

Abstract
Vatalanib is an oral vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitor and everolimus inhibits mammalian target of rapamycin (mTOR). A phase Ib study of vatalanib and everolimus was performed in patients with advanced solid tumors to determine the maximum tolerated dose (MTD) of the combination. Although treatment at the full therapeutic dose for both agents was not feasible, evidence of efficacy and long-term tolerability was demonstrated in some patients. This suggests that with dose adjustments, combination therapy with certain VEGFR and mTOR inhibitors may be possible and efficacious, particularly in renal cell carcinoma (RCC).
Background
Vatalanib is an oral vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitor (TKI), whereas everolimus inhibits mammalian target of rapamycin (mTOR). Combination therapy with VEGFR and mTOR inhibitors has not been well tolerated to date but may have efficacy in renal cell carcinoma (RCC).
Patients and Methods
A phase Ib study of vatalanib and everolimus was performed in patients with advanced solid tumors to determine the maximum tolerated dose (MTD), safety, and tolerability of the combination. A dose-expansion cohort of 20 patients with metastatic RCC was studied to further define toxicity and preliminary efficacy in patients with RCC.
Results
We evaluated 32 patients over 3 dose levels and a dose-expansion cohort. The most common toxicities of any grade were proteinuria, fatigue, hypertriglyceridemia, nausea, and vomiting. Dose-limiting toxicities (DLTs) included severe hypertension, diarrhea, neutropenia, mucositis, and fatigue. The MTD for the combination was vatalanib 1000 mg daily and everolimus 5 mg daily. In all patients, median overall survival (OS) was 16.3 months. In patients with RCC, median progression-free survival (PFS) was 5.8 months, and OS was 16.5 months. OS was significantly better in treatment-naive patients (25.1 months) compared with patients who had received previous vascular endothelial growth factor (VEGF)-targeted therapy (6.3 months). Seven of 24 (29.2%) evaluable patients demonstrated a partial response, and an additional 15 patients exhibited stable disease. Long-term tolerability (> 1 year) was demonstrated in 19% of patients.
Conclusion
Relevant doses of vatalanib and everolimus were achieved in combination, with expected toxicities. A substantial number of patients with RCC achieved an objective response in the treatment-naive setting, with prolonged tolerability and survival. Further comparative phase II/III studies of specifically targeted VEGF and mTOR inhibitor combinations may be warranted in patients with RCC.
Keywords: Clinical trial, mTOR inhibitor, Renal cell carcinoma, VEGFR inhibitor
Introduction
Targeted therapies that inhibit the vascular endothelial growth factor (VEGF) pathway have demonstrated clinical benefit in patients with many solid tumor types, including colorectal,1 breast,2-4 lung,5,6 hepatocellular,7 thyroid,8 and renal cancers,9-11 as well as brain tumors.12 Inhibition of the VEGF pathway is thought to elicit antitumor effects primarily through disruption of tumor blood flow, altering existing vascular permeability to enhance chemotherapy delivery, blocking new vessel formation, or angiogenesis.13,14 Approved VEGF-targeted strategies include bevacizumab, a neutralizing antibody to all isoforms of VEGF-A15; ziv-aflibercept, a recombinant VEGFR fusion protein16; as well as multitargeted small molecules sunitinib, sorafenib, pazopanib, axitinib, and regorafenib, which function in part as tyrosine kinase receptor inhibitors (TKIs) of VEGF receptors (VEGFRs).9,11,17-19
Vatalanib (PTK787/ZK 222584) is an orally active small-molecule TKI VEGFR that has demonstrated both preclinical and clinical efficacy in solid tumors.20-22 In phase I/II testing, vatalanib has been relatively well tolerated as a single agent, with expected class effect toxicities including hypertension and proteinuria, as well as gastrointestinal toxicities (nausea, diarrhea) and fatigue. Combination studies with vatalanib have been limited. A phase I study with bevacizumab showed significant proteinuria and hypertension, and further development was not recommended.23 A phase I study in combination with imatinib showed good tolerability with some signals of efficacy.24 Vatalanib has demonstrated radiographic evidence of a decrease in tumor blood flow at a daily dose of 1000 mg or more in several clinical trials, whereas tolerable dosing has been said to be as high as 2000 mg daily.21,25 In colorectal cancer, however, vatalanib dosed at 1250 mg daily was studied with oxaliplatin-based chemotherapy in both first-line and advanced settings and provided no benefit over placebo.26,27
mTOR is an intracellular serine/threonine kinase that is activated by several signal transduction pathways, including PI3 kinase/AKT and Ras/Raf/ERK, as well as metabolic and nutrient stress signals.28 In cancers, dysregulation of mTOR signaling results in cellular proliferation, survival, and angiogenesis.29 Consequently, inhibition of mTOR has antitumor effects by inhibiting several of these downstream processes. Of special note, mTOR inhibition is thought to inhibit the activity of hypoxia-inducible factor (HIF).30 Temsirolimus was the first inhibitor of mTOR approved for the treatment of patients with advanced RCC, based on a phase III study in patients with untreated poor-risk RCC, and demonstrated a significant overall survival (OS) advantage over interferon alfa.31 Everolimus is an orally absorbed macrolide that, like all rapalogs, functions to bind intracellular mTOR and its chaperone FKBP12 in an inactive state, inhibiting activation in a mechanism of action similar to that of rapamycin.32 In a phase III registration study, everolimus demonstrated significant improvement in progression-free survival (PFS) over placebo in patients with metastatic RCC who were previously treated with a TKI VEGFR; it was well tolerated in treated patients.33 Everolimus also has activity in some cases of breast cancer,34 pancreatic neuroendocrine tumors,35 and tuberous sclerosis–associated brain tumors.36 Common side effects include rash, fatigue, stomatitis, anemia, leukopenia, thrombocytopenia, and hyperglycemia. Uncommon side effects include infection, diarrhea, nausea, vomiting, hair and nail changes, and pneumonitis. Severe adverse events (AEs) are rare, and only anemia, lymphopenia, and hyperglycemia occurred as grade 3/4 toxicities with > 5% frequency. Based on this safety profile, treatment with everolimus in combination with other targeted agents is under evaluation in multiple settings.
