

Abstract
This first-in-human study examined the safety and pharmacokinetics of ch-mAb7F9, an anti-methamphetamine monoclonal antibody, in healthy volunteers. Single, escalating doses of ch-mAb7F9 over the range of 0.2 to 20 mg/kg were administered to 42 subjects who were followed for 147 d. Safety was measured by physical examinations, adverse events, vital signs, electrocardiograms, and clinical laboratory testing. Serum ch-mAb7F9 concentration and immunogenicity analyses were performed. There were no serious adverse reactions or discontinuations from the study due to adverse events. No trends emerged in the frequency, relatedness, or severity of adverse events with increased dose or between active and placebo treated subjects. Ch-mAb7F9 displayed expected IgG pharmacokinetic parameters, including a half-life of 17–19 d in the 3 highest dose groups and volume of distribution of 5–6 L, suggesting the antibody is confined primarily to the vascular compartment. Four (12.5%) of the 32 subjects receiving ch-mAb7F9 were confirmed to have developed a human anti-chimeric antibody response by the end of the study; however, this response did not appear to be dose related. Overall, no apparent safety or tolerability concerns were identified; a maximum tolerated dose was not reached in this Phase 1 study. Ch-mAb7F9 therefore appears safe for human administration.
Keywords: addiction, chimeric antibody, first in human, healthy volunteers, methamphetamine, monoclonal antibody, pharmacokinetics
Abbreviations
- AE
adverse event
- AUC(0-inf)
area under the concentration-time curve from 0 to infinity
- Cmax
maximum concentration
- CTCAE
Common Terminology Criteria for Adverse Events
- CL
clearance
- ECG
electrocardiogram
- FDA
Food and Drug Administration
- GLP
good laboratory practice
- HACA
human anti-chimeric antibodies
- KD
dissociation constant
- mAb
monoclonal antibody
- METH
(+)methamphetamine
- t1/2
apparent terminal half-life
- Vd
volume of distribution
Introduction
There are no medications approved by the US Food and Drug Administration (FDA) for the treatment of methamphetamine (METH) abuse and dependence disorders, which is a substantial barrier to successful treatment. Anti-METH monoclonal antibody (mAb) medications are a potentially groundbreaking pharmacologic methodology for treating METH use and addiction. Chimeric anti-METH mAb, ch-mAb7F9 (IgG2κ; METH KD = 7 nM), is the first anti-METH mAb to be developed and successfully tested in non-METH-using human volunteers.
MAbs and active vaccines against METH, cocaine, and nicotine have all shown potential for reducing central nervous system (CNS) effects such as horizontal locomotion and self-administration in animal models.1-5 Anti-METH mAbs reside in the blood and extracellular fluid compartments where they bind METH with such high affinity that they rapidly redistribute METH from its sites of action in the brain and other organs into the blood stream.6-8
MAbs offer specific advantages as a drug abuse therapy. Compared with other immunotherapies such as vaccines, mAbs necessarily do not require the function of the user's immune system to work, making them appropriate for immunosuppressed patients, including those with HIV/AIDS or autoimmune conditions. The protection against the effects of METH afforded by mAbs is immediate and does not require the 6–12 weeks needed for vaccines to establish an immune response. Most importantly, a mAb will have the same general characteristics, i.e., affinity for METH, in every patient. MAbs are also advantageous over small molecule medications because mAbs are not likely to be addictive or interrupt normal neurotransmitter function, and they have a much longer half-life, which improves patient compliance.
MAbs could also be effective adjunctive therapies in established behavioral treatment paradigms such as contingency management because they should not interfere with learning of lifestyle changes. Integration of ch-mAb7F9 into these programs is expected to improve the drug user's decision-making ability. For instance, reducing the value of METH use through reduction of pleasurable or reinforcing effects may help widen the gap between the reward for abstinence and the consequences of continued use (see related discussion in refs 9,10). As suggested by preclinical efficacy studies, an anti-METH mAb is likely to reduce the peak and duration of the high,11 thereby reducing the value of METH use. Given the larger difference between the value of the abstinence reward vs. the value of the high, the choice for abstinence may be easier to make.
No other anti-METH mAb has progressed as far as ch-mAb7F9 in clinical development. The potential human efficacy of ch‑mAb7F9 is demonstrated by important in vivo preclinical studies with its murine parent antibody, mAb7F9, which show that it is long-acting,12 reduces METH-induced locomotor activity in rats in an overdose model,11 and decreases METH-induced locomotor activity in rats 2 weeks after repeated mAb treatment was discontinued.13 The development of ch-mAb7F9 from mAb7F9, preclinical studies demonstrating that ch-mAb7F9 retains the specificity and ability to alter METH pharmacokinetics like mAb7F9, and GLP preclinical safety testing results were reported previously.14 Together these data supported the initiation of this first-in-human study of ch‑mAb7F9.
This was a Phase 1, double-blind, randomized, placebo-controlled, ascending single-dose safety and tolerability study of ch-mAb7F9 administered as an intravenous infusion to healthy volunteers. Forty-two subjects were treated in 5 dose groups ranging from 0.2 to 20 mg/kg ch‑mAb7F9, with 10 of the 42 subjects receiving normal saline as a placebo control. Subjects were followed for 147 d after dosing with regular safety assessments and for pharmacokinetic and immunogenicity analyses.
