

Abstract
Most research to date has focused on epigenetic modifications in the nuclear genome, with little attention devoted to mitochondrial DNA (mtDNA). Placental mtDNA content has been shown to respond to environmental exposures that induce oxidative stress, including airborne particulate matter (PM). Damaged or non-functioning mitochondria are specifically degraded through mitophagy, exemplified by lower mtDNA content, and could be primed by epigenetic modifications in the mtDNA. We studied placental mtDNA methylation in the context of the early life exposome. We investigated placental tissue from 381 mother-newborn pairs that were enrolled in the ENVIRONAGE birth cohort. We determined mtDNA methylation by bisulfite-pyrosequencing in 2 regions, i.e., the D-loop control region and 12S rRNA (MT-RNR1), and measured mtDNA content by qPCR. PM2.5 exposure was calculated for each participant's home address using a dispersion model. An interquartile range (IQR) increment in PM2.5 exposure over the entire pregnancy was positively associated with mtDNA methylation (MT-RNR1: +0.91%, P = 0.01 and D-loop: +0.21%, P = 0.05) and inversely associated with mtDNA content (relative change of −15.60%, P = 0.001) in placental tissue. mtDNA methylation was estimated to mediate 54% [P = 0.01 (MT-RNR1)] and 27% [P = 0.06 (D-loop)] of the inverse association between PM2.5 exposure and mtDNA content. This study provides new insight into the mechanisms of altered mitochondrial function in the early life environment. Epigenetic modifications in the mitochondrial genome, especially in the MT-RNR1 region, substantially mediate the association between PM2.5 exposure during gestation and placental mtDNA content, which could reflect signs of mitophagy and mitochondrial death.
Keywords: air pollution, DNA methylation, epigenetics, mitochondria, mitochondrial DNA content, mitochondrial DNA methylation, particulate matter, placenta
Abbreviations
- 12S rRNA
Mitochondrial ribosomal 12S rRNA
- D-loop
Displacement loop
- IQR
Interquartile range
- mtDNA
Mitochondrial DNA
- mtDNMT1
Mitochondrial isoform of nuclear-encoded DNA methyltransferase enzyme 1
- MT-RNR1
Mitochondrial region RNR1
- PM
Particulate matter
Introduction
Prenatal life is an important window of susceptibility to adverse effects of environmental exposures. In epidemiological studies, it is well established that exposure to airborne particulate matter (PM) and tobacco smoke during gestation increases the risk of low birth weight and preterm birth.1-3 While the specific mechanisms behind fetal programming are still poorly understood, a large body of evidence suggests the human placenta as a main target organ since it serves as the gatekeeper between mother and fetus.4 Indeed, the placenta is the first organ to be fully developed during pregnancy. It is in contact with all nutritional, hormonal, and other environmental stress factors throughout pregnancy. During its 9 months' lifespan, the human placenta records morphological, functional, genetic, and epigenetic information, representing thus a molecular “footprint” that can reveal the impact of in utero exposures on the fetus.5
The placenta is a unique vascular organ that requires a constant and abundant source of energy. Mitochondria are intracellular organelles providing cellular energy through the production of adenosine-5′-triphosphate (ATP) via oxidative phosphorylation. In humans, these powerhouses contain multiple copies of double stranded, circular mitochondrial DNA (mtDNA). Mitochondria, and associated mtDNA copies, may change in number or mass due to mitophagy, a specific form of autophagy in which dysfunctional or damaged mitochondria are removed and, thus, is critical for maintaining proper cellular function.6,7 Analyzing mitochondrial mass by qPCR (i.e., mtDNA content) has been used as a quantitative method for specifically monitoring the last step of the degradation process of mitophagy in mammalian cells.8 As such, alterations in mtDNA content are a marker of mitochondrial damage and dysfunction9 and has been identified as an etiological determinant in a variety of human diseases, such as diabetes, obesity, cardiovascular disease, and cancer.10 Changes in fetal mtDNA content may represent a biological effect along the path linking air pollution to effects on the fetus, such as birth weight. Previously, we showed a lower mtDNA content in placental tissue in association with in utero exposure to PM10 (PM with aerodynamic diameter ≤ 10 µm),11 which reflects signs of mitophagy and mitochondrial death.
Most research to date has focused on epigenetic modifications in the nuclear genome, with little attention devoted to mtDNA. Not up until the discovery of a mitochondrial isoform of nuclear-encoded DNA methyltransferase enzyme 1 (mtDNMT1) by Shock et al.,12 there was controversy and inconclusive evidence of DNA methylation in the mitochondrial genome. Although mtDNA methylation, by means of 5-methylcytosine and 5-hydroxymethylcytosine, has been increasingly well established after this breakthrough and led to several significant disease-related studies,13,14 such as cervical15 and colorectal cancer,16 non-alcoholic fatty liver disease,17 and aging,18 there are still conflicting reports in the literature regarding the existence and function of mtDNA methylation.19,20 Indeed, the functional consequences of this epigenetic mark are still under investigation but emerging evidence suggests a role in transcription modification of the mitochondrial genome.16,17 For example, Bellizzi et al.21 confirmed that mtDNA is methylated at the displacement loop (D-loop), a crucial promoter region from which mtDNA replication and transcription are initiated. Furthermore, some authors have suggested that aberrant DNA methylation in the mitochondrial RNR1 (MT-RNR1) sequence, encoding for the mitochondrial ribosomal 12S rRNA (12S rRNA), could affect normal function and integrity of the mitochondrial ribosome.22 Structural changes of 12S rRNA lead to instability of the small subunit of the ribosome and abolishes translation of mtDNA-encoded RNAs into proteins.23 Therefore, it is plausible that epigenetic modifications at important hotspots in the mtDNA can regulate replication and/or transcription of the mtDNA.
