

Abstract
The main purpose of newborn screening is to diagnose genetic, metabolic, and other inherited disorders, at their earliest to start treatment before the clinical manifestations become evident. Understanding and tracing the biochemical data obtained from tandem mass spectrometry is vital for early diagnosis of metabolic diseases associated with such disorders. Accordingly, it is important to focus on the entire diagnostic process, including differential and confirmatory diagnostic options, and the major factors that influence the results of biochemical analysis. Compared to regular biochemical testing, this is a complex process carried out by a medical physician specialist. It is comprised of an integrated program requiring multidisciplinary approach such as, pediatric specialist, expert scientist, clinical laboratory technician, and nutritionist. Tandem mass spectrometry is a powerful tool to improve screening of newborns for diverse metabolic diseases. It is likely to be used to analyze other treatable disorders or significantly improve existing newborn tests to allow broad scale and precise testing. This new era of various screening programs, new treatments, and the availability of detection technology will prove to be beneficial for the future generations.
Keywords: Tandem mass spectrometry, Inherited metabolic disease, Early diagnosis, Differential diagnosis, Newborn screening
Introduction
Inborn errors of metabolism are a heterogeneous group of metabolic disorders caused by an enzyme defect in a metabolic pathway, leading to dysfunction of metabolism and the accumulation of toxic intermediate metabolites. Newborn screening test (NST), introduced in the 1960s by scientist Robert Guthrie, is used to screen newborn and infants for metabolic disorders using classical methods that includes Guthrie assay, bacterial inhibition assay, and enzyme immune assay. Later, tandem mass spectrometry had been introduced as combined with fast atom bombardment and electrospray ionization1,2,3,4,5,6,7) recently with high resolution chromatography8) and direct analysis in real time9) mass spectrometry for NST1,2,3,4,5,6,7,8,9,10). For the recent two decades, tandem mass spectrometry has been applied to screen over 60 different defined metabolic disorders, thus enabling their early diagnosis and treatment2,11,12). The specificity and sensitivity of tandem mass spectrometry method could be up to 99.995% and 99%, respectively, for most organic acidemias, amino acid disorders, and fatty acid oxidation disorders as well as extended program comprosing of Fabry, Pompe, and mucopolysaccharidosis-I13,14,15). Though the technical advances achieved in mass spectrometry have facilitated the use of dozens of various biochemical markers, special care needs to be taken in interpreting output data of the tested marker metabolites because false interpreting can lead to a misdiagnosis12,16,17,18) to metabolic disorders more thoroughly, and speed up the process of early diagnosis and treatment12,16,17,18).
Metabolites as a targeted marker on dried blood-spot and inborn metabolic diseases
One abnormal finding in tandem mass spectrometry points to several different metabolic diseases as shown in Table 119,20,21). One of the reasons for such abnormal result is that the acylcarnitine isomers cannot be differentiated by tandem mass spectrometry. Medication can also trigger changes in the body that mimic a metabolic disease. For these reasons, tandem mass spectrometry is often performed for initial mass screening, and second alternate tests, such as urine organic acid analysis and plasma amino acid analysis, are additionally required for the confirmatory follow-up diagnostic test19,20,21).
Table 1. Metabolites and suspicious metabolic disease.
