into account the specifications of the device and the unique physical and chemical properties of the mAb molecule.
Materials and Methods
Materials
The human polyclonal immunoglobulin was purchased from Europa Bioproducts. The monoclonal antibodies were full human IgG1 and IgG4, which are currently under development. It was dissolved in sterile water to obtain a concentration of 45 mg/ml (in phosphate-buffered saline, PBS). PBS pH 7.2 (4.2 mM phosphate buffer; 155 mM NaCl) was purchased from Gibco-Thermo Fisher and used for polyclonal immunoglobulin dialysis with 20K MWCO Slide-A-Lyzer Dialysis Cassettes from Thermo Fisher Scientific. All solutions were passed through a syringe filter with 0.22 μm pores from Sarstedt and protein concentration was determined by measuring absorption at 280 nm with a Nanodrop 2000 from Thermo Fisher Scientific. Nile Red, PS-20, PS-80, and B-35 were purchased from Sigma Aldrich. All solutions were stored at 4 °C until nebulization. Both the human polyclonal immunoglobulin and the monoclonal antibody samples were treated exactly the same to analyze aggregates generation following nebulization.
Mesh nebulizers
We used three vibrating-mesh devices currently available on the market. The Omron NE-U22VMicroAir nebulizer (Omron) (static mesh nebulizer) consists of a piezoelectric element that vibrates a transducer horn, which pulses fluid through a mesh, creating the aerosol droplets. The Aerogen Solo aerosol generator (Aerogen), a commercially available PARI eFlow nebulizer and a customized PARI device (PARI Pharma GmbH) (vibrating mesh nebulizers) produce an aerosol through a perforated mesh activated by an annular piezoelectric element. The passive vibrations of the Omron mesh result in the liquid being broken up into larger droplets than those that are generated by the active vibrations of the mesh in the Aerogen Solo device and the PARI eFlow device. This improves the conversion of the nebulizer fluid into smaller droplets. The Omron mesh nebulizer, with its passive vibration system, has an output rate only half that of the active mesh technology of the Aerogen Solo or PARI eFlow. All experiments were performed with the Omron NE-U22VMicroAir, Aerogen Solo and PARI eFlow nebulizers. The starting volume in all studies was 2 ml, except for the Omron nebulizer, for which a larger volume was used (4 ml) due to the loss of aerosol in the environment during collection after nebulization. The aerosol generated by nebulization was collected and analyzed (DLS, fluorescence microscopy). Samples were then stored at –80 °C until SEC analysis.
We determined the aerosol characteristics of isotonic NaCl solution, as a reference nebulization fluid. The VMD obtained was 6.4 ± 0.1 μm (mean ± SD, n = 3) for the Omron, 5.1 ± 0.1 μm (mean ± SD, n = 4) for the Aerogen Solo, and 4.1 ± 0.2 μm (mean ± SD, n = 3) for the PARI eFlow nebulizer.
Fluorescence microscopy
Before and after nebulization, immunoglobulin G polyclonal antibody samples were stained with Nile Red and observed under a fluorescence microscope. Briefly, we diluted 0.25 mM Nile Red stock solution in DMSO to obtain a 10 μM Nile Red solution in water. We then added 2 μl of this solution to 18 μl of protein solution. Immediately after staining, the protein solution was placed on microscope slides from RS France (76 mm × 26 mm × 1.1 mm). Aggregates were counted with the grid option of an AMG Evos FL microscope (Saint Aubin). At least six different squares of the grid were counted and the analysis was carried three times for each sample. Results are expressed as numbers of aggregates per ml of solution.
Dynamic light scattering
Dynamic light scattering experiments were performed at 25 °C, with a Dynapro Nanostar® instrument, using a 659 nm/100 mW laser and a 90° detection angle (Wyatt Technology, Europe GmbH), to determine the size distribution profile of the solutions before and after nebulization. For each sample, we performed 10 acquisitions of 7 s each per measurement and PBS was used as the reference solvent. The results were evaluated with Dynamics® (7.1.5.6) software and are reported as the diameter and percentage polydispersity of the detected particles. Samples for which fewer than 70% of acquisitions were successful were considered “not evaluable,” as recommended by the manufacturer. The dynamic data filter was set as follows: baseline limit ± 0.04 and maximum SOS 1000. Results are expressed as the percentage, in mass, of the monomer and other species, the hydrodynamic diameter of species (nm).