Whether exposure to PM during gestation influences methylation patterns of the mitochondrial genome in placental tissue is not known. In the framework of the ENVIRONAGE birth cohort (ENVIRonmental influence ON early AGEing), we hypothesized that, in addition to mtDNA content, methylation at the D-loop and MT-RNR1 region (Fig. 1) in placental tissue is associated with airborne PM2.5 exposure during gestation. To elucidate possible mechanisms that regulate mitochondrial function, we also examined mtDNA methylation as potential mediator between early life exposure to airborne PM and placental mtDNA content.
Figure 1.

Location of mitochondrial targets in the human mitochondrial genome. The double stranded, circular mtDNA (16569 bp) contains 37 genes, specifying 13 polypeptides (blue boxes), 2 rRNAs, i.e., 12S rRNA and 16S rRNA (orange boxes), and 22 tRNAs (yellow boxes). The single letter codon associated with the yellow box refers to the amino acid. The D-loop region (green box) contains the origin of replication and is the place where promoters initiate transcription of the H-strand. The target location for DNA methylation analysis in the D-loop and MT-RNR1 region are denoted by the black arcs, with magnified pyrograms. The red arcs depict the targets for mtDNA content analysis (MTF3212/R3319 and ND1). NADH dehydrogenase (ND); Cytochrome c oxidase (COX); F1F0-ATP synthase (ATPase); Cytochrome b (Cyt b).
Results
Participant's characteristics and exposure levels
Demographic and pregnancy-related characteristics of the 381 mother-newborn pairs are reported in Table 1. Briefly, mean maternal age was 29.0 y (range: 18–42 y) and more than 50% of the mothers were highly educated. Pre-gestational body mass index (BMI) of the participating mothers averaged 24.3 (SD = 4.5) kg/m2. Most women (66.9%; n = 255) never smoked cigarettes and 65 women (17.1%) stopped smoking before pregnancy (median with 25th – 75th percentile: 1.5 (0–5) years), whereas 61 women (16.0%) reported to have smoked during pregnancy (on average 7.8 cigarettes per day). The newborns, 193 of which were girls (50.7%), had a mean gestational age of 39.2 weeks (range: 35–42) and comprised 200 (52.5%) primiparous and 141 (37.0%) secundiparous. About 90% (n = 332) of the newborns were Europeans of Caucasian ethnicity. Birth weight averaged 3431 (425) grams. Nineteen (5.0%) newborns were delivered with a caesarian section.
Table 1.
Characteristics of mother-newborn pairs (n = 381)
| Characteristics | Mean ± SD or range and number (%) | |
|---|---|---|
| Maternal | ||
| Age, y | 29.0 | (18–42) |
| Pre-gestational BMI, kg/m2 | 24.3 | ± 4.5 |
| Maternal education | ||
| Low | 47 | (12.7%) |
| Middle | 132 | (34.7%) |
| High | 200 | (52.6%) |
| Smoking | ||
| Never-smoker | 255 | (66.9%) |
| Past-smoker | 65 | (17.1%) |
| Smoker | 61 | (16.0%) |
| Parity | ||
| 1 | 200 | (52.5%) |
| 2 | 141 | (37.0%) |
| ≥ 3 | 40 | (10.5%) |
| Mode of delivery | ||
| Caesarian section | 19 | (5.0%) |
| Newborn | ||
| Gender | ||
| Female | 193 | (50.7%) |
| Ethnicity | ||
| European | 332 | (87.1%) |
| Non-European | 49 | (12.9%) |
| Gestational age, w | 39.2 | (35–42) |
| Season at conception | ||
| Winter | 90 | (23.6%) |
| Spring | 87 | (22.8%) |
| Summer | 118 | (31.0%) |
| Fall | 86 | (22.6%) |
| Apgar score after 5 min | ||
| ≤ 8 | 22 | (5.7%) |
| 9 | 110 | (28.9%) |
| 10 | 249 | (65.4%) |
| Birth weight, g | 3431 | ± 425 |
| MT-RNR1 mtDNA methylation, %* | 9.54 | ± 4.2 |
| D-loop mtDNA methylation, %* | 3.60 | ± 1.28 (n = 356) |
| mtDNA content*,# | 1.04 | (0.45–2.45) |
Measured in placental tissue.
Geometric mean with 10th–90th percentiles.
Table 2 displays the daily outdoor PM2.5 exposure levels averaged for the entire pregnancy and each of the 3 trimesters of pregnancy. Average (interquartile range (IQR): 25th-75th percentile) trimester-specific PM2.5 exposure was 16.0 (11.8–19.6) μg/m3 for the first trimester, 16.9 (12.2–20.4) μg/m3 for the second trimester, 17.3 (11.9–21.7) μg/m3 for the third trimester, and 16.7 (15.2–18.2) μg/m3 for the entire pregnancy.
Table 2.
Exposure characteristics (n = 381)
| Exposure variable | Mean | ± SD | 25th percentile | 75th percentile |
|---|---|---|---|---|
| PM2.5, µg/m3 | ||||
| 1st trimester | 16.0 | ± 5.3 | 11.8 | 19.6 |
| 2nd trimester | 16.9 | ± 5.0 | 12.2 | 20.4 |
| 3rd trimester | 17.3 | ± 5.8 | 11.9 | 21.7 |
| Entire pregnancy | 16.7 | ± 2.3 | 15.2 | 18.2 |
Placental mtDNA methylation and mtDNA content in association with PM2.5 exposure during gestation
We found a positive association of placental mtDNA methylation (MT-RNR1 and D-loop, separately as well as combined) with PM2.5 exposure during the entire gestation, which was independent of gender, age, gestational age, smoking, maternal education, parity, ethnicity, and season at conception (Table 3). The association was most pronounced in the first trimester of pregnancy (MT-RNR1: 1.27%, 95% CI: 0.23 to 2.32%; D-loop: 0.44%, 95% CI: 0.12 to 0.75%; n = 356 for an IQR increment of PM2.5 levels).
