

Abstract
Pharmacokinetic (PK) testing of a humanized (κI, VH3 framework) and affinity matured anti-hepatitis C virus E2-glycoprotein (HCV-E2) antibody (hu5B3.κ1VH3.v3) in rats revealed unexpected fast clearance (34.9 mL/day/kg). This antibody binds to the rat recycling receptor FcRn as expected for a human IgG1 antibody and does not display non-specific binding to baculovirus particles in an assay that is correlated with fast clearance in cynomolgus monkey. The antigen is not expressed in rat so target-dependent clearance does not contribute to PK. Removal of the affinity maturation changes (hu5B3.κ1VH3.v1) did not restore normal clearance. The antibody was re-humanized on a κ4, VH1 framework and the non-affinity matured version (hu5B3.κ4VH1.v1) was shown to have normal clearance (8.5 mL/day/kg). Since the change in framework results in a lower pI, primarily due to more negative charge on the κ4 template, the effect of additional charge variation on antibody PK was tested by incorporating substitutions obtained through phage display affinity maturation of hu5B3.κ1VH3.v1. A variant having a pI of 8.61 gave very fast clearance (140 mL/day/kg) whereas a molecule with pI of 6.10 gave slow clearance (5.8 mL/kg/day). Both antibodies exhibited comparable binding to rat FcRn, but biodistribution experiments showed that the high pI variant was catabolized in liver and spleen. These results suggest antibody charge can have an effect on PK through alterations in antibody catabolism independent of FcRn-mediated recycling. Furthermore, introduction of affinity maturation changes into the lower pI framework yielded a candidate with PK and virus neutralization properties suitable for clinical development.
Keywords: antibody pharmacokinetics, FcRn recycling, antibody catabolism, isoelectric point, humanized
Introduction
Hepatitis C virus (HCV), a member of the Flaviviridae family of viruses, is a major cause of chronic hepatitis and hepatocellular carcinoma.1,2 The HCV genome is a positive strand ∼9.6 kb RNA molecule consisting of a single open reading frame (ORF) that encodes a polyprotein of ∼3000 amino acids in length. Post-translational processing yields at least ten different proteins: core, envelope proteins E1 and E2, p7, and non-structural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B.1,3,4 The E1-E2 glycoprotein heterodimer is essential for HCV entry into hepatocytes. To date, at least four host entry factors have been identified: CD81,5 scavenger receptor B type I (SR-BI),6 occludin (OCLN),7,8 and claudin 1 (CLDN1).9 HCV E2 glycoprotein has been demonstrated to bind CD81, SR-BI, and CLDN1.
Antibodies that bind E2 and block interaction with the cellular factors could have therapeutic potential in the treatment of HCV-associated disorders, especially in the liver transplant setting. Previously, we described two antibodies that bind to a highly conserved epitope on E2 (E2412–423) that are broadly neutralizing across multiple HCV genotypes.10 Both antibodies originated from mouse hybridomas, were humanized, and required affinity maturation to achieve the in vitro virus neutralization potency expected to support monthly, subcutaneous dosing for treatment of chronic HCV-infected patients. We report here that one of these higher affinity antibodies, hu5B3.κ1VH3.v3, gave surprisingly fast clearance in rodents. Since HCV does not infect rodents, these animals do not express the E2 antigen and thus fast clearance cannot be related to target binding. Fast clearance did not result from the amino acid changes introduced through affinity maturation, but appears to be a consequence of the charge on the antibody from the framework used for humanization. Re-humanization of the light chain onto a more negatively charged human framework restores normal clearance, thus enabling development of viable clinical candidates with enhanced neutralization potency.
Results
Murine antibody 5B3 was humanized on a κ1 light chain human variable domain and VH3 subgroup heavy chain human variable domain framework and affinity matured as previously described10 to generate hu5B3.κ1VH3.v3. The pharmacokinetic (PK) profile observed for hu5B3.κ1VH3.v3 (huIgG1 format) following a single intravenous (IV) bolus dose of 5 mg/kg in Sprague-Dawley rats is shown in Figure 1 with PK parameters summarized in Table 1. Clearance (34.9 ± 5.0 mL/day/kg) was considerably faster than the range (4.8–14.6 mL/day/kg) observed for a panel of human IgG1 antibodies in rat.11 Fast clearance (Fig. 1; Table 1) was also observed for hu5B3.κ1VH3.v1, which has the two affinity maturation changes in complementarity-determining region (CDR)-L2, His-54 and Ala-55, reverted back to the parental Gln-54 and Gly-55. A re-examination of the murine 5B3 variable domain sequences (Fig. 2) suggested better homology with human variable domains of the κ4 and VH1 subgroups compared with the κ1, VH3 subgroups used for hu5B3.κ1VH3.v1,3. For example, there are only 20 amino acid changes in the VL framework between m5B3 and a κ4 consensus VL compared with 27 between m5B3 and a κI consensus VL framework. On the VH domain, m5B3 differs from consensus human VH1 and VH3 at 23 and 32 positions, respectively.
Figure 1.
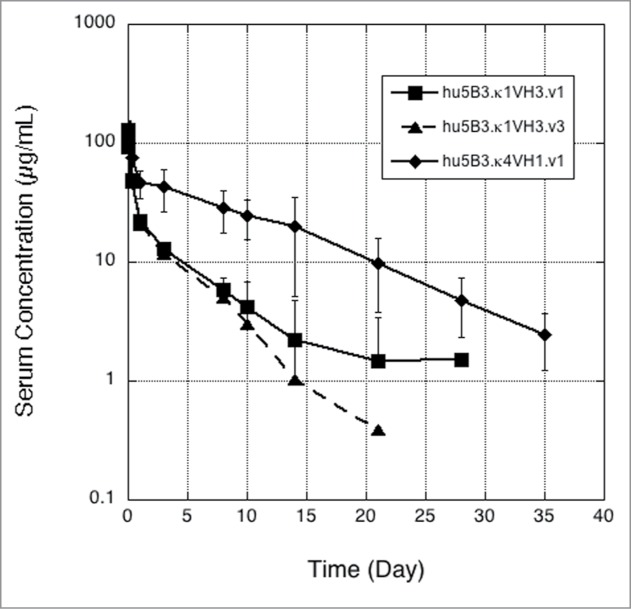
Single dose IV pharmacokinetics of 5B3 framework variants k1VH3 (v1 or v3) and k4VH1 (v1) in SD rats. Error bars are standard deviation (n = 4 except n = 3 for day 10, and n = 2 for day 21, of k1VH3.v3).