We investigated the safety and tolerability of combining vatalanib with everolimus for several reasons. Both VEGFR inhibition and mTOR inhibition demonstrate preclinical antiangiogenic properties that may be additive or synergistic. 37,38 In addition, activation of VEGFR may result in signal transduction through the PI3 kinase/AKT/mTOR pathway; therefore, inhibition of both VEGFR and mTOR could result in a sequential or vertical inhibition of this pathway.39 For example, in a gastric cancer model, the combination of vatalanib and everolimus had superior efficacy than either agent alone.40 Third, treatment with a TKI VEGFR has been shown to induce increased plasma concentrations of VEGF that may be mediated by HIF. 41-45 Given that inhibitory strategies of VEGFR and mTOR have demonstrated clinical benefit in patients with RCC, we included an expanded cohort of patients with RCC to further define the safety and tolerability in this select patient population and to evaluate preliminary efficacy.
With respect to dosing, in phase I studies vatalanib has been dosed either daily or twice daily; however, phase II and III studies focused on once daily dosing. Therefore, at the time of the study design, we chose once daily dosing in combination with everolimus, with the rationale that the prolonged half-life of everolimus would offer some antiproliferative activity even if vatalanib exposures were intermittent, given its short half-life of 3 to 6 hours.22
Patients and Methods
Patients
Patients eligible for this study included adults (≥ 18 years) with histologically confirmed nonhematologic malignancy and radiographically measurable metastatic disease for which there is no standard therapy. For patients with prostate cancer, biochemical evidence of disease (ie, elevated prostate-specific antigen [PSA]) in the setting of nonmeasurable bone metastases was allowed. Patients were required to have good performance status (Karnofsky performance status ≥ 70) and acceptable bone marrow, kidney, and liver function (absolute neutrophil count ≥ 1500/mm3, platelet count ≥ 100,000/mm3, hemoglobin value ≥ 9 g/dL, creatinine clearance > 40 mL/min, bilirubin level ≤ 1.5 times the upper limit of normal, and aspartate aminotransferase and alanine aminotransferase levels ≤ 2.5 times the upper limit of normal). Total cholesterol was required to be < 300 mg/dL and triglyceride levels < 350 mg/dL. Previous chemotherapy (> 4 weeks; > 6 weeks for mitomycin C, nitrosureas, or antibody therapy), radiotherapy, biological/immunotherapy or surgery (> 4 weeks) were permitted provided that there was resolution of all toxicities of Common Terminology Criteria for Adverse Events grade ≤ 1.
The dose-expansion portion of the study was limited to patients with unresectable or metastatic renal cell carcinoma (RCC) and 1 or more sites of measurable disease by Response Evaluation in Solid Tumors (RECIST) criteria. Exclusion criteria included concurrent malignancies other than superficial nonmelanomatous skin cancer, superficial bladder cancer, or nonmetastatic prostate cancer. After the US Food and Drug Administration's approval of sorafenib and sunitinib for the treatment of advanced RCC in January 2006, the study was amended to allow previous treatment with these agents as well as bevacizumab.
Additional exclusion criteria included active central nervous system disease, liver disease, pleural effusion, ascites, uncontrolled hypertension, history of labile hypertension, or history of poor compliance with an antihypertensive regimen. Patients with uncontrolled cardiac disease (angina, congestive heart failure, arrhythmia) or myocardial infarction (≤ 6 months); uncontrolled diabetes, infections, or hyperlipidemia; interstitial pneumonia or extensive symptomatic interstitial lung fibrosis; chronic renal disease; or gastrointestinal disease that could affect absorption of the study drug were also excluded. In addition, participants were excluded if they had received other investigational drugs ≤ 4 weeks before starting study treatment. Use of warfarin or oral anticoagulants metabolized by the cytochrome P450 system was not permitted, whereas heparin and low-molecular-weight heparin were allowed. Patients with human immunodeficiency virus infection and other severe acute or chronic medical or psychiatric conditions were also excluded. Participants agreed to use effective birth control during and for at least 6 months after completion of the study treatment.
All patients provided written informed consent. The study was approved by the Institutional Review Board of the Duke University Medical Center and was conducted in accordance with the Declaration of Helsinki.
Study Design
The study was a single-arm, open-label phase Ib trial conducted at the Duke University Medical Center. The dose escalation portion assessed the combination of vatalanib and everolimus (both supplied by Novartis Pharmaceuticals, East Hanover, NJ) for potential drug pharmacokinetic interactions, safety and tolerability, and determination of the maximum tolerated dose (MTD) and appropriate doses for additional combination studies. Each drug was given on a daily oral schedule, with escalation of vatalanib in 250-mg increments from 1000 to 1500 mg (3 patients per cohort) and everolimus at 5 mg or 10 mg. Vatalanib was begun on day 1 of the 28-day cycle, whereas everolimus was begun on day 15 to allow assessment of vatalanib pharmacokinetics at steady state, both alone and in combination with everolimus. In subsequent cycles, patients received both drugs from day 1 until discontinuation for disease progression, AEs, or the investigator's discretion. Patients were monitored for AEs every 2 weeks using the National Cancer Institute Common Toxicity Criteria, version 3.0. Dose-limiting toxicity (DLT) was defined as any nonhematologic grade ≥ 3 event (except hematuria and acute renal failure that was defined as grade ≥ 2), grade 3 neutropenia or thrombocytopenia lasting ≥ 7 days, or any grade 4 hematologic toxicity occurring within the first 42 days of treatment (28 days of combined treatment). In the absence of DLT in ≥ 2 of 3 patients, the cohort was expanded to 6 patients, and in the absence of DLT in ≥ 2 of 6 patients, the dose was escalated to the next level.
In the expanded cohort of patients with metastatic renal cell cancer, the safety and tolerability of the combination at the recommended phase II dose was assessed, and a preliminary assessment of response to this regimen was investigated using RECIST, version 1.0.46
Pharmacokinetic Assessments
Pharmacokinetic (PK) sampling for vatalanib was performed on days 14 (before initiating everolimus) and 29 (± 3 days) in cycle 1 with collections before dosing and 1, 2, 3, 4, 6, 8, and 24 hours after dosing, using a peripheral intravenous line and heparin lock system. Everolimus PK sampling occurred only on day 29 at the same time points just noted. Everolimus PK analyses were performed in ethylenediaminetetraacetic acid plasma; those for vatalanib were performed in heparinized plasma, and the plasma samples were stored frozen until analysis. The time to peak concentration and value of the maximum plasma concentration were determined by visual inspection of the plasma concentration vs. time data for each analyte of interest. The area under the plasma concentration (AUC) vs. time curve was determined for each dosing interval of 24 hours using the linear trapezoidal rule. The steady-state plasma concentration was determined by dividing the AUC by the dosing interval (24 hours). The terminal phase rate constant, and half-life was obtained by applying linear regression to the natural log-transformed concentration vs. time data in the terminal phase. The oral clearance was calculated by dividing the daily dose by the AUC over the 24-hour dosing interval. The area volume of distribution after oral dosing was calculated as the quotient CL/λz.