Table 3.
PM2.5 exposure during the different periods of pregnancy in association with placental mtDNA methylation and placental mtDNA content
| First trimester |
Second trimester |
Third trimester |
Entire pregnancy |
|||||
|---|---|---|---|---|---|---|---|---|
| Variable | β | (95% CI) | β | (95% CI) | β | (95% CI) | β | (95% CI) |
| mtDNA methylation* | ||||||||
| MT-RNR1, % | 1.27 | (0.23 to 2.32)† | 0.19 | (−0.80 to 1.16) | 1.04 | (−0.20 to 1.86) | 0.91 | (0.56 to 4.18)† |
| D-loop, %# | 0.44 | (0.12 to 0.75)† | 0.09 | (−0.22 to 0.39) | 0.04 | (−0.29 to 0.36) | 0.21 | (−0.003 to 1.02) |
| Combined, % | 0.75 | (0.16 to 1.34)† | 0.10 | (−0.47 to 0.65) | 0.46 | (−0.23 to 0.96) | 0.47 | (0.20 to 2.23)† |
| mtDNA content, %** | −7.57 | (−20.78 to 7.86) | −15.19 | (−28.34 to 0.38) | −23.58 | (−36.27 to −8.37)† | −15.60 | (−23.92 to −6.38)† |
β represents an absolute change in percentage methylation for an IQR increment of PM2.5 exposure (µg/m3).
β represents a relative change in placental mtDNA content for an IQR increment of PM2.5 exposure (µg/m3).
Data available for 356 individuals.
P-value significantly lower than 0.05. All models are adjusted for gender, age, gestational age, smoking, maternal education, parity, ethnicity, and season at conception.
Furthermore, when adjusting for the aforementioned covariates, a lower placental mtDNA content of −15.60% (95% CI: −23.92 to −6.38%) was observed for an IQR increment of PM2.5 exposure over the entire pregnancy, and the association was most pronounced in the third trimester (−23.58%, 95% CI: −36.27 to −8.37%) (Table 3).
Correlation of placental mtDNA content and mtDNA methylation
To explore the functional significance of the association of mtDNA methylation with exposure to PM2.5, we evaluated the correlation between placental mtDNA content, a measure of damaged mitochondria, and mtDNA methylation. Newborns with lower mtDNA content in placental tissue exhibited higher levels of methylation in both mitochondrial regions as shown in Figure 2, and remained significant after adjustment for gender, age, gestational age, smoking, maternal education, parity, ethnicity, and season at conception (MT-RNR1: β = −0.04 ± 0.002, P < 0.0001; D-loop: β = −0.10 ± 0.01, P < 0.0001; n = 356).
Figure 2.
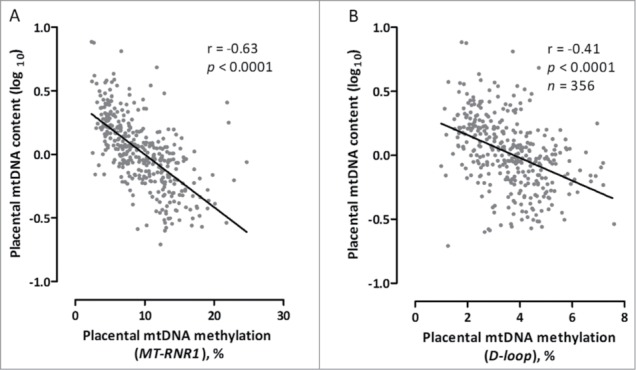
Correlation of mtDNA methylation and mtDNA content in placental tissue. Placental mtDNA content is log10-transformed. Methylation levels are indicated as absolute percentage levels. (A) MT-RNR1 methylation levels. (B) D-loop methylation levels.
Mediating effects of mtDNA methylation on the association of PM2.5 exposure and mtDNA content
We used mediation analysis to test if the negative association between placental mtDNA content and PM2.5 exposure during gestation was mediated by increased mtDNA methylation in both mitochondrial regions. All assumptions for mediation analysis were satisfied and we used average methylation levels of MT-RNR1 and D-loop separately. We considered the exposure averaged over the entire pregnancy because of the discrepancy in trimester effect of PM2.5 exposure on mtDNA methylation (first trimester) and mtDNA content (third trimester). Estimates of the proportion of mediation indicated that placental MT-RNR1 and D-loop methylation mediated respectively 54% (95% CI: 31 to 60%, P = 0.01) and 27% (95% CI: −3 to 36%, P = 0.06) of the inverse association between PM2.5 exposure and placental mtDNA content (Fig. 3).
Figure 3.
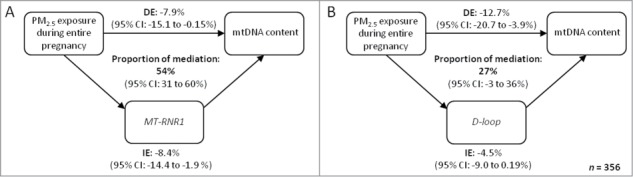
Estimated proportion of effects of PM2.5 exposure during gestation on mtDNA content mediated through changes in mtDNA methylation. The figure displays placental mtDNA methylation as mediator [(A) MT-RNR1; (B) D-loop], the estimates of indirect effect (IE), the estimates of the direct effect (DE), and proportion of mediation (IE/DE+IE). The effects are relative changes in placental mtDNA content for an IQR increment of PM2.5 exposure. The mediation model was adjusted for gender, age, gestational age, smoking, maternal education, parity, ethnicity, and season at conception.
Sensitivity analysis
In a sensitivity analysis, we repeated the main statistical analysis after additional adjustment of caesarian section but this did not alter the reported association between PM2.5 exposure and mtDNA content or mtDNA methylation. In addition, we repeated all statistical analyses while excluding preterm born infants (< 37 weeks, n = 13) and also excluding women who smoked during pregnancy (n = 61). This did not alter the reported associations between mtDNA content or mtDNA methylation and PM2.5 exposures (Table S1).