Table 1.
Properties of humanized 5B3 variants
| Antibody | Residue changes in CDRs | Relative EC50 HCV pp (H77) | Relative EC50 HCV pp (Con1) | pI | Rat FcRn KD (μM) | Clearance (mL/day/kg) |
|---|---|---|---|---|---|---|
| hu5B3.κ1VH3.v1 | 1.0 ± 0.6 | 1.0 ± 0.3 | 7.95 | 0.7 | 29.5 ± 6.6 | |
| hu5B3.κ1VH3.v3 | L2-Q54H, G55A | 0.9 ± 0.3 | 0.5 ± 0.3 | 7.96 | 1.0 | 34.9 ± 5.0 |
| hu5B3.κ4VH1.v1 | 1.0 | 0.8 | 6.71 | 1.0 | 8.5 ± 3.9 | |
| hu5B3.κ4VH3.v1 | 0.8 | 1.5 | ND | ND | 9.8 ± 2.4 | |
| hu5B3.κ1VH3. pI8.61 | L3-E93R; H2-Q61K | ND | ND | 8.61 | 1.1 | 140 ± 27 |
| hu5B3.k4VH1. pI6.10 | L1-N27dD; H3-K100aE | ND | ND | 6.10 | 0.8 | 5.8 ± 2.7 |
| hu5B3.k4VH1.HA | L1-Y28W; H3-Y98F,K100aE | 0.02 | 0.01 | 6.24 | 0.9 | 8.0 ± 2.1 |
Relative EC50 values were calculated by dividing the EC50 value of the variant by the value measured for hu5B3.κ1VH3.v1 on genotype 1a H77 (0.018 ± 0.01 μg/mL) or genotype 1b Con1 (1.1 ± 0.3 μg/mL) HCVpp. The pI values for the variants were determined by using imaged capillary isoelectric focusing (iCIEF) as described in the methods. ND = not determined.
Figure 2.

Alignment of m5B3 to human germline consensus sequence.
5B3 was re-humanized on a consensus κ4, VH1 framework12 and incorporated the same key mouse framework residues at Vernier positions as described for the κ1, VH3 scaffold.10 As shown in Table 1, hu5B3.κ4VH1.v1 retained virus neutralization potency comparable to hu5B3.κ1VH3.v1. Determination of PK parameters (Fig. 1; Table 1) indicated that the re-humanized version had slower clearance (8.5 mL/day/kg) in SD rats. Both the hu5B3.κ1VH3.v1 and hu5B3.κ4VH1.v1 bound to rat FcRn with similar affinity at low pH, and fast release kinetics at neutral pH (data not shown), suggesting that differences in clearance are not a consequence of altered FcRn binding (Table 1). A humanized version retaining the VH3 framework (hu5B3.κ4VH3.v1) also showed normal clearance, suggesting the light chain framework is contributing to the clearance effect. As shown by differential scanning calorimetry on protein solutions buffered at pH 5 (Fig. S1), the Fab portions of hu5B3.κ1VH3.v1, hu5B3.κ1VH3.v3, and hu5B3.κ4VH1.v1 all gave melting temperatures (Tm) greater than 72 °C. This suggests that the clearance differences observed are not due to conformational instability of the faster clearing antibodies.
Both the consensus VH1 and consensus VH3 frameworks contribute a net of two positively charged residues to the humanized antibody. In contrast, the consensus κ4 framework has a net of three more negatively charged residues than the consensus κ1 framework. This suggests that the more negative charge on the κ4 humanized antibody results in slower clearance. To further explore this hypothesis, we chose to perform PK studies with 5B3 variants having additional charged residue substitutions. For this purpose, we desired CDR substitutions with minimal effects on antigen binding such that antigen-based ELISA could be used for antibody quantitation in serum samples. We used a combination of alanine-scanning mutagenesis and phage display affinity maturation to identify potential sites of charge substitution. First, we displayed single-site alanine mutants of hu5B3.κ1VH3.v1 on the surface of M13 bacteriophage and measured binding to soluble E2 protein derived from two viral genotypes. As shown in Table 2, many sites in the CDRs appeared to be substitutable since there was little effect on binding to either genotype from replacement with an alanine residue. A few CDR residues, for example CDR-H3 Y100c, showed a large reduction in binding to both genotype E2s when replaced with alanine. Notably, alanine substitution of CDR-H3 residue Tyr98 produced a differential effect with larger loss in binding for genotype 1b than for genotype 1a, E2 protein. A few of the substitutable positions were selected for replacement with charged residues because these sites are not in contact with an epitope peptide in the co-crystal structure,10 are solvent accessible polar or charged residues, and varied residues are found at these positions in human germline genes. Residues CDR-L1 Asn27d, CDR-L3 Glu93, and CDR-H2 Gln61were chosen for replacement with Asp, Arg, and Lys, respectively. These sites are not in contact with peptide in the structure (Fig. 3A) of the hu5B3.κ1VH3.v3: E2412–423 peptide complex.
Table 2.
