

Abstract
Mcl-1, a pro-survival member of the Bcl-2 protein family, is an attractive target for cancer therapy. We have recently identified the natural product marinopyrrole A (maritoclax) as a novel small molecule Mcl-1 inhibitor. Here, we describe the structure-activity relationship study of pyoluteorin derivatives based on maritoclax. To date, we synthesized over 30 derivatives of maritoclax and evaluated their inhibitory actions and cytotoxicity toward Mcl-1-dependent cell lines. As a result, several functional groups were identified in the pyoluteorin motif that significantly potentiate biological activity. A number of such derivatives, KS04 and KS18, interacted with Mcl-1 in a conserved fashion according to NMR spectroscopy and molecular modeling. KS04 and KS18 induced apoptosis selectively in Mcl-1-dependent but not Bcl-2-dependent K562 cells through selective Mcl-1 down-regulation, and synergistically enhanced apoptosis in combination with ABT-737. Moreover, the intraperitoneal administration of KS18 (10 mg/kg/d) and ABT-737 (20 mg/kg/d) significantly suppressed the growth of ABT-737-resistant HL-60 xenografts in nude mice without apparent toxicity. Overall, we identified the pharmacophore of pyoluteorin derivatives that act as potent and promising Mcl-1 antagonists against Mcl-1-dependent hematological cancers.
Keywords: Apoptosis, Bcl-2 family, leukemia, lymphoma, multiple myeloma, Mcl-1, maritoclax, pyoluteorin
Abbreviations
- ABTR
ABT-737 resistant
- AML
Acute Myeloid Leukemia
- AUCinf
area under curve extrapolated to time infinity
- AUClast
area under curve until last observed timepoint
- Bcl-2
B-cell lymphoma-2
- BH3
Bcl-2 homology domain 3
- CHX
Cycloheximide
- CL
rate of plasma clearance
- Cmax
maximal plasma concentration
- EC50
Half maximal effective concentration
- i.p.
Intraperitoneal
- IRES
Internal ribosome entry site
- LD50
median lethal dose
- Mcl-1
Myeloid cell leukemia-1
- MTD
Maximal tolerated dose
- NMR
Nuclear magnetic resonance
- PARP
Poly (ADP-ribose) polymerase
- SAR
Structure-activity relationship
- T1/2
plasma half-life
- Tmax
time to maximal plasma concentration
- VD
Volume of distribution
Introduction
The B-cell lymphoma-2 (Bcl-2) protein family consists of both pro-apoptotic and anti-apoptotic members to regulate mitochondrial apoptosis. The anti-apoptotic members, including Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1, sequester both direct (i.e., Bid, Bim, Puma) and indirect (e.g., Bad, Bik, Bmf, Noxa) activator BH3-only proteins to maintain homeostasis. The release of direct activator BH3-only proteins from the anti-apoptotic Bcl-2 family proteins results in the oligomerization of multi-domain pro-apoptotic Bax and Bak proteins to induce mitochondrial outer membrane permeabilization, releasing factors such as cytochrome C and Smac to execute caspase activation and apoptosis.1
Hematological malignancies such as leukemias, lymphomas, and myelomas are projected to account for 9.4% of all cancer deaths in the USA for 2014,2 and the up-regulation of anti-apoptotic Bcl-2 family proteins is frequently observed. The t(14;18)(q32;q21) chromosomal translocation occurs in the majority of follicular lymphoma cases to drive Bcl-2 upregulation.3 Bcl-2 and Bcl-xL are also up-regulated in chronic lymphocytic leukemia4 and multiple myeloma.5 Furthermore, Bcl-2 up-regulation is associated with poor prognosis and chemo-resistance in acute myeloid leukemia (AML), B-cell lymphoma, and multiple myeloma.6-8 Highly selective Bcl-2 inhibitor ABT-737, and its analogs ABT-263 and ABT-199 developed by Abbott Laboratories show both pre-clinical and clinical efficacy against a number of hematological cancers.1,9-12 However, Mcl-1-dependent cancers are resistant to selective Bcl-2 antagonism. For instance, subsets of multiple myeloma could be Mcl-1-dependent and resist ABT-737 treatment.10,13,14 The survival of AML cells was also shown to be largely Mcl-1-dependent.15 Furthermore, prolonged treatment of ABT-737 to leukemic cells could cause acquired resistance, often through Mcl-1 upregulation.9 A selective small molecule Mcl-1 antagonist would thus be promising both as a single agent and in combination for the treatment of hematological cancers.
Through a natural compound library screen, we identified a small molecule, marinopyrrole A (maritoclax), a novel bispyrrole compound which was selectively cytotoxic to Mcl-1-dependent but not Bcl-2 dependent cancer cells.16 Maritoclax could activate Bax/Bak-dependent and caspase-dependent apoptotic machinery subsequent of proteasome-mediated Mcl-1 downregulation. Based on both NMR and computational docking studies, maritoclax is hypothesized to bind to a pocket near the C-terminus of the BH3-binding groove on Mcl-1.16 Unlike other BH3 peptides, the C-terminal region of Noxa BH3 domain uniquely induces Mcl-1 degradation.17 Maritoclax was therefore postulated to destabilize Mcl-1 by mimicking Noxa tail binding. Maritoclax has demonstrated efficacy against Mcl-1-dependent melanoma and AML cells through Mcl-1 down-regulation, inducing apoptosis both as a single agent and synergistically with ABT-737.18,19
Although maritoclax was promising against Mcl-1-dependent cancers, the compound demonstrated micromolar potency toward sensitive cells and was lipophilic. Therefore, structure-activity relationship (SAR) studies of its chemical derivatives in hematological cancer models could precisely define the pharmacophore, and generate compounds with greater solubility and potency. In this study, we synthesized over 30 pyoluteorin derivatives based on maritoclax, several of which demonstrated equal or greater potency against Mcl-1-dependent cell lines compared to maritoclax. Key pharmacophore sites which induce activity cliffs for Mcl-1-dependent cytotoxicity were identified on the pyoluteorin motif. Accordingly, 2 of these compounds, KS04 and KS18, physically interacted with Mcl-1 in a conserved manner. Both compounds induced selective Mcl-1 downregulation and subsequent Bak-dependent cytotoxicity alone and in synergy with ABT-737. KS18 demonstrated selectivity toward cancer cells, and overcame stroma-mediated drug resistance, while exhibiting less toxicity than daunorubicin against bone marrow cells. Finally, intraperitoneal (i.p.) administration of KS18 as a single agent and synergistic co-treatment with ABT-737 significantly reduced ABT-737-resistant (ABTR) HL60 xenograft tumor volumes in nude mice.
