

Abstract
Mutations in the human gene encoding contactin-associated protein-like 2 (CNTNAP2) have been strongly associated with autism spectrum disorders (ASDs). Cntnap2−/− mice recapitulate major features of ASD, including social impairment, reduced vocalizations, and repetitive behavior. In addition, Cntnap2−/− mice show reduced cortical neuronal synchrony and develop spontaneous seizures throughout adulthood. As suggested for other forms of ASDs, this phenotype could reflect some form of synaptic dysregulation. However, the impact of lifelong deletion of CNTNAP2 on synaptic function in the brain remains unknown. To address this issue, we have assessed excitatory and inhibitory synaptic transmission in acute hippocampal slices of Cntnap2−/− mice. We found that although excitatory transmission was mostly normal, inhibition onto CA1 pyramidal cells was altered in Cntnap2−/− mice. Specifically, putative perisomatic, but not dendritic, evoked IPSCs were significantly reduced in these mice. Whereas both inhibitory short-term plasticity and miniature IPSC frequency and amplitude were normal in Cntnap2−/− mice, we found an unexpected increase in the frequency of spontaneous, action potential-driven IPSCs. Altered hippocampal inhibition could account for the behavioral phenotype Cntnap2−/− mice present later in life. Overall, our findings that Cntnap2 deletion selectively impairs perisomatic hippocampal inhibition while sparing excitation provide additional support for synaptic dysfunction as a common mechanism underlying ASDs.
SIGNIFICANCE STATEMENT The gene encoding contactin-associated protein-like 2 (CNTNAP2) stands out as one the first genes to have both rare and common mutations strongly associated with ASDs. Whereas Cntnap2−/− mice appear to recapitulate core behavioral endophenotypes (e.g., social impairment, language deficits, and repetitive behavior), the cellular and circuit bases of this phenotype remain poorly understood. Here, we report that synaptic inhibition specifically onto the perisomatic compartment of CA1 pyramidal cells is impaired, whereas dendritic inhibition and excitatory transmission remain basically unchanged in Cntnap2−/− mice. These findings provide additional support to the notion that disruption of inhibition, in particular perisomatic inhibition onto principal cells, could play a critical role in ASDs.
Keywords: CASPR2, cell adhesion, E/I balance, epilepsy, perisomatic inhibition, synaptopathy
Introduction
Autism spectrum disorders (ASDs) are neurodevelopmental disorders associated with both genetic and environmental risk factors. Among the large number of identified candidate genes, the human gene encoding contactin-associated protein-like 2 (CNTNAP2) stands out as one the first genes to have both rare and common mutations strongly associated with ASDs (Peñagarikano and Geschwind, 2012; Rodenas-Cuadrado et al., 2014). In addition, mutations in CNTNAP2 have also been linked to intellectual disability, epilepsy, schizophrenia, attention deficit hyperactivity disorder, learning disability, and language impairment (for review, see Rodenas-Cuadrado et al., 2014). CNTNAP2 encodes CASPR2, a neuronal cell-adhesion molecule whose function in the brain is poorly understood. In search of a common neural mechanism to ASDs and other neurodevelopmental disorders, several studies rely on animal models carrying relevant genetic mutations. Cntnap2−/− mice appear to reproduce all three core behavioral endophenotypes of ASDs: social impairment, language deficits, and repetitive behavior (Peñagarikano et al., 2011). In addition, these mice show reduced neuronal synchrony in neocortex and develop spontaneous seizures throughout adulthood. Remarkably, the cellular and circuit bases of this phenotype remain unknown.