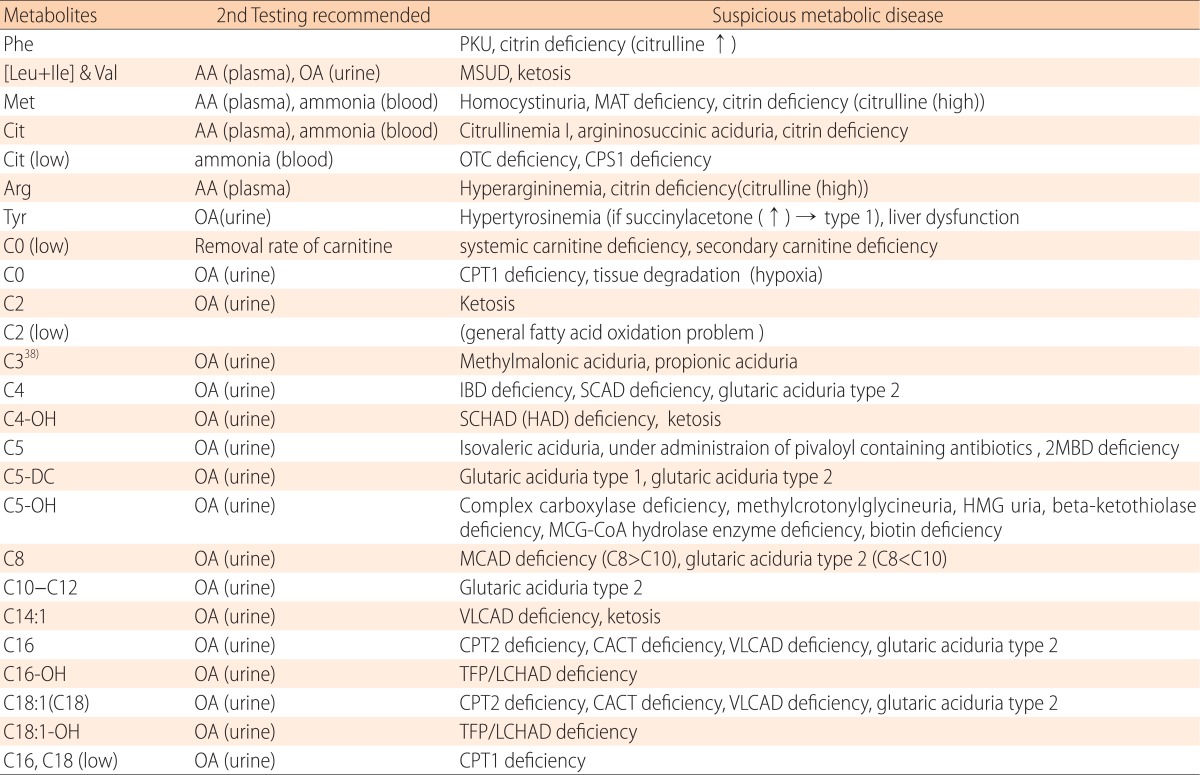
PKU, phenylketonuria; AA, amino acid analysis; OA, organic acid analysis; MSUD, maple syrup urine disease; MAT, methionine adenosyl transferase ; OTC, ornithine transcarbamylase; CPS1, carbamoyl phosphate synthetase I; IBD, isobutyryl CoA dehydrogenase; SCAD, short chain acyl-CoA dehydrogenase; SCHAD, short chain 3-hydroxyacyl-CoA dehydrogenase; 2MBD, 2-methylbutyryl-CoA dehydrogenase; HMG, 3-hydroxy-3-methyl-glutaryl-CoA reductase; MCG, methylcrotonylglycine; MCAD, medium-chain acyl-coenzyme A dehydrogenase; VLCAD, very long-chain acyl CoA dehydrogenase; CPT, carnitine palmitoyltransferase; CACT, carnitine acylcarnitine translocase; TFP, trifunctional protein; LCHAD, long-chain 3-hydroxyacyl-CoA dehydrogenase.
Acylcarnitine as a marker for fatty acid oxidation disorder and organic aciduria
It is necessary to differentiate between systemic carnitine deficiency (carnitine uptake deficiency, CUD) and secondary carnitine deficiency in case of low free carnitine (C0)18,20). CUD is characterized by increased renal excretion and low concentrations of C0 and all acylcarnitines. CUD is a congenital deficiency of carnitine/organic cation transporter 2 characterized by defects in the beta-oxidation pathway due to low carnitine levels. Though the diagnostic marker for CUD is decreased C0 value, the possibility of false negative cannot be ruled out, especially in long-stored samples due to increase in C0 values over time. However, there is a report that systemic carnitine deficiency is hardly influenced by hydrolysis because of the low level of the total acylcarnitine19,22). On the other hand, there are cases where secondary carnitine deficiency is accompanied by increase of a specific acylcarnitine (increase of 3-hydroxyisovaleylcarnitine [C5-OH], C5, and C3 in methylcrotonylglycinuria, isovaleric aciduria, and methylmalonic aciduria, respectively). While screening newborns if the mother has systemic carnitine deficiency, the newborn exhibits low C0 concentration even when unaffected by the metabolic disease. Moreover, it has been reported that if the mother is affected by glutaric aciduria type 1, secondary carnitine deficiency can occur and the newborn can also show low C0 concentration. In many organic aciduria patients, secondary free carnitine deficiency occurs with elevation of specific acylcarnitine. For this reason, long-stored samples are likely to fail to confirm secondary carnitine deficiency due to the ease of hydrolytic degradation22).