Results
Pyoluteorin derivatives antagonize Mcl-1
Maritoclax is a chimeric molecule belonging to the distinct class of bispyrrole compounds. However, the cytotoxic activities of the (+) and (−) enantiomers did not differ significantly between each other or the racemic mixture.19 Thus, a single pyrrole moiety may be sufficient for Mcl-1 antagonism. We therefore synthesized a series of compounds, KS01-KS31, bearing a single pyrrole moiety. Interestingly, compounds which mimicked the single pyrrole moiety of maritoclax were structural analogs of pyoluteorin (Table 1). Pyoluteorin is a natural occurring antibiotic, herbicidal, and antifungal agent for which anti-cancer activities have not been previously described.20 One such pyoluteorin derivative, KS04, demonstrated potency comparable to that of maritoclax toward the U937 AML cell line (Table 1).16 KS17 is a pro-drug of KS04 through the esterification of the hydroxyl group at the Y position on the pyoluteorin motif, and therefore similar to KS04 in potency to U937 cells. Compared to the parental compound maritoclax, KS04 contained a halogen substitution at the X position. On the other hand, KS02, which completely mimics the single pyrrole moiety of maritoclax, was weakly potent. Therefore, while a single pyrrole moiety was sufficient for Mcl-1 antagonism, substitutions at additional sites on the pyoluteorin motif could enhance Mcl-1-dependent cytotoxicity. We therefore performed additional substitutions on the KS04 motif in attempt to further enhance cytotoxicity. A bromo substitution at the Z group in KS06 or a methyl substitution at the X group in KS13 yielded decreased potency. The addition of hydroxyl or methoxy groups at R1, R2, and R3 sites in KS07-KS12 and KS14 all reduced the potency of pyoluteorin derivatives. A halogen or p-PhCl substitution at the R3 site in KS18–20, KS24, and KS27, on the other hand, generated an activity cliff of pyoluteorin derivatives with sub-micromolar EC50 against U937 cells.
Table 1.
Chemical structures of maritoclax derivatives bearing a pyoluteorin motif. Viability of U937 cells treated with the indicated compounds over 48 hours were used to calculate the EC50 values. For additional compounds that deviate from the pyoluteorin motif, see Table S1
| Pyoluteorin Motif | Compound | X | Y | Z | R1 | R2 | R3 | EC50 (μM) |
|---|---|---|---|---|---|---|---|---|
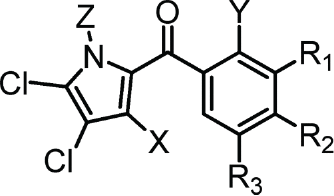 |
KS01 | ‒H | ‒OCH3 | ‒H | ‒H | ‒H | ‒H | > 50 |
| KS02 | ‒H | ‒OH | ‒H | ‒H | ‒H | ‒H | 19 | |
| KS03 | ‒Br | ‒OCH3 | ‒H | ‒H | ‒H | ‒H | > 50 | |
| KS04 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒H | 1.7 | |
| KS05 | ‒Br | ‒OCH3 | ‒Br | ‒H | ‒H | ‒H | 13 | |
| KS06 | ‒Br | ‒OH | ‒Br | ‒H | ‒H | ‒H | 11 | |
| KS07 | ‒H | ‒OCH3 | ‒H | ‒OCH3 | ‒OCH3 | ‒H | > 50 | |
| KS08 | ‒H | ‒OH | ‒H | ‒OCH3 | ‒OCH3 | ‒H | 30 | |
| KS09 | ‒H | ‒OH | ‒H | ‒OH | ‒OH | ‒H | 33 | |
| KS11 | Br | ‒OCH3 | ‒H | ‒OCH3 | ‒OCH3 | ‒H | 22 | |
| KS12 | Br | ‒OH | ‒H | ‒OH | ‒OCH3 | ‒H | 5.0 | |
| KS13 | ‒CH3 | ‒OH | ‒H | ‒H | ‒H | ‒H | 13 | |
| KS14 | ‒Br | ‒OH | ‒H | ‒H | ‒OH | ‒H | 30 | |
| KS17 | ‒Br | ‒OCOCH3 | ‒H | ‒H | ‒H | ‒H | 1.8 | |
| KS18 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒Cl | 0.5 | |
| KS18a | ‒Br | ‒OPO3H2 | ‒H | ‒H | ‒H | ‒Cl | 3.3 | |
| KS19 | ‒Br | ‒OCOCH3 | ‒H | ‒H | ‒H | ‒Cl | 0.5 | |
| KS20 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒F | 0.7 | |
| KS21 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒OH | 13 | |
| KS22 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒CH3 | 3.1 | |
| KS23 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒CH2CH3 | 2.5 | |
| KS24 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒p‒PhCl | 0.6 | |
| KS27 | ‒Br | ‒OH | ‒H | ‒H | ‒H | ‒Br | 0.8 |
As KS18 was a highly potent compound against the U937 cell line, pro-drugs were generated that could improve in vivo distribution and solubility through esterification or phosphorylation of the hydroxyl group in compounds KS19 and KS18a respectively at the Y site. KS18 and KS19 exhibited similar potencies against U937 cells, whereas KS18a was less potent. The substitution of the R3 site with a large hydrophobic group in KS31 decreased its potency toward U937 cells (Table S1).
We screened a number of promising pyoluteorin derivatives against a panel of hematological cancer cell lines, controlled with maritoclax, ABT-737, and daunorubicin (Table 2). The potencies of KS04 and its pro-drug KS17 against the panel of cell lines were highly similar to that of maritoclax, overcoming both multi-drug resistance and ABT-737 resistance (ABTR). Compounds KS18 and its pro-drug KS19, KS20, and KS24 were consistently more potent than maritoclax in sensitive cell lines. Furthermore, pyoluteorin derivatives were potent against multiple myeloma cell lines such NCI-H929, which was resistant to ABT-737 treatment. Importantly, Bcl-2-dependent K562 (K562/Bcl-2-IRES-Bim) and Bax/Bak-deficient Jurkat (JurkatΔBak) cells were resistant to pyoluteorin derivatives (Table 2).
Table 2.