Size exclusion chromatography
SEC was performed on an eAlliance 2695 (Waters) equipped with a Wyatt miniDawn Treos and Optilab T-rEx (Wyatt Technology Europe GmbH). A SHODEX KW803 column (300 mm × 7.8 mm) was used. We injected 50 μg of each sample onto the column and separation was performed at a flow rate of 0.7 ml/minute. The elution buffer was PBS pH 7.2. UV detection was performed at 280 nm, with the PDA 2998 apparatus (Waters), whereas multiple-angle laser light-scattering (MALLS) detection was performed with a three-angle (43.6°; 90.0°; 136.4°) miniDawn Treos detector (Wyatt Technology Europe) operating with a 60-mW solid-state laser at 658 nm. The molecular weights corresponding to the antibody peaks were calculated with Astra software version 6.0.6 (Wyatt Technology Europe). A dn/dc of 0.185 (ml/g) and a second virial coefficient of 0 were used. Molecular weights were calculated with the Zimm equation. Results are expressed as the percentage of monomers and of other species.
Output determination
The total aerosol output of the nebulizers was determined by gravimetric methods involving the weighing of the nebulizer before and after each nebulization experiment. We assumed that the difference in weight obtained provided an accurate measurement of the amount of aerosol generated. This weight difference and the density were used to calculate the aerosol output rate in ml/minute. All experiments were performed at room temperature (20–25 °C) and ambient relative humidity (40–60%).
Viscosity
We determined the viscosity of the fluids prepared for nebulization, with a capillary viscometer of the Micro Ostwald type (Schott). All measurements were performed at room temperature (21 °C ± 1 °C) to ensure that the system was at thermal and mechanical equilibrium. The samples were held in the container cell for 10 min before the start of each run. The viscosity of each solution was calculated as the mean of five measurements.
Determination of droplet size
The VMD of the aerosol was determined with a laser diffraction technique on a Spraytec™ instrument (Malvern Instruments Ltd.). Laser diffraction is a well-established technique for determining the size of the aerosol droplets generated by nebulizers. The VMD measured is the median size of a spherical particle with the same volume as the particle in question. We nebulized 2 ml of each solution. Nebulizers were held in the normal position recommended by the manufacturer (mouthpiece, T piece or not) and were connected to the inhalation cell (Malvern Instruments Ltd.). The aerosol droplets were aspirated by a constant suction pump at an airflow rate of 20 l/minute, on the opposite side of the aerosol beam. All experiments were performed at room temperature (20–25 °C) and ambient relative humidity (40–60%).
Preparation of solutions for nebulization
The antibody was diluted to different concentrations (2, 5, 10, 15, 30, and 40 mg/ml) in a PBS buffer pH 7.2, with and without surfactants (PS-20, PS-80 and B-35) at various concentrations (from 0.00001% to 0.01% [w/v]).The minimum and maximum concentrations were fixed on the basis of the results obtained in a preliminary study. The minimum concentration had no effect on antibody aggregation and the maximum concentration had no effect on aerosol performance.
All solutions were characterized before nebulization, by DLS for the determination of medium-sized aggregates (<1 μm), by SEC-UV-MALLS-RI for the proportion of monomers/dimers and small aggregates (<0.1 μm), by fluorescence microscopy for large aggregates (>2 μm), and with a capillary viscometer.