If muscle tissue proteolysis occurs in carnitine palmitoyltransferase I (CPT1) deficiency and hypoxic shock, large quantity of C0 contained in the tissue is released into the blood, thus, increasing the blood free carnitine (C0) concentration. This is very often concurrent with the elevation of C3 and C4. The characteristic of CPT1 deficiency are increased C0 concentration, decreased long-chain acylcarnitine, and increased C0/[C16+C18] ratio. However, samples unaffected with CPT1 deficiency can present a pattern similar to that of CPT1 deficiency due to hydrolysis of acylcarnitine22,23).
In case of concurrent increase in 3-hydroybutyryl carnitine (C4-OH) and C2, the odds are high that 3-hydroxylactate also increases, and ketosis can be suspected. Since C2 levels increase in short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD)/3-hydroxyacyl-CoA dehydrogenase (HAD) deficiency, increased C4-OH/C2 ratio is used as a marker for screen SCHAD deficiency. Additionally, by the time SCHAD deficiency is manifested, C6-OH increases concurrently. However, care should be taken when a nonderivatization method is used in tandem mass spectrometric analysis it is expressed by the increase of glutarylcarnitine (C5-DC) instead of elevation of C6-OH22,23,24).
An elevation of C5 is indicative of the presence of one of these four isomers; isovalerylcarnitine, 2-methylbutyrylcarnitine, pivaloylcarnitine, and valerylcarnitine. Isovalerylcarnitine increases in isovaleryl aciduria, and so does 2-methylbutyryl-CoA dehydrogenase deficiency 2-methylbutyrylcarnitine16,25,26,27). A long-term administration of oral antibiotics and some parenteral antimicrobials, it leads to increase in pivaloylcarnitine. They can be differentiated by urine organic acid analysis, whereby slight increase of C5 can occur for unclear reasons23,25).
High C5-DC level requires a differential diagnosis (glutaric acidemia types I and II) by urine organic acid analysis. Newborn patient with glutaric aciduria type 1 excretes abnormal quantities of glutaric acid and 3-hydroxyglutaric acid whereas glutaric aciduria type 2, 2-hydroxyglutaric acid. However, elevation of glutaric acid excretion in infants and young children is not always clearly noticeable in glutaric aciduria type 1. When ketone increases, C5-DC levels may be slightly elevated23).
If C5-OH increases, diagnosis is simple because urine exhibits a distinctively abnormal metabolite excretion in urine organic acid analysis. If a preterm infant is fed special formula devoid of biotin, C5-OH increases due to biotin deficiency. While biotin injection can rapidly lower the serum C5-OH level, C5-OH accumulated in the red blood cells cannot transfuse out and C5-OH level continues to rise depending on red blood cell survival time. The urine organic acid analysis of a preterm infant with biotin deficiency reveals the elevated level of 3-hydroxyisovaleric acid. If the mother has methylcrotonylglycinuria, the maternal C5-OH is transfused to the fetus and gets accumulated in the red blood cells. In such a case, C5-OH accumulated in the red blood cells is detected in the dried blood-spot analysis, resulting in a false positive result25). For confirmatory diagnosis, maternal urine organic acid analysis is necessary23).
C5-OH increases at an extremely low rate in mild beta-ketothiolase deficiency unless symptoms are acute.
In most of such cases, mothers are not aware of their pathological conditions. They usually exhibit mild symptoms or remain asymptomatic depending on environmental factors during their childhood and youth. Although found through newborn screening the maternal blood free carnitine level is very low, symptoms may or may not manifest depending on any given occasion. Hypocarnitinemia may be prevented by carnitine intake as needed23).