The EC50 values of the indicated cell lines treated with the indicated compounds over 48 hours, *28 hours, **24 hours
| EC50 (μM) | Maritoclax | KS04 | KS17 | KS18 | KS19 | KS20 | KS24 | ABT-737 | Daunorubicin |
|---|---|---|---|---|---|---|---|---|---|
| K562/Mcl-1-IRES-Bim | 0.99 | 1.41 | 1.13 | 0.60* | 0.41 | 0.55 | 0.58 | 9.3** | |
| K562/Bcl-2-IRES-Bim | >20 | >25 | >20 | >50* | 0.35** | ||||
| Jurkat | 2.53 | 2.97 | 1.15 | 1.21 | 1.68 | 0.66 | |||
| JurkatΔBak | >15 | >20 | >25 | >25 | >25 | >50 | |||
| HL60/VCR | 1.67 | 1.35 | 1.17 | 0.69 | 0.66 | 0.71 | >50 | 30.41 | |
| HL60/ABTR | 2.15 | 3.03 | 1.63 | 0.73 | 0.80 | 0.83 | 1.83 | >100 | 12.17 |
| Kasumi-1 | 1.80 | 6.70 | 1.47 | 0.01 | |||||
| Kasumi-1/ABT | 2.15 | 2.91 | 0.70 | 0.51 | 0.46 | ||||
| THP-1 | 6.31 | 3.74 | 1.96 | 1.27 | 0.065 | ||||
| U937 | 1.23 | 1.66 | 1.77 | 0.54 | 0.50 | 0.67 | 0.57 | 5.29 | 0.86 |
| C1498 | 1.66 | 1.12 | 0.70 | 0.85 | 0.68 | 6.13 | 1.92 | ||
| RPMI 8226 | 2.68 | 1.87 | 0.94 | 0.72 | 0.58 | 0.57 | 0.25 | 0.23 | |
| MM.1S | 3.72 | 1.92 | 1.71 | 0.79 | 0.68 | 0.69 | 0.40 | 0.12 | |
| NCI-H929 | 1.47 | 1.00 | 1.30 | 0.72 | 0.74 | 0.73 | 15.21 | 0.24 | |
| U266 | 2.15 | 2.04 | 0.90 | 0.68 |
Pyoluteorin derivatives interact with Mcl-1
Four BH3 binding pockets p1, p2, p3, and p4 on Mcl-1 has been identified, corresponding to positions of E74, L78, I81, and V85 of mouse NoxaB (mNoxaB) BH3 domain, respectively.21 Noxa binding to Mcl-1 induces Mcl-1 degradation, and the amino acid sequence LRQKLL in the tail region of mNoxaB BH3 helix is considered essential for inducing degradation.17 NMR structures17 show amino acid residues K84, R88, and N93 of mNoxaB can form hydrogen bonds with Mcl-1 residues G308, E306, and E298 and F299 backbone carbonyl groups. These hydrogen bonds allow the C-terminus of mNoxaB BH3 helix to attach to the unfolded tail of Mcl-1 helix 8.17,21 However, the NMR structures17 contain different conformations of helix 8 tail region, and these specific hydrogen bonds may not hold in some of the conformations.
Maritoclax was previously shown to physically interact with Mcl-1 by NMR spectroscopy.16 In order to determine whether pyoluteorin derivatives bearing a single pyrrole moiety similarly interact with Mcl-1, we titrated 15N-labeled Mcl-1 with KS04 and KS18 for analysis through NMR spectroscopy. Both KS04 and KS18 appeared to interact with Mcl-1, apparent due to average chemical shift differences on the Mcl-1 spectra (Fig. 1A). Average chemical shift changes greater than 0.05 were observed for residues E161, K175, G198, T207, R214, L216, K219, E221, V224, S228, R229, V234, G243, I245, V246, T247, S250, F251, V255, N263, L279, R281, K289, H301, V302, L305, and E306. Among those, residues S228, G243, I245, S250, F251, V246, V255, L279, and E306 were also found to have changed significantly when titrated with maritoclax, suggesting that maritoclax, KS04, and KS18 could interact with a conserved site on Mcl-1.16 Indeed, the mapping of residues which demonstrated significant average chemical shift differences on the NMR structure of Mcl-1 indicates that maritoclax, KS04, and KS18 could mimic Noxa in its interaction with Mcl-1 (Fig. 1B). The conserved residues undergoing chemical shift differences upon addition of maritoclax, KS04, and KS18 reside almost exactly at BH3 binding pockets p2 and p4. Additional residues which demonstrated significant chemical shift differences for both KS04 and KS18 reside along the BH3 binding groove at pockets p1 and p3, as well as outside the BH3 binding pockets which might be due to conformational changes upon ligand binding. NMR spectroscopy data collectively suggest that maritoclax, KS04, and KS18 interact with Mcl-1 in a similar manner.
Figure 1.

Pyoluteorin derivatives bind to and induce Mcl-1 degradation. (A) Plot of average chemical shift differences in the spectra of 15N-labeled Mcl-1 upon titration with KS04 (top) and KS18 (bottom). (B) Pockets p2 (left, light blue) and p4 (left, dark red) are indicated on the NMR structure of Mcl-1 complexed with Noxa. The same NMR structure of Mcl-1 is also indicated with residues which demonstrate significant average chemical shift differences according to NMR spectroscopy for all of maritoclax, KS04, and KS18 (right, blue), both KS04 and KS18 (right, red), KS04 alone (right, light green), and KS18 alone (right, dark green). (C) The structure of KS04 and KS18 computationally docked to Mcl-1 by GLIDE. Blue and red colors of the molecular surface represent positive and negative potentials respectively. The carbon atoms of KS04 are colored gray and those of KS18 green. Chlorine atoms are colored dark green, bromine atom purple, oxygen atoms red, nitrogen blue, and polar hydrogen white. Non-polar hydrogen atoms are not shown. Left: The pyrrole group of the KS compounds docked in the p4 pocket of the molecular surface of Mcl-1 with their phenol group extended to the tail region of Mcl-1 helix 8. Right: Hydrogen bonding and amino acid residues near the predicated binding site of KS compounds. Mcl-1 backbone is represented by red to purple ribbons from N-terminus to C-terminus. Two hydrogen bonds between KS compounds and Mcl-1 are shown as yellow dotted lines. (D) U937 cells were treated with vehicle, 2.5 μM KS04 or 2 μM KS18 for 1 hour before adding 10 μg/ml CHX and collecting cells at the indicated times. Protein levels were detected by immunoblotting and quantified by densitometry.
KS04 and KS18 also demonstrated binding differences. KS04 selectively caused significant average chemical shift changes greater than 0.05 on residues A171, A208, F235, F254, H258, V262, V288, V297, and G307; and KS18 caused significant chemical shift changes on residues D154, E182, G211, K225, W293, and E298. Interestingly, residue K225 on Mcl-1 was significantly shifted following titration with KS18, but the same spectral peak for this residue remained completely unchanged following titration with KS04 (Fig. 1A).
The experimental observation that KS04 and KS18 might interact with Mcl-1 at BH3 binding pockets p2 and p4 in NMR studies led to computational studies for structural insights in their putative binding pocket. When KS04 and KS18 were docked to Mcl-1, their docking poses were very similar (Fig. 1C). Two possible binding regions were observed for both compounds, one is centered at p2 pocket between helices 4 and 5 in contact with helix 3, and the other centered at p4 pocket between helices 5 and 8 in contact with helix 2, similar to observations from NMR spectroscopy. The binding pocket p2 is more hydrophobic compared to p4, binding with L78 of mNoxaB BH3 domain. According to the docking results, the pyrrole group of KS compounds is located in p4 pocket. The hydroxyl oxygen located in the phenol group forms a hydrogen bond with N204 at the end of helix 2 and the hydroxyl hydrogen forming a hydrogen bond with G308 of the Mcl-1 helix 8 tail region. Among residues with chemical shifts greater than 0.05 ppm with KS04, F235 is near the end of helix 4, H258 is near the end of helix 5, and V297 is located within helix 8. G307 is located near the C-terminus flexible region. Among residues with chemical shifts greater than 0.05 ppm with KS18, both W293 and E298 are on helix 8, supporting KS18 binding near the end of the helix 8. D154 is at the beginning of helix 1 and its chemical shift could be the result of the N-terminal flexibility upon ligand binding. K225 is located on helix 4, and G211 is located on helix 5. However, these residues which specifically interact with either compound face away from the BH3 binding pockets, suggesting a difference in conformational change resulting from a conserved interaction site on Mcl-1. As mentioned previously, the binding site of KS04 and KS18 near pocket p4 may not be the only possible site, but the close interaction of these KS compounds with the tail region of Mcl-1 helix 8, similar to that of mNoxaB BH3 C-terminus, suggests that binding to Mcl-1 helix 8 tail region may be related to Mcl-1 degradation in cells.