Alanine-scan of hu5B3. κ1VH3.v1
| Mutant | GT1a-E2 | GT1b-E2 |
|---|---|---|
| H1-T28A | 9.9 | 4.2 |
| H1-T30A | 1.1 | 1.4 |
| H1-N31A | 1.3 | 1.7 |
| H1-Y32A | 2.3 | 3.2 |
| H1-W33A | 5.5 | 8.0 |
| H2-Y52A | 1.2 | 1.4 |
| H2-F56A | 2.6 | 3.5 |
| H2-N58A | 5.6 | 6.1 |
| H2-Q61A | 1.4 | 1.6 |
| H3-Y98A | 2.0 | 7.5 |
| H3-K100aA | 1.1 | 1.2 |
| H3-Y100cA | 11.3 | 8.2 |
| L1-E27A | 1.2 | 1.4 |
| L1-N27dA | 1.2 | 1.4 |
| L1-Y28A | 3.3 | 5 |
| L1-I30A | 2.6 | 2.8 |
| L1-F32A | 12.5 | 7.6 |
| L2-S50A | 3.5 | 3.9 |
| L2-N53A | 1.4 | 1.4 |
| L2-H54A | 1.2 | 1.2 |
| L2-S56A | 1.2 | 1.4 |
| L3-S91A | 2.4 | 2.7 |
| L3-K92A | 1.3 | 1.3 |
| L3-E93A | 1.4 | 1.4 |
| L3-Y96A | 6.1 | 8.8 |
Fold change in IC50 value relative to wild-type for competition of hu5B3.κIVH3 mutant phage binding with soluble E2 protein.
Figure 3.
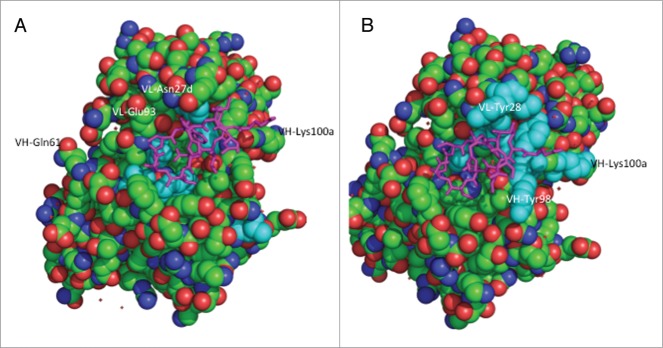
Structure of hu5B3.v3 (space filling) in complex with epitope peptide (magenta tube).10 (A) Residues with greater than 5-fold loss in binding to HCV-gt1a-E2 when substituted with alanine colored cyan. (B) Residues included in affinity maturation library colored cyan.
Further sites of charge residue substitution were identified during the process of affinity maturation of hu5B3.κIVH3.v1 using phage display technology. A cluster of residues forming one end of the peptide binding pocket (Fig. 3B) were targeted for combinatorial mutagenesis. A library containing variants of residues VL-Y28, I30, F32, S91, Y96, and VH-Y98, Y99, K100a, Y100c was constructed. Tyr and Phe positions VL-F32, Y96 and VH-Y98, Y99, Y100c were randomized with a limited diversity codon (TWC) specifying only Tyr or Phe, whereas full randomization was achieved for the four other sites using NNK codons. As shown in Table 3, clones selected from the library and shown to have improved binding to E2 proteins had substitutions at positions VL-28 and 30, and VH-98 and 100a. In particular, a Trp substitution at VL-28, and Phe and Glu substitutions at VH-98 and 100a, respectively, appeared to result in improved binding. The K100aE substitution was appealing for further charge studies since it resulted in a net charge change of –2.
Table 3.
Variants selected using phage display
| VL-residue |
VH-residue |
Phage IC50 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | 28 | 30 | 32 | 91 | 96 | 98 | 99 | 100a | 100c | GT1a | GT1b |
| WT | Y | I | F | S | Y | Y | Y | K | Y | 1 | 1 |
| HA1 | W | L | F | S | Y | Y | Y | I | Y | 20 | 4 |
| HA2 | Y | F | F | S | Y | Y | F | P | Y | 6 | 2 |
| HA3 | W | A | F | S | Y | Y | Y | P | Y | 2 | 8 |
| HA4 | F | L | F | S | Y | Y | Y | H | Y | 3 | 10 |
| HA5 | Y | V | F | S | Y | Y | Y | T | Y | 3 | 10 |
| HA6 | W | I | F | S | Y | F | Y | E | Y | 6 | 5 |
| HA7 | F | I | F | S | Y | F | Y | P | F | 2 | 2 |
Phage IC50 are shown as fold decrease relative to WT.
Charge variant IgGs were constructed by installing additional amino acid changes in the κ1, VH3 or κ4, VH1 framework antibodies. Addition of CDR-L1 N27dD and CDR-H3 K100aE to hu5B3.κ4VH1.v1 generated an antibody with additional negative charge and low measured pI (Table 1). A high pI variant was produced by incorporating CDR-H2 Q61K and CDR-L3 E93R into hu5B3.κ1VH3.v1. A variant with improved activity (hu5B3.κ4VH1.HA) including the substitutions CDR-L1 Y28W, CDR-H3 Y98F and K100aE, in hu5B3.κ4VH1.v1 was also constructed. As shown in Table 1, hu5B3.κ4VH1.HA had improved potency, ∼50-fold on H77 (genotype 1a) and 100-fold on Con1 (genotype 1b) HCV pseudoparticle (HCVpp) assays, for virus neutralization.
Upon single dose IV administration in rats, hu5B3.κ4VH1.pI6.10 and hu5B3.κ1VH3.pI8.61 gave very different PK profiles (Fig. 4). The high pI variant had a clearance of 140 ml/day/kg, about 20-fold faster clearance than the low pI variant. According to a two compartmental analysis (Table S1), the pI 8.61 variant had a larger distribution phase, larger volume of distribution at steady-state, and ∼3-fold faster terminal half-life compared with the pI 6.10 variant. Faster clearance was not explained by alteration in rat FcRn binding since surface plasmon resonance measurements indicate similar FcRn-binding affinity for these variants (Table 1). A comparison of all variants indicates that clearance is dependent on the measured pI for this antibody series (Fig. 5A) with slower clearance associated with lower pI values. Although these variants also differ in hydrophobicity of the Fv portion calculated from the amino acid sequence, and there is a trend of decreased clearance with increasing calculated hydrophobicity, the clearance value is not as strongly correlated with hydrophobicity (Fig. 5B) compared with pI (Fig. 5A). Consistent with the measured pI, the clearance of the potency improved variant (hu5B3.κ4VH1.HA) is in the acceptable range.