Synaptic dysfunction and neural circuit deficits are thought to play a crucial role in the pathophysiology of ASDs (Zoghbi and Bear, 2012; Delorme et al., 2013). That some candidate genes for ASDs encode for synaptic cell-adhesion molecules (Betancur et al., 2009) further supports the hypothesis that synaptic pathology underlies the ASD phenotype. Moreover, synaptic dysfunction has been found in several animal models of syndromic forms of ASDs (Zoghbi and Bear, 2012; Delorme et al., 2013), and shifts in the balance between synaptic excitation and inhibition have emerged as common mechanisms across different ASD animal models (Gogolla et al., 2009; Ramamoorthi and Lin, 2011; Coghlan et al., 2012; Nelson and Valakh, 2015). Cntnap2 mRNA is widely expressed in the brain (Bakkaloglu et al., 2008), and a recent study reported CASPR2 expression in dendritic spines, soma, and axons of cortical cultured neurons (Varea et al., 2015). In addition, CASPR2 is enriched in the synaptic plasma membrane fraction of rat forebrain homogenates (Bakkaloglu et al., 2008). Consistent with these observations, two studies reported synaptic phenotypes in CASPR2-deficient, cortical cultured neurons. Acute knockdown of Cntnap2 was associated with significant deficits in dendritic arborization, spine development, and global synaptic function (Anderson et al., 2012), and Cntnap2 knock-out neurons displayed reduced spine density and levels of the GluA1 subunit of AMPA receptors in dendritic spines (Varea et al., 2015). However, the consequences of lifelong deletion of CNTNAP2 on synaptic function in the brain have not yet been investigated. In the present study, we have assessed excitatory and inhibitory synaptic transmission in acute hippocampal slices of Cntnap2−/− mice. Our findings show that excitatory synaptic transmission was mostly normal, whereas inhibition was impaired. Altered inhibition may contribute to the behavioral phenotypes of Cntnap2−/− mice and ultimately to ASD and other neurodevelopmental disorders associated with CNTNAP2 mutations.
Materials and Methods
Animals.
Experiments were conducted blind to genotype in adult (postnatal days 50–60) male and female Cntnap2+/+ and Cntnap2−/− littermates obtained from heterozygous crossings (C57BL/6J background), and genotype was confirmed post hoc by PCR using the following primers: wild-type BSA51, 5′-TGCTGCTGCCAGCCCAGGAACTGG-3′, and BSA52, 5′-TCAGAGTTGATACCCGAGCGCC-3′; knock-out BSA359, 5′-AGCAGCCGATTGTCTGTTGT-3′, and BSA360, 5′-CTCACCCAATCTCACAAACAAG-3′. Mice were kept in a 12 h light/dark cycle with ad libitum access to food and water. All animal procedures were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee and adhered to National Institutes of Health guidelines.
Hippocampal slice preparation.
Animals were deeply anesthetized with isoflurane and killed by decapitation. The brain was removed and quickly placed in ice-cold cutting solution (in mm: 215 sucrose, 2.5 KCl, 20 glucose, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, and 4 MgSO4, equilibrated with 95% O2/5% CO2). Four hundred-micrometer-thick transverse hippocampal slices were prepared with a VT1200S microtome (Leica) and immediately transferred to a chamber containing a mixture of cutting solution and regular artificial CSF (ACSF; 1:1) at room temperature for 30 min. This medium was gradually switched to 100% ACSF (in mm: 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose, equilibrated with 95% O2/5% CO2). Slices were allowed to recover at room temperature for at least 1.5 h before being transferred to a submerged recording chamber (TC-344B; Warner Instruments) and perfused with ACSF at 2 ml/min.
Electrophysiology.
Recordings were performed using a MultiClamp 700B amplifier (Molecular Devices). For extracellular field recordings, a patch-type micropipette was filled with 1 m NaCl. For voltage-clamp experiments, CA1 pyramidal cells were blind-patched with a micropipette (3–4 MΩ) filled with an intracellular recording solution containing the following (in mm): 123 cesium gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, and 10 glucose, pH 7.2 (280–290 mOsm). To assess cell stability, series and input resistances were monitored with a 5 mV, 80 ms hyperpolarizing test pulse, and cells with >15% change in series resistance were excluded from analysis. To elicit synaptic responses, paired, monopolar square-wave voltage 200-μs-wide pulses were delivered through a DS2A stimulus isolator (Digitimer) connected to a broken tip patch-type micropipette filled with ACSF. A stimulating pipette (Figs. 1, 2) was placed in the middle third of stratum radiatum (st. radiatum) in CA1, 200 μm away from the recording electrode. To evoke perisomatic inhibition, the stimulating pipette was placed in the middle of stratum pyramidale (st. pyramidale) 200 μm away from the recorded cell. To evoke dendritic inhibition, the stimulating pipette was placed in stratum radiatum (outer half), 300–400 μm away from the recorded cell.
Figure 1.