Accordingly, KS04 and KS18 decreased Mcl-1 protein half-life from 178 minutes to 21 and 20 minutes, respectively, in U937 cells treated with CHX (Fig. 1D). Our data cumulatively suggest that pyoluteorin derivatives KS04 and KS18 interact with Mcl-1 in a similar mechanism compared to maritoclax, by destabilizing Mcl-1 protein levels through physical interaction with Mcl-1.
Pyoluteorin derivatives cause Mcl-1-dependent cell death in hematological cancer cell lines
We determined whether pyoluteorin derivatives could induce Mcl-1-dependent apoptosis in hematological cancer cell lines. KS04 and KS18 were at least 10-fold more selective against K562/Mcl-1-IRES-Bim compared to K562/Bcl-2-IRES-Bim cell lines (Fig. 2A, B). Correspondingly, KS04 induced selective Mcl-1 down-regulation in the K562/Mcl-1-IRES-Bim cell line over 72 hours, leading to induction of apoptosis (Fig. 2D). However, other Bcl-2 family proteins remained largely unchanged in both K562/Mcl-1-IRES-Bim and K562/Bcl-2-IRES-Bim cell lines, suggesting that KS04 induced Mcl-1-dependent apoptosis. Similarly, KS18 and KS24 induced concentration-dependent degradation of Mcl-1 followed by caspase-3 and PARP cleavage (Fig. 2E, F).
Figure 2.

Pyoluteorin derivatives induce Mcl-1-dependent cell death through Bax and Bak in hematological cancer cell lines. Viabilities of K562/Mcl-1-IRES-Bim and K562/Bcl-2-IRES-Bim cells were measured after treatment with the indicated concentrations of KS04 (A) and KS18 (B) for 48 hours. Error bars = SD of 3 replicates. (C) Parental and ΔBak Jurkat cells were treated with the indicated concentrations of KS18 for 48 hours. (D) K562/Mcl-1-IRES-Bim and K562/Bcl-2-IRES-Bim cells were treated with 2 μM KS04 over the indicated times be analyzed through immunoblotting. K562/Mcl-1-IRES-Bim cells were treated with the indicated concentrations of KS18 (E) and KS24 (F) for 18 and 24 hours respectively and analyzed by immunoblotting.
As antagonism of Mcl-1 leads to the activation and oligomerization of Bax and Bak to initiate apoptosis, the cytotoxic effects of selective Mcl-1 inhibitors should require Bax and Bak. We confirmed that the Jurkat/ΔBak cell line that constitutively lacks Bak22 was highly resistant to KS18 treatment at an EC50 above 25 μM, unlike parental Jurkat cells with an EC50 of 1.15 μM (Fig. 2C).
KS04 and KS18 can overcome Mcl-1-mediated ABT-737 resistance in hematologic cancer cell lines
Mcl-1 up-regulation is a major mechanism by which cancer cells gain resistance to selective Bcl-2 inhibitors.9 We previously generated a number of ABT-737 resistant (ABTR) leukemic cell lines HL60/ABTR, Kasumi-1/ABTR, and the KG1a/ABTR cell lines through prolonged culture with sub-optimal concentrations of ABT-737. All of these ABTR cell lines demonstrated significant Mcl-1 upregulation.19 Expectedly, pyoluteorin derivatives demonstrated equal or greater potency toward HL60/ABTR and Kasumi-1/ABTR cells compared to maritoclax, which was previously shown to overcome Mcl-1-mediated drug resistance (Table 2).19 The Kasumi-1/ABTR cell line was twice as sensitive to KS04 and KS18 compared to the parental Kasumi-1 cell line. The mechanism of cell death was confirmed through immunoblotting, where KS04 and KS18 were able to induce the selective downregulation of Mcl-1 in the HL60/ABTR cell line (Fig. 3A). While down-regulated Mcl-1 was apparent at 12 hours in both cases, caspases-3 and PARP cleavage only occurred between 24 and 48 hours, suggesting that apoptosis induction is a downstream event of Mcl-1 down-regulation.
Figure 3.

KS04 and KS18 overcome Mcl-1-mediated ABT-737 resistance in hematological cancer cell lines. (A) HL60/ABTR cells were treated with 2 μM KS04 and 1.5 μM KS18 over the indicated times and collected for analysis through immunoblotting. (B) Viability (top) of KG1a/ABTR cells was measured after treatment with the indicated concentrations ABT-737 with vehicle or a sub-optimal concentration of 1.6 μM KS04 for 48 hours. Error bars = SD of 3 replicates. Combination index between ABT-737 and KS04 was calculated based on viability data (bottom). Combination index <1 signifies synergy. (C) HL60/ABTR cells were treated with the indicated concentrations of ABT-737, KS18, or co-treatment at 1:1 ratio to the final indicated concentration over 48 hours to measure viability (top). Error bars = SD of 3 replicates. Combination index between ABT-737 and KS18 was calculated based on the viability data (bottom). (D) HL60/ABTR cells were treated with the indicated concentrations of KS18, ABT-737, or co-treatment at 1:1 ratio to the final indicated concentration for 48 hours and subjected to immunoblotting.
The addition of a sub-optimal concentration of KS04 and ABT-737 to the KG1a/ABTR cell line synergistically induced cell death (Fig. 3B). Similarly, the combinatorial treatment of KS18 and ABT-737 also synergistically killed HL60/ABTR cells (Fig. 3C). Although KS18 alone induced Mcl-1 protein downregulation in HL60/ABTR cells, combination with ABT-737 enhanced caspase-3 and PARP cleavage at lower concentrations of either compound (Fig. 3D). These data collectively suggest that pyoluteorin derivatives could overcome ABT-737 resistance through selective Mcl-1 down-regulation.
KS04 and KS18 overcome stroma-mediated drug resistance and are less toxic than daunorubicin to stroma cells
For hematological malignancies which originate from the bone marrow, the in vitro culture of these cancer cells with bone marrow stroma can mimic the bone marrow microenvironment, resulting in decreased proliferation and resistance to chemotherapy.23-25 Mcl-1 up-regulation has been implicated in this process.26 Therefore, we cultured U937 cells constitutively expressing luciferase (U937-luc) alone or with the human bone marrow stroma cell line HS-5 under the treatment of KS04, KS18, or daunorubicin (Fig. 4A). Expectedly, U937-luc cells cultured with stroma gained resistance to daunorubicin, but remained sensitive to KS04 and KS18 treatment. However, HS-5 stroma cells were more resistant to KS04 and KS18 compared to U937 leukemia cells, suggesting the existence of a putative therapeutic window.
Figure 4.