Increase in C6, C8, C10, with C8 most markedly, indicates medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency28,29,30). Slightly elevated C8 of a neonate is also presented in MCAD deficiency carrier. If C10 is higher than C8, glutaric aciduria type 2 is suspected. Elevated levels of C8 and C10 also appear in preterm infants fed with preterm formula in the form of medium-chain triglyceride (MCT) oil25), which can be confirmed with enzyme activity assay and gene analysis22).
Specific diagnostic marker, C14:1 carnitine increases in very long-chain acyl CoA dehydrogenase (VLCAD) deficiency. Extremely severe case of VLCAD deficiency is associated with more marked increase in C16 and C18:1 than in C14:1 in both serum and dried blood-spot analysis. Carnitine palmitoyl transferase 2 (CPT2) deficiency and carnitine acylcarnitine translocase (CACT) deficiency involves elevated C16 and C18:131), but no change in C14:1 levels, and can thus be easily differentiated from VLCAD deficiency. C14:1 is slightly elevated in ketosis as well, which involves increase in C2, thus lowering the C14:1/C2 ratio. For confirmatory diagnosis, enzyme activity assay and gene analysis should be performed22).
An elevation of C16 and C18:1 (or C18) point towards CPT 2 deficiency and CACT deficiency. Since certain levels of C16 and C18:1 are found in blood-spot even in healthy conditions, reflecting the red blood cell concentration, only a remarkable elevation can be considered to be abnormal31). Increase in C14:1 along with considerably elevated C16 and C18:1 levels is indicative of VLCAD deficiency, and not CPT 2 or CACT deficiency. If C2 level is increased along with slightly elevated C16 and C18:1 levels, odds for ketosis are very much high22).
Low level of C16 and C18:1 (C18) in dried blood-spot analysis is indicative of CPT1 deficiency. Decreased C16 and C18:1 (C18) levels can also be found in neonates who have recovered from extremely ill conditions such as suspended animation, but the phenomenon is transient and the levels are often normalized in re-examination22).
An elevation of C16-OH and C18:1-OH is a condition found in mitochondrial trifunctional protein (TFP)/long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. In LCHAD deficiency C18-OH level increases as well as, at a higher rate, C18:1-OH increases. Patients with severe type of TFP/LCHAD deficiency exhibit significantly elevated C16-OH and C18:1-OH levels, but the degree of increase fluctuates, and severity cannot be predicted only by the degree of increase of C18:1-OH levels. If C2 is increased along with C16-OH and C18:1-OH, it is most likely caused due to ketosis6,18,22,23,32).
Amino acids as a marker for amino acid metabolism disorders
An elevated level of tyrosine detected in newborn screening by tandem mass spectrometry is usually due to transient neonatal hypertyrosinemia. To differentiate, tyrosinemia type 1 can be diagnosed by presence of succinylacetone in serum or urine34). Although the patient has tyrosinemia type 1, tyrosine concentration does not always high levels as in neonate32).
An elevated phenylalanine (Phe) is indicative of phenylketonuria33). Additionally, newborns and infants with neonatal intrahepatic cholestasis caused by citrine deficiency may have elevated Phe levels concomitant with increase in citrulline. Phe may increase along with tyrosine and methionine levels, secondary to liver dysfunction3).
An elevated citrulline appears in citrullinemia type 1, argininosuccinic aciduria, and citrine deficiency. Argininosuccinic acid increases concurrently during argininosuccinic aciduria in urine. Patients with neonatal intrahepatic cholestasis caused by citrine deficiency have an elevated amino acid level, besides citrulline, accompanied by occasional increase in serum galactose32).
Low serum citrulline concentration is associated with ornithine transcarbamylase (OTC) deficiency34) and carbamoyl phosphate synthetase I (CPS1) deficiency concomitant with hyperammonemia. However, in newborn screening by tandem mass spectrometry, these diseases have been reported even in cases with normal citrulline concentration32).
An elevated leucine, isoleucine, and valine appear in maple syrup urine disease (MSUD)35). If it is concomitant with C2 increase, odds are high that it is due to the ketosis-related expedited catabolism. For differential diagnosis, serum alloisoleucine analysis and urine organic acid analysis can be used, the presence of alloisoleucine is essential in MSUD. Enzyme activities could be measured for confirmatory diagnosis32).