The associations in the main analysis between the first trimester PM2.5 exposure with the D-loop and MT-RNR1 region differed by CpG position in magnitude and significance. For example, methylation levels at position 2 and 3 of the D-loop region were strongest associated with the first trimester PM2.5 exposure whereas the CpG positions of the MT-RNR1 region differed in magnitude and significance (Table S2).
Neither methylation levels of the MT-RNR1 nor the D-loop region were associated with low birth weight (MT-RNR1: P = 0.58 and D-loop: P = 0.57) or small for gestational age (MT-RNR1: P = 0.66 and D-loop: P = 0.72) (Table S3).
Discussion
The key finding of our study is that placental mtDNA methylation is positively associated with PM2.5 exposure during gestation, especially in the first trimester of pregnancy. Further, these epigenetic modifications in the mitochondria substantially mediate the association between PM2.5 exposure during gestation and placental mtDNA content, demonstrating an intermediate mechanism of mtDNA alterations that could reflect mitophagy and mitochondrial death. To date, epigenetic mitochondrial modifications in the context of exposures in the early life environment have not been reported.
Epigenetic changes, of which DNA methylation is the most commonly characterized, can occur throughout the course of life, but much of the epigenome is already established in germ cells and embryos as it appears to be particularly important for the regulation of embryonic growth and placental development.25 Up until recently, it was thought that epigenetic modifications could only occur in nuclear DNA and not in mtDNA. Despite the discovery of the mtDNMT1 enzyme by Shock et al.12 and studies demonstrating mtDNA methylation in humans,21,22,26 mammals,21,23 and cultured cells,21,27 there are still controversial studies reporting complete absence of mtDNA methylation in human cells lines19 or questioning the clinical usefulness of this biological phenomenon.20 Nevertheless, the rationale of studying mtDNA methylation is underscored by its role in the etiology of a variety of human diseases13,15-17 and aging.18 Growing evidence suggests mitochondrial epigenetics as a novel mechanism to understand the pathobiology of diseases with a mitochondrial dysfunction involvement.14,17 An important hotspot for epigenetic regulation in the mitochondrial genome is the displacement loop or D-loop.21 DNA methylation levels in this control region are suggested to play an important role in modulating either replication or transcription of mtDNA since nearly the entire mitochondrial genome transcribes from this region.28 In tumor and corresponding noncancerous tissues from colorectal cancer patients, Feng et al.16 observed a correlation between D-loop methylation and expression of ND2, a subunit of NADH encoded by mtDNA, which is a rate-limiting enzyme of the oxidative phosphorylation system. In addition, the methylation status of ND6, which is another crucial subunit for complex I assembly, significantly impacts the transcriptional regulation of the gene,17 an effect probably dependent on the enhanced expression of DNMT1.12 Our other studied gene MT-RNR1 encodes for the 12S rRNA protein, which is critical for normal function and integrity of the mitochondrial ribosome. Methylation of MT-RNR1 may cause malfunction of mitochondrial ribosomes and abolished translation of mtDNA-encoded RNAs into proteins.23 A preliminary study in human blood showed that the methylation status of mitochondrial MT-RNR1 gene decreased during aging.29
Epigenetic modifications represent a potential link between adverse insults and altered fetal development. Previously, we have shown that PM2.5 exposure during pregnancy affects global DNA methylation levels in placental tissue,30 while in this study, we investigated the impact of PM2.5 exposure upon epigenetics of placental mitochondria. Using the bisulfite-pyrosequencing approach, we were able to detect subtle differences in methylation levels of specific regions in the mitochondrial genome (Fig. 1). Our observations in newborns are partially in accordance with steel workers. Byun and colleagues22 found a significant positive association between metal-rich PM1 and DNA methylation at the MT-RNR1 sequence, but not at the D-loop, in blood leukocytes of steel workers, whereas we found positive associations of PM2.5 exposure with methylation at both mitochondrial regions in placental tissue. It is noteworthy to mention that we observed an inverse correlation between placental mtDNA methylation and mtDNA content in contrast to a positive correlation observed in blood leukocytes.22 Previous investigations have shown that mtDNA content is variable and fluctuates during aging,31,32 under the influence of different environmental factors,33 and the tissue investigated,11,31,34 as well as different effects from recent versus long-term exposures. Nevertheless, we found similar results when we compared placental mtDNA content between mothers who continued smoking vs. non-smoking pregnant mothers (i.e., lower mtDNA content in smokers; relative difference of −21.98%, 95% CI: −41.10 to −0.86%, P = 0.01). Considering smoking as a personal form of intensive exposure to particulate air pollution, this consistency with ambient air pollution adds confidence to the causal interpretation of our association between PM2.5 exposure and placental mtDNA content. In addition, a study that employed side-by-side epigenome-wide methylation and gene expression arrays in placentas of smokers, showed a significant correlation between methylation and gene expression of 438 genes that were involved in mitochondrial dysfunction, oxidative phosphorylation, and hypoxia pathways.5