Results
The primary objective of this first human study was to determine the safety and tolerability of single, ascending intravenous doses of ch-mAb7F9 in healthy subjects via physical examinations, adverse events, vital signs, electrocardiograms, and clinical laboratory testing. The secondary objectives were to characterize the pharmacokinetics of ch‑mAb7F9 and immune response by measurement of antibody serum concentrations and human anti-ch-mAb7F9 antibody serum titers.
Subject demographics and baseline characteristics
A summary of subject demographics and baseline characteristics is presented by treatment group in Table 1. There was a disproportionate number of male subjects in the 2 lower treatment groups. Similarly, there was a higher number of white compared to black subjects in the 0.6 and 2 mg/kg treatment groups. All other demographic and baseline characteristics were similar between treatments.
Table 1.
Subject demographic and baseline characteristics by treatment. All dosed subjects are included
| ch-mAb7F9 |
|||||||
|---|---|---|---|---|---|---|---|
| Variable/ Category | Placebo N = 10 | 0.2 mg/kg N = 6 | 0.6 mg/kg N = 6 | 2 mg/kg N = 7a | 6 mg/kg N = 6 | 20 mg/kg N = 7 | All Subjects N = 42 |
| Sex | |||||||
| Male | 6 (60.0%) | 5 (83.3%) | 5 (83.3%) | 4 (57.1%) | 2 (33.3%) | 3 (42.9%) | 25 (59.5%) |
| Female | 4 (40.0%) | 1 (16.7%) | 1 (16.7%) | 3 (42.9%) | 4 (66.7%) | 4 (57.1%) | 17 (40.5%) |
| Race | |||||||
| White | 3 (30.0%) | 3 (50.0%) | 5 (83.3%) | 6 (85.7%) | 3 (50.0%) | 2 (28.6%) | 22 (52.4%) |
| Black/AA | 6 (60.0%) | 3 (50.0%) | 1 (16.7%) | 1 (14.3%) | 3 (50.0%) | 3 (42.9%) | 17 (40.5%) |
| Asian | 0 | 0 | 0 | 0 | 0 | 1 (14.3%) | 1 (2.4%) |
| NH/other Pacific Islander | 0 | 0 | 0 | 0 | 0 | 1 (14.3%) | 1 (2.4%) |
| AI or Alaskan Native | 1 (10.0) | 0 | 0 | 0 | 0 | 0 | 1 (2.4%) |
| Ethnicity | |||||||
| Not Hispanic | 9 (90.0%) | 5 (83.3%) | 6 (100.0%) | 7 (100.0%) | 6 (100.0%) | 7 (100.0%) | 40 (95.2%) |
| Hispanic | 1 (10.0%) | 1 (16.7%) | 0 | 0 | 0 | 0 | 2 (4.8%) |
| Age (years) | |||||||
| Mean | 30 | 24 | 26 | 22 | 32 | 26 | 27 |
| SD | 10 | 3 | 6 | 4 | 12 | 7 | 8 |
| Range | 20–44 | 20–28 | 20–36 | 19–28 | 18–46 | 19–36 | 18–46 |
Notes: AA = African American; AI = American Indian; NH = Native Hawaiian; SD = standard deviation. aInfusion was stopped/interrupted for one subject due to an adverse event of infusion reaction, but the subject's characteristics are included in the 2 mg/kg group column and totals.
Safety results
There were no deaths, serious adverse events during the study, or discontinuations from the study due to treatment-emergent adverse events (AEs). Overall, 40 (95%) subjects experienced at least 1 AE during the study. There were no apparent trends in the frequency, relatedness, or severity of AEs with increased dose or between active- and placebo-treated subjects. The most frequently reported AEs (≥4 [12.5%] over all active-treated subjects) during the study were increased blood creatine phosphokinase, upper respiratory tract infection, decreased hemoglobin, headache, increased aspartate aminotransferase and alanine aminotransferase, proteinuria, decreased white blood cell count, and nasal congestion (Table 2). All other AEs were reported in 4 or fewer active-treatment subjects. AEs considered by the investigator to be related to study medication were limited to single events in the 2 mg/kg treatment group and included infusion reaction, bronchospasm, and proteinuria. Treatment-emergent AEs were reported for 20 subjects overall who had associated changes in physical examination findings following dosing; all of these AEs were considered unlikely to be related to study medication.
Table 2.