Conditions influencing numeric value of metabolites
In quantifying targeted metabolites using tandem mass spectrometry, many factors influence the results of acylcarnitine and amino acids7,18,31). In newborn screening, level of metabolites can be moderately changed in accordance with changed biological metabolic process by medical treatment or influenced by specimen status based on various external factors32,36).
As for sample storage environment if samples are stored in high temperature with humidified conditions, levels of measured metabolites are likely to yield inaccurate values. Especially under such environments level of free carnitine tend to increase and acylcarnitine decrease. Altered metabolite value can be minimized by storing the dried blood samples in a freezer (or refrigerator), preferably in an airtight bag. If possible fresh samples should be used except for unavoidable cases such as postmortem examination after a sudden death37,38).
Regarding effect of pH ayclcarnitine becomes more unstable in a high pH than in low pH environment. In urine samples stored prolonged period, the pH tends to exceed pH >8.0, the influence of pH should be accounted for. Excessive hemolysis increase in long-chain acylcarnitine level due to excessive hemolysis37). Hemolysis induced by "squeezing blood collection" does not pose problems, but samples with excessive hemolysis can yield findings similar to long-chain fatty acid metabolism disorder38).
Short-chain acylcarnitine (C-C4) is more prone to hydrolysis than long- (C12-C18) and medium-chain (C5-C10) acylcarnitine species25). Therefore, propionic acidemia and methylmalonic acidemis, for which C3 acts as biomarker, may not be detected in dried blood spot stored at room temperature for over one year. In contrast, long-chain fatty acid metabolism disorder was diagnosable without difficulties in blood-spot samples stored for over three years32,37,38).
The measured values of C6, C8, C10, and C12 increase while administering MCT oil. The ratio of C8/C10 can be used to differentiate such increases due to MCAD deficiency. Many reported cases proved that urine organic acid analysis is efficient for differentiation between MCT oil and MCAD deficiency32,37).
Some total parenteral nutrition (TPN) products are rich in branched chain amino acids, leading to test values similar to that in MSUD20). For differential diagnosis, amino acid ratios, such as Phe/Tyr, Met/Phe, and Leu/Ala ratios, are important markers32). In some cases, detailed secondary confirmatory testing is required. Most importantly, specimen should be collected before beginning parenteral nutrition as much as possible. Moreover, TPN contains substances similar to C8 and C12, whose levels may look elevated2). Since TPN does not contain carnitine, babies under a long-term parenteral nutrition therapy can develop carnitine deficiency25).
Some antibiotics such as ampicillin and cefotaxime can induce increase in the level of acylcarnitines (C5, C14:1, and C16:1-OH). The level of C5 can increase by consuming antibiotics containing the pivaloyl functional group, such as cefcapenepivoxil, cefditorenpivoxil, and cefteram pivoxil24). There have been cases in which C5 levels increased after the administration of ciberestat. These components can be differentiated by urine organic acid analysis32,38).
If the mother has a carrier for inborn metabolic disorder, blood-spot result of her healthy neonate can exhibit abnormal findings in newborn screening. Reported metabolic diseases like these cases are associated with asymptomatic systemic carnitine deficiency, methylcrotonylglycinuria, 3-methylglutaconic aciduria, glutaric aciduraia type 1, and vitamin B12 deficiency. Neonates from vegetarian mothers show decreased free carnitine levels due to the mother's status of low carnitine level resulted from vegetarianism32,39).
In addition, other conditions influencing numeric value of metabolites are in a poorly fed neonate and cholestasis neonate38,39,40).
Newborns with low birth weight often exhibit transient neonatal tyrosinemia. In preterm and low-birth weight newborns, implications of measured values are often unclear and no defined reference values have been established so far39). When interpreting the test values for preterm and low-birth weight newborns, it should also be borne in mind that nonspecific pathological manifestations can also affect measured value15,18,39).