Figure 4.
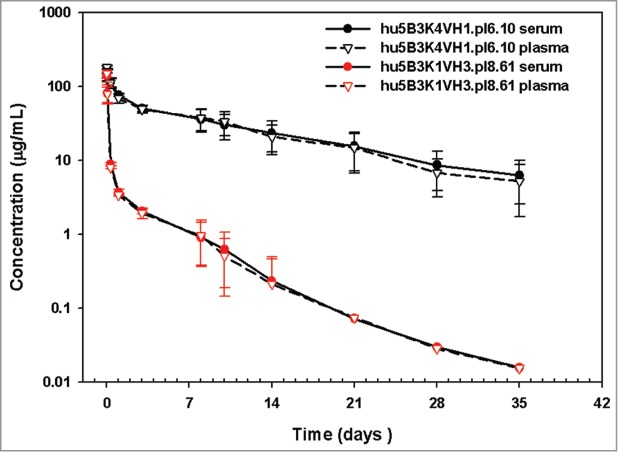
Pharmacokinetics of 5B3 pI variants in SD rats.
Figure 5.
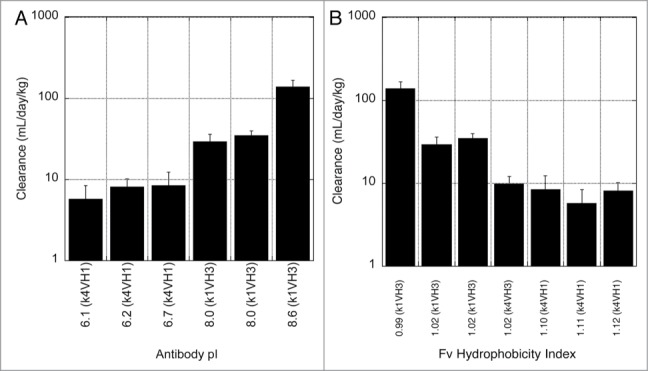
Dependence of clearance in rats on antibody pI (A) or hydrophobicity calculated for the Fv portion (B). Hydrophobic index was calculated from the ratio of hydrophobic to hydrophilic amino acids using the Eisenberg and McLachlan hydrophobicity scale.26
The PK curve shape observed for the pI 8.61 antibody, having a large distribution phase and shorter terminal half-life, is consistent with a high rate of catabolism in tissues. However, other mechanisms could explain this curve shape and we can't completely exclude an anti-therapeutic antibody (ATA) response (Fig. S2) as causing the shorter terminal half-life. We used a differential radiolabeling method to compare the biodistribution of the pI 6.10 and pI 8.61 antibodies. Molecules were conjugated to either a non-residualizing probe ([125I]) or a residualizing probe [111In]-DOTA. Labeling of antibodies with radiometals results in a different cellular retention of the radioactive species relative to iodine labeling. For these antibodies in rats, which do not express the antigen, the antibodies undergo non-specific, fluid-phase endocytosis and lysosomal degradation. However, cellular efflux of the radiolabel does not readily occur for [111In]-DOTA-labeled antibodies. Unlike [125I], which diffuses out of the cell (as iodotyrosine or free iodide) following proteolysis, In-111-DOTA−lysine cannot easily cross the plasma membrane due to its charge and polarity. Because of its tendency to become intracellularly trapped, In-111-DOTA is referred to as a residualizing label. As shown in Figure 6A, the low pI variant exhibits highly similar biodistribution and relative abundance in different organs regardless of whether it is labeled with [125I] or [111In]-DOTA. By contrast, the high pI variant distribution profile is highly dependent on the label used (Fig. 6B). High levels of antibody degradation are detected in spleen and liver by residualization of the [111In]-DOTA probe, while low amounts of antibody are detected in any of the examined tissues when labeled with the non-residualizing [125I] probe, consistent with increased whole-body clearance of the high, but not the low, pI variant with liver and spleen being the major sites of catabolism (Fig. 6C, D). Fast plasma clearance, reflecting sequestration and degradation, is observed (Fig. 7) for the high pI variant regardless of label, whereas the low pI variant exhibits slow clearance when detected by either label.
Figure 6.
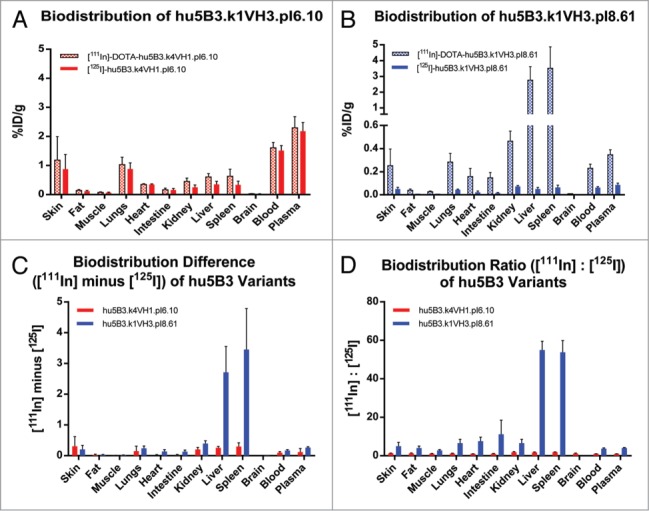
Tissue distribution of 111In and 125I labeled 5B3 variants in SD rats. (A) Low pI variant hu5B3.κ4VH1.pI6.10, and (B) high pI variant hu5B3.κ1VH3.pI8.61. (C) Difference between amounts of 111In and 125I labeled 5B3 variants detected in various tissues. (D) Ratio of label in various tissues.