Number (%) of subjects with most frequently reported (≥5 % of all active-treated subjects) treatment-emergent adverse events by preferred term and treatment
| ch-mAb7F9 |
|||||||
|---|---|---|---|---|---|---|---|
| Preferred Term | Placebo N = 10 | 0.2 mg/kg N = 6 | 0.6 mg/kg N = 6 | 2 mg/kg N = 6a | 6 mg/kg N = 6 | 20 mg/kg N = 7 | All Active-treated N = 32a |
| Blood CPK Increased | 4 (40.0) | 3 (50.0) | 2 (33.3) | 2 (33.3) | 1 (16.7) | 4 (57.1) | 12 (37.5) |
| URI | 2 (20.0) | 0 | 1 (16.7) | 4 (66.7) | 2 (33.3) | 2 (28.6) | 10 (31.3) |
| Hemoglobin Decreased | 5 (50.0) | 2 (33.3) | 2 (33.3) | 2 (33.3) | 1 (16.7) | 3 (42.9) | 10 (31.3) |
| Headache | 2 (20.0) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 0 | 2 (28.6) | 6 (18.8) |
| AST Increased | 2 (20.0) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 0 | 2 (28.6) | 6 (18.8) |
| Proteinuria | 2 (20.0) | 1 (16.7) | 0 | 2 (33.3) | 2 (33.3) | 1 (14.3) | 6 (18.8) |
| ALT Increased | 2 (20.0) | 2 (33.3) | 0 | 1 (16.7) | 0 | 2 (28.6) | 5 (15.6) |
| WBC Count Decreased | 2 (20.0) | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (28.6) | 4 (12.5) |
| Nasal Congestion | 2 (20.0) | 0 | 0 | 2 (33.3) | 2 (33.3) | 0 | 4 (12.5) |
| Lipase Increased | 2 (20.0) | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 3 (9.4) |
| Abdominal Pain | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (6.3) |
| Toothache | 1 (10.0) | 0 | 0 | 0 | 1 (16.7) | 1 (14.3) | 2 (6.3) |
| Gastroenteritis | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (14.3) | 2 (6.3) |
| Urinary Tract Infection | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 2 (6.3) |
| Vaginitis Bacterial | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 2 (6.3) |
| Viral Infection | 1 (10.0) | 0 | 1 (16.7) | 0 | 0 | 1 (14.3) | 2 (6.3) |
| Laceration | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 2 (6.3) |
| Sunburn | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 2 (6.3) |
| Blood Calcium Decreased | 2 (20.0) | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 2 (6.3) |
| Blood LDH Increased | 1 (10.0) | 1 (16.7) | 0 | 0 | 0 | 1 (14.3) | 2 (6.3) |
| Oropharyngeal Pain | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (14.3) | 2 (6.3) |
| Blood Potassium Decreased | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (14.3) | 2 (6.3) |
| Platelet Count Decreased | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 2 (6.3) |
Notes: ALT = alanine aminotransferase; AST = aspartate aminotransferase; CPK = creatine phosphokinase; LDH = lactate dehydrogenase; URI = upper respiratory tract infection; WBC = white blood cell. aInfusion was stopped/interrupted for one subject due to an adverse event of infusion reaction; therefore, this subject was excluded for the 2 mg/kg group and included in the ‘All Active-treated Subjects’ group.
Of 216 reported AEs, corresponding Common Terminology Criteria for Adverse Events (CTCAE) severity was graded as follows: 3 events as Grade 4 (life-threatening), 6 events as Grade 3 (severe), 47 events as Grade 2 (moderate), and 160 events as Grade 1 (mild). There was no apparent trend in increased CTCAE grade severity with increased dose or between active- and placebo-treated subjects. The Grade 4 AEs (2 active, 1 placebo) were all elevations in blood creatine phosphokinase levels that lasted approximately 7 d and were considered unrelated to the ch-mAb7F9; all resolved with no treatment.
Of the 3 events in 2 subjects considered by the Investigator as related to study medication, the infusion reaction (Grade 3) and bronchospasm (Grade 2) occurred in the same subject. The infusion reaction began 72 minutes after the initiation of the infusion with the complaint of ‘very slight itching’ in the back of the throat. No other symptoms were noted upon examination, vital signs were stable, and the infusion continued. At 20 minutes after the infusion reaction onset, a single wheeze was noted when the subject took a deep breath. The infusion was then stopped; about half the dose had been administered. A separate subject experienced an AE of proteinuria (Grade 1) considered related to the study drug. Both subjects with related AEs were in the same dose group (2 mg/kg ch‑mAb7F9); thus increasing the dose did not result in more AEs considered related to study medication.
Overall, no trends or clinically meaningful changes were observed in most clinical laboratory analytes throughout the study. Transient increases in mean creatine phosphokinase values were noted in all treatment groups. Treatment-emergent AEs associated with abnormal laboratory values in 4 (12.5%) or more overall active-treated subjects included increased blood creatine phosphokinase, decreased hemoglobin, increased aspartate aminotransferase and alanine aminotransferase, decreased white blood cell, and proteinuria; however, the frequency of events was similar or greater in placebo-treated subjects compared with active-treated subjects overall.
There were no trends or clinically meaningful changes noted in mean or median vital sign data throughout the study. Changes in vital signs that were reported as treatment-emergent AEs included 1 event each of presyncope (0.2 mg/kg) and hypertension (placebo) and were considered by the investigator as moderate in severity and not related to study medication.
There were no trends or clinically meaningful changes in mean or median 12-lead electrocardiogram (ECG) values. However, a slight increase in frequency of clinically noteworthy 12‑lead ECG values was observed with increased dose, although the frequency was highest in the placebo group. There were no subjects with QT, QTc, QTcB, or QTcF values above 480 ms or with increases following dosing in QT, QTc, QTcB, or QTcF greater than 60 ms. No AEs were reported for abnormal 12‑lead ECG findings.
Pharmacokinetic results
Individual ch-mAb7F9 serum concentrations were quantifiable for 126 d and 147 d in more than half the subjects in the 2 higher dose groups (6 mg/kg and 20 mg/kg), respectively. In the semi-logarithmic presentation (Fig. 1), the terminal portion of the concentration-time profiles for the 3 higher doses appear to be parallel, providing evidence that the rate of elimination was similar across these dose groups. The terminal portion of the concentration-time profile for the 2 lower doses is somewhat steeper compared to that of the 3 higher doses. With these lower doses, the serum concentrations fell below the limits of quantitation early in the time course and the terminal portion of the concentration-time profile may not be adequately defined.
Figure 1.