The mitochondrion is an important environmental biosensor and the findings of our study brings us one step closer to unravel the relevance of mtDNA content and mtDNA methylation to exposure-related human diseases.14,35 Our data indicate that mtDNA methylation, especially at the MT-RNR1 region, might be a method by which mitochondrial biogenesis and function is regulated. We observed that the association between PM2.5 exposure and mtDNA methylation is most pronounced in the first trimester and the association between PM2.5 exposure and mtDNA content most pronounced in the third trimester of pregnancy. Given the functional relevance of the D-loop region and the 12S rRNA, encoded by MT-RNR1, in controlling replication, transcription, and translation of the mtDNA, we postulate that aberrant mtDNA methylation at these hotspots could interfere with mtDNA biogenesis. This premise holds true when we consider the results of our mediation analysis indicating that a substantial proportion of the association between PM2.5 exposure and placental mtDNA content was mediated by mtDNA methylation and, therefore, strengthens the evidence of an intermediate mechanism of mtDNA alterations. Our findings of lower mtDNA content in placental tissue in association with prenatal PM exposure might reflect increased mitophagy and mitochondrial death, or it could indicate a decline in mitochondrial biogenesis due to demethylation of the mtDNA-specific polymerase gamma A (POLGA)36 or a reduction in levels of PPARGC1A (peroxisome proliferator-activated receptorg-coactivator1α), the master regulator of mitochondrial biogenesis.37 Furthermore, depletion of mtDNA results in significant changes in the methylation pattern of a number of nuclear genes,38 indicating a possible effect of mitochondrial dysfunction on the epigenetic landscape of the nuclear genome.39
We acknowledge several limitations in the present study. Although we reported effects on 2 mtDNA regions separately and performed a combined analysis of these 2 regions, we cannot extrapolate these findings to the entire mitochondrial genome. We selected 2 regions with potential functional impacts, including a promoter region and a key rRNA sequence (Fig. 1). Future work is warranted to determine whether DNA methylation and hydroxymethylation in other regions of the mtDNA, such as ND2 and ND6, which have a biological significance for mitochondrial function, are specifically sensitive to air pollutants, and whether these changes are linked to functional alterations in expression of mitochondrial genes. Secondly, we postulate that aberrant mtDNA methylation could interfere with mtDNA biogenesis. However, because our measurements were performed at birth, we cannot ascertain the temporal sequence, although our proposed biological model seems plausible. Indeed, in epidemiological settings, even by use of mediation analysis, the biological direction of observed associations cannot be determined. Lastly, we can only speculate about the potential health consequences of altered mtDNA methylation and mtDNA content in placental tissue in response to PM2.5 exposure and, therefore, a follow-up study is warranted. Of note, a preliminary study showed differential mtDNA methylation in growth restricted placentas and underscores a possible functional relevance of mtDNA methylation in the placenta.40 In our study, we did not find an association between altered placental mtDNA methylation and adverse pregnancy outcomes such as low birth weight or small for gestational age.
In summary, our study indicates mtDNA methylation as an early molecular event involved, at least partially, with a decrease in placental mtDNA content, which might reflect mitophagy, in association with prenatal exposure to PM2.5. Although concise pathological roles of mitochondrial methylation during development need to be further elucidated, our findings open a new area to the molecular epidemiological understanding of mitochondrial alterations in early life.
Materials and Methods
Study population
We recruited 400 mother-newborn pairs (only singletons) from the ongoing ENVIRONAGE birth cohort as previously described.11 The study participants lived in the Flemish region of Belgium. The placenta could not be collected from 10 newborns, DNA yield was insufficient for 4 placentas and 5 newborns had missing data for PM exposure (lived outside Belgium). Therefore, the final sample included 381 newborns. Written informed consent was obtained from all participating mothers when they arrived at the hospital for delivery and was in accordance with procedures approved by the Ethical Committee of Hasselt University and East-Limburg Hospital in Genk. The study was conducted according to the principles outlined in the Helsinki Declaration for investigation of human subjects. The participation rate of eligible mothers (able to fill out a Dutch language questionnaire) was 56% and enrolment was spread equally over all seasons of the year. Study questionnaires were completed in the postnatal ward after delivery and provided detailed information on age, place of residence, maternal education, occupation, pre-gestational BMI, smoking status, use of medication, alcohol consumption, parity, mode of delivery (vaginal or caesarian section), and newborn's ethnicity. Past-smokers were defined as those who had quit before pregnancy and smokers as having smoked before and during pregnancy. Maternal education was coded as low (no diploma or primary school), middle (high school) or high (college or university degree). Perinatal parameters such as birth date, gestational age, newborn's gender, birth weight and length, Apgar score, and ultrasonographic data were collected after birth.
Exposure measurement
For each mother's residential address, we interpolated the regional background levels of PM2.5 (micrograms per cubic meter) using a spatial temporal interpolation method (Kriging)41 that uses pollution data collected in the official fixed site monitoring network (n = 34) and land cover data obtained from satellite images (Corine land cover data set) in combination with a dispersion model (dispersion modeling described by Lefebvre et al.42,43). This model chain provides daily PM2.5 values using data from the Belgian telemetric air quality network, point sources and line sources which are then interpolated to a high-resolution receptor grid. In the Flemish region of Belgium, the interpolation tool explained more than 80% of the temporal and spatial variability (R2).44 The exposure during the entire pregnancy was calculated as the mean of all pregnancy days. The date of conception was estimated based on ultrasound data. We explored potential critical windows of exposures during pregnancy using daily mean PM2.5 concentrations averaged over various periods. The exposure windows of interest included each of the 3 trimesters of pregnancy, with trimesters being defined as: 1–13 weeks (first trimester), 14–26 weeks (second trimester), and 27 weeks to delivery (third trimester). Address changes during the period of pregnancy were taken into account when calculating the exposure windows (n = 38; 9.9%).
Placental sampling
Placentas were obtained in the delivery room and deep-frozen within 10 minutes. As described previously,30 we took villous tissue (1 to 2 cm3) at a fixed location from the fetal side of the placenta, approximately 1–1.5 cm below the chorio-amniotic membrane, and preserved the biopsies at –80°C. Chorio-amniotic membrane contamination was avoided by careful visual examination and dissection.