Figure 7.
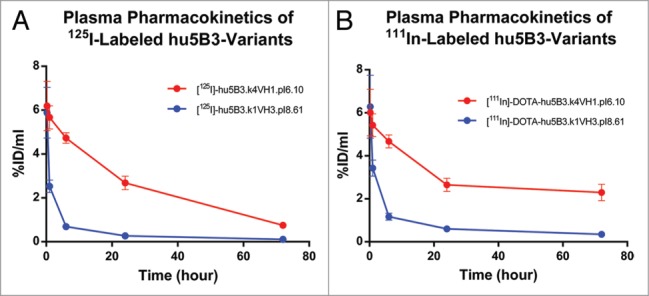
Plasma concentration vs. time profiles for 5B3 variants.
Discussion
Here, we have shown that clearance in rats of IgG1, κ isotype antibodies can be influenced by the choice of framework for humanization. Faster clearance does not appear to arise from alterations in binding to rat FcRn, or instability of the humanized antibody as assessed by DSC (Fig. S1). We have not detected instability in serum for this series of antibodies or off-target binding in vitro using an assay of non-specific binding that has been correlated with clearance in monkeys13 (Fig. S3). Unintended specific binding has been proposed to explain the fast clearance observed for an anti-FGFR4 antibody in rodents14 and an anti- amyloid β antibody in monkeys.15 Given that similar levels of antibody are detected for both serum and plasma samples (Fig. 4), it appears unlikely that unintended but specific off-target binding to a serum protein is the explanation for the fast clearance.
Although we cannot eliminate changes in hydrophobicity as a factor in determining clearance differences, clearance does appear to correlate better with the charge on the variable domains. The tighter association with pI is in part due to the fact we have an experimental measure of pI whereas the hydrophobicity is only calculated, and the calculation only uses sequence and not structural information. The antibody humanized on the κ4, VH1 framework has a lower pI, mostly due to the more negatively charged κ4 framework, and slower clearance compared with the κ1, VH3 humanized antibody. The distribution of charge on hu5B3. κ4VH1.v1 is different from hu5B3. κ1VH3.v3 or bevacizumab, having a negative charge patch on the VL domain and smaller patches of positive charge (Fig. S4), but it is difficult to parse out the relative roles of charge distribution vs. total net charge regarding clearance. Sampei et al.16 found a positive charge patch on an anti-FIXa antibody that was implicated as causing fast clearance. Modulating the charge patch with addition of an acidic residue resulted in decreased clearance. Further negative charge addition to the κ4, VH1 5B3 antibody slows clearance, whereas an increase in positive charge in the κ1, VH3 5B3 antibody results in faster clearance. These results suggest that antibodies with high pI values and suboptimal PK properties may be improved through negative charge addition to the variable domains, independent of FcRn binding. This can be accomplished through use of an alternative human framework. Conversion to an alternative framework can result in retained target binding and would be expected to have low impact on immunogenicity in humans. Alternatively, the pI of the κ1, VH3 antibody could have been decreased, with a potential to slow clearance, through substitution in the κ1 CDR or framework regions with negatively charged residues. In addition to the CDR substitutions noted in Table 1, point mutations in the κ1 framework region (K42Q; S60D) could increase the negative charge. Since the sequences of the framework regions are generally more strongly conserved than the CDR regions, point mutations in the framework could carry a higher risk of immunogenicity.
A dependence of clearance determined in mice on antibody pI was previously observed for a series of humanized IgG4, κ isotype antibodies.17 Igawa et al. used a slightly more narrow pI range (7.2–9.2 vs. 6.1–8.6), and reported a smaller change in the clearance effects (4.3-fold vs. ∼20-fold) compared with our study. We clearly demonstrated here that the major contribution of pI variation on 5B3 clearance differences is a faster distribution phase (Figs. 1 and 4). This is also consistent with the larger volume of distribution and greater AUC 0–10 observed for those 5B3 variants with faster clearance (Table S1). Of note, these 5B3 variants have comparable C0, which means that the distribution differences appeared immediately after dosing. The biodistribution studies suggest this rapid distribution phase reflects antibody catabolism in highly perfused tissues. It is unclear whether the larger clearance effect we observed is related to the isotype difference, resulting in a different charge asymmetry, or reflects a species difference in clearance. Indeed, Igawa et al. observed a smaller effect for clearance measured in cynomolgus monkey.
It was proposed17 that the reduced elimination of the lower pI variants resulted from a decreased amount of fluid phase pinocytosis due to the negative charge on the antibody causing a decrease in binding to cells. In addition, the increased positive charge on the high pI antibody at the lower pH of lysosomes could result in a faster rate of degradation when the antibody is internalized via pinocytosis. The labeling experiments we have performed don't distinguish between these two mechanisms and both may be involved. In both cases, one would expect the site(s) of increased degradation to be metabolically active or highly vascularized organs, which we have observed as the high pI variant is preferentially sequestered and degraded in the liver and spleen relative to the low pI variant. However, examination of the ratio of [111In] signal to [125I] signal for each molecule makes it clear that the high pI variant is more highly degraded in several other tissues, including the lung, heart, intestine, and kidney (Fig. 6D).
It was surprising that the antibody humanized on the κI, VH3 framework (hu5B3.κIVH3.v1) gave relatively fast clearance (29.5 mL/day/kg) in rats. Bevacizumab, also humanized on a κI, VH3 scaffold, and having a pI of 8.2 gives a clearance of 7.0 mL/day/kg in rats. For a panel of 13 antibodies humanized on a κI, VH3 scaffold, clearance in rats is not correlated with pI (Fig. S5). Similarly, for a larger panel of antibodies, clearance in cynomolgus monkey was not correlated with pI.13 The net difference in charged residues in the light chain CDR regions between 5B3 and bevacizumab is zero. Therefore, it seems unlikely that the benefit of change to κ4 framework for clearance of 5B3 reflects an incompatibility between the light chain CDR residues and the κ1 framework. A comparison to other antibodies humanized on the VH3 framework indicates that 5B3 has more negative charge (–2 – –4) in CDR-H2 and more positive (+1 – +2) charge in framework region 3 of the humanized antibody. It is unclear if and how this charge distribution on the heavy chain would combine with light chain framework changes to affect clearance.