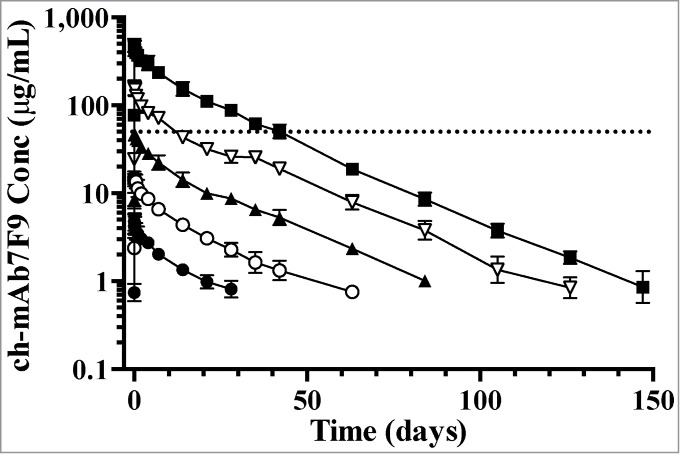
Mean (SD) ch-mAb7F9 serum concentration-time profiles by treatment (days 1 through 147). Dose groups shown received 0.2 (closed circles), 0.6 (open circles), 2 (closed triangles), 6 (open triangles), and 20 (closed squares) mg/kg ch-mAb7F9. Data from the one subject in the 2 mg/kg group that received only half a dose are not included; data from dropout subjects and replacements are included where available. Sample results reported as below the limit of quantitation were omitted. Only points that are the average of 3 or more quantified samples are shown. The dotted line marks 50 μg/mL, the predicted serum concentration needed for efficacy.
Pharmacokinetic parameters are summarized by dose group in Table 3. A 100-fold increase in dose from 0.2 mg/kg to 20 mg/kg resulted in an approximate 120-fold increase in mean AUC(0-inf) and an approximate 100-fold increase in mean Cmax across this same range of doses. Dose proportionality was assessed based on whether the 90% confidence interval constructed for the estimate of the slope of the linear relationship between exposure and dose was contained within the interval (0.95, 1.05). For Cmax this criterion was met, thus demonstrating this parameter increased in a dose-proportional manner across this range of doses. The 90% confidence interval for the slope for Cmax was (0.979, 1.05). The 90% confidence interval for this slope for AUC(0-inf) (1.02, 1.07) fell just above the upper limit of this interval. Although the statistical criteria were not met for this parameter, a visual inspection of mean AUC(0-inf) versus dose shows this parameter increased in a general dose‑proportional manner as well.
Table 3.
Summary of ch-mAb7F9 pharmacokinetic parameters by treatment
| AUC(0-inf) (μg*h/mL) | AUC(0-last) (μg*h/mL) | Cmax (μg/mL) | tmaxa (h) | t1/2 (h) | CL (L/h) | Vd (L) | |
|---|---|---|---|---|---|---|---|
| 0.2 mg/kg | |||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean | 1400 | 1070 | 5.21 | 3.18 (2.03, 4.00) | 341 | 0.0104 | 5.01 |
| SD | 200 | 214 | 1.82 | — | 74.1 | 0.00173 | 0.652 |
| 0.6 mg/kg | |||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean | 4620 | 4160 | 15.5 | 2.25 (2.25, 6.00) | 383 | 0.0101 | 5.38 |
| SD | 714 | 690 | 2.33 | — | 128 | 0.00204 | 1.73 |
| 2 mg/kg | |||||||
| n | 5 | 5 | 6 | 6 | 5 | 5 | 5 |
| Mean | 16800 | 16300 | 50.6 | 3.52 (2.25, 6.00) | 432 | 0.00794 | 5.03 |
| SD | 1520 | 1500 | 3.43 | — | 57.4 | 0.00134 | 1.43 |
| 6 mg/kg | |||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean | 53500 | 53000 | 178 | 3.50 (2.00, 6.00) | 418 | 0.00855 | 5.14 |
| SD | 4640 | 4640 | 16.5 | — | 51.5 | 0.00109 | 0.743 |
| 20 mg/kg | |||||||
| n | 6 | 6 | 7 | 7 | 6 | 6 | 6 |
| Mean | 166000 | 165000 | 517 | 2.28 (2.00, 4.02) | 460 | 0.00896 | 5.88 |
| SD | 17700 | 17500 | 89.3 | — | 42.0 | 0.00273 | 1.51 |
Note(s): n = number of subjects; SD = standard deviation. aOnly median (minimum, maximum) are presented for tmax.
The half-life values observed in the 2 lowest dose groups were shorter than those observed in the 3 higher dose groups. Serum concentrations in these 2 lower dose groups fell below the limits of quantitation much earlier in the concentration-time course when compared to the 3 higher dose groups, and likely resulted in an inadequate estimation of the elimination rate constant for the 2 lower dose groups. This was associated with an underestimation of AUC(0-inf) in the lower dose groups.
Immunogenicity results
None of the subjects randomized to placebo developed detectable human anti-chimeric antibodies (HACA) during the study. One subject's predose sample screened positive for HACA, but the titer was not greater than the low positive control. By the end-of-study assessment, 4 (12.5%) of the 32 subjects who received ch-mAb7F9 were confirmed positive for HACA based on titer levels greater than the low positive control (Table 4). The confirmed development of HACA did not appear to be dose related, as there was one subject in each of the 2 lowest active dose groups and one subject in each of the 2 highest active dose groups.
Table 4.