The blood test is generally performed when a baby is 24 to 48 hours old after enough feeding. This timing is very important because certain conditions may go undetected if the blood sample is drawn before 24 hours of age. Newborn screening does not confirm a baby has a certain condition. If a positive screen is detected, parents will be notified immediately and follow-up testing will be done.
Conclusions
Three decades have passed since the first newborn screening has started in South Korea in 1985. To fulfill the goal of newborn screening, early diagnosis and treatment of metabolic diseases, integrated interdisciplinary collaboration between specialist pediatricians and expert scientist of related fields is essential, especially for accurate measurements and interpretations of biochemical data obtained by tandem mass spectrometry. Thereby reflecting each patient's meticulously examined physical status, enables customized differential diagnosis of inherited metabolic disorders.
Acknowledgments
This study was supported by the Duksung Women's University Research Grant 2015, Seoul, South Korea.
Footnotes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Chace DH, DiPerna JC, Mitchell BL, Sgroi B, Hofman LF, Naylor EW. Electrospray tandem mass spectrometry for analysis of acylcarnitines in dried postmortem blood specimens collected at autopsy from infants with unexplained cause of death. Clin Chem. 2001;47:1166–1182. [PubMed] [Google Scholar]
- 2.Chace DH, DiPerna JC, Naylor EW. Laboratory integration and utilization of tandem mass spectrometry in neonatal screening: a model for clinical mass spectrometry in the next millennium. Acta Paediatr Suppl. 1999;88:45–47. doi: 10.1111/j.1651-2227.1999.tb01156.x. [DOI] [PubMed] [Google Scholar]
- 3.Chace DH, Sherwin JE, Hillman SL, Lorey F, Cunningham GC. Use of phenylalanine-to-tyrosine ratio determined by tandem mass spectrometry to improve newborn screening for phenylketonuria of early discharge specimens collected in the first 24 hours. Clin Chem. 1998;44:2405–2409. [PubMed] [Google Scholar]
- 4.Chen WH, Hsieh SL, Hsu KP, Chen HP, Su XY, Tseng YJ, et al. Web-based newborn screening system for metabolic diseases: machine learning versus clinicians. J Med Internet Res. 2013;15:e98. doi: 10.2196/jmir.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall PL, Marquardt G, McHugh DM, Currier RJ, Tang H, Stoway SD, et al. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med. 2014;16:889–895. doi: 10.1038/gim.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.la Marca G. Mass spectrometry in clinical chemistry: the case of newborn screening. J Pharm Biomed Anal. 2014;101:174–182. doi: 10.1016/j.jpba.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 7.Clinical and Laboratory Standard Institute. Newborn screening by tandem mass spectrometry; Approved guideline. Wayne (PA): Clinical and Laboratory Standard Institute; 2010. [Google Scholar]
- 8.Miller JH, 4th, Poston PA, Karnes HT. A quantitative method for acylcarnitines and amino acids using high resolution chromatography and tandem mass spectrometry in newborn screening dried blood spot analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;903:142–149. doi: 10.1016/j.jchromb.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang C, Zhu H, Cai Z, Song F, Liu Z, Liu S. Newborn screening of phenylketonuria using direct analysis in real time (DART) mass spectrometry. Anal Bioanal Chem. 2013;405:3159–3164. doi: 10.1007/s00216-013-6713-8. [DOI] [PubMed] [Google Scholar]
- 10.Dott M, Chace D, Fierro M, Kalas TA, Hannon WH, Williams J, et al. Metabolic disorders detectable by tandem mass spectrometry and unexpected early childhood mortality: a population-based study. Am J Med Genet A. 2006;140:837–842. doi: 10.1002/ajmg.a.31180. [DOI] [PubMed] [Google Scholar]