The change to κ4, VH1 framework facilitates development of potency improved variants since acceptable clearance is obtained for this framework. Although affinity improved variants were discovered through phage display on the κ1, VH3 framework, they are translatable to the κ4, VH1 template. Molecular modeling suggests that the murine CDRs are equally compatible with the κ1, κ4 and VH1, VH3 scaffolds. These frameworks can support proper CDR conformation, as shown by the equivalent potency of hu5B3.κ1VH3.v1 and hu5B3.κ4VH1.v1, and high thermal stability of both antibodies (Fig. S1), such that changes in solvent-accessible CDR residues would be expected to have similar effects in the different frameworks. Further assessment of the development capability of the potency improved variant will require pre-clinical studies in a species, such as cynomolgus monkey, that is generally accepted as a better predictor of expected behavior in humans. It will be interesting to determine if the charge hypothesis extends to PK properties in primates.
Materials and Methods
Indium-111 (111InCl3 in 0.05 HCl) and di[125I]-Bolton Hunter Reagent (N-succinimidyl-3-(4-hydroxy-3-[125I]iodophenyl)propionate) were purchased from Perkin Elmer. DOTA-NHS ester (C20H31N5O10•HPF6•CF3CO2H; DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; NHS = N-hydroxysuccinimidyl) was purchased from Macrocyclics, Inc. Dimethyl sulfoxide anhydrous and H2O Trace Select Ultratrace analysis were purchased from Sigma Aldrich, Inc. Amicon Ultra-4 centrifugal filter unit with Ultracel-50 kDa was purchased from EMD Millipore. Indium (III) chloride tetrahydrate (InCl3•4H2O) was purchased from Strem Chemicals, Inc. NAP-5 column was purchased from GE Healthcare.
Sorting and analysis of phage-displayed antibody libraries
Binding clones were selected by incubating the phage display libraries with 1 and 0.1 nM biotinylated HCV-E2a or HCV-E2b protein in successive rounds of selection, and then competed with either 100 nM HCV-E2a /E2b or hu5B3.κ1VH3.v1 IgG to reduce enrichment of clones with weaker affinity to HCV-E2 protein. Clones bound to biotinylated target were captured on ELISA plates coated with neutravidin, washed and eluted in 10 mM HCl for 10 min at room temperature. The eluted phage was neutralized with 1/10 volume of 1 M Tris pH 8.0 and used to infect E. coli for amplification for the next round of selection. Clones from the second round of selection were sequenced to determine mutations that are frequent in selected phage.
Competitive Phage ELISA
A competitive phage ELISA18 was used to estimate the affinity of phage-displayed anti-HCV variants. A fixed, subsaturating concentration of phage was preincubated for 2 h with serial dilutions of HCV-E2 protein and then transferred to HCV-E2-coated Maxisorp plates (96well, Nunc). After 15 min incubation, the plates were washed with PBT buffer (PBS containing 0.02% Tween-20) and incubated 30 min with horseradish peroxidase (HRP)/anti-M13 antibody conjugate (1:5000 dilution in PBT buffer) (Pharmacia). The plates were washed, developed for 15 min with 3,3′,5,5′-Tetramethyl-benzidine/H2O2 peroxidase (TMB) substrate (Kirkegaard and Perry Laboratories, Inc.), quenched with 1.0 M H3PO4 and read spectrophotometrically at 450 nm. The binding affinities were estimated as IC50 values defined as the concentration of HCV-E2 protein that blocked 50% of phage binding to immobilized HCV-E2.
Antibody expression and purification
5B3 was humanized as described for hu5B3.κ1VH3.v310 except that consensus κ4, VH1 frameworks were used to generate hu5B3.κ4VH1.v1. Genes for the hu5B3.κ4VH1.v1 antibody heavy and light chain variable domains were synthesized (Blue Heron Inc.). Additional sequence variants were generated using oligonucleotide-directed mutagenesis as described.19 All variants were produced as full-length human IgG1 antibodies by transient expression in CHO cells followed by immunoaffinity purification essentially as described previously.10,19
Imaged Capillary Isoelectric Focusing
Imaged capillary isoelectric focusing was performed on an iCE280 Analyzer (ProteinSimple, Toronto, ON). The separation was performed using a 50 mm long, 100 μm inner diameter × 200 μm outer diameter silica capillary coated with fluorocarbon (Protein Simple).
The anolyte and catholyte were 80 mM phosphoric acid and 100 mM NaOH, respectively, each prepared with 0.1% methylcellulose. IgG samples were prepared by diluting in water containing carboxypeptidase (CpB) enzyme at a ratio of 100:1 (w/w) and incubated at 37 °C for 20 min to remove C-terminal lysine residues. An ampholyte mixture comprised of 3.1% (v/v) Pharmalytes (GE Life Sciences), 2.5 M urea, 0.1% (v/v) methylcellulose, and pI 5.5 and 9.77 markers (Protein Simple) was combined with CpB-treated antibody to obtain a final concentration of 0.25 mg/mL. Final solutions were mixed and centrifuged at 11200-g for 2 min. Samples were introduced to the capillary through a Prince autoinjector and focused for 1 and 10 min at 1.5 kV and 3.0 kV, respectively. Electropherogram were imaged with the optical absorption detector operated at 280 nm.