Normal excitatory/inhibitory balance and synaptic input-to-spike coupling in Cntnap2−/− mice. A, Representative traces (left; 20 averaged responses in each trace) of isolated inhibitory (IPSC; upward) and excitatory (downward; EPSC) synaptic currents recorded from CA1 pyramidal cells at 0 and −55 mV, respectively. Synaptic responses were elicited by electrical stimulation in st. radiatum and under conditions of intact excitation and inhibition. Summary plot (right) of the EPSC/IPSC ratio shows no significant difference between wild-type (+/+) and knock-out (−/−) littermates (wild-type: 0.29 ± 0.07; knock-out: 0.29 ± 0.04; p > 0.7). B, Input/output curve (right) showing no significant difference in the slope of the fEPSP and the subsequent population spike at a range of stimulation intensities between wild-type (+/+) and knock-out (−/−) littermates (p > 0.05 for all values). Representative traces (left; 10 averaged responses in each trace) of extracellular field recordings in st. pyramidale after electrical stimulation in st. radiatum are shown. Data are presented as mean ± SEM; numbers in parentheses represent animals and experiments (cell or slices), respectively.
Figure 2.

Excitation is mostly normal in the hippocampus of Cntnap2−/− mice. A, Input/output curve showing no difference in fEPSP slope versus fiber volley amplitude between wild-type (+/+) and knock-out (−/−) littermates. Representative traces (left) and a summary plot (right) are shown. B, Paired-pulse ratio of fEPSP responses elicited at various interstimulus intervals revealed no difference between genotypes. C, Synaptic responses evoked by a 25 stimuli, 14 Hz train. Normalized fEPSPs (left) and a summary plot (right) are shown. No difference was observed between wild-type (+/+) and knock-out (−/−) littermates (p > 0.05). D, mEPSC frequency and amplitude recorded from CA1 pyramidal cells (representative traces on right) were not different between wild-type (+/+) and knock-out (−/−) littermates (frequency: wild type, 0.91 ± 0.14 Hz; knock-out, 1.44 ± 0.31 Hz; p > 0.07; amplitude: wild type, 12.7 ± 0.37 pA; knock-out, 12.4 ± 0.19 pA; p > 0.4). E, The ratio between NMDAR-EPSCs (upward) and AMPAR-EPSCs (downward) showed a modest but significant decrease in the knock-out (−/−) compared with wild-type (+/+) littermates (*p < 0.05). F, High-frequency stimulation-induced LTP (100 pulses, 100 Hz, 20 s apart, in the presence of 50 μm picrotoxin, three times) was normal in knock-out (−/−) versus wild-type (+/+) littermates (p > 0.7 at 30–40 min after induction). Representative fEPSP traces (left) correspond to the numbers in the time course plot (right). Data are presented as mean ± SEM; numbers in parentheses represent animals and experiments (cells or slices), respectively.
Field recordings and the excitation/inhibition ratio were performed in the absence of any drugs, with the exception of long-term potentiation (LTP) experiments that were performed in the presence of the GABAA receptor (GABAAR) antagonist picrotoxin (50 μm). IPSCs were monitored in the continuous presence of the NMDA receptor (NMDAR) antagonist d-APV (25 μm) and AMPA receptor (AMPAR) antagonist 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX; 10 μm). AMPAR-mediated EPSCs were recorded in picrotoxin (50 μm) while voltage clamping at −60 mV, and NBQX was subsequently added to the bath to record NMDAR-EPSCs while holding the cell at +40 mV. Recordings of miniature EPSCs (mEPSCs; Vh = −60 mV) and miniature IPSCs (mIPSCs; Vh = 0 mV) were made in the presence of the sodium channel blocker tetrodotoxin (500 nm), at 32°C, in addition to the synaptic blockers of inhibition and excitation, respectively. With the exception of the above-specified experiments, all other recordings were performed at 25°C. LTP was induced with three trains of high-frequency stimulation (100 pulses at 100 Hz each) applied 20 s apart, and synaptic stimulation was performed at 0.05 Hz. d-APV and NBQX were obtained from the National Institute of Mental Health Chemical Synthesis and Drug Supply Program. All salts and other drugs were obtained from Sigma. Wild-type and knock-out groups were compared for statistical significance using the Mann–Whitney test (nonparametric data) or unpaired, two-tailed Student's t test (parametric data). Normality of datasets was determined using the D'Agostino–Pearson's omnibus K2 normality test. The two-sample Kolmogorov–Smirnov test was used to compare distributions.