- 11.Rinaldo P, Hahn S, Matern D. Clinical biochemical genetics in the twenty-first century. Acta Paediatr Suppl. 2004;93:22–26. doi: 10.1111/j.1651-2227.2004.tb03051.x. [DOI] [PubMed] [Google Scholar]
- 12.Walter JH, Patterson A, Till J, Besley GT, Fleming G, Henderson MJ. Bloodspot acylcarnitine and amino acid analysis in cord blood samples: efficacy and reference data from a large cohort study. J Inherit Metab Dis. 2009;32:95–101. doi: 10.1007/s10545-008-1047-y. [DOI] [PubMed] [Google Scholar]
- 13.Duffey TA, Bellamy G, Elliott S, Fox AC, Glass M, Turecek F, et al. A tandem mass spectrometry triplex assay for the detection of Fabry, Pompe, and mucopolysaccharidosis-I (Hurler) Clin Chem. 2010;56:1854–1861. doi: 10.1373/clinchem.2010.152009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coman D, Bhattacharya K. Extended newborn screening: an update for the general paediatrician. J Paediatr Child Health. 2012;48:E68–E72. doi: 10.1111/j.1440-1754.2011.02199.x. [DOI] [PubMed] [Google Scholar]
- 15.Mak CM, Lee HC, Chan AY, Lam CW. Inborn errors of metabolism and expanded newborn screening: review and update. Crit Rev Clin Lab Sci. 2013;50:142–162. doi: 10.3109/10408363.2013.847896. [DOI] [PubMed] [Google Scholar]
- 16.Levy PA. An overview of newborn screening. J Dev Behav Pediatr. 2010;31:622–631. doi: 10.1097/DBP.0b013e3181eedf01. [DOI] [PubMed] [Google Scholar]
- 17.Rebollido-Fernandez MM, Castineiras DE, Boveda MD, Couce ML, Cocho JA, Fraga JM. Development of electrospray ionization tandem mass spectrometry methods for the study of a high number of urine markers of inborn errors of metabolism. Rapid Commun Mass Spectrom. 2012;26:2131–2144. doi: 10.1002/rcm.6325. [DOI] [PubMed] [Google Scholar]
- 18.Eddy M, Gottesman GS. Newborn metabolic screening and related pitfalls. Mo Med. 2009;106:234–240. [PubMed] [Google Scholar]
- 19.Rashed MS, Bucknall MP, Little D, Awad A, Jacob M, Alamoudi M, et al. Screening blood spots for inborn errors of metabolism by electrospray tandem mass spectrometry with a microplate batch process and a computer algorithm for automated flagging of abnormal profiles. Clin Chem. 1997;43:1129–1141. [PubMed] [Google Scholar]
- 20.Scolamiero E, Cozzolino C, Albano L, Ansalone A, Caterino M, Corbo G, et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol Biosyst. 2015;11:1525–1535. doi: 10.1039/c4mb00729h. [DOI] [PubMed] [Google Scholar]
- 21.Selim LA, Hassan SA, Salem F, Orabi A, Hassan FA, El-Mougy F, et al. Selective screening for inborn errors of metabolism by tandem mass spectrometry in Egyptian children: a 5 year report. Clin Biochem. 2014;47:823–828. doi: 10.1016/j.clinbiochem.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Waisbren SE, Landau Y, Wilson J, Vockley J. Neuropsychological outcomes in fatty acid oxidation disorders: 85 cases detected by newborn screening. Dev Disabil Res Rev. 2013;17:260–268. doi: 10.1002/ddrr.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marquardt G, Currier R, McHugh DM, Gavrilov D, Magera MJ, Matern D, et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet Med. 2012;14:648–655. doi: 10.1038/gim.2012.2. [DOI] [PubMed] [Google Scholar]
- 24.Abdenur JE, Chamoles NA, Guinle AE, Schenone AB, Fuertes AN. Diagnosis of isovaleric acidaemia by tandem mass spectrometry: false positive result due to pivaloylcarnitine in a newborn screening programme. J Inherit Metab Dis. 1998;21:624–630. doi: 10.1023/a:1005424331822. [DOI] [PubMed] [Google Scholar]