Neutralization assay
Neutralization assays using HCVpp were performed as described previously.20,21
Surface plasmon resonance and rat FcRn affinity measurements
Soluble rat FcRn (rFcRn) was expressed and purified as reported previously.22 The binding affinity of rFcRn for hIgG1 anti-gD was measured at pH 5.8 by surface plasmon resonance using a Biacore T-200TM instrument (GE Healthcare, Piscataway, NJ). The anti-HCV.E2 variants were immobilized onto a CM5 Series S sensor chip and 12 serial dilutions of rFcRn were injected for 60 s at a flow rate of 50 μL/min. Following each rFcRn injection the surface was regenerated by two 30 s injections of pH 8.0 buffer to dissociate bound rFcRn. Raw sensorgram data were reduced and referenced using the Scrubber II software package (BioLogic Software) and fit to a simple 1:1 binding model under equilibrium conditions.
Pharmacokinetic study in SD rats
Pharmacokinetic (PK) studies in Sprague-Dawley (SD) rats were performed following animal protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Genentech, Inc. SD female rats (n = 4/group) received a single intravenous (IV) anti-HCV-E2 antibody dose of 5 mg/kg. Blood samples were collected from the tail vein at the following time-points: 15min, 1, 8, and 24 h; and 3, 8, 10, 14, 21, 28, and 35 d, and processed to collect serum and/or plasma for subsequent bioanalysis (see below).
Bioanalysis of serum samples from pharmacokinetic studies
Serum samples from rats were analyzed for anti-E2 antibody concentrations using a quantitative enzyme-linked immunosorbent assay (ELISA). The ELISA utilized a soluble E2 capture agent and a goat anti-human IgG Fc-horseradish-peroxidase (HRP) detection agent for humanized 5B3 variants. The minimum quantifiable concentrations were 0.0625 ug/ml for hu5B3.κ1VH3.v1, and 0.0031 ug/ml for other variants. Anti-therapeutic antibody (ATA) was not measured in this study
Pharmacokinetic data analysis
Serum concentration-time profiles were used to estimate the following PK parameters in rats using non-compartmental analysis (WinNonlin, version 5.2.1; Pharsight Corporation): the area under the serum concentration-time curve (AUC) until day 10 (AUCDays0–10), concentrations at Time = 0 following an IV bolus dose (C0). In addition, two-compartmental analyses (WinNonlin, version 5.2.1; Pharsight Corporation) were used to estimate clearance (CL) and other PK parameters (not reported here). PK parameters are reported as mean ± standard deviation for n = 4 animals per group. Nominal sample collection times and doses were used in the data analysis. Values below the limit of detection (LOD) were not used in PK analysis or for graphical or summary purposes. The time points that showed potential ATA impacts were removed from the data analysis in the two-compartmental analysis.
DOTA Conjugation
Anti-E2 h5B3 charged variants of pI = 6.10 and pI = 8.61 were labeled with DOTA for 111In complexation by random modification of lysine residues as previously described.23 Anti-E2 h5B3 charged variants of pI = 6.10 and pI = 8.61 were conjugated at a ratio of 5 mol equivalent of DOTA-NHS ester to 1 mol of antibody. A 2-μL aliquot of 37.33 mM solution of DOTA-NHS in DMSO was added to anti-E2 h5B3 charged variants (pI = 6.10, pI = 8.61; 1.67 and 1.94 mg, respectively) in 0.5 mL of borate buffer, pH 8.5.
Radiochemistry
DOTA-conjugated anti-E2 hB3 charged variants were radiolabeled with 111In as previously described with a slight modification.23 To a conical polypropylene vial was added 300 μCi of 111In, 8.5 μL of 0.3 M ammonium acetate buffer, and 8.5 μL of DOTA conjugated h5B3 variant (pI = 6.10 or pI = 8.61; 0.128 and 0.125 mg, respectively). At the end of the reaction, 2 μL of 4.26 mM of the naturally occurring isotope, 115InCl3, was added to saturate unoccupied DOTA (and neutralize its negative charge) with constant mixing (350 rpm) for 10 min. The reaction was then terminated by adding a 5 μL of 50 mM EDTA challenge solution followed by an additional of 75 μL of 0.3 M ammonium acetate buffer, pH 7.0. Radiochemical yields were 70.0% and 80.3%, respectively.
Radioiodination of anti-E2 h5B3 charged variants of pI = 6.10 and pI = 8.61 was performed by using the Bolton Hunter method.24 Immediately prior to use, 12 μL of di[125I]-Bolton Hunter Reagent was gently evaporated to dryness with a gentle stream of N2. A 75 μg aliquot of each mAb in 25 mM borate buffer, pH 8.5 was then added to the reaction. After 15 min reaction at 25 °C with constant mixing (350 rpm), the reaction mixtures were purified using NAP5 column pre-equilibrated in PBS. Radiochemical yields of 85.1% and 70.2% were obtained for anti-E2 h5B3 charged variants of pI = 6.10 and pI = 8.61, respectively.
Purity of the radiolabeled mAbs was assessed by size-exclusion radio-HPLC (isocratic, PBS, 0.5 mL/min) on an Agilent 1100 series HPLC system coupled with a Raytest Gabi Star radioactive flow monitor running ChemStation software. The column was a Biosep-SEC S-3000 column, 200 mm × 7.8 mm, 5 μm (Phenomenex). The mobile phase was PBS, and the flow rate was 0.5 mL/min for 32 min (isocratic).