DLS analysis before nebulization revealed that >99.7% of the antibody corresponded to a single peak of particles with a median diameter of ∼11 nm and a polydispersity percentage of 15 to 25%, in all solutions prepared. SEC-UV-MALLS-RI showed the proportion of dimers and small aggregates to lie in the following range: 88–92% monomers/8–12% dimers and small aggregates. The proportion of dimers and small aggregates could explain the polydispersity values obtained with DLS. Finally, the number of large aggregates observed on fluorescence microscopy was 9729–20720/ml.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by the French National Research Agency as part of the “Investissements d’avenir” program, under Grant Agreement LabEx MAbImprove: ANR- 10-LABX-53. It was also supported by grants provided by Région Centre (Stabiomed and PCMAB) and the DGA (Direction Générale des Armées – AeroRiMac grant). We also thank Julie Sappa, of Alex Edelman and Associates, for assistance with the edition of this manuscript.
References
- 1. Scolnik PA. mAbs: a business perspective. MAbs 2009; 1:179-84; PMID:20061824; http://dx.doi.org/ 10.4161/mabs.1.2.7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reichert JM. Antibodies to watch in 2014. MAbs 2014; 6:5-14; PMID:24284914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 4. Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, Maleeff BE, Eustis S, Schwartz LW, Tsui P, Appelbaum ER, et al. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy Clin Immunol 2001; 108:250-7; PMID:11496242; http://dx.doi.org/ 10.1067/mai.2001.116576 [DOI] [PubMed] [Google Scholar]
- 5. Stokes JR. Promising future therapies for asthma. Int Immunopharmacol 2014; (Forthcoming); PMID:24957689; http://dx.doi.org/ 10.1016/j.intimp.2014.05.032 [DOI] [PubMed] [Google Scholar]
- 6. Antoniu SA, Cojocaru I. Pitrakinra for asthma. Expert Opin Biol Ther 2010; 10:1609-15; PMID:20923253; http://dx.doi.org/ 10.1517/14712598.2010.524203 [DOI] [PubMed] [Google Scholar]
- 7. Lightwood D, O’Dowd V, Carrington B, Veverka V, Carr MD, Tservistas M, Henry AJ, Smith B, Tyson K, Lamour S, et al. The discovery, engineering and characterisation of a highly potent anti-human IL-13 fab fragment designed for administration by inhalation. J Mol Biol 2013; 425:577-93; PMID:23219467; http://dx.doi.org/ 10.1016/j.jmb.2012.11.036 [DOI] [PubMed] [Google Scholar]
- 8. Maillet A, Congy-Jolivet N, Le Guellec S, Vecellio L, Hamard S, Courty Y, Courtois A, Gauthier F, Diot P, Thibault G, et al. Aerodynamical, immunological and pharmacological properties of the anticancer antibody cetuximab following nebulization. Pharm Res 2008; 25:1318-26; PMID:18030605; http://dx.doi.org/ 10.1007/s11095-007-9481-3 [DOI] [PubMed] [Google Scholar]
- 9. Maillet A, Guilleminault L, Lemarié E, Lerondel S, Azzopardi N, Montharu J, Congy-Jolivet N, Reverdiau P, Legrain B, Parent C, et al. The airways, a novel route for delivering monoclonal antibodies to treat lung tumors. Pharm Res 2011; 28:2147-56; PMID:21491145; http://dx.doi.org/ 10.1007/s11095-011-0442-5 [DOI] [PubMed] [Google Scholar]
- 10. Münster AM, Bendstrup E, Jensen JI, Gram J. Jet and ultrasonic nebulization of single chain urokinase plasminogen activator (scu-PA). J Aerosol Med 2000; 13:325-33; PMID:11262439; http://dx.doi.org/ 10.1089/jam.2000.13.325 [DOI] [PubMed] [Google Scholar]