DNA methylation analysis
Genomic DNA was isolated from placental tissue (n = 381) using the QIAamp DNA mini kit (Qiagen, Inc.., Venlo, the Netherlands). We performed DNA methylation analysis by highly quantitative bisulfite-PCR pyrosequencing as previously described in detail.24 Briefly, bisulfite conversions were performed using 1 µg of extracted genomic DNA with the EZ-96 DNA methylation Gold kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. We interrogated CpG sites within specific regions of the mitochondrial genome (MT-RNR1 and D-loop) as described by Byun et al.22 and Janssen et al.24 (Fig. 1). Detailed information regarding primer sequences is given in Table S4. PCR amplification of regions of interest prior to pyrosequencing was performed in a total reaction volume of 30 µl, using 15 µl GoTaq Hot Start Green Master Mix (Promega, Madison, WI, USA), 10 pmol forward primer, 10 pmol reverse primer, 1 µl bisulfite-treated genomic DNA and water. PCR products were purified and sequenced by pyrosequencing using the PyroMark Q96 MD Pyrosequencing System (Qiagen, Inc., Germantown, MD, USA). The degree of methylation was expressed as the percentage of methylated cytosines over the sum of methylated and unmethylated cytosines. Samples were run in duplicate on 2 different plates from which the average methylation levels were used. The coefficient of variation for MT-RNR1 was 3.7% and for D-loop 5.8%. The efficiency of the bisulfite-conversion process was assessed using non-CpG cytosine residues within the sequence. The between- and within-placenta variability, exemplified by the intra-class correlation coefficient, was evaluated in a subset of 19 placentas and was 58% vs. 42% (P = 0.009) for MT-RNR1 and 61% vs. 39% (P = 0.01) for the D-loop region.24
mtDNA content analysis
mtDNA content was measured in placental tissue (n = 381) by determining the ratio of 2 mitochondrial gene copy numbers (MTF3212/R3319 and ND1) (Fig. 1) to 2 single-copy nuclear control genes (RPLP0 and ACTB) using a quantitative real-time polymerase chain reaction (qPCR) assay as previously described11 but with small modification. Briefly, 2.5 µl diluted genomic DNA (5 ng/µl) was added to 7.5 µl mastermix consisting of Fast SYBR® Green I dye 2 × (5 µl/reaction), forward and reverse primer (each 0.3 µl/reaction), and RNase free water (1.9 µl/reaction) to a final volume of 10 µl per reaction. Primers (Table S5) were diluted to a final concentration of 300 nM in the master mix. Samples were run in triplicate in 384-well format. Real-time PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with following thermal cycling profile: 20 s at 95°C (activation), followed by 40 cycles of 1 s at 95°C (denaturation), and 20 s at 60°C (annealing/extension), ending with melting curve analysis (15 s at 95°C, 15 s at 60°C, 15 s at 95°C). qBase software (Biogazelle, Zwijnaarde, BE) automatically averages triplicate measurements that pass quality control and normalizes the data to nuclear reference genes while correcting for run-to-run differences.45
Statistical analysis
For database management and statistical analysis, we used the SAS software program (version 9.2; SAS Institute Inc., Cary, NC, USA). mtDNA content was log10-transformed to improve normality. We measured methylation levels of 2 and 3 CpG sites for the MT-RNR1 and D-loop region respectively (Fig. 1). Pearson correlation coefficients were used to assess correlations between adjacent CpG sites within one region (MT-RNR1 or D-loop) and to assess the correlation between mtDNA content and mtDNA methylation. Within each region, the associations with individual CpG positions were similar in size and direction (r ≥ 0.90, Fig. S1) and we assumed equal correlation between CpG positions in our models. We used 2 statistical modeling approaches to interpret mtDNA methylation data: 1) with mixed-effect models, we took into account each CpG dinucleotide position of both regions (combination of MT-RNR1 and D-loop) to obtain a robust estimate of the average effect on DNA methylation in the mitochondrial genome; 2) with mixed-effect models, we took into account each CpG dinucleotide position of either MT-RNR1 or D-loop to obtain an estimate of each region separately. We performed multiple linear regression to determine the effect-size of PM2.5 exposure during pregnancy on placental mtDNA content. All adjusted models were controlled for a priori chosen variables including gender, maternal age, gestational age, smoking, maternal education, parity, ethnicity, and season at conception. β regression coefficients represent a relative (mtDNA content) or absolute (mtDNA methylation) percentage change for an increment between the IQR (25th-75th percentile) in the independent variable. The Shapiro-Wilk statistic and Q-Q plots of the residuals were used to test the assumptions of all linear models.
We used mediation analysis to investigate potential mechanisms that underlie the association between the exposure variable (PM2.5) and outcome variable (mtDNA content) by examining how they relate to a third intermediate variable, the mediator (mtDNA methylation).46 We accomplished this by decomposing the total effect into direct effects (exposure effect on outcome at a fixed level of the mediator) and indirect effects (exposure effect on outcome that operate through mediator levels). Mediation analysis usually requires a significant relation of the outcome to the exposure, a significant relation of the outcome to the mediator and a significant relation of the mediator to the exposure. This is based on the a priori assumption that a mediated effect is biologically plausible. For the mediation analysis, we considered PM2.5 exposure during the entire pregnancy and average methylation levels of each mitochondrial region. We used SAS macros provided by Valeri et al.46
In a sensitivity analysis, we assessed the association between PM2.5 exposure during pregnancy and mtDNA methylation or mtDNA content while excluding preterm births (< 37 weeks) or women who smoked during pregnancy. In addition, we examined whether the PM2.5 exposure associations we observed in the main analysis were specific to certain CpG positions within the 2 mitochondrial regions. Finally, we investigated whether mtDNA methylation was associated with adverse pregnancy outcomes such as low birth weight using linear regression models and small for gestational age using logistic regression models.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the participating women, as well as Anja Moors and Anneleen Staelens of the maternity ward, midwives, and the staff of the clinical laboratory of East-Limburg Hospital in Genk. We thank Dr. Linda Valeri for her help with the mediation analysis.