Biodistribution and Pharmacokinetics Studies
All experimental animal studies were conducted according to protocols that were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) of Genentech Laboratory Animal Research (LAR). Female Sprague-Dawley rats in a weight range of 150 – 225 g at the initiation of the study were randomly assigned to 2 groups (n = 5 per group), and administered an intravenous bolus, via femoral cannula, consisting of a mixture of 125I- and 111In-labeled h5B3 tracer (5 μCi of each) together with the respective unmodified antibody (pI = 6.10 or pI = 8.61) for a total dose of 5 mg/kg. Blood samples were collected from each animal via jugular cannula at 15 min, 1 h, 6 h, 1 d and via cardiac puncture at 3 d to derive plasma and total conjugated antibody concentration. At the end of the 3 d time point, tissues were harvested for analysis as previously described.25
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Tom Patapoff and Vikas Sharma for help with calculations of Fv hydrophobicity, Jeff Lutman for contributions to supplemental figures, and Saileta Prabhu and Leslie Dickmann for critical review of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Bradley DW. Studies of non-A, non-B hepatitis and characterization of the hepatitis C virus in chimpanzees. Curr Top Microbiol Immunol 2000; 242:1-23; PMID:10592653; http://dx.doi.org/ 10.1007/978-3-642-59605-6_1 [DOI] [PubMed] [Google Scholar]
- 2. Trowbridge R, Gowans EJ. Identification of novel sequences at the 5′ terminus of the hepatitis C virus genome. J Viral Hepat 1998; 5:95-8; PMID:9572033; http://dx.doi.org/ 10.1046/j.1365-2893.1998.00090.x [DOI] [PubMed] [Google Scholar]
- 3. Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631-48; PMID:10859368 [DOI] [PubMed] [Google Scholar]
- 4. Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol 2000; 242:55-84; PMID:10592656; http://dx.doi.org/ 10.1007/978-3-642-59605-6_4 [DOI] [PubMed] [Google Scholar]
- 5. Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. . Binding of hepatitis C virus to CD81. Science 1998; 282:938-41; PMID:9794763; http://dx.doi.org/ 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 6. Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 2002; 21:5017-25; PMID:12356718; http://dx.doi.org/ 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benedicto I, Molina-Jiménez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernández A, Aldabe R, López-Cabrera M, et al. . The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol 2009; 83:8012-20; PMID:19515778; http://dx.doi.org/ 10.1128/JVI.00038-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009; 457:882-6; PMID:19182773; http://dx.doi.org/ 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007; 446:801-5; PMID:17325668; http://dx.doi.org/ 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 10. Pantua H, Diao J, Ultsch M, Hazen M, Mathieu M, McCutcheon K, Takeda K, Date S, Cheung TK, Phung Q, et al. . Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol 2013; 425:1899-914; PMID:23458406; http://dx.doi.org/ 10.1016/j.jmb.2013.02.025 [DOI] [PubMed] [Google Scholar]
- 11. Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 2011; 3:61-6; PMID:20962582; http://dx.doi.org/ 10.4161/mabs.3.1.13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dennis MS. CDR repair: A novel approach to antibody humanization. In: Shire SJ, Gombotz W, Becthold-Peters K, Andya J, eds. Current trends in monoclonal antibody development and manufacturing. New York, New York: Springer, 2010:9-28. [Google Scholar]
- 13. Hötzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. . A strategy for risk mitigation of antibodies with fast clearance. MAbs 2012; 4:753-60; PMID:23778268; http://dx.doi.org/ 10.4161/mabs.22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bumbaca D, Wong A, Drake E, Reyes AE, 2nd, Lin BC, Stephan JP, Desnoyers L, Shen BQ, Dennis MS. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs 2011; 3:376-86; PMID:21540647; http://dx.doi.org/ 10.4161/mabs.3.4.15786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vugmeyster Y, Szklut P, Wensel D, Ross J, Xu X, Awwad M, Gill D, Tchistiakov L, Warner G. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm Res 2011; 28:1696-706; PMID:21424161; http://dx.doi.org/ 10.1007/s11095-011-0405-x [DOI] [PubMed] [Google Scholar]
- 16. Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. . Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One 2013; 8:e57479; PMID:23468998; http://dx.doi.org/ 10.1371/journal.pone.0057479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. . Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; http://dx.doi.org/ 10.1093/protein/gzq009 [DOI] [PubMed] [Google Scholar]
- 18. Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol 2000; 328:333-63; PMID:11075354; http://dx.doi.org/ 10.1016/S0076-6879(00)28406-1 [DOI] [PubMed] [Google Scholar]
- 19. Kelley RF, Meng YG. Methods to engineer and identify IgG1 variants with improved FcRn binding or effector function. Methods Mol Biol 2012; 901:277-93; PMID:22723108; http://dx.doi.org/ 10.1007/978-1-61779-931-0_18 [DOI] [PubMed] [Google Scholar]
- 20. Diao J, Pantua H, Ngu H, Komuves L, Diehl L, Schaefer G, Kapadia SB. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol 2012; 86:10935-49; PMID:22855500; http://dx.doi.org/ 10.1128/JVI.00750-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hötzel I, Chiang V, Diao J, Pantua H, Maun HR, Kapadia SB. Efficient production of antibodies against a mammalian integral membrane protein by phage display. Protein Eng Des Sel 2011; 24:679-89; PMID:21810920; http://dx.doi.org/ 10.1093/protein/gzr039 [DOI] [PubMed] [Google Scholar]
- 22. Martin WL, West AP Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell 2001; 7:867-77; PMID:11336709; http://dx.doi.org/ 10.1016/S1097-2765(01)00230-1 [DOI] [PubMed] [Google Scholar]
- 23. Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, Fourie A, Chuh J, Koppada N, Saad O, et al. . Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem 2011; 22:1994-2004; PMID:21913715; http://dx.doi.org/ 10.1021/bc200212a [DOI] [PubMed] [Google Scholar]
- 24. Bolton AE, Hunter WM. The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acylating agent. Biochem J 1973; 133:529-39; PMID:4733239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boswell CA, Mundo EE, Zhang C, Stainton SL, Yu SF, Lacap JA, Mao W, Kozak KR, Fourie A, Polakis P, et al. . Differential effects of predosing on tumor and tissue uptake of an 111In-labeled anti-TENB2 antibody-drug conjugate. J Nucl Med 2012; 53:1454-61; PMID:22872740; http://dx.doi.org/ 10.2967/jnumed.112.103168 [DOI] [PubMed] [Google Scholar]
- 26. Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature 1986; 319:199-203; PMID:3945310; http://dx.doi.org/ 10.1038/319199a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.