Peripheral Blood Mononuclear Cell Isolation and S6RP Analysis
Blood was drawn into ethylenediaminetetraacetic acid in blood collection tubes from patients at baseline, after 1 month of treatment (day 29), and at the end of study (EOS). Peripheral blood mononuclear cells (PBMCs) were isolated within 1 hour of collection by density gradient centrifugation using Ficoll-Hypaque (GE Healthcare, Uppsala, Sweden). After centrifugation, PBMCs were collected, washed twice with phosphate-buffered saline, flash frozen, and kept at − 80°C until analyzed.
PBMCs were resuspended in lysis buffer and homogenized using the FastPrep system (Qbiogene, Inc, Carlsbad, CA) at 4°C. Cell lysates (50 μg) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis on 4% to 20% gradient gels (Novex, Inc, Wadsworth, OH), transferred to nitrocellulose membranes, and probed using specific primary antibodies against phospho S6 ribosomal protein (Ser235/236) (Cell Signaling Technology, Beverly, MA) and S6 ribosomal protein (54D2) (Cell Signaling Technology). β-actin was also quantified as a normalization control. Visualization was performed using the appropriate fluorescent-labeled secondary antibody (Alexa Fluor 680 [molecular probes, Life Technologies, Carlsbad, CA] and IRDye800 [Rockland, Inc, Philadelphia, PA]) and analyzed using the Odyssey Imaging system from Li-Cor Biosciences (Lincoln, Nebraska). All assays were performed in duplicate, and data reflect the average of 2 separate experiments.
Tumor Response Evaluation
Tumor volume was measured at baseline (≤ 3 weeks before the start of the study) by computed tomography or magnetic resonance imaging, and responses were assessed after every 2 cycles (8 weeks) using RECIST, version 1.0.46
Statistical Analyses
Sample size for the dose-escalation portion of the study was determined by toxicity using a 3 + 3 design as described earlier. An arbitrarily determined dose expansion of 20 patients with metastatic RCC was performed to further describe adverse event rates at the MTD and to preliminarily explore efficacy end points. The primary end point of this study was the determination of a recommended phase II dose based on an assessment of safety and tolerability over multiple dose levels. Secondary efficacy end points of the dose-expansion phase of this study were overall survival (OS), PFS, and the percentage of patients with a partial response (PR), stable disease, or progressive disease. Patients were grouped according to previous VEGF inhibitor treatment for analysis. OS and PFS in the overall population were calculated using the Kaplan-Meier method. OS was defined as the time from the date of study treatment to date of death from any cause, and patients alive as of the last follow-up had OS censored at the last follow-up date. PFS was defined as the time from the date of study treatment initiation to the date of death from any cause or date of first documented progression. Patients alive whose disease had not progressed as of the last follow-up had PFS censored at the last follow-up date. OS and PFS estimates across subgroups were analyzed in an exploratory fashion using Kaplan-Meier estimates and compared using the log-rank test.
Results
Patient Demographics
From February 2005 through June 2007, 32 patients were enrolled in this phase Ib study; 14 patients were enrolled in the dose-escalation stage and 18 patients with metastatic RCC were enrolled in the dose-expansion stage. In addition, there were 2 patients with metastatic RCC treated at the MTD in the dose-escalation stage included in the dose-expansion analysis. At the date of analysis, disease in all patients had progressed, and 30 of 32 total patients had died. Demographics of the patient population are shown in Table 1. Briefly, the dose escalation portion included patients with anal cancer (n = 1), colon cancer (n = 1), lung cancer (n = 1), pancreatic cancer (n = 2), prostate cancer (n = 4), and RCC (n = 5). All patients had metastatic disease, and US Food and Drug Administration–approved therapeutic options had failed in all. For the dose-expansion stage, enrollment was limited to those with metastatic RCC, including patients who had received previous treatment with a VEGF-targeted therapy. No patients had received previous mTOR inhibition. Combining the dose-escalation and dose-expansion patients, a total of 20 patients with RCC were treated at the recommended dose of vatalanib 1000 mg daily and everolimus 5 mg daily. Ten of 20 (50%) patients had received previous treatment with at least 1 VEGF-targeted therapy.
Table 1. Summary of Patient Demographics and Cancer Characteristics From Those Treated With Vatalanib and Everolimus.
| Demographics | |
|---|---|
| Total No. Patients | 32 |
| Age, years, median (range) | 59 (38-74) |
| Sex M/F | 19/13 |
| Race, White/Other | 29/3 |
| Diagnosis | Total (%) |
| Prostate | 4 (12.5) |
| Lung | 1 (3.1) |
| Pancreas | 2 (6.2) |
| Colon | 1 (3.1) |
| Anal | 1 (3.1) |
| Renal | 23 (72.0) |
| Renal Cell Carcinoma Patient Demographics (n = 23) | |
| Histologic Type | Total (%) |
| Clear cell | 18 (78) |
| Other | 5 (22) |
| Motzer Risk Criteria | |
| Low risk | 1 (4.3) |
| Intermediate risk | 20 (87.0) |
| Poor risk | 2 (8.7) |
| Previous Therapy | |
| Cytokine treatment only | 1 (4.3) |
| VEGF inhibition only | 4 (17.4) |
| Cytokine treatment and VEGF inhibition | 6 (26.1) |
| No previous systemic therapy | 12 (52.2%) |
Abbreviation: VEGF = vascular endothelial growth factor.
Dose-Limiting Toxicity and Adverse Events
Patients were treated over 3 dose levels in the dose-escalation stage of the study, and AEs were seen across the dose levels. The first dose level contained vatalanib dosed at 1000 mg daily and everolimus dosed at 5 mg daily. The second patient treated at this dose level experienced a DLT (grade 4 hypertension), and the cohort was expanded to 6 patients, with no further DLTs. Dose escalation was carried out in the subsequent cohort of 3 patients: vatalanib 1250 mg daily and everolimus 5 mg daily. DLTs were seen in 1 of the first 3 patients (grade 3 neutropenia) and 2 of 6 patients treated at this dose level overall (including grade 3 mucositis, fatigue); therefore, the study was amended to include an intermediate dose level (level 2a) of vatalanib 1000 mg and everolimus 10 mg. Two of 2 patients treated at this dose level demonstrated DLTs (grade 3 fatigue/weakness, grade 3 diarrhea/dehydration), so vatalanib 1000 mg daily and everolimus 5 mg daily was established as the MTD for the dose expansion.