- 25.De T, Kruthika-Vinod TP, Nagaraja D, Christopher R. Postnatal variations in blood free and acylcarnitines. J Clin Lab Anal. 2011;25:126–129. doi: 10.1002/jcla.20445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janzen N, Steuerwald U, Sander S, Terhardt M, Peter M, Sander J. UPLC-MS/MS analysis of C5-acylcarnitines in dried blood spots. Clin Chim Acta. 2013;421:41–45. doi: 10.1016/j.cca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Forni S, Fu X, Palmer SE, Sweetman L. Rapid determination of C4-acylcarnitine and C5-acylcarnitine isomers in plasma and dried blood spots by UPLC-MS/MS as a second tier test following flow-injection MS/MS acylcarnitine profile analysis. Mol Genet Metab. 2010;101:25–32. doi: 10.1016/j.ymgme.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Blois B, Riddell C, Dooley K, Dyack S. Newborns with C8-acylcarnitine level over the 90th centile have an increased frequency of the common MCAD 985A>G mutation. J Inherit Metab Dis. 2005;28:551–556. doi: 10.1007/s10545-005-0551-6. [DOI] [PubMed] [Google Scholar]
- 29.Najdekr L, Gardlo A, Madrova L, Friedecky D, Janeckova H, Correa ES, et al. Oxidized phosphatidylcholines suggest oxidative stress in patients with medium-chain acyl-CoA dehydrogenase deficiency. Talanta. 2015;139:62–66. doi: 10.1016/j.talanta.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 30.Aksglaede L, Christensen M, Olesen JH, Duno M, Olsen RK, Andresen BS, et al. Abnormal newborn screening in a healthy infant of a mother with undiagnosed medium-chain acyl-CoA dehydrogenase deficiency. JIMD Rep. 2015;23:67–70. doi: 10.1007/8904_2015_428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fingerhut R, Ensenauer R, Roschinger W, Arnecke R, Olgemoller B, Roscher AA. Stability of acylcarnitines and free carnitine in dried blood samples: implications for retrospective diagnosis of inborn errors of metabolism and neonatal screening for carnitine transporter deficiency. Anal Chem. 2009;81:3571–3575. doi: 10.1021/ac8022235. [DOI] [PubMed] [Google Scholar]
- 32.Ozben T. Expanded newborn screening and confirmatory follow-up testing for inborn errors of metabolism detected by tandem mass spectrometry. Clin Chem Lab Med. 2013;51:157–176. doi: 10.1515/cclm-2012-0472. [DOI] [PubMed] [Google Scholar]
- 33.Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104(Suppl):S2–S9. doi: 10.1016/j.ymgme.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 34.Janzen N, Terhardt M, Sander S, Demirkol M, Gokcay G, Peter M, et al. Towards newborn screening for ornithine transcarbamylase deficiency: fast non-chromatographic orotic acid quantification from dried blood spots by tandem mass spectrometry. Clin Chim Acta. 2014;430:28–32. doi: 10.1016/j.cca.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Fingerhut R. Recall rate and positive predictive value of MSUD screening is not influenced by hydroxyproline. Eur J Pediatr. 2009;168:599–604. doi: 10.1007/s00431-008-0804-0. [DOI] [PubMed] [Google Scholar]
- 36.Sun A, Lam C, Wong DA. Expanded newborn screening for inborn errors of metabolism: overview and outcomes. Adv Pediatr. 2012;59:209–245. doi: 10.1016/j.yapd.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 37.Simonsen H, Jensen UG. Technical aspects of neonatal screening using tandem mass spectrometry. Report from the 4th meeting of the International Society for Neonatal Screening. Acta Paediatr Suppl. 1999;88:52–54. doi: 10.1111/j.1651-2227.1999.tb01158.x. [DOI] [PubMed] [Google Scholar]
- 38.Teodoro-Morrison T, Kyriakopoulou L, Chen YK, Raizman JE, Bevilacqua V, Chan MK, et al. Dynamic biological changes in metabolic disease biomarkers in childhood and adolescence: a CALIPER study of healthy community children. Clin Biochem. 2015;48:828–836. doi: 10.1016/j.clinbiochem.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Oladipo OO, Weindel AL, Saunders AN, Dietzen DJ. Impact of premature birth and critical illness on neonatal range of plasma amino acid concentrations determined by LC-MS/MS. Mol Genet Metab. 2011;104:476–479. doi: 10.1016/j.ymgme.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Turgeon CT, Magera MJ, Cuthbert CD, Loken PR, Gavrilov DK, Tortorelli S, et al. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem. 2010;56:1686–1695. doi: 10.1373/clinchem.2010.148957. [DOI] [PubMed] [Google Scholar]