Funding
The ENVIRONAGE birth cohort is supported by the EU Program “Ideas” (ERC-2012-StG 310898), by the Flemish Scientific Fund (FWO, N1516112/G.0.873.11.N.10), and Bijzonder Onderzoeks Fonds of Hasselt University (BOF). This work was also supported by funding from the National Institute of Environmental Health Sciences (R01ES021733, R01ES021357, and R21ES022694–01A1).
Supplemental Material
Supplemental data for this article can be found on the publisher's website.
References
- 1.Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, Gehring U, Glinianaia SV, Gouveia N, Ha EH, et al.. Maternal exposure to particulate air pollution and term birth weight: a multi-country evaluation of effect and heterogeneity. Environ Health Perspect 2013; 121:267-373; PMID:23384584; http://dx.doi.org/ 10.1289/ehp.1205575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sram RJ, Binkova B, Dejmek J, Bobak M. Ambient air pollution and pregnancy outcomes: a review of the literature. Environ Health Perspect 2005; 113:375-82; PMID:15811825; http://dx.doi.org/ 10.1289/ehp.6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox B, Martens E, Nemery B, Vangronsveld J, Nawrot TS. Impact of a stepwise introduction of smoke-free legislation on the rate of preterm births: analysis of routinely collected birth data. BMJ 2013; 346:f441; PMID:23412829; http://dx.doi.org/ 10.1136/bmj.f441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myllynen P, Pasanen M, Pelkonen O. Human placenta: a human organ for developmental toxicology research and biomonitoring. Placenta 2005; 26:361-71; PMID:15850640; http://dx.doi.org/ 10.1016/j.placenta.2004.09.006 [DOI] [PubMed] [Google Scholar]
- 5.Suter M, Ma J, Harris AS, Patterson L, Brown KA, Shope C, Showalter L, Abramovici A, Aagaard-Tillery KM. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011; 6:1284-94; PMID:21937876; http://dx.doi.org/ 10.4161/epi.6.11.17819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res Int 2014; 2014:Article ID 238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stern S, Adiseshaiah P, Crist R. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Particle and Fibre Toxicology 2012; 9:20; PMID:22697169; http://dx.doi.org/ 10.1186/1743-8977-9-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012; 393:547-64; PMID:22944659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al.. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011; 470:359-65; PMID:21307849; http://dx.doi.org/ 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen 2010; 51:440-50; PMID:20544884 [DOI] [PubMed] [Google Scholar]
- 11.Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, Fierens F, Penders J, Vangronsveld J, Gyselaers W, et al.. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect 2012; 120:1346-52; PMID:22626541; http://dx.doi.org/ 10.1289/ehp.1104458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA 2011; 108:3630-5; PMID:21321201; http://dx.doi.org/ 10.1073/pnas.1012311108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab 2013; 110:25-34; PMID:23920043; http://dx.doi.org/ 10.1016/j.ymgme.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 14.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, et al.. Mitochondria, Energetics, Epigenetics, and Cellular Responses to Stress. Environ Health Perspect 2014; 122:1271-8; PMID:25127496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecologic Oncol 2012; 121:59-63; http://dx.doi.org/ 10.1016/j.ygyno.2011.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng S, Xiong L, Ji Z, Cheng W, Yang H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol Med Rep 2012; 6:125-30; PMID:22505229 [DOI] [PubMed] [Google Scholar]
- 17.Pirola CJ, Gianotti TF, Burgueño AL, Rey Funes M, Loidl CF, Mallardi P, Martino JS, Castaño GO, Sookoian S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013; 62:1356-63; PMID:22879518; http://dx.doi.org/ 10.1136/gutjnl-2012-302962 [DOI] [PubMed] [Google Scholar]
- 18.Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging 2012; 33:2881-91; PMID:22445327; http://dx.doi.org/ 10.1016/j.neurobiolaging.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong EE, Okitsu CY, Smith AD, Hsieh C-L. Regionally Specific and Genome-Wide Analyses Conclusively Demonstrate the Absence of CpG Methylation in Human Mitochondrial DNA. Mol Cell Biol 2013; 33:2683-90; PMID:23671186; http://dx.doi.org/ 10.1128/MCB.00220-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maekawa M, Taniguchi T, Higashi H, Sugimura H, Sugano K, Kanno T. Methylation of Mitochondrial DNA Is Not a Useful Marker for Cancer Detection. Clin Chem 2004; 50:1480-1; PMID:15277367; http://dx.doi.org/ 10.1373/clinchem.2004.035139 [DOI] [PubMed] [Google Scholar]
- 21.Bellizzi D, D'Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res 2013; 20:537-47; PMID:23804556; http://dx.doi.org/ 10.1093/dnares/dst029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, Bertazzi P, Baccarelli A. Effects of airborne pollutants on mitochondrial DNA Methylation. Particle Fibre Toxicol 2013; 10:18; PMID:23656717; http://dx.doi.org/ 10.1186/1743-8977-10-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metodiev MD, Lesko N, Park CB, Cámara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA Is Necessary for In Vivo Stability of the Small Subunit of the Mammalian Mitochondrial Ribosome. Cell Metab 2009; 9:386-97; PMID:19356719; http://dx.doi.org/ 10.1016/j.cmet.2009.03.001 [DOI] [PubMed] [Google Scholar]