An expanded cohort of vatalanib dosed at 1000 mg daily and everolimus dosed at 5 mg daily was explored in 18 patients to better define the toxicity and safety profile of this combination and to explore preliminary efficacy end points. Two additional patients with metastatic RCC were treated at the MTD in the dose-escalation stage and were included in the dose-expansion analysis. Overall, the regimen was manageable, although dose reductions were necessary in 4 patients, and 8 patients required discontinuation for AEs. The AE profile was predictable based on the side effect profiles of the 2 agents. Cumulative (any cycle) grade 3 and 4 AEs seen in the study population and their frequency are listed in Table 2. Fatigue was the most common serious toxicity (22.6%) and was managed with dose reduction of both drugs, as were diarrhea (9.7%), thrombocytopenia (6.5%), and mucositis (3.2%). Hypertriglyceridemia (9.7%), hypercholesterolemia (6.5%), and rash (6.5%) were managed with dose reduction of everolimus only, whereas hypertension (3.2%) and neutropenia (3.2%) were managed with reduction of the vatalanib dose alone. Late-onset toxicities (> 1 year of treatment) included grade 2 hypertension (n = 2), diarrhea (n = 1), nausea/vomiting (n = 1), and grade 4 reversible posterior leukoencephalopathy syndrome (n = 1).
Table 2. Summary of Grade 3 and Grade 4 Adverse Events in Patients Treated With Vatalanib and Everolimus From Both the Dose-Escalation and Dose-Expansion Cohorts.
| Adverse Event | No. (%) | Grade |
|---|---|---|
| Fatigue | 7 (22.6) | 3 |
| Hypertriglyceridemia | 3 (9.7) | 3 |
| Diarrhea | 3 (9.7) | 3 |
| Anorexia | 2 (6.5) | 3 |
| Hypercholesterolemia | 2 (6.5) | 3/4 |
| Rash | 2 (6.5) | 3 |
| Thrombocytopenia | 2 (6.5) | 3 |
| Arrhythmia | 1 (3.2) | 3 |
| Hypercalcemia | 1 (3.2) | 3 |
| Elevated Alkaline Phosphatase | 1 (3.2) | 3 |
| Mucositis | 1 (3.2) | 3 |
| Nausea | 1 (3.2) | 3 |
| Neutropenia | 1 (3.2) | 3 |
| Pain, Bone | 1 (3.2) | 3 |
| Pain, Feet | 1 (3.2) | 3 |
| Hypertension | 1 (3.2) | 4 |
| Leukoencephalopathy | 1 (3.2) | 4 |
| Asthenia | 1 (3.2) | 3 |
| Infection, Calf | 1 (3.2) | 3 |
Clinical Efficacy
Tumor response by RECIST criteria was seen in individual patients with various solid tumor types. For instance, confirmed partial responses were seen in patients with pancreatic neuroendocrine cancer (n = 1), pancreatic adenocarcinoma (n = 1), and RCC (n = 5). Several additional patients demonstrated long periods of stable disease (> 4 months) with minor tumor regressions. Two of 4 patients with metastatic castration-resistant, chemotherapy-refractory prostate cancer demonstrated stable disease for > 6 months with minor PSA reduction. Tumor responses were seen at all dosing levels.
OS for the study is shown in Figure 1A and demonstrates a median OS of 16.3 months. In the expanded cohort, we treated 18 patients with metastatic RCC, in addition to 2 patients with RCC from the dose-escalation cohort treated at the same doses, 10 of whom had previously received VEGF-targeted therapy. The median OS in the RCC dose-expansion cohort was 16.5 months. An exploratory analysis revealed that patients with RCC whose disease had progressed despite previous treatment with a VEGF-targeted therapy had reduced OS and PFS. As shown in Figure 1B, the median OS for patients with RCC who had received previous VEGF-targeted therapy was 6.3 months, and the OS for the previously untreated RCC cohort was 25.1 months. This cohort of patients with RCC treated at the MTD demonstrated a median PFS of 5.8 months (95% confidence interval [CI], 2.8-25.1), with the Kaplan-Meier curve shown in Figure 1C. The PFS for patients with RCC who had received previous VEGF-targeted therapy was 4.6 months (95% CI, 1.8-6.4) and the PFS for previously untreated patients with RCC was 25.1 months (95% CI, 1.9-43.7), as illustrated in Figure 1D.
Figure 1.
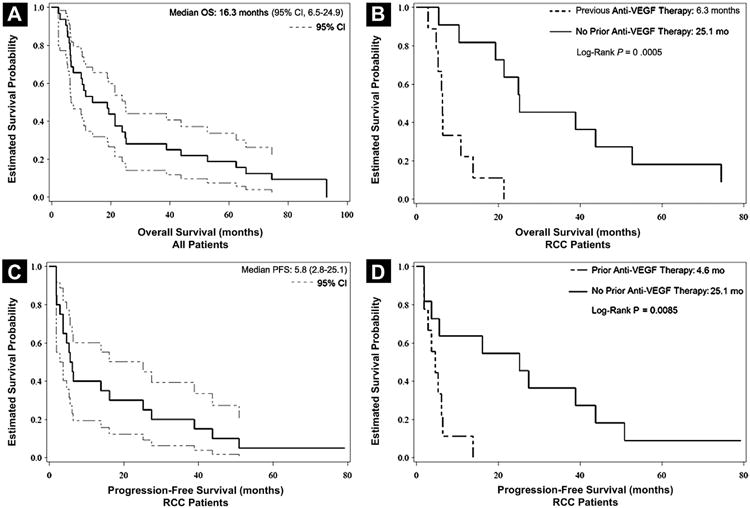
Survival and Progression-Free Survival (PFS) in Patients Treated With Vatalanib and Everolimus. (A) Overall Survival (OS) in all Patients (n = 32). Dashed Lines Represent 95% Confidence Intervals (CIs). Median Survival Time is 16.3 Months (95% CI, 6.5-24.9). (B) OS Comparing Patients With Renal Cell Carcinoma (RCC) in the Dose-Expansion Cohort who had Received Previous Vascular Endothelial Growth Factor (VEGF) Therapy (n = 10) vs. Those who had Received no Previous VEGF Therapy (n = 10). Median OS in Patients With Previous VEGF Therapy was 6.3 Months vs. 25.1 Months in Patients With no Previous VEGF Therapy (log-Rank P = .0005). (C) PFS in Patients With RCC Treated at the Maximum Tolerated Dose (MTD) (n = 20). Dashed Lines Represent 95% CI. Median PFS was 5.8 Months (95% CI, 2.8-25.1). (D) PFS Comparing Patients With RCC in the Dose-Expansion Cohort who had Received Previous VEGF Therapy (n = 10) and Those who had Received no Previous VEGF Therapy (n = 10). Median PFS in Patients who had Received Previous VEGF Therapy is 4.6 Months vs. 25.1 Months in Patients who had not Received Previous VEGF Therapy (log-Rank P = .0085)
A waterfall plot illustrating tumor response is shown in Figure 2. Twenty-four of 32 participants were evaluable for response. Three patients were not included in the plot because of detectable PSA levels and nonmeasurable bony metastases. By PSA determination, imaging, and clinical criteria, 2 of those men were classified as having stable disease and 1 as having disease progression. As illustrated in the plot, the majority of evaluable patients had stable disease, although 7 PRs were seen.