Results
Cntnap2 deletion does not affect excitatory/inhibitory balance or pyramidal cell output
A change in the excitatory/inhibitory (E/I) balance is a likely mechanism underlying ASDs (Gogolla et al., 2009; Ramamoorthi and Lin, 2011; Coghlan et al., 2012; Nelson and Valakh, 2015). As an initial approach to evaluate synaptic function in Cntnap2−/− mice, we assessed E/I balance in acute hippocampal slices from these mice. Stimulation in st. radiatum generates a compound EPSC/IPSC in CA1 pyramidal cells (this mode of stimulation mainly recruits excitatory and inhibitory inputs onto the apical dendrites), and each component can be isolated by holding the recorded cell at the Cl− (−55 mV) and Na+ (0 mV) reversal potentials, respectively. The resulting ratio between the peak amplitude of both components was comparable between Cntnap2+/+ and Cntnap2−/− mice (Fig. 1A), suggesting no significant changes in E/I balance in these mice when measured through activation of putative dendritic inputs. We next assessed the excitatory synaptic input-to-spike (ES) coupling, i.e., the relationship between the synaptic response [measured as extracellular field EPSP (fEPSP)] generated after stimulation in st. radiatum, and its ability to drive pyramidal cells to fire (measured as the amplitude of the population spike amplitude). No significant differences were observed across genotypes (Fig. 1B), suggesting normal dendritic computation in the absence of CASPR2. The lack of change in these parameters raises the possibility that more subtle alterations in neural circuits may underlie the behavioral deficits observed in these mice. To explore this possibility, we next examined excitatory and inhibitory synaptic transmission in isolation.
Excitatory transmission is mostly normal in the CA1 area of Cntnap2−/− mice
Recent studies in cultured cortical neurons suggest that CASPR2 plays a role in glutamatergic transmission (Anderson et al., 2012; Varea et al., 2015). To test this possibility in Cntnap2−/− mice, fEPSPs in CA1 were elicited in st. radiatum by applying increasing intensities of electrical stimulation. To our surprise, analysis of the input/output function, measured as fEPSP slope versus fiber volley amplitude, revealed no significant difference between Cntnap2+/+ and Cntnap2−/− mice (Fig. 2A). We also estimated release probability by monitoring the paired-pulse ratio (PPR) at various interstimulus intervals (Fig. 2B) and synaptic depression in response to a stimulus train (25 stimuli, 14 Hz; Fig. 2C), but we found no significant differences between genotypes. To assess quantal excitatory transmission, we performed whole-cell recordings from CA1 pyramidal cells and monitored mEPSCs. Again, no differences in amplitude or frequency of spontaneous mEPSCs were found between Cntnap2+/+ and Cntnap2−/− littermates (Fig. 2D). Together, our results show that deletion of Cntnap2 does not significantly affect basal excitatory synaptic transmission.
A disrupted maturation of excitatory synapses is a common mechanism in neurodevelopmental disorders (Penzes et al., 2013), and such disruption is commonly assessed by monitoring the relative contribution of the AMPAR- and NMDAR-mediated components of synaptic transmission. To examine whether a similar mechanism could be linked to CASPR2 deletion, we monitored the NMDA/AMPA ratio in Cntnap2−/− mice and found a modest decrease in these mice compared with wild-type littermates (Fig. 2E). Changes in NMDAR subunit composition, manifested as changes in NMDAR-EPSC decay, could be altered in Cntnap2−/− mice. However, we found that the NMDAR-EPSC decay was identical in wild-type versus knock-out mice (p > 0.5). Furthermore, a shift in the NMDA/AMPA ratio could translate into deficits on NMDAR-mediated LTP. To explore that possibility, we performed field recordings in st. radiatum in CA1 but observed no significant differences between genotypes (Fig. 2F). Thus, beyond a modest change in the NMDA/AMPA ratio, CASPR2 deletion does not seem to significantly disrupt maturation or plasticity at excitatory synapses.