KS04 and KS18 are less toxic to healthy bone marrow cells compared to daunorubicin. (A) U937-luc cells alone or co-cultured with HS-5 stromal cells, or HS-5 stroma cells alone, were treated with the indicated concentrations of daunorubicin (left), KS04 (middle), and KS18 (right) over 48 hours. Viability of single culture cells and the viability of U937-luc cells in the co-culture were determined. Error bars = SD of 3 replicates. (B) Primary mouse bone marrow was treated with maritoclax, KS04, KS18, ABT-737, and daunorubicin over 24 hours to measure viability (errors = SEM of 5 independent experiments). (C) Primary mouse bone marrow seeded to methylcellulose medium supplemented with growth factors were treated with the indicated concentrations of KS18 and daunorubicin for 7 days, and the number of haematopoietic cell colonies were counted.
A previous study demonstrated that Mcl-1 is essential for hematopoiesis in the bone marrow, raising the concern that pharmacologic inhibitors of Mcl-1 could be too toxic for the treatment of hematological malignancies.27 We therefore evaluated the toxicity of pyoluteorin derivatives to primary bone marrow cells from healthy C57BL/6J mice. In a series of independent 24 hour viability studies, both ABT-737 and daunorubicin killed significantly more primary mouse bone marrow cells compared to maritoclax, KS04, or KS18 (Fig. 4B). KS18 appeared to be more toxic to the primary bone marrow cells compared to KS04. However, the increased toxicity correlated the increased potency of KS18. Whereas KS18 demonstrated 3.4-fold lower EC50 compared to KS04 in U937 cells (Table 1), KS18 was only 2.5-fold more toxic to bone marrow cells. Although bone marrow cells were more sensitive to KS18 treatment based on the EC50 value alone, the margin of safety may have instead increased compared to KS04.
We evaluated the toxicity of KS18 and daunorubicin against primary mouse bone marrow haematopoietic progenitor cells by incubating bone marrow cells in methylcellulose medium supplemented with stem cell factor, IL-3, IL-6, and erythropoietin (Fig. 4C). Daunorubicin was acutely toxic to bone marrow haematopoietic progenitor cells, as no colonies were observed at any of the concentrations tested. KS18, however, was markedly less toxic. Even after 7 d of continuous incubation with the compound, more than 60% of colonies remained at 1 μM. KS18 demonstrated an apparent 7-day EC50 of 1.62 μM toward haematopoietic colonies.
KS18 synergizes with ABT-737 to reduce HL60/ABTR xenograft tumor growth in female athymic nude mice
Given promising in vitro results suggesting the existence of a therapeutic window for KS18, the in vivo toxicity of the compound was evaluated. We administered different doses of KS18 to female athymic nude mice both intraperitoneally (i.p.) and orally (p.o.) to female athymic nude mice. The maximum tolerated dose (MTD) of once daily i.p. administration was 10 mg/kg and a median lethal dose (LD50) was 15 mg/kg. The compound demonstrated a p.o. MTD of 20 mg/kg and LD50 of greater than 30 mg/kg.
One of the major goals for synthesising maritoclax derivatives was to improve its solubility and concentration in peripheral blood. We therefore determined the pharmacokinetics of maritoclax (10 mg/kg) and KS18 (5 mg/kg) by i.p. administration in Balb/c mice (Table 3; Table S2). Maritoclax administration demonstrated favorable half-life (T1/2) in mice plasma at 3.47 hours, reaching a maximum concentration (Cmax) corresponding to 3.01 μM. The large volume of distribution (VD) confirmed that maritoclax was lipophilic and was likely significantly distributed to tissues, a feature that may be undesirable for the treatment of hematologic malignancies. On the other hand, KS18 indeed demonstrated a markedly lower VD and a 10-fold higher Cmax at 37.2 μM. KS18 was able to reach well above its therapeutic concentration, suggesting that KS18 would exert its effects at this dose in vivo. However, the compound demonstrated a lower T1/2 at 2.78 hours, suggesting that the more hydrophilic KS18 could undergo renal clearance more rapidly.
Table 3.
The pharmacokinetic parameters of maritoclax and KS18 by intraperitoneal injection in female BALB/c mice
| Maritoclax (i.p.) 10 mg/kg | KS18 (i.p.) 5 mg/kg | |
|---|---|---|
| T1/2 (hours) | 3.47 | 2.78 |
| Tmax (hours) | 2.0 | 0.50 |
| Cmax (ng/mL) | 1536.16 | 14268.36 |
| AUClast (hr*ng/mL) | 5408.65 | 14311.68 |
| AUCINF_OBS (hr*ng/mL) | 5418.96 | 14321.47 |
| VD (mL/kg) | 9239.86 | 1401.30 |
| CL (mL/hour/kg) | 1845.37 | 349.13 |
The in vivo efficacy of KS18 was then evaluated in athymic nude mice xenografted with HL60/ABTR tumors. After tumor staging, animals were treated with vehicle, ABT-737, KS18, or their combination by daily i.p. administration for 14 consecutive days. As the cells were ABT-737 resistant, the tumors were not responsive to ABT-737 treatment (Fig. 5A). On the other hand, KS18 alone caused a significant reduction in HL60/ABTR tumor volumes. The combination treatment of ABT-737 and KS18 synergistically reduced xenograft tumor sizes as calculated by their combination index (Fig. 5A). However, weight loss in these treated mice did not extend beyond 10% of their initial weights. We also subjected 5 mice from the co-treatment group to histopathological examination after 14 d of continuous drug administration. We were not able to detect any signs of acute toxicity due to drug treatment in the brain, heart, lungs, liver, kidneys, or spleen (data not shown).
Figure 5.
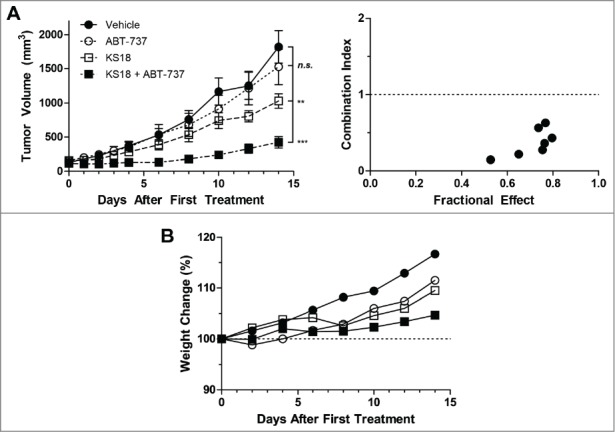
KS18 and ABT-737 synergize to reduce HL60/ABTR xenograft tumor growth in athymic nude mice. (A) Female athymic nude mice bearing HL60/ABTR xenograft tumors were treated with vehicle, ABT-737 (20 mg/kg), KS18 (10 mg/kg), or both ABT-737 and KS18 at 20 mg/kg and 10 mg/kg respectively, for 14 consecutive days after tumor staging. Tumor volumes were measured every 2 days. Error bars = SEM of 10–20 xenograft tumors. n.s. = not statistically significant, **P <0.005, ***P <0.0005 by Student's T-test of tumor volumes at day 14. Combination index between ABT-737 and KS18 was calculated based on perecent tumor size of vehicle control at each day (right). Combination index <1 signifies synergy. (B) The average body weights of mice bearing HL60/ABTR xenograft tumors expressed as a percentage of day 0.