Figure 2. Waterfall Plot to Evaluate Tumor Response.
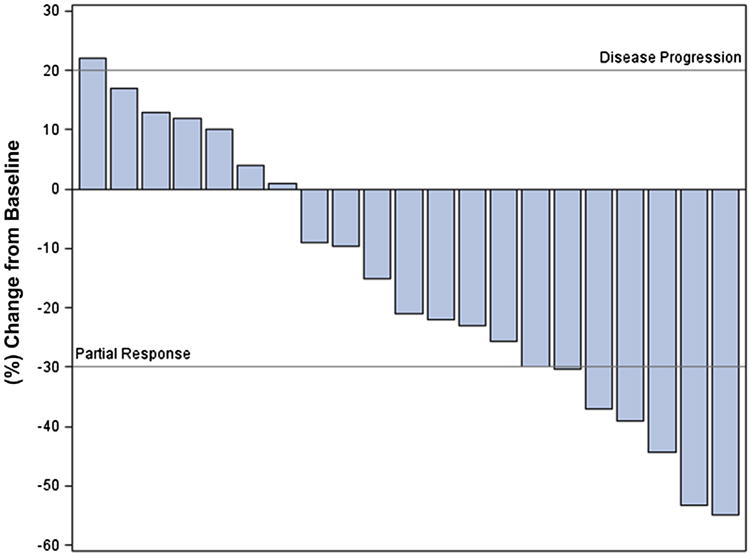
In patients with RCC, tumor response rates varied according to whether or not patients had received previous treatment with VEGF-targeted therapy. Eighteen of 23 patients with RCC from both the escalation and expansion cohorts were evaluable for response. For patients with RCC who had received no previous VEGF-targeted treatment, a PR defined as a ≥ 30% decrease in tumor size per RECIST measurements, was seen in 5 patients, with a median OS of 65.7 months for these responders. By contrast, no patients with previous VEGF-targeted therapy demonstrated a PR, although 3 patients demonstrated some degree of tumor regression characterized by a 1% to 29% decrease in RECIST measurements.
Long-term tolerability and response were seen in a significant subset of patients throughout the entire cohort. Of the 32 patients treated in this study, 6 patients remained on treatment for > 1 year (19%) and 3 patients remained on treatment for > 2 years (9%), all demonstrating a PR. This subset of long-term responders demonstrated no obvious differences from the other patients, except that they had not had previous exposure to VEGF-targeted therapy.
PK Assessments
Exploratory PK assessments of vatalanib and everolimus and the effect of everolimus on vatalanib were undertaken. Patients were treated with a 2-week lead-in of vatalanib alone and then started on everolimus. As such, PK sampling was performed on patients for assessment of vatalanib at steady state alone and with everolimus at steady state. As demonstrated in Table 3, there appeared to be no substantial effect of everolimus on the PK parameters of vatalanib in terms of the maximum or steady-state plasma concentrations. The peak concentration of vatalanib was achieved in 2 hours, regardless of everolimus exposure, and the half-life for vatalanib alone was 6.3 hours and was 5.7 hours for vatalanib after exposure to everolimus. The systemic exposure of vatalanib (AUC) was also not significantly affected by the addition of everolimus. Overall, mean plasma concentrations over time were not affected and were consistent with previous reports of vatalanib and everolimus PK parameters from previous phase I studies (Fig. 3).22,47,48
Table 3. Exploratory Pharmacokinetic Summary From Patients Receiving Vatalanib Alone (Day 14), Vatalanib After Everolimus Started (Day 29), and Everolimus With Vatalanib (Day 29).
| Agent | Vatalanib Alone | Vatalanib + Everolimus | Everolimus |
|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | |
| t1/2(h) | 6.32 ± 4.46 | 5.71 ± 3.06 | 14.0 ± 2.5 |
| tmax(h) | 2.00 (1.08-6.08)a | 2.08 (1.00-4.08)a | 1.31 (0.73-17.7)a |
| Cssmax(ng/mL) | 4825 ± 2910 | 3963 ± 1861 | 48.5 ± 14.8 |
| AUC (ng·h/mL) | 26,384 ± 13,201 | 21,626 ± 9778 | 377 ±117 |
| Cssavg(ng/mL) | 1099 ± 551 | 901 ± 403 | 15.7 ± 4.9 |
| CLss/F (L/h) | 53.7 ± 38.7 | 53.7 ± 38.7 | 14.3 ± 3.7 |
| Vz/F (L) | 488 ± 481 | 488 ± 481 | 216 ± 74 |
Abbreviations: AUC = area under the plasma concentration vs. time curve; CL/F = oral clearance; Cssavg = steady-state plasma concentration; Cssmax = value of maximum plasma concentration; t1/2 = terminal phase rate constant and half-life; tmax = time to peak concentration; Vz/F = area volume of distribution after oral dosing.
Median.
Figure 3.