Inhibition is altered in an input-specific manner in the hippocampus of Cntnap2−/− mice
The observation that Cntnap2−/− mice display hyperactivity, seizures, and a reduced number of interneurons (Peñagarikano et al., 2011) suggests that synaptic inhibition could be altered in the absence of CASPR2. To directly test this possibility, we analyzed the input/output function of evoked IPSCs by activating putative perisomatic or dendritic inputs onto CA1 pyramidal cells (see Materials and Methods). “Perisomatic” inputs likely include an axon initial segment, soma, and proximal dendritic inputs (Freund and Katona, 2007). Consistent with this interpretation, the rise time of putative perisomatic IPSCs was significantly faster than that of dendritic IPSCs (perisomatic: 1.96 ± 0.21 ms, n = 23 cells; dendritic: 7.06 ± 0.26 ms, n = 15; p < 0.0001, Mann–Whitney test). We found that perisomatic but not dendritic IPSCs showed a significant rightward shift in the input/output curve of Cntnap2−/− mice (Fig. 3A,B), indicating a selective impairment of a subpopulation of inhibitory inputs. In contrast, the decay kinetics of evoked IPSCs (putative perisomatic inputs), a parameter that can be affected by the GABAAR subunit composition, was unchanged between Cntnap2+/+ and Cntnap2−/− littermates (wild type: 27.2 ± 2.1 ms, n = 23; knock-out: 26.2 ± 1.6 ms, n = 22; p > 0.5). To determine whether the deficit in perisomatic inhibition could be attributable to a reduction in transmitter release, we assessed presynaptic function by delivering paired stimulation in st. pyramidale at various interstimulus intervals and by applying a train of 25 stimuli at 14 Hz. Analysis of PPR and burst stimulation revealed a similar profile in both Cntnap2+/+ and Cntnap2−/− littermates (Fig. 3C,D). Thus, it is unlikely that a reduction in transmitter release can account for the selective deficit in perisomatic inhibition observed in Cntnap2−/− mice.
Figure 3.

Inhibition is altered in CA1 area of Cntnap2−/− mice. A, B, Input/output curve (right) and representative traces (left; 20 averaged responses in each trace) of perisomatic (A) and dendritic (B) IPSCs evoked in CA1 pyramidal cells by electrical stimulation in st. pyramidale or st. radiatum, respectively. The amplitude of perisomatic IPSCs was significantly reduced (*p < 0.05; **p < 0.01; ***p < 0.001) in the knock-out (−/−) compared with wild-type (+/+) littermates. C, Paired-pulse ratio of perisomatic IPSCs at various interstimulus intervals (left, representative traces; right, summary plot) showed no differences between genotypes. D, Perisomatic IPSCs evoked by a 25 stimuli, 14 Hz train (left, representative traces; right, summary plot) also showed no difference between genotypes.
Given the differential impact of Cntnap2 deletion on putative perisomatic versus dendritic inhibition, we sought to determine cell-wide inhibitory transmission by recording spontaneous inhibitory events from CA1 pyramidal cells. Intriguingly, we observed a significant increase in the frequency of spontaneous, action potential-driven IPSCs (sIPSCs) in Cntnap2−/− mice, whereas sIPSC amplitude and rise-time kinetics remained unchanged (Fig. 4A). No significant difference in CA1 pyramidal cell input resistance was observed between Cntnap2+/+ and Cntnap2−/− littermates (155.4 ± 8.7 and 169.1 ± 6.7 MΩ, respectively; n = 27 cells each group; p = 0.21). We next monitored mIPSCs but found no significant difference in either mIPSC frequency or amplitude between genotypes (Fig. 4B). However, the rise time of these events was significantly slower in Cntnap2−/− mice (p < 0.0001, Kolmogorov–Smirnov test; Fig. 4B), suggesting a reduction in the relative contribution of somatic versus dendritic inputs.
Figure 4.