Discussion
Upregulated Mcl-1 contributes to survival and chemo-resistance in many hematological cancers.9,13-15 We previously identified a small molecule inhibitor of Mcl-1, maritoclax, which demonstrated in vitro and in vivo efficacy toward melanoma and AML.16,18,19 However, maritoclax was lipophilic and might be sequestered into fatty compartments in vivo, as evidenced by its large volume of distribution (Table 3). We therefore sought to optimize maritoclax with improved solubility and potency toward Mcl-1-dependent cancer cells, as well as to optimize pharmacokinetic parameters to improve the in vivo therapeutic effect. To this purpose, we synthesized a library of small molecules, KS01-KS31, in order to identify the pharmacophores responsible for Mcl-1 inhibition and cytotoxicity (Table 1; Table S1). Based on structure-activity guided synthesis of small molecule derivatives, several compounds bearing a single pyoluteorin motif with nearly half the molecular weight of maritoclax were identified to be Mcl-1 antagonists.
Pyoluteorin is a naturally-occurring small molecule synthesized by fluorescent Pseudomonas. This compound is currently under investigation for its antibiotic, antifungal, and herbicidal activities.20 We identified a number of pyoluteorin analogs that behaved as Mcl-1 inhibitors possessing sub-micromolar potency toward Mcl-1 dependent hematological cancer cells. Based on SAR studies, our data indicates that a bromo substitution at position X, a hydroxyl functional group at Y, as well as a halogen functional group at R3 of the phenol group are necessary for potent Mcl-1 antagonism (Table 1). Substitutions at the Z, R1, and R2 sites consistently reduced or abrogated pyoluteorin derivative potency, suggesting that larger functional groups could result in steric hindrance, weakening the interaction between Mcl-1 and pyoluteorin derivatives.
A series of pyoluteorin derivatives were eventually identified to have enhanced solubility, potency, and selectivity toward Mcl-1-dependent hematological cancer cells. The first group of compounds KS04 and KS17 were identified to demonstrate similar potency and mechanism of action compared to maritoclax. Further studies led to the synthesis of KS18-20, KS24, and KS27 with enhanced potency. Among these, we chose the lead molecules KS04 and KS18 for detailed analysis as Mcl-1 inhibitors. NMR spectroscopy and molecular modeling collectively suggest that KS04 and KS18 may interact with BH3 binding pockets p2 or p4, mimicking Noxa binding to Mcl-1. As selective Bcl-2 family inhibitors should depend on Bax/Bak oligomerization of apoptosis induction, we confirmed that Jurkat/ΔBak cells were highly resistant to both KS04 and KS18 treatment compared to parental Jurkat cells (Fig. 2C). Our data collectively indicate that pyoluteorin derivatives such as KS04 and KS18 interact with Mcl-1 to induce Mcl-1-dependent apoptosis.
Selective Bcl-2 inhibitors are promising for the treatment of Bcl-2 dependent hematological cancers. However, Mcl-1 upregulation is a major mechanism of resistance and remains a major hurdle in selective Bcl-2 inhibitor therapy.9 Pharmacologic Mcl-1 downregulation can sensitize these resistant cells to selective Bcl-2 inhibition, increasing their therapeutic benefit.16,18,19,28,29 Accordingly, KS04 and KS18 synergized with ABT-737 to induce apoptosis in hematological cancer cells through Mcl-1 down-regulation (Fig. 3). KS04 and KS18 overcame Mcl-1-dependent drug-resistance and sensitized these cells to selective Bcl-2 inhibitor treatment.
Mcl-1 was reported to be necessary for the survival of haematopoietic stem cells,27 yet a therapeutic window was suggested to exist in another genetic Mcl-1 knock-out mouse model.15 In our studies with pyoluteorin derivatives, KS04 and KS18 were less toxic than daunorubicin or ABT-737 to healthy bone marrow cells (Fig. 4). Additionally, the i.p. administration of KS18 alone to female athymic nude mice significantly reduced tumor sizes compared to vehicle treated controls, but did not cause apparent toxicity upon histopathological examination (Fig. 5). Our studies affirm that a therapeutic window could potentially exist for the in vivo treatment of small molecule inhibitors of Mcl-1.
In conclusion, a number of pyoluteorin derivatives based on maritoclax were synthesized for SAR studies as Mcl-1 inhibitors. We identified several compounds which demonstrate sub-micromolar potency against Mcl-1-dependent hematological cancer cell lines. Among these, KS04 and KS18 induced Mcl-1 downregulation and subsequent Bax/Bak-dependent apoptosis alone or synergistically with ABT-737. KS18 was significantly less toxic than daunorubicin against primary mouse bone marrow and haematopoietic progenitor cells. KS18 is bioavailable in mice, and its i.p. administration in female athymic nude mice significantly reduced HL60/ABTR xenograft tumor volumes both as a single agent, and in synergistic combination with ABT-737. We were able to describe pharmacophore sites for which a hydroxyl group and a halogen group generate activity cliffs for Mcl-1 antagonism. Our data collectively suggest that pyoluteorin derivatives such as KS04 and KS18 are Mcl-1 inhibitors, possessing potent anti-cancer activity against Mcl-1-dependent cancers both in vitro and in vivo.
Materials and Methods
Synthesis of pyoluteorin derivatives
See online Supplemental Materials for methods and materials of the synthesis of pyoluteorin derivatives.
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy with 15N-labeled sample of Mcl-1ΔNC2321 titrated with KS04 or KS18 were completed as described.16 Briefly, 5 μL or 12.5 μL of 10 mM KS04 in DMSO, or 5 μL of 10 mM KS18 in DMSO, was added to the 15N-labeled Mcl-1 sample. A 15N-1H HSQC spectrum was collected on this sample with standard Bruker pulse program and parameters. NMR data was collected after each addition of either compound. Mcl-1 residues were assigned based on previously published peak-lists.30 Residues which experienced an average chemical shift difference greater than 0.08 were colored on the NMR structure of Mcl-1ΔNC23 complexed with NoxaA (2ROD.pdb, NoxaA hidden). Average chemical shift differences were calculated according to the following formula:
Molecular modeling
KS04 and KS18 docking was carried out using the GLIDE (Grid-based Ligand Docking from Energetics) program31 from Schrödinger, L.L.C. OPLS-2005 force field32 was applied in the GLIDE program. The optimal binding geometry for each model was obtained by utilization of Monte Carlo sampling techniques coupled with energy minimization. GLIDE uses a scoring method based on ChemScore33 but with additional terms added for greater accuracy. The NMR solution structures of mouse Mcl-1 in complex with mouse NoxaB (2JM6.pdb)17 were used for docking KS compounds to Mcl-1.
Antibodies and compounds
Antibodies were obtained from the following sources: human polyclonal Mcl-134; human polyclonal Bcl-234; monoclonal Bcl-xL (Sigma B9429); polyclonal Bim (Sigma B7929); monoclonal β-actin (Sigma A5441); polyclonal PARP (Cell Signaling #9542); polyclonal Cleaved Caspase-3 (Cell Signaling #9661); polyclonal Bax (Santa Cruz sc-493); polyclonal Bak (Millipore 06-536). Racemic maritoclax was synthesized as previously described.35 Cycloheximide (CHX) and daunorubicin were obtained from Sigma (C6798 and D8809 respectively). ABT-737 was obtained from Abott Laboratories.