Number of subjects screening positive and negative for HACA and confirmed positive for HACA by titer
| ch-mAb7F9 (mg/kg) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | 0.2 | 0.6 | 2 | 6 | 20 | All Active | ||
| Study Day | Outcome | N = 10 | N = 6 | N = 6 | N = 6a | N = 6 | N = 7 | N = 32a |
| End of Study | Positive Screen | 0 (0.0%) | 1 (16.7%) | 1 (16.7%) | 1 (16.7%) | 3 (50.0%) | 2 (28.6%) | 8 (25.0%) |
| Negative Screen | 9 (90%) | 5 (83.3%) | 5 (83.3%) | 4 (66.7%) | 3 (50.0%) | 4 (57.1%) | 22 (68.8%) | |
| Not Done | 1 (10.0%) | 0 (0.0%) | 0 (0.0%) | 1 (16.7%) | 0 (0.0%) | 1 (14.3%) | 2 (6.3%) | |
| Confirmed Positive by Titer | — | 1 (16.7%) | 1 (16.7%) | 0 (0.0%) | 1 (16.7%) | 1 (14.3%) | 4 (12.5%) | |
Notes: The end-of-study sample was collected on Day 126 for 29 active subjects and 9 placebo subjects, on Day 84 for 1 active subject, and an end-of-study sample was not obtained in 2 active subjects and 1 placebo subject. aInfusion was stopped/interrupted for one subject due to an adverse event of infusion reaction; therefore, this subject was excluded for the 2 mg/kg group and included in the ‘All Active-treated Subjects’ group.
Whether the development of HACA altered the pharmacokinetics of ch‑mAb7F9 in a clinically meaningful way is not clear from the study findings due to the small study population. However, 3 of the 4 antibody-positive subjects had the highest rate of systemic clearance and lowest level of overall exposure [AUC(0‑inf)] in their respective treatment groups. Systemic clearance and AUC(0-inf) values for the 1 remaining antibody-positive subject were consistent with the antibody-negative subjects within the respective dose group.
Discussion
This first-in-human study was a randomized, double-blind, placebo-controlled, single-ascending-dose safety, tolerability, and pharmacokinetic study in healthy adult subjects. In this study, single intravenous doses of ch-mAb7F9 or placebo were given to different groups of adult subjects. This ascending-dose design, with enrollment of subjects into higher dose groups only after the lower dose group was determined to be safe and well-tolerated, is a standard design for evaluation of multiple dose levels in Phase 1 studies because this design helps minimize risk to subjects receiving an investigational new drug. Inclusion and exclusion criteria were designed to maximize the safety of the subjects and ensure that stimulant users who were not seeking treatment for methamphetamine abuse were not exposed.
Doses of ch-mAb7F9 administered in this study were selected based on effective doses of murine anti-METH antibodies in preclinical rat studies, physiologic differences in cerebral blood flow between humans and rats, and on calculations of the amount needed to neutralize typical METH doses. It is predicted that 20 mg/kg will be effective in humans in part because it is more than sufficient to bind the METH in the blood after a 30 mg dose (given intravenously over 2 min), which results in a METH Cmax of ∼110 ng/mL.15 The concentration of ch‑mAb7F9 required to bind this much METH is ∼50 μg/mL. Evaluation of pharmacokinetic data from this Phase 1 study indicates that ch-mAb7F9 concentrations remain above 50 μg/ml for 1 to 2 weeks after 6 mg/kg, and for about 5 weeks after 20 mg/kg (Fig. 1).
Similar mAb doses have been used previously in human studies of antibodies intended to bind other non-endogenous targets. Raxibacumab, a human IgG1λ against the protective antigen of B. anthracis, has been dosed up to 40 mg/kg in healthy subjects.16,17 The chimeric IgG1κ, cαStx2, which binds a subunit of the E. coli protein Stx2, has been administered at 10 mg/kg.18 Two mAbs intended to treat sepsis from staphylococci are the humanized IgG1κ tebifazumab,19 which binds clumping factor A and the chimeric IgG1 pagibaximab,20 which binds lipoteichoic acid. Both have been dosed in healthy subjects up to 20 and 10 mg/kg, respectively. Safety studies of these mAbs suggest that, like ch-mAb7F9, they were safe and well tolerated. In each study, the AEs were typically mild, transient, and not mAb dose-related. Headache was the most common event reported. The development of human anti-mAb antibodies was reported as undetected for tefibazumab,19 but 4 of 17 subjects (24%) dosed with cαStx2 developed HACA,18 though like the anti-ch-mAb7F9 reported here, the response was not dose related.
Compared to these same mAbs, ch-mAb7F9 pharmacokinetic parameters are also generally similar. Like ch-mAb7F9, raxibacumab and tefibazumab both have half-lives of approximately 3 weeks.17,19 Pagibaximab is somewhat longer at 33 days20 and cαStx2 is shorter at 8.6 d18 The long ch‑mAb7F9 elimination half-life could be a substantial improvement over many small molecule medications used to treat METH users. Because of its 18-day half-life, ch‑mAb7F9 dosing should be no more frequent than every 3 weeks, which will increase compliance for motivated patients. Additionally, the volume of distribution was reported for 3 of the mAbs and ranged from approximately 5.25–7 L, which is also similar to that reported here for ch‑mAb7F9. As expected based on the intentional design of ch-mAb7F9 with IgG2 human constant domains, ch‑mAb7F9 has typical human IgG pharmacokinetic parameters. Altogether, these results suggest that ch-mAb7F9 is safe and can be maintained at predicted levels needed for efficacy; thus, the clinical evidence supports further development of ch-mAb7F9.