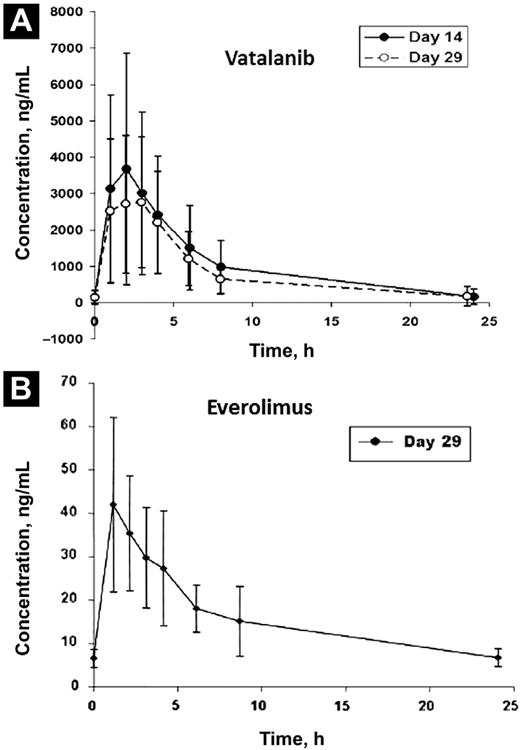
Mean Plasma Concentration vs. Time for Vatalanib and Everolimus. (A) Pharmacokinetic (PK) Sampling for Vatalanib Performed on Cycle 1 day 14 and Cycle 1 day 29. (B) PK Sampling for Everolimus Performed on Cycle 1 day 29 With Peak Concentrations at 2 Hours
Pharmacodynamic Assessment of S6 Ribosomal Protein
We evaluated the pharmacodynamic effect of everolimus on the phosphorylation of S6 ribosomal protein in the PBMCs of patients to document target inhibition. As shown in Figure 4A, the phospho/total S6RP ratio was measured at baseline, after 1 month of treatment, and at EOS if available. Over the initial treatment period (baseline to 1 month), no mean change was observed in the phospho/total ratio of S6RP, but a 2.4-fold increase was observed in phospho/total S6RP levels from baseline to EOS. Additionally, this effect was linked to OS. Although not statistically significant, there was a trend indicating that patients with increased phospho/total S6RP had better survival outcomes (P = .069; hazard ratio, 0.569). The change from baseline to 1 month of treatment may represent the initial steady-state drug effects, whereas the change from baseline to EOS may represent the overall change in the phosphorylation of S6RP while treatment was ongoing. Interestingly, when patients were dichotomized into groups of either increasing or decreasing phospho/total S6RP levels, survival differences were noted (Fig. 4B). It should be noted that the EOS time points varied across patients, and the time that patients received treatment may be related to host or tumor factors, or both, as well as treatment and treatment interaction with these factors. Additionally, because of the small sample size, the P values should be considered descriptive only.
Figure 4.
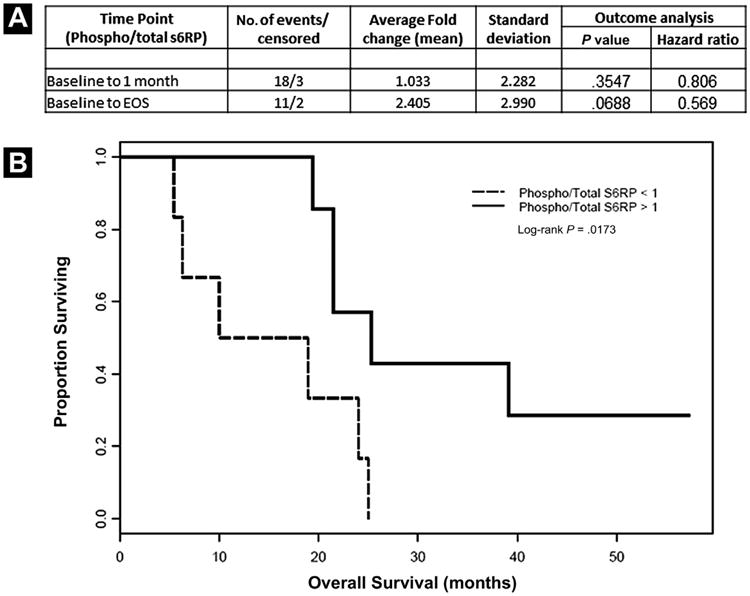
(A) Pharmacodynamic Effect of Everolimus on the Phosphorylation of S6 Ribosomal Protein. Phosphorylated S6RP was Evaluated in Peripheral Blood Mononuclear Cells (PBMCs) of Patients at Baseline, at the end of 1 Month of Treatment, and at the end of Study (EOS). The Data are Presented as the Ratio of Phosphorylated S6RP/Total S6RP (Phospho/Total S6RP). (B) Kaplan-Meier Plot of Patients From Baseline to EOS (n = 13), Dichotomized by Increasing vs. Decreasing Levels of Phosphorylated S6RP/Total S6RP
Discussion
Based on the rationale that VEGF and mTOR inhibition may synergistically repress angiogenesis and tumor growth, we undertook a phase Ib trial of vatalanib and everolimus in patients with advanced cancers, particularly RCC. To gain insight into the tolerability of this combination, we included an expansion cohort in patients with advanced RCC, with the expectation that some patients would demonstrate a durable response and provide information on the long-term safety and tolerability of this combination. DLTs did restrict the combination to doses less than the individual MTDs for each agent; however, both drugs were able to be given within their biologically active dose ranges, and an acceptable dose level was achieved at which antitumor activity was observed. Although the expected toxicities of each agent were seen, fatigue was the most common limiting factor.
Roughly one third of patients in the expansion cohort stopped treatment prematurely because of toxicity, which is higher than expected with either agent alone. However, although many patients required a dose reduction of 1 or both agents, overall tolerability was reasonable, especially in the subset of responding patients. In patients maintained beyond 1 year of therapy, there was 1 case of grade 4 reversible posterior leukoencephalopathy syndrome in the setting of a hypertensive crisis, highlighting the need for continued close blood pressure monitoring. By contrast, several reports combining either sunitinib or sorafenib with mTOR inhibitors have demonstrated higher frequencies and earlier onset of toxicities, including grade 3 or 4 mucositis, hand-foot syndrome, and thrombocytopenia.49,50 Likewise, in a combination study of bevacizumab and everolimus in RCC, proteinuria and mucositis were the most common limiting factors.51 Most studies have not clearly evaluated the potential PK interactions between these classes of agents. In this study, the lead-in period of vatalanib monotherapy established that there was no increased drug exposure to vatalanib with the subsequent addition of everolimus. Similarly, the PK of everolimus in the setting of vatalanib exposure is consistent with the published PK data for single-agent everolimus,47 suggesting that combination therapy does not significantly affect the exposure of either drug. Although other TKIs need to be tested for PK interaction with mTOR inhibitors such as everolimus, these results suggest that drug-drug interactions may not be a limiting factor to combination strategies.