Cell culture and transfection
The HL60/ABTR, Kasumi-1/ABTR, and KG1a/ABTR cell lines were generated by incubation with sub-optimal concentrations of ABT-737 as previously described.19 The K562/Mcl-1-IRES-Bim and K562/Bcl-2-IRES-Bim cell lines were generated by retroviral transduction as previously described.16 Jurkat, JurkatΔBak, HL60, HL60/ABTR, HL60/VCR, THP-1, U937, C1498, RPMI 8226, and MM.1S cell lines were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). The U266 cell line was maintained in RPMI-1640 medium with 15% FBS. The Kasumi-1, Kasumi-1/ABTR, KG-1, KG1a, and KG1a/ABTR cell lines were maintained in RPMI-1640 medium with 20% FBS. The NCI-H929 cell line was maintained in RPMI-1640 medium with 10% FBS, 2 mM glutamine, and 0.05 mM 2-mercaptoethanol. All cell lines were maintained with 100 units/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B (Cellgro) and cultured at 37˚C with 5% CO2 unless otherwise stated. The U937-luc cell line in co-culture studies with the HS-5 cell line were previously described,19 and the specific viability of leukemic cells were measured through luminescence immediately following the addition of 150 μg/mL D-luciferin. Immunoblotting was done as previously described after cells were lysed in 1% CHAPS or RIPA lysis buffer.16
Cell viability assay
Cell viability was determined following 48 hours of treatment unless otherwise specified by the indicated compounds in medium supplemented with 10% FBS by measuring intracellular ATP levels with the CellTiter Glo Luminescent Cell Viability Assay kit (Promega G7571) according to the manufacturer's recommendations.
In vitro culture of primary mouse bone marrow
The bone marrow was collected from femur and tibia of C57BL/6J mice, and washed twice in IMDM medium. 1 × 106 viable cells/mL were seeded in IMDM medium with 10% FBS, and treated immediately for cell viability assay over 24 hours.
Primary mouse bone marrow colony formation assay
The bone marrow was collected from the femur and tibia of female C57BL/6J mice, washed twice in IMDM medium, and viable cells by trypan blue staining were seeded at 2 × 105 cells/mL in methylcellulose medium with recombinant cytokines (MethoCult GF M3434, StemCell Technologies) according to the manufacturer's recommendations with the indicated compounds (in 0.25% DMSO) or 0.25% DMSO control for 7 d before counting colonies.
Pharmacokinetics of maritoclax and pyoluteorin derivatives in Balb/c mice
All pharmacokinetics studies were completed by GenScript (Piscataway, NJ 08854, USA). In brief, 30 female Balb/c mice between 20–22 g were used for each treatment group. Maritoclax or KS18 was administered in PTD solvent36 with ≤1% DMSO by i.p., and 3 mice were sacrificed at each time point: 0, 15, and 30 minutes, and 1, 2, 4, 6, 8, and 24 hours after drug administration to collect blood samples. The compounds in plasma samples were then analyzed by a validated LC-MS/MS method. For detailed methods, see Supplementary Methods.
Toxicity of KS18 to female athymic nude mice
Animal studies were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committees at the Penn State University College of Medicine. Female athymic nude (NCI Athymic NCr-nu/nu #01B74) mice were obtained from Jackson Laboratories. Four 6 weeks old mice were injected with KS18 dissolved in PTD solvent36 with <3% DMSO i.p. or orally with oral gavage to determine the maximum tolerated dose (MTD), defined as the maximum dose of KS18 that the animals received without causing mortality or greater than 10% loss in body weight, as well as the median lethal dose (LD50), defined as the dose for which 50% of treated animals were moribund.
HL60/ABTR xenograft tumor model in female athymic nude mice
A total of 60 female athymic nude mice were subcutaneously transplanted with 5 × 106 of HL60/ABTR cells in 200 μL PBS with 50% Matrigel (BD Biosciences) on either or both flanks at 6 weeks of age. Tumor volumes were measured by electronic caliper and calculated with the following formula:
Xenografted mice bearing tumors at ∼150 mm3 were randomly assigned to the following treatment groups: vehicle control (100 μL PTD); ABT-737 (20 mg/kg body weight); KS18 (10 mg/kg); and ABT-737 (20 mg/kg) + KS18 (10 mg/kg). ABT-737 and KS18 were formulated in PTD solvent with <3% DMSO and administered i.p. daily to animals for up to 17 consecutive days. Tumor volumes and body weights were monitored every 2 d until 14 d following after initial tumor staging. A total of 83 xenografted tumors were analyzed (n = 29, 10, 27, 17 corresponding to the control, ABT-737, KS18 and ABT-737 + KS18, respectively). Animals bearing tumors larger than 1500 mm3 or over 20 mm in diameter were immediately euthanized. 17 days following initial tumor staging, 5 mice treated with ABT-737 + KS18 were subjected to histopathological examination of major organs including the brain, heart, lung, liver, kidneys, and spleen.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego California USA, www.graphpad.com). EC50 calculations for viability were calculated through non-linear regression with normalized data assuming variable slope. Synergy was calculated by the CompuSyn software (ComboSyn Inc., Paramus, NJ).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Gerard Grosveld (St Jude Children's Research Hospital) for the MSCV-Luciferase-IRES-YFP construct; Dr Mark Hinds (Walter and Eliza Hall Institute of Medical Research) for the backbone and side-chain resonance assignments of the mouse Mcl-1 protein; Dr Hannah Rabinowich (University of Pittsburgh) for the JurkatΔBak cell line; Dr Timothy Cooper (Penn State College of Medicine) for the histopathological examination of mouse tissues.
Funding
This work was supported by the Lois High Berstler Endowment Fund and the Four Diamonds Fund of Penn State College of Medicine.
Supplemental Materials
Supplemental materials for this article are available online at the publisher's website.