There are potential risks to moving forward with testing of ch-mAb7F9 in METH-abusing patients. One possibility is that ch-mAb7F9-treated subjects will attempt to surmount the binding capacity of ch-mAb7F9 for METH by self-administering larger doses of METH, which may accumulate in the blood or tissues and result in unexpected peripheral toxicity. Further, the safety of ch-mAb7F9-METH complexes remains to be studied thoroughly in GLP toxicology studies. Significant discussions with FDA about such concerns have resulted in an outline of nonclinical studies necessary to support the initiation of the next clinical study, a proposed safety study in METH-using subjects of the interaction between ch-mAb7F9 and METH.
Following completion of the required nonclinical studies, a risk mitigation plan will be developed to maximize the safety of the planned Phase 1b study as much as possible. In this later study, inpatient METH-using subjects will receive a single dose of ch-mAb7F9 followed by a series of METH challenges at weekly intervals. The primary objective will be to determine the safety of the interaction between METH and ch‑mAb7F9.
These early studies of a therapeutic mAb provide results that may be broadly applicable to other immunotherapies in development for treating drug abuse. For instance, studies of mAbs can define minimum effective serum antibody concentrations and possibly affinity values, yielding specific targets for the design of effective active vaccines. However, because of ongoing improvements in technology, costs of production for a mAb therapy are likely to decrease, enhancing the feasibility of this approach. By showing proof-of-concept and outlining a development pathway with a high affinity anti-METH mAb, these and the upcoming studies of ch-mAb7F9 will further enable development of immunotherapies for treating METH use and improve the options for treating this unmet need.
Methods
Study design
This Phase 1, double-blind, randomized, placebo-controlled, ascending intravenous single-dose study was approved by the MidLands Independent Review Board (protocol number TOA62936, clinicaltrials.gov identifier NCT01603147). The study was conducted at Quintiles Phase One Services in Overland Park, KS, USA in compliance with the Good Clinical Practice guidelines.
The study comprised a screening period (Days −22 to −2), a 4-day/3-night residential period, and subsequent outpatient visits on the day after discharge (Day 4), weekly (±1 day) from Days 7 to 42 (Days 7, 14, 21, 28, 35, and 42), and then every 3 weeks (±3 days) to Day 147 (Days 63, 84, 105, 126, and 147). Safety and pharmacokinetic assessments were performed during the first 48 hours after the start of the infusion, after which subjects were discharged from the study center. They returned for follow-up safety and pharmacokinetic evaluations on the aforementioned outpatient-visit days and were discharged from the study following completion of Day 147 assessments.
Participants
Eligible subjects were healthy adults, aged 18 to 50 years, inclusive, with a screening body mass index of 18.5 to 30.5 kg/m2, inclusive, and a body weight of 50 to 100 kg, inclusive. Key exclusion criteria were history of treatment with a mAb in the past year, history of severe allergy to any medications or asthma, and any history of stimulant use or abuse.
Test and control articles
The test article was anti-methamphetamine chimeric mAb 7F9 (ch‑mAb7F9) produced under Good Manufacturing Practices by Catalent Pharma Solutions using a Chinese hamster ovary cell line described previously.14 Ch-mAb7F9 was supplied as a 20 mg/mL solution in 10 mL vials and stored at 2–8°C. The antibody was diluted for intravenous administration using normal saline to a volume of 224 mL. Placebo treatment was normal saline.
Treatment regimen
The study comprised a 21-day screening period, a 4-day/3-night in-house study period, and outpatient visits from discharge to Day 147. The ch-mAb7F9 was given via intravenous infusion on the morning of Day 1, and the subjects were discharged on Day 3, 48 hours after the ch‑mAb7F9 infusion.
Subjects received a single dose of ch-mAb7F9 administered intravenously over 2 hours at progressively increasing infusion rates to achieve a 0.2, 0.6, 2, 6, or 20 mg/kg dose. Two subjects (1 active [sentinel]/1 placebo [control]) in each cohort received their dose before the other subjects. Dosing of subsequent subjects in the cohort did not proceed for 48 hours, to allow the investigator and study personnel to perform safety assessments on each cohort's first 2 dosed subjects. Dosing of each of the remaining subjects in the dose group occurred one at a time, with dosing in each subject separated by a minimum of 24 hours.
Randomization
In total, 40 eligible subjects were sequentially enrolled into 1 of 5 study groups. Prior to the beginning of dosing for each group, the 8 subjects in each group were randomly assigned to a block of 2, and a block of 6. In the block of 2, 1 subject was given placebo (normal saline) by random assignment and the other the active dose for that group. In the block of 6, 1 subject was assigned to placebo, and the other 5 subjects were assigned to the active dose. Thus, 2 control subjects were included in each group. At least 1 representative from each sex in each study group was required.
Blinding
The Investigator, Study Coordinator, personnel providing care to the subjects and administering the study product, and subjects themselves were blinded to the product (active agent or placebo) throughout the duration of the study. As the diluted ch-mAb7F9 was the same in appearance as the placebo, the blind was maintained during the administration procedure. Three subjects were lost to follow up and therefore unblinded early for replacement purposes; 2 additional subjects were unblinded for safety reasons due to AEs.