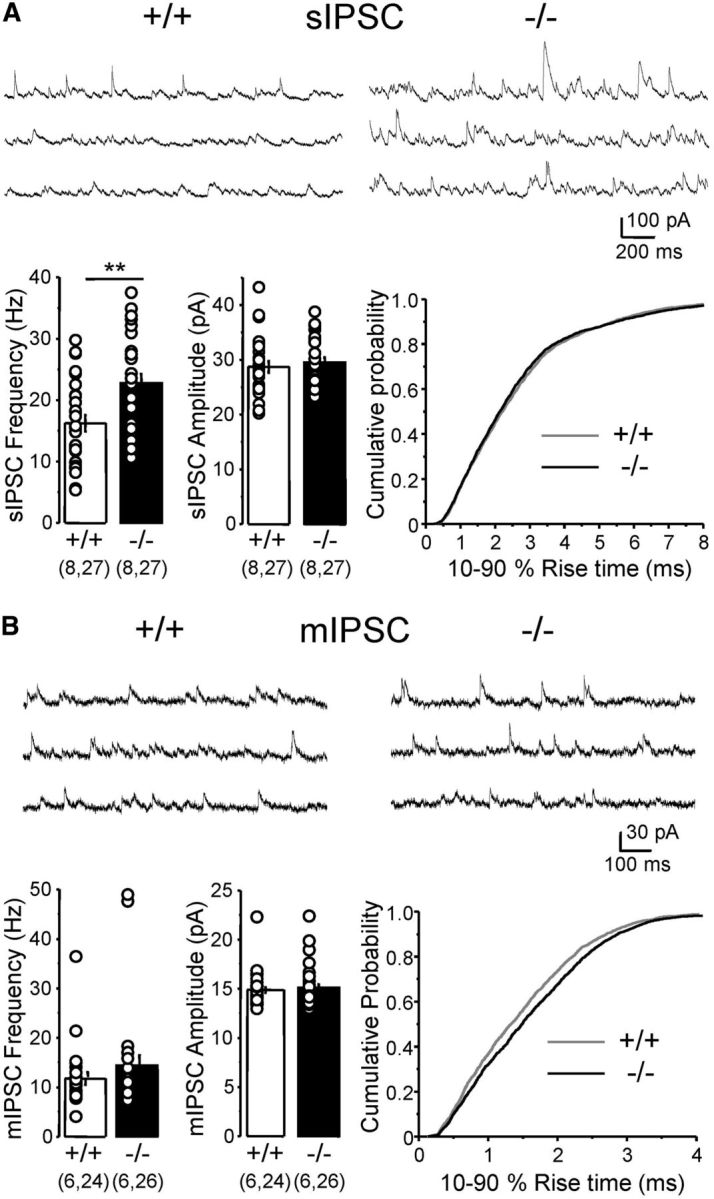
Altered spontaneous inhibitory activity in Cntnap2−/− mice. A, Spontaneous, action potential-driven IPSCs recorded from CA1 pyramidal cells (sIPSC; representative traces on top). sIPSC frequency, but not amplitude, was significantly increased in knock-out (−/−) versus wild-type (+/+) littermates (frequency: wild type, 16.2 ± 1.3 Hz; knock-out, 22.7 ± 1.5 Hz; **p < 0.01; amplitude: wild type, 28.7 ± 1.1 pA; knock-out, 29.5 ± 1.0 pA; p > 0.5). A cumulative plot of sIPSC rise time (bottom right) is shown. B, mIPSC frequency and amplitude recorded from CA1 pyramidal cells (representative traces on top) were not significantly different between wild-type (+/+) and knock-out (−/−) littermates (frequency: wild type, 11.8 ± 1.3 Hz; knock-out, 14.4 ± 2.1 Hz; p > 0.29; amplitude: wild type, 14.9 ± 0.3 pA; knock-out, 15.1 ± 0.4 pA; p > 0.3). The rise time of mIPSCs was slower in Cntnap2−/− mice (bottom right; wild type: 1.48 ± 0.02 ms, n = 2125 events; knock-out: 1.63 ± 0.02 ms, n = 2732 events). Data are presented as mean ± SEM; numbers in parentheses represent animals and cells, respectively.
Discussion
The main finding of this study is a selective impairment of inhibition in the CA1 area of Cntnap2−/− mice. Although excitatory transmission was mostly normal, the inhibitory deficit was restricted to perisomatic inputs. In addition, we found a significant increase in spontaneous, action potential-driven inhibitory synaptic events (sIPSCs). To our knowledge, these findings provide the first evidence for functional synaptic impairments in the Cntnap2−/− mouse and suggest that subtle, yet significant, changes in inhibition may underlie behavioral deficits in CNTNAP2-related ASDs.