References
- 1. Liu Q, Wang HG. Anti-cancer drug discovery and development: Bcl-2 family small molecule inhibitors. Commun Integr Biol 2012; 5:557-65; PMID:23336025; http://dx.doi.org/ 10.4161/cib.21554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: Cancer J Clin 2014; 64:9-29; PMID:24399786 [DOI] [PubMed] [Google Scholar]
- 3. Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984; 226:1097-9; PMID:6093263; http://dx.doi.org/ 10.1126/science.6093263 [DOI] [PubMed] [Google Scholar]
- 4. Schena M, Larsson LG, Gottardi D, Gaidano G, Carlsson M, Nilsson K, Caligaris-Cappio F. Growth- and differentiation-associated expression of bcl-2 in B-chronic lymphocytic leukemia cells. Blood 1992; 79:2981-9; PMID:1375120 [PubMed] [Google Scholar]
- 5. Harada N, Hata H, Yoshida M, Soniki T, Nagasaki A, Kuribayashi N, Kimura T, Matsuzaki H, Mitsuya H. Expression of Bcl-2 family of proteins in fresh myeloma cells. Leukemia: Off J Leukemia Soc Am, Leukemia Res Fund, UK 1998; 12:1817-20; PMID:9823959; http://dx.doi.org/ 10.1038/sj.leu.2401168 [DOI] [PubMed] [Google Scholar]
- 6. Tu Y, Xu FH, Liu J, Vescio R, Berenson J, Fady C, Lichtenstein A. Upregulated expression of BCL-2 in multiple myeloma cells induced by exposure to doxorubicin, etoposide, and hydrogen peroxide. Blood 1996; 88:1805-12; PMID:8781438 [PubMed] [Google Scholar]
- 7. Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud JP, Guyotat D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993; 81:3091-6; PMID:7684624 [PubMed] [Google Scholar]
- 8. Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, Lynch J, Hans CP, Weisenburger DD, Greiner TC, et al. . BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol 2006; 24:961-8; PMID:16418494; http://dx.doi.org/ 10.1200/JCO.2005.03.4264 [DOI] [PubMed] [Google Scholar]
- 9. Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, et al. . Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10:375-88; PMID:17097560; http://dx.doi.org/ 10.1016/j.ccr.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 10. Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S, Avet-Loiseau H, Bataille R, Moreau P, Le Gouill S, et al. . ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood 2011; 118:3901-10; PMID:21835956; http://dx.doi.org/ 10.1182/blood-2010-11-317438 [DOI] [PubMed] [Google Scholar]
- 11. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. . ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19:202-8; PMID:23291630; http://dx.doi.org/ 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 12. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al. . Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014; 4:362-75; PMID:24346116; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia: Off J Leukemia Soc Am, Leukemia Res Fund, UK 2005; 19:1248-52; PMID:15902294; http://dx.doi.org/ 10.1038/sj.leu.2403784 [DOI] [PubMed] [Google Scholar]
- 14. Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002; 100:194-9; PMID:12070027; http://dx.doi.org/ 10.1182/blood.V100.1.194 [DOI] [PubMed] [Google Scholar]
- 15. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, et al. . Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 2012; 26:120-5; PMID:22279045; http://dx.doi.org/ 10.1101/gad.182980.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doi K, Li R, Sung SS, Wu H, Liu Y, Manieri W, Krishnegowda G, Awwad A, Dewey A, Liu X, et al. . Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem 2012; 287:10224-35; PMID:22311987; http://dx.doi.org/ 10.1074/jbc.M111.334532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Nat Acad Sci U S A 2007; 104:6217-22; PMID:17389404; http://dx.doi.org/ 10.1073/pnas.0701297104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pandey MK, Gowda K, Doi K, Sharma AK, Wang HG, Amin S. Proteasomal degradation of Mcl-1 by maritoclax induces apoptosis and enhances the efficacy of ABT-737 in melanoma cells. PloS One 2013; 8:e78570; PMID:24223823; http://dx.doi.org/ 10.1371/journal.pone.0078570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doi K, Liu Q, Gowda K, Barth BM, Claxton D, Amin S, Loughran TP, Jr, Wang HG. Maritoclax induces apoptosis in acute myeloid leukemia cells with elevated Mcl-1 expression. Cancer Biol Ther 2014; 15:1077-86; PMID:24842334; http://dx.doi.org/ 10.4161/cbt.29186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J, Wang W, Lu X, Xu Y, Zhang X. The stability and degradation of a new biological pesticide, pyoluteorin. Pest Manage Sci 2010; 66:248-52; PMID:19834883; http://dx.doi.org/ 10.1002/ps.1856 [DOI] [PubMed] [Google Scholar]
- 21. Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol 2008; 380:958-71; PMID:18589438; http://dx.doi.org/ 10.1016/j.jmb.2008.05.071 [DOI] [PubMed] [Google Scholar]
- 22. Wang GQ, Gastman BR, Wieckowski E, Goldstein LA, Gambotto A, Kim TH, Fang B, Rabinovitz A, Yin XM, Rabinowich H. A role for mitochondrial Bak in apoptotic response to anticancer drugs. J Biol Chem 2001; 276:34307-17; PMID:11447222; http://dx.doi.org/ 10.1074/jbc.M103526200 [DOI] [PubMed] [Google Scholar]
- 23. Garrido SM, Appelbaum FR, Willman CL, Banker DE. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp Hematol 2001; 29:448-57; PMID:11301185; http://dx.doi.org/ 10.1016/S0301-472X(01)00612-9 [DOI] [PubMed] [Google Scholar]
- 24. Yamamoto-Sugitani M, Kuroda J, Ashihara E, Nagoshi H, Kobayashi T, Matsumoto Y, Sasaki N, Shimura Y, Kiyota M, Nakayama R, et al. . Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc Nat Acad Sci U S A 2011; 108:17468-73; PMID:21987825; http://dx.doi.org/ 10.1073/pnas.1111138108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheung WC, Van Ness B. The bone marrow stromal microenvironment influences myeloma therapeutic response in vitro. Leukemia: Off J Leukemia Soc Am, Leukemia Res Fund, UK 2001; 15:264-71; PMID:11236942; http://dx.doi.org/ 10.1038/sj.leu.2402022 [DOI] [PubMed] [Google Scholar]
- 26. Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood 2009; 113:149-53; PMID:18836097; http://dx.doi.org/ 10.1182/blood-2008-02-138560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307:1101-4; PMID:15718471; http://dx.doi.org/ 10.1126/science.1106114 [DOI] [PubMed] [Google Scholar]
- 28. Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67:782-91; PMID:17234790; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3964 [DOI] [PubMed] [Google Scholar]
- 29. Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, Maiga S, Bene MC, Moreau P, Pellat-Deceunynck C, et al. . The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia: Off J Leukemia Soc Am, Leukemia Res Fund, UK 2014; 28:210-2; PMID:23860449; http://dx.doi.org/ 10.1038/leu.2013.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem 2005; 280:4738-44; PMID:15550399; http://dx.doi.org/ 10.1074/jbc.M411434200 [DOI] [PubMed] [Google Scholar]
- 31. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. . Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004; 47:1739-49; PMID:15027865; http://dx.doi.org/ 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- 32. Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996; 118:11225-36; http://dx.doi.org/ 10.1021/ja9621760 [DOI] [Google Scholar]
- 33. Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput-Aided Mol Des 1997; 11:425-45; PMID:9385547; http://dx.doi.org/ 10.1023/A:1007996124545 [DOI] [PubMed] [Google Scholar]
- 34. Krajewski S, Bodrug S, Gascoyne R, Berean K, Krajewska M, Reed JC. Immunohistochemical analysis of Mcl-1 and Bcl-2 proteins in normal and neoplastic lymph nodes. Am J Pathol 1994; 145:515-25; PMID:8080035 [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng C, Pan L, Chen Y, Song H, Qin Y, Li R. Total synthesis of (+−)-marinopyrrole A and its library as potential antibiotic and anticancer agents. J Comb Chem 2010; 12:541-7; PMID:20429575; http://dx.doi.org/ 10.1021/cc100052j [DOI] [PubMed] [Google Scholar]
- 36. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. . An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677-81; PMID:15902208; http://dx.doi.org/ 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.