- 24.Janssen BG, Byun HM, Cox B, Gyselaers W, Izzi B, Baccarelli AA, Nawrot TS. Variation of DNA methylation in candidate age-related targets on the mitochondrial-telomere axis in cord blood and placenta. Placenta 2014; 35:665-72; PMID:25047690; http://dx.doi.org/ 10.1016/j.placenta.2014.06.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, et al.. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002; 417:945-8; PMID:12087403; http://dx.doi.org/ 10.1038/nature00819 [DOI] [PubMed] [Google Scholar]
- 26.O'Sullivan M, Rutland P, Lucas D, Ashton E, Hendricks S, Rahman S, Bitner-Glindzicz M. Mitochondrial m.1584A 12S m62A rRNA methylation in families with m.1555A>G associated hearing loss. Hum Mol Genet 2015; 24:1036-44; PMID:25305075; http://dx.doi.org/ 10.1093/hmg/ddu518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rebelo AP, Williams SL, Moraes CT. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res 2009; 37:6701-15; PMID:19740762; http://dx.doi.org/ 10.1093/nar/gkp727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aloni Y, Attardi G. Expression of the mitochondrial genome in HeLa cells. II. Evidence for complete transcription of mitochondrial DNA. J Mol Biol 1971; 55:251-67; PMID:5548607; http://dx.doi.org/ 10.1016/0022-2836(71)90195-1 [DOI] [PubMed] [Google Scholar]
- 29.Giordano M, Cristiani C, Crocco P, D'Aquila P, De Rango F, Pisani T, Scafone T, Tallaro F, Rose G, Passarino G, et al.. Methylation of the human mitochondrial 12S rRNA gene is correlated with aging. 12th FISV Congress. Rome, Italy, 2012:77. [Google Scholar]
- 30.Janssen BG, Godderis L, Pieters N, Poels K, Kiciński M, Cuypers A, Fierens F, Penders J, Plusquin M, Gyselaers W, et al.. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol 2013; 10:22; PMID:23742113; http://dx.doi.org/ 10.1186/1743-8977-10-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cassano P, Sciancalepore AG, Lezza AM, Leeuwenburgh C, Cantatore P, Gadaleta MN. Tissue-specific effect of age and caloric restriction diet on mitochondrial DNA content. Rejuvenation Res 2006; 9:211-4; PMID:16706645; http://dx.doi.org/ 10.1089/rej.2006.9.211 [DOI] [PubMed] [Google Scholar]
- 32.Lee HC, Lu CY, Fahn HJ, Wei YH. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. Febs Letters 1998; 441:292-6; PMID:9883902; http://dx.doi.org/ 10.1016/S0014-5793(98)01564-6 [DOI] [PubMed] [Google Scholar]
- 33.Meyer JN, Leung MC, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, Bess AS. Mitochondria as a target of environmental toxicants. Toxicol Sci 2013; 134:1-17; PMID:23629515; http://dx.doi.org/ 10.1093/toxsci/kft102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andreu AL, Martinez R, Marti R, Garcia-Arumi E. Quantification of mitochondrial DNA copy number: pre-analytical factors. Mitochondrion 2009; 9:242-6; PMID:19272467; http://dx.doi.org/ 10.1016/j.mito.2009.02.006 [DOI] [PubMed] [Google Scholar]
- 35.Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol 2012; 41:177-87; PMID:22287136; http://dx.doi.org/ 10.1093/ije/dyr232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee W, Johnson J, Gough DJ, Donoghue J, Cagnone GLM, Vaghjiani V, Brown KA, Johns TG, John JC.. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis 2015; 6:e1664; PMID:25719248; http://dx.doi.org/ 10.1038/cddis.2015.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome Proliferator-activated Receptor γ Co-activator α (PGC-1α) and Sirtuin 1 (SIRT1) Reside in Mitochondria: Possible direct function in mitochondrial biogenesis. J Biol Chem 2010; 285:21590-9; PMID:20448046; http://dx.doi.org/ 10.1074/jbc.M109.070169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther 2008; 7:1182-90; PMID:18458531; http://dx.doi.org/ 10.4161/cbt.7.8.6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minocherhomji S, Tollefsbol TO, Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012; 7:326-34; PMID:22419065; http://dx.doi.org/ 10.4161/epi.19547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strong N, Lambertini L, Ma Y, Stone J. Differential mitochondrial DNA methylation in growth restricted placentas. Am J Obstet Gynecologic 2013; 208:S192; http://dx.doi.org/ 10.1016/j.ajog.2012.10.607 [DOI] [Google Scholar]
- 41.Janssen S, Dumont G, Fierens F, Mensink C. Spatial interpolation of air pollution measurements using CORINE land cover data. Atmospheric Environment 2008; 42:4884-903; http://dx.doi.org/ 10.1016/j.atmosenv.2008.02.043 [DOI] [Google Scholar]
- 42.Lefebvre W, Degrawe B, Beckx C, Vanhulsel M, Kochan B, Bellemans T, Janssens D, Wets G, Janssen S, de Vlieger I, et al.. Presentation and evaluation of an integrated model chain to respond to traffic- and health-related policy questions. Environ Model Software 2013; 40:160-70; http://dx.doi.org/ 10.1016/j.envsoft.2012.09.003 [DOI] [Google Scholar]
- 43.Lefebvre W, Vercauteren J, Schrooten L, Janssen S, Degraeuwe B, Maenhaut W, de Vlieger I, Vankerkom J, Cosemans G, Mensink C, et al.. Validation of the MIMOSA-AURORA-IFDM model chain for policy support: Modeling concentrations of elemental carbon in Flanders. Atmospheric Environ 2011; 45:6705-13; http://dx.doi.org/ 10.1016/j.atmosenv.2011.08.033 [DOI] [Google Scholar]
- 44.Maiheu B, Veldeman B, Viaene P, De Ridde RK, Lauwaet D, Smeets N, Deutsch F, Janssen S. Identifying the best available large-scale concentration maps for air quality in Belgium. 2012. 67-99. http://www.milieurapport.be/Upload/main/0_onderzoeksrapporten/2013/Eindrapport_Concentratiekaarten_29_01_2013_TW.pdf. [Google Scholar]
- 45.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007; 8:R19; PMID:17291332; http://dx.doi.org/ 10.1186/gb-2007-8-2-r19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods 2013; 18:137-50; PMID:23379553; http://dx.doi.org/ 10.1037/a0031034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.