Evidence from both patient studies and animal models strongly suggests that dysfunctional GABAergic signaling plays a major role in ASDs (Pizzarelli and Cherubini, 2011; Ramamoorthi and Lin, 2011; Coghlan et al., 2012). Both failure in interneuron migration and impaired functional inhibition might contribute to the behavioral phenotypes of ASD. An E/I imbalance with loss of GABAergic function is a common feature of neurodevelopmental disorders, including ASD (Gogolla et al., 2009; Ramamoorthi and Lin, 2011; Coghlan et al., 2012; Nelson and Valakh, 2015). Hippocampal interneurons can be segregated into two major classes, i.e., perisomatic or dendritic, according to the pyramidal cell compartment that they innervate (Miles et al., 1996; Freund and Katona, 2007). Dendritic inhibition is thought to control CA1 pyramidal cell firing by shunting incoming synaptic excitation, whereas perisomatic inhibition controls the local generation of action potentials (Miles et al., 1996). We found that putative perisomatic but not dendritic evoked IPSCs were reduced in Cntnap2−/− mice. It is unlikely that a reduction in GABA release could account for this phenotype given that short-term plasticity remained unchanged in these mice. Because the perisomatic compartment of pyramidal cells is highly innervated by PV+ interneurons (Hu et al., 2014), our findings could be explained by a reduced number PV+ interneurons in the hippocampus of the Cntnap2−/− mice, as reported previously (Peñagarikano et al., 2011). Interestingly, PV+ interneurons critically control neuronal oscillations (Sohal et al., 2009), an impairment of which has been associated with ASD (Uhlhaas and Singer, 2012). In particular, a selective impairment of PV+ basket cell-mediated inhibition in CA1 was reported in the ASD-associated point mutation in neuroligin 3, R451C knock-in mice (Földy et al., 2013), and deletion of the cell-adhesion molecule neuroligin 2 is associated with a loss of postsynaptic specializations specifically at perisomatic inhibitory synapses (Poulopoulos et al., 2009). Furthermore, a reduction in molecular markers for inhibitory synapses in the perisomatic compartment has recently been observed in mouse models of ASD (Gogolla et al., 2014). Thus, our findings provide additional evidence that impaired perisomatic inhibition could be a common alteration in ASDs.
We also found an unexpected increase in the frequency of spontaneous, action potential-driven inhibitory events impinging onto CA1 pyramidal cells (Fig. 4A). This synaptic phenotype could be attributable to an increase in interneuron excitability, either as an indirect compensatory effect or as a direct consequence of CASPR2 deletion. Whether the increase in sIPCS frequency arises from a specific interneuron type remains to be determined. Of note, a recent study showed that CASPR2 autoantibodies associated with limbic encephalitis in humans mainly target axons of inhibitory hippocampal neurons in culture, in particular GABAergic axons surrounding cell bodies (Pinatel et al., 2015). These observations raise the possibility that interneuron axonal function (e.g., excitability) could be altered in Cntnap2−/− mice. Although CASPR2 contributes to clustering voltage-gated potassium channels in the peripheral nervous system (Poliak et al., 2003), evidence for a similar mechanism in the CNS is lacking, and the direct effect of Cntnap2 deletion on interneuron excitability remains to be tested.
Recent studies in cultured cortical neurons have shown a global reduction of synaptic function in RNAi-mediated Cntnap2 knock-down neurons (Anderson et al., 2012) and reduced dendritic spine density and GluA1 localization in the dendritic spines of Cntnap2 knock-out neurons (Varea et al., 2015). These observations suggest that CASPR2 could play a role in glutamatergic transmission. However, our results using Cntnap2−/− mice revealed no major phenotype at excitatory synapses. Although we found a modest decrease in the NMDA/AMPA ratio, the mechanism and functional relevance of this change are unclear. Cntnap2−/− mice also showed normal ES coupling and E/I ratio, which is consistent with normal input/output function at Schaffer collaterals-to-CA1 synapses (Fig. 2A) and normal dendritic inhibition (Fig. 3B). Given the neurodevelopmental nature of ASD, and that most human ASD-related mutations in CNTNAP2 result in nonfunctional proteins that are not tethered to the cellular membrane (Falivelli et al., 2012), we argue that the Cntnap2−/− mouse phenotype likely provides more relevant insights on the mechanisms underlying ASD.
How exactly Cntnap2 deletion leads to altered inhibition is unknown. Defects in neural migration found in Cntnap2−/− mice (Peñagarikano et al., 2011) could account for the reduced number of interneurons. In addition, the deficit in dendritic arborization and spine development observed in CASPR2-deficient neurons suggests that this protein may play an organizational role in developing neurons (Anderson et al., 2012; Varea et al., 2015). Finally, CASPR2 may participate in a neuroglial junction multimolecular complex (Poliak et al., 2001) and by this means contribute to optimal neuroglial interaction, which is required for normal neuronal migration.