Pharmacokinetics
Serial blood samples for the measurement of ch-mAb7F9 were collected predose, at specified time points during the 48 hours after the start of the infusion, and on specified days from Day 4 through 147. Serum concentrations of ch-mAb7F9 were determined by means of a validated, sensitive, and specific enzyme-linked immunosorbent assay. Pharmacokinetic parameters were derived using noncompartmental methods with Phoenix® WinNonlin® 6.3 (Pharsight Corp., Mountain View, CA). All pharmacokinetic computations were performed using Phoenix® WinNonlin® 6.3 or SAS® Version 9.2 (SAS Institute, Inc., Cary, NC). Primary pharmacokinetic parameters included maximum observed serum concentration (Cmax), time of Cmax (tmax), area under the serum concentration-time curve from zero (predose) to time of last measurable concentration [AUC(0-last)], area under the concentration-time curve from zero (predose) extrapolated to infinite time [AUC(0-inf)], apparent terminal half-life (t1/2), systemic clearance (CL), and volume of distribution (Vd).
Immunogenicity
Blood samples were screened for the measurement of human anti-chimeric antibody (HACA) using a validated qualitative electrochemiluminescent method. Positives were confirmed by drug competition testing and titered. Subjects were considered to have developed a HACA response only if the titer was above the low positive control value.
Safety assessments
Safety was evaluated by adverse event and concomitant medication monitoring; review of data from routine clinical laboratory tests (including blood chemistry, hematology, coagulation, urinalysis, troponin I, and cytokine levels); vital signs (including systolic and diastolic blood pressure, pulse and respiration rate, oral body temperature, and pulse oximetry); 12-lead electrocardiograms and telemetry; and physical examinations.
All AEs were coded to body system and preferred term using the Medical Dictionary for Regulatory Activities, (MedDRA) Version 15.0. The intensity of AEs was assessed according to the definitions in the CTCAE Version 4.0. Common Terminology Criteria for AE grades were used in the summary of AEs; however, for AEs that were not defined in the CTCAE, the intensity as determined by the investigator was used.
Statistical methods
Pharmacokinetic serum concentrations were summarized using descriptive statistics (available data, mean, standard deviation, percent coefficient of variation, median, minimum, and maximum) and graphic displays, as appropriate. Dose proportionality of the primary pharmacokinetic parameters was assessed using graphical methods and statistically using a power model approach. Concentrations below the limit of quantitation were treated as zero for the computation of descriptive statistics. Concentrations assigned a value of missing were omitted from the calculation of descriptive statistics.
Human anti-chimeric antibody screening results data were summarized categorically as negative, positive, or not done. If quantifiable, titer results were summarized for positive antibody results.
All safety assessments, including adverse events, clinical laboratory evaluations, vital signs, 12-lead electrocardiograms, telemetry results, and physical examination findings were listed, and where appropriate, summarized using descriptive statistics.
Disclosure of Potential Conflicts of Interest
SMO and WBG are full time faculty members at the University of Arkansas for Medical Sciences and RLH is a full time faculty member at the University of Arkansas. They have financial interests in and serve as Chief Scientific Officer (SMO), Chief Medical Officer (WBG), and Vice President of Biopharmaceutics (RLH) for InterveXion Therapeutics, LLC (Little Rock, AR), a pharmaceutical biotechnology company focused on treating human drug addiction with antibody-based therapy.
Acknowledgments
The authors thank the subjects for their participation in this study. In addition, the authors would also like to thank the staff at Quintiles Phase One Services for their very capable conduct and reporting of this first in human study (especially Sara Rothermich and Hal Galbraith) and those at Charles River Laboratories, Reno, NV for the PK and immunogenicity sample analysis (especially Sharif Rumjahn and Kelly Colletti).
Funding
This work was supported by grants from the National Institute on Drug Abuse (R01DA031944) and the Translational Research Institute (UL1TR000039) through the National Center for Research Resources and National Center for Advancing Translational Sciences of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Fox BS, Kantak KM, Edwards MA, Black KM, Bollinger BK, Botka AJ, French TL, Thompson TL, Schad VC, Greenstein JL, et al. Efficacy of a therapeutic cocaine vaccine in rodent models. Nat Med 1996; 2:1129-32; PMID:8837612; http://dx.doi.org/ 10.1038/nm1096-1129 [DOI] [PubMed] [Google Scholar]
- 2.Carrera MR, Ashley JA, Zhou B, Wirsching P, Koob GF, Janda KD. Cocaine vaccines: antibody protection against relapse in a rat model. Proc Natl Acad Sci USA 2000; 97:6202-6; PMID:10823960; http://dx.doi.org/ 10.1073/pnas.97.11.6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LeSage MG, Keyler DE, Hieda Y, Collins G, Burroughs D, Le C, Pentel PR. Effects of a nicotine conjugate vaccine on the acquisition and maintenance of nicotine self-administration in rats. Psychopharmacology 2005; 184:409-16; PMID:15991003; http://dx.doi.org/ 10.1007/s00213-005-0027-2 [DOI] [PubMed] [Google Scholar]