We were unable to achieve the full monotherapy doses for both drugs in this combination; however, we believe these agents were still potent in their target effects. A recent study demonstrated that the combination of sorafenib and everolimus, even at reduced doses, offered impressive objective response results not typically seen with either agent alone, even at full doses.52
In our study, increases in blood pressure were seen as a likely on-target effect of vatalanib. Although phospho-S6 ribosomal protein (pS6RP) levels in PBMCs were measured to verify that everolimus was modulating its target, the pS6RP changes in PBMCs as described here suggest that pS6RP levels in nontumor cells may not be an accurate readout of everolimus effect. A previous study similarly showed nonsignificant changes in pS6RP in PBMCs isolated from patients before and after treatment with everolimus.53 Although pS6RP in PBMCs treated exogeneously with everolimus may be a useful in vitro assay,54 the pS6RP levels in tumor cells from patients are likely a more accurate assessment of rapalog activity.55 Alternatively, the higher phospho/total S6RP ratios noted at the end of our study may be accurate and may represent a resistance mechanism whereby alternate signaling leads to S6RP activation.
We surmise that inhibition with a cell surface TKI and downstream mTOR modulation may result in greater “off- target” effects, accounting for the increased toxicity, rather than changes in drug exposure or metabolism. If so, the toxicity profile combining VEGFR TKIs with mTOR inhibition may be limited by the off-target profile of the VEGFR TKI. As such, multitargeted TKIs, such as sunitinib and sorafenib, may have a limited therapeutic index to prevent synergistic off-target toxicities, whereas next-generation VEGFR TKIs may have a target profile more amenable to such combinations. An alternative explanation is that “on-target” pharmacodynamic actions of the combined mTOR and potent VEGF inhibitors lead to earlier onset and more severe toxicity, perhaps as a result of overlapping side effect profiles (ie, fatigue, diarrhea, mucositis). Patient variability likely also factors into tolerance, because there was a subset of patients who responded and tolerated treatment well for > 1 year.
During the study, regulatory approval for the treatment of patients with advanced RCC with sunitinib or sorafenib led us to modify our eligibility requirements to allow patients who had previously received 1 or more of these therapies. There appeared to be differences in both the objective response rates and the OS between the cohorts of patients with and those without previous exposure to VEGF-targeted agents. There was a substantial subset of untreated patients with RCC who exhibited prolonged PFS of > 1 year. Given the small numbers and the wide CIs for PFS rates, we caution any further conclusions, but we hypothesize that there may be a subset of previously untreated patients who would tolerate and benefit from a first-line combination strategy. Predictive factors could include clinical, disease-related, or pharmacogenomic markers. For example, lactate dehydrogenase has recently been shown to be a predictive biomarker for response to mTOR inhibition in patients with poor-risk RCC and may warrant further exploration.56 Similarly, patients with colorectal cancer and a high lactate dehydrogenase level treated with vatalanib had a longer PFS than those treated with placebo, suggesting a potential biomarker for VEGF-targeted therapy.57 In the future, evaluation of other VEGFR TKIs with mTOR inhibitors should include pharmacogenetic and tumor biomarker analyses to prospectively define the subset of patients who can tolerate and benefit from combination approaches.
Conclusion
Combination therapy with the VEGFR TKI vatalanib and the mTOR inhibitor everolimus was possible in clinically active doses and was tolerable and effective in a subset of patients with RCC over prolonged periods. There were no inherent PK limitations to these specific agents, and toxicities were predictable and additive because the individual MTD for each drug was not achievable. However, further development of vatalanib as an anticancer agent has been halted, and therefore if combination therapy is to be further pursued, it must be done with an alternative agent. VEGFR TKIs or VEGF inhibitors that demonstrate a well-defined on-target profile with few off-target effects, such as axitinib or pazopanib, may have a therapeutic index for combination with mTOR inhibitors such as everolimus and should be tested for safety and comparative efficacy against the sequential single-agent strategy currently used in clinical practice.
Clinical Practice Points.
VEGFR and mTOR inhibition are the cornerstones of the treatment paradigm for metastatic RCC today.
We conducted a dose-escalation phase Ib study of an oral VEGFR-targeted TKI with an oral mTOR inhibitor and a dose-expansion cohort in patients with advanced RCC and found modest clinical activity.
These results support the concept that combinations of a TKI and an mTOR inhibitor may be used in patients with RCC for long-term disease control.
Acknowledgments
We wish to thank the patients and their families for participation in this study.
This work was supported in part by Novartis Pharmaceuticals USA (East Hanover, NJ).
AJA serves as consultant for Novartis and Bayer and obtains research support from Novartis and Pfizer. DJG serves as a consultant for AVEO, Astellas, Bayer, BMS, Exelixis, Genentech/Roche, Novartis, and Pfizer and obtains research support from BMS, GSK, Novartis, and Pfizer. HIH serves as a consultant for and obtains research support from Novartis. ABN serves as a consultant for GlaxoSmithKline and Novartis and obtains research support from Amgen, F. Hoffman-La Roche, Pfizer, and Tracon Pharmaceuticals. SYW has served on advisory boards for Teva Pharmaceuticals and Astellas/Medivation.
Footnotes
This work was presented in part at the 2007 Annual Meeting of the American Society of Clinical Oncology.
Disclosure: All other authors have stated that they have no conflicts of interest.