- 11. Safdar A, Shelburne SA, Evans SE, Dickey BF. Inhaled therapeutics for prevention and treatment of pneumonia. Expert Opin Drug Saf 2009; 8:435-49; PMID:19538104; http://dx.doi.org/ 10.1517/14740330903036083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Satoh H, Tazawa R, Sakakibara T, Ohkouchi S, Ebina M, Miki M, Nakata K, Nukiwa T. Bilateral peripheral infiltrates refractory to immunosuppressants were diagnosed as autoimmune pulmonary alveolar proteinosis and improved by inhalation of granulocyte/macrophage-colony stimulating factor. Intern Med 2012; 51:1737-42; PMID:22790136; http://dx.doi.org/ 10.2169/internalmedicine.51.6093 [DOI] [PubMed] [Google Scholar]
- 13. Steckel H, Eskandar F, Witthohn K. Effect of excipients on the stability and aerosol performance of nebulized aviscumine. J Aerosol Med 2003; 16:417-32; PMID:14977432; http://dx.doi.org/ 10.1089/089426803772455677 [DOI] [PubMed] [Google Scholar]
- 14. Thipphawong J. Inhaled cytokines and cytokine antagonists. Adv Drug Deliv Rev 2006; 58:1089-105; PMID:17023089; http://dx.doi.org/ 10.1016/j.addr.2006.07.014 [DOI] [PubMed] [Google Scholar]
- 15. Shoyele SA, Slowey A. Prospects of formulating proteins/peptides as aerosols for pulmonary drug delivery. Int J Pharm 2006; 314:1-8; PMID:16563674; http://dx.doi.org/ 10.1016/j.ijpharm.2006.02.014 [DOI] [PubMed] [Google Scholar]
- 16. Wang W, Roberts CJ. Aggregation of therapeutic proteins. Hoboken, N.J.: Wiley, 2010. [Google Scholar]
- 17. Hertel S, Pohl T, Friess W, Winter G. Prediction of protein degradation during vibrating mesh nebulization via a high throughput screening method. Eur J Pharm Biopharm 2014; 87:386-94; PMID:24709473; http://dx.doi.org/ 10.1016/j.ejpb.2014.03.020 [DOI] [PubMed] [Google Scholar]
- 18. Hertel S, Pohl T, Friess W, Winter G. That's cool! - Nebulization of thermolabile proteins with a cooled vibrating mesh nebulizer. Eur J Pharm Biopharm 2014; 87:357-65; PMID:24662437; http://dx.doi.org/ 10.1016/j.ejpb.2014.03.001 [DOI] [PubMed] [Google Scholar]
- 19. Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci 2007; 96:1-26; PMID:16998873; http://dx.doi.org/ 10.1002/jps.20727 [DOI] [PubMed] [Google Scholar]
- 20. Dolovich MB, Dhand R. Aerosol drug delivery: developments in device design and clinical use. Lancet 2011; 377:1032-45; PMID:21036392; http://dx.doi.org/ 10.1016/S0140-6736(10)60926-9 [DOI] [PubMed] [Google Scholar]
- 21. Waldrep JC, Dhand R. Advanced nebulizer designs employing vibrating mesh/aperture plate technologies for aerosol generation. Curr Drug Deliv 2008; 5:114-9; PMID:18393813; http://dx.doi.org/ 10.2174/156720108783954815 [DOI] [PubMed] [Google Scholar]
- 22. Pilcer G, Amighi K. Formulation strategy and use of excipients in pulmonary drug delivery. Int J Pharm 2010; 392:1-19; PMID:20223286; http://dx.doi.org/ 10.1016/j.ijpharm.2010.03.017 [DOI] [PubMed] [Google Scholar]
- 23. Mahler HC, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci 2009; 98:2909-34; PMID:18823031; http://dx.doi.org/ 10.1002/jps.21566 [DOI] [PubMed] [Google Scholar]
- 24. Randolph TW, Jones LS. Surfactant-protein interactions. Pharm Biotechnol 2002; 13:159-75; PMID:11987751; http://dx.doi.org/ 10.1007/978-1-4615-0557-0_7 [DOI] [PubMed] [Google Scholar]
- 25. Nema S, Washkuhn RJ, Brendel RJ. Excipients and their use in injectable products. PDA J Pharm Sci Technol 1997; 51:166-71; PMID:9277127 [PubMed] [Google Scholar]
- 26. Kreilgaard L, Jones LS, Randolph TW, Frokjaer S, Flink JM, Manning MC, Carpenter JF. Effect of Tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant human factor XIII. J Pharm Sci 1998; 87:1597-603; PMID:10189273 [DOI] [PubMed] [Google Scholar]