In conclusion, we report a selective inhibitory phenotype in the hippocampus of Cntnap2−/− mice indicating that CNTNAP2 may be required for normal inhibitory interneuron development and function. Our findings also highlight the impairment of perisomatic inhibition as a potential common mechanism underlying ASDs.
Footnotes
This work was supported by National Institutes of Health Grants R01-MH081935 and R01-DA17392 and a pilot grant from the Rose F. Kennedy Intellectual and Developmental Disabilities Research Center to P.E.C. We thank Dr. Brett Abrahams (Albert Einstein College of Medicine, Bronx, NY) for the Cntnap2+/− mice. We thank all members of the Castillo laboratory for help with experiments and valuable discussions and Hannah Monday for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Anderson et al., 2012.Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Südhof TC. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci U S A. 2012;109:18120–18125. doi: 10.1073/pnas.1216398109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkaloglu et al., 2008.Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, Tanriover G, Abrahams BS, Duvall JA, Robbins EM, Geschwind DH, Biederer T, Gunel M, Lifton RP, State MW. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur et al., 2009.Betancur C, Sakurai T, Buxbaum JD. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009;32:402–412. doi: 10.1016/j.tins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan et al., 2012.Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev. 2012;36:2044–2055. doi: 10.1016/j.neubiorev.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme et al., 2013.Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med. 2013;19:685–694. doi: 10.1038/nm.3193. [DOI] [PubMed] [Google Scholar]
- Falivelli et al., 2012.Falivelli G, De Jaco A, Favaloro FL, Kim H, Wilson J, Dubi N, Ellisman MH, Abrahams BS, Taylor P, Comoletti D. Inherited genetic variants in autism-related CNTNAP2 show perturbed trafficking and ATF6 activation. Hum Mol Genet. 2012;21:4761–4773. doi: 10.1093/hmg/dds320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy et al., 2013.Földy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund and Katona, 2007.Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Gogolla et al., 2009.Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord. 2009;1:172–181. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla et al., 2014.Gogolla N, Takesian AE, Feng G, Fagiolini M, Hensch TK. Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron. 2014;83:894–905. doi: 10.1016/j.neuron.2014.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu et al., 2014.Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345:1255263. doi: 10.1126/science.1255263. [DOI] [PubMed] [Google Scholar]
- Miles et al., 1996.Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/S0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Nelson and Valakh, 2015.Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87:684–698. doi: 10.1016/j.neuron.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano and Geschwind, 2012.Peñagarikano O, Geschwind DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med. 2012;18:156–163. doi: 10.1016/j.molmed.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano et al., 2011.Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes et al., 2013.Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem. 2013;126:165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinatel et al., 2015.Pinatel D, Hivert B, Boucraut J, Saint-Martin M, Rogemond V, Zoupi L, Karagogeos D, Honnorat J, Faivre-Sarrailh C. Inhibitory axons are targeted in hippocampal cell culture by anti-Caspr2 autoantibodies associated with limbic encephalitis. Front Cell Neurosci. 2015;9:265. doi: 10.3389/fncel.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzarelli and Cherubini, 2011.Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011;2011:297153. doi: 10.1155/2011/297153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak et al., 2001.Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, Peles E. Localization of Caspr2 in myelinated nerves depends on axon–glia interactions and the generation of barriers along the axon. J Neurosci. 2001;21:7568–7575. doi: 10.1523/JNEUROSCI.21-19-07568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak et al., 2003.Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos et al., 2009.Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Ramamoorthi and Lin, 2011.Ramamoorthi K, Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol Med. 2011;17:452–462. doi: 10.1016/j.molmed.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas-Cuadrado et al., 2014.Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. 2014;22:171–178. doi: 10.1038/ejhg.2013.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal et al., 2009.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas and Singer, 2012.Uhlhaas PJ, Singer W. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron. 2012;75:963–980. doi: 10.1016/j.neuron.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Varea et al., 2015.Varea O, Martin-de-Saavedra MD, Kopeikina KJ, Schürmann B, Fleming HJ, Fawcett-Patel JM, Bach A, Jang S, Peles E, Kim E, Penzes P. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc Natl Acad Sci U S A. 2015;112:6176–6181. doi: 10.1073/pnas.1423205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi and Bear, 2012.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4:a009886. doi: 10.1101/cshperspect.a009886. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]