References
- 1.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 2.Burstein HJ, Elias AD, Rugo HS, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26:1810–6. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 3.Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 4.Miller KD, Chap LI, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–9. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 5.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 6.Socinski MA, Novello S, Brahmer JR, et al. Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:650–6. doi: 10.1200/JCO.2007.13.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 8.Wells SA, Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 10.Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 11.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 12.Desjardins A, Reardon DA, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res. 2008;14:7068–73. doi: 10.1158/1078-0432.CCR-08-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 14.Jain RK. Normalization of tumor vasculature: an emerging concept in anti-angiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 16.Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499–506. doi: 10.1200/JCO.2012.42.8201. [DOI] [PubMed] [Google Scholar]
- 17.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–8. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 18.Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–9. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 19.Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–12. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 20.Drevs J, Hofmann I, Hugenschmidt H, et al. Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000;60:4819–24. [PubMed] [Google Scholar]
- 21.Morgan B, Thomas AL, Drevs J, et al. Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: results from two phase I studies. J Clin Oncol. 2003;21:3955–64. doi: 10.1200/JCO.2003.08.092. [DOI] [PubMed] [Google Scholar]
- 22.Thomas AL, Morgan B, Horsfield MA, et al. Phase I study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of PTK787/ZK 222584 administered twice daily in patients with advanced cancer. J Clin Oncol. 2005;23:4162–71. doi: 10.1200/JCO.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 23.Jones SF, Spigel DR, Yardley DA, 3rd, et al. A phase I trial of vatalanib (PTK/ZK) in combination with bevacizumab in patients with refractory and/or advanced malignancies. Clin Adv Hematol Oncol. 2011;9:845–52. [PubMed] [Google Scholar]
- 24.Spigel D, Jones S, Hainsworth J, et al. A phase I trial to determine the safety of imatinib in combination with vatalanib in patients with advanced malignancies. Cancer Invest. 2011;29:308–12. doi: 10.3109/07357907.2011.568567. [DOI] [PubMed] [Google Scholar]
- 25.de Bazelaire C, Alsop DC, George D, et al. Magnetic resonance imaging-measured blood flow change after antiangiogenic therapy with PTK787/ZK 222584 correlates with clinical outcome in metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:5548–54. doi: 10.1158/1078-0432.CCR-08-0417. [DOI] [PubMed] [Google Scholar]
- 26.Hecht JR, Trarbach T, Hainsworth JD, et al. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J Clin Oncol. 2011;29:1997–2003. doi: 10.1200/JCO.2010.29.4496. [DOI] [PubMed] [Google Scholar]
- 27.Van Cutsem E, Bajetta E, Valle J, et al. Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol. 2011;29:2004–10. doi: 10.1200/JCO.2010.29.5436. [DOI] [PubMed] [Google Scholar]
- 28.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Baldo P, Cecco S, Giacomin E, et al. mTOR pathway and mTOR inhibitors as agents for cancer therapy. Curr Cancer Drug Targets. 2008;8:647–65. doi: 10.2174/156800908786733513. [DOI] [PubMed] [Google Scholar]
- 30.Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 31.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 32.Garcia JA, Danielpour D. Mammalian target of rapamycin inhibition as a therapeutic strategy in the management of urologic malignancies. Mol Cancer Ther. 2008;7:1347–54. doi: 10.1158/1535-7163.MCT-07-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 34.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 37.Lane HA, Wood JM, McSheehy PM, et al. mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin Cancer Res. 2009;15:1612–22. doi: 10.1158/1078-0432.CCR-08-2057. [DOI] [PubMed] [Google Scholar]
- 38.O'Reilly T, Lane HA, Wood JM, et al. Everolimus and PTK/ZK show synergistic growth inhibition in the orthotopic BL16/BL6 murine melanoma model. Cancer Chemother Pharmacol. 2011;67:193–200. doi: 10.1007/s00280-010-1307-z. [DOI] [PubMed] [Google Scholar]
- 39.Kim WY, Kaelin WG., Jr Molecular pathways in renal cell carcinoma—rationale for targeted treatment. Semin Oncol. 2006;33:588–95. doi: 10.1053/j.seminoncol.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Jaeger-Lansky A, Cejka D, Ying L, et al. Effects of vatalanib on tumor growth can be potentiated by mTOR blockade in vivo. Cancer Biol Ther. 2010;9:919–27. doi: 10.4161/cbt.9.11.11805. [DOI] [PubMed] [Google Scholar]
- 41.Abraham RT. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr Top Microbiol Immunol. 2004;279:299–319. doi: 10.1007/978-3-642-18930-2_18. [DOI] [PubMed] [Google Scholar]
- 42.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–60. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 43.Humar R, Kiefer FN, Berns H, et al. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002:771–80. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 44.Treins C, Giorgetti-Peraldi S, Murdaca S, et al. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–81. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 45.Zundel W, Schindler C, Haas-Kogan D, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–3. [PMC free article] [PubMed] [Google Scholar]
- 46.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 47.O'Donnell A, Faivre S, Burris HA, 3rd, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588–95. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 48.Thomas AL, Trarbach T, Bartel C, et al. A phase IB, open-label dose-escalating study of the oral angiogenesis inhibitor PTK787/ZK 222584 (PTK/ZK), in combination with FOLFOX4 chemotherapy in patients with advanced colorectal cancer. Ann Oncol. 2007;18:782–8. doi: 10.1093/annonc/mdl469. [DOI] [PubMed] [Google Scholar]
- 49.Patel PH, Senico PL, Curiel RE, et al. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2009;7:24–7. doi: 10.3816/CGC.2009.n.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sosman J, Puzanov I. Combination targeted therapy in advanced renal cell carcinoma. Cancer. 2009;115:2368–75. doi: 10.1002/cncr.24234. [DOI] [PubMed] [Google Scholar]
- 51.Hainsworth JD, Spigel DR, Burris HA, 3rd, et al. Phase II trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J Clin Oncol. 2010;28:2131–6. doi: 10.1200/JCO.2009.26.3152. [DOI] [PubMed] [Google Scholar]
- 52.Harzstark AL, Small EJ, Weinberg VK, et al. A phase 1 study of everolimus and sorafenib for metastatic clear cell renal cell carcinoma. Cancer. 2011;117:4194–200. doi: 10.1002/cncr.25931. [DOI] [PubMed] [Google Scholar]
- 53.Fouladi M, Laningham F, Wu J, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol. 2007;25:4806–12. doi: 10.1200/JCO.2007.11.4017. [DOI] [PubMed] [Google Scholar]
- 54.Boulay A, Zumstein-Mecker S, Stephan C, et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004;64:252–61. doi: 10.1158/0008-5472.can-3554-2. [DOI] [PubMed] [Google Scholar]
- 55.Armstrong AJ, Netto GJ, Rudek MA, et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin Cancer Res. 2010;16:3057–66. doi: 10.1158/1078-0432.CCR-10-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Armstrong AJ, George DJ, Halabi S. Serum lactate dehydrogenase predicts for overall survival benefit in patients with metastatic renal cell carcinoma treated with inhibition of mammalian target of rapamycin. J Clin Oncol. 2012;30:3402–7. doi: 10.1200/JCO.2011.40.9631. [DOI] [PubMed] [Google Scholar]
- 57.Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin Cancer Res. 2011;17:4892–900. doi: 10.1158/1078-0432.CCR-10-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]