- 4.Byrnes-Blake KA, Laurenzana EM, Landes RD, Gentry WB, Owens SM. Monoclonal IgG affinity and treatment time alters antagonism of (+)-methamphetamine effects in rats. Eur J Pharmacol 2005; 521:86-94; PMID:16182279; http://dx.doi.org/ 10.1016/j.ejphar.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 5.Norman AB, Norman MK, Buesing WR, Tabet MR, Tsibulsky VL, Ball WJ. The effect of a chimeric humanmurine anti-cocaine monoclonal antibody on cocaine self-administration in rats. J Pharmacol Exp Ther 2009; 328:873-81; PMID:19088302; http://dx.doi.org/ 10.1124/jpet.108.146407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes-Blake KA, Laurenzana EM, Carroll FI, Abraham P, Gentry WB, Landes RD, Owens SM. Pharmacodynamic mechanisms of monoclonal antibody-based antagonism of (+)-methamphetamine in rats. Eur J Pharmacol 2003; 461:119-28; PMID:12586207; http://dx.doi.org/ 10.1016/S0014-2999(03)01313-X [DOI] [PubMed] [Google Scholar]
- 7.Laurenzana EM, Hendrickson HP, Carpenter D, Peterson EC, Gentry WB, West M, Che Y, Carroll FI, Owens SM. Functional and biological determinants affecting the duration of action and efficacy of anti-(+)-methamphetamine monoclonal antibodies in rats. Vaccine 2009; 27:7011-20; PMID:19800446; http://dx.doi.org/ 10.1016/j.vaccine.2014.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gentry WB, Ruedi-Bettschen D, Owens SM. Anti-(+)-methamphetamine monoclonal antibody antagonists designed to prevent the progression of human diseases of addiction. Clin Pharmacol Ther 2010; 88:390-3; PMID:20668443; http://dx.doi.org/ 10.1038/clpt.2010.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gowin JL, Stewart JL, May AC, Ball TM, Wittmann M, Tapert SF, Paulus MP. Altered cingulate and insular cortex activation during risk-taking in methamphetamine dependence: losses lose impact. Addiction 2014; 109:237-47; PMID:24033715; http://dx.doi.org/ 10.1111/add.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoptaw S. Commentary on Gowin et al. (2014): Brain is behavior—methamphetamine dependence and recovery–Shoptaw–2014–Addiction–Wiley Online Library. Addiction 2014; 109:248-9; PMID:24422617; http://dx.doi.org/ 10.1111/add.12442 [DOI] [PubMed] [Google Scholar]
- 11.Laurenzana EM, Stevens MW, Frank JC, Hambuchen MD, Hendrickson HP, White SJ, Williams DK, Owens SM, Gentry WB. Pharmacological effects of two anti-methamphetamine monoclonal antibodies: Supporting data for lead candidate selection for clinical development. Hum Vaccin Immunother 2014; 10; PMID:25003991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Owens SM, Atchley WT, Hambuchen MD, Peterson EC, Gentry WB. Monoclonal antibodies as pharmacokinetic antagonists for the treatment of (+)-methamphetamine addiction. CNS Neurol Disord Drug Targets 2011; 10:892-8; PMID:22229314; http://dx.doi.org/ 10.2174/187152711799219370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hambuchen MD, Ruedi-Bettschen D, Williams DK, Hendrickson H, Owens SM. Treatment of rats with an anti-(+)-methamphetamine monoclonal antibody shortens the duration of action of repeated (+)-methamphetamine challenges over a one month period. Vaccine 2014; pii: S0264-410X:01284-5; in press; PMID:25252196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens MW, Tawney RL, West CM, Kight AD, Henry RL, Owens SM, Gentry WB. Preclinical characterization of an anti-methamphetamine monoclonal antibody for human use. MAbs 2014; 6:547-55; PMID:24492290; http://dx.doi.org/ 10.4161/mabs.27620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruickshank CC, Dyer KR. A review of the clinical pharmacology of methamphetamine. Addiction 2009; 104:1085-99; PMID:19426289; http://dx.doi.org/ 10.1111/j.1360-0443.2009.02564.x [DOI] [PubMed] [Google Scholar]
- 16.Subramanian GM, Cronin PW, Poley G, Weinstein A, Stoughton SM, Zhong J, Ou Y, Zmuda JF, Osborn BL, Freimuth WW. A Phase 1 Study of PAmAb, a Fully Human Monoclonal Antibody against Bacillus anthracis Protective Antigen, in Healthy Volunteers. Clin Infect Dis 2005; 41:12-20; PMID:15937757; http://dx.doi.org/ 10.1086/430708 [DOI] [PubMed] [Google Scholar]
- 17.Migone T-S, Subramanian GM, Zhong J, Healey LM, Corey A, Devalaraja M, Lo L, Ullrich S, Zimmerman J, Chen A, et al. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med 2009; 361:135-44; PMID:19587338; http://dx.doi.org/ 10.1056/NEJMoa0810603 [DOI] [PubMed] [Google Scholar]
- 18.Dowling TC, Chavaillaz PA, Young DG, Melton-Celsa A, O’Brien A, Thuning-Roberson C, Edelman R, Tacket CO. Phase 1 safety and pharmacokinetic study of chimeric murine-human monoclonal antibody c alpha Stx2 administered intravenously to healthy adult volunteers. Antimicrob Agents Chemother 2005; 49:1808-12; PMID:15855500; http://dx.doi.org/ 10.1128/AAC.49.5.1808-1812.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reilley S, Wenzel E, Reynolds L, Bennett B, Patti JM, Hetherington S. Open-label, dose escalation study of the safety and pharmacokinetic profile of tefibazumab in healthy volunteers. Antimicrob Agents Chemother 2005; 49:959-62; PMID:15728889; http://dx.doi.org/ 10.1128/AAC.49.3.959-962.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisman LE, Fischer GW, Thackray HM, Johnson KE, Schuman RF, Mandy GT, Stratton BE, Adams KM, Kramer WG, Mond JJ. Safety and pharmacokinetics of a chimerized anti-lipoteichoic acid monoclonal antibody in healthy adults. Int Immunopharmacol 2009; 9:639-44; PMID:19268719; http://dx.doi.org/ 10.1016/j.intimp.2009.02.008 [DOI] [PubMed] [Google Scholar]