

Abstract
Brain metastasis is a major contributor to cancer mortality, yet, the genetic changes underlying the development of this capacity remain poorly understood. RASSF proteins are a family of tumor suppressors that often suffer epigenetic inactivation during tumorigenesis. However, their epigenetic status in brain metastases has not been well characterized. We have examined the promoter methylation of the classical RASSF members (RASSF1A-RASSF6) in a panel of metastatic brain tumor samples. RASSF1A and RASSF2 have been shown to undergo promoter methylation at high frequency in primary lung and breast tumors and in brain metastases. Other members exhibited little or no methylation in these tumors. In examining melanoma metastases, however, we found that RASSF6 exhibits the highest frequency of inactivation in melanoma and in melanoma brain metastases. Most melanomas are driven by an activating mutation in B-Raf. Introduction of RASSF6 into a B-RafV600E-containing metastatic melanoma cell line inhibited its ability to invade through collagen and suppressed MAPK pathway activation and AKT. RASSF6 also appears to increase the association of mutant B-Raf and MST1, providing a potential mechanism by which RASSF6 is able to suppress MAPK activation. Thus, we have identified a novel potential role for RASSF6 in melanoma development. Promoter methylation leading to reduced expression of RASSF6 may play an important role in melanoma development and may contribute to brain metastases.
Keywords: brain metastases, invasion, melanoma, MAPK, Ras, RASSF
Introduction
Malignant melanoma arises from melanocytes or cellular derivatives of melanocytes.1 Approximately 13,000 deaths a year are due to melanoma in the USA.2 Brain metastases in advanced melanoma are frequent and are a major cause of death in the disease.3 Genomic studies have identified oncogenic mutations in the B-Raf gene as the most common driver of melanoma.4 B-Raf is a Serine/Threonine kinase that stimulates the MAPK pathway by phosphorylating and activating MEK.5 The most common activating mutation is the V600E mutation that serves to constitutively activate the kinase.4 Thus, the MAPK pathway is hyperactivated and cells remain in a highly proliferative state. Further studies have shown that mutant B-Raf activity is also a key requirement for extravasation.6
As activated B-Raf is so important in melanoma, a series of B-Raf inhibitors have been developed. Initial clinical studies have shown striking effects of the inhibitors in melanoma patients, with the majority of patients showing tumor shrinkage. However, the development of resistance to the drugs as single agents is common7; therefore, their overall impact on long-term survival remains limited. A greater understanding of the genetic and epigenetic defects that collaborate with B-Raf to drive transformation and metastasis would facilitate the development of targeted combination therapy.
RASSF proteins are members of a family of tumor suppressors.8 They modulate pro-apoptotic signaling pathways and are frequently inactivated in human tumors by promoter methylation. The family members all contain a domain with a SARAH motif that can bind the pro-apoptotic kinases MST1 and MST2.8 RASSF1A and RASSF5 have been reported to activate the MST kinases.9 Curiously, Ikeda et al. showed that the RASSF6 family member actually inhibits MST kinase activity,10 making RASSF6 somewhat unique. MST kinases have a number of target proteins but perhaps the most important signaling pathway that they modulate is referred to as the Hippo pathway.11
We have examined a series of brain metastases from different tumor types. RASSF1A and RASSF2 were subjected to epigenetic inactivation at high levels in lung and breast brain metastases, as measured by promoter methylation, while RASSF6 was not. However, the RASSF6 promoter was methylated at much higher frequencies than RASSF1A or RASSF2 in melanoma brain metastases, suggesting a particular role for RASSF6 in melanoma. Previously, only RASSF10, an N-terminal RASSF member, has been found to be epigenetically downregulated in melanoma.12 No studies have examined the functional consequences of RASSF6 expression in melanoma cells. When we introduced RASSF6 into the B-RafV600E driven metastatic melanoma A375 cell line, we observed little difference in growth rate but a marked decrease in the ability of the cells to invade through collagen gels. Molecular analysis showed that the activity of both the MAPK pathway and AKT was suppressed in the RASSF6-transfected cells. Additionally, RASSF6 appears to increase the association of mutant B-Raf and MST1, potentially providing a mechanism for RASSF6 ability to modulate MAPK activation. Thus, we identify an unanticipated role for RASSF6 loss in the development of melanoma and melanoma brain metastases.
Results
Epigenetic inactivation of RASSF genes in brain metastases
The methylation status of the classical RASSF family members, RASSF1A-RASSF6 was examined across a panel of 123 brain metastases originating from primary lung tumors (n = 40), primary breast tumors (n = 40) and primary melanomas (n = 43) (Fig. 1). We identified no methylation in RASSF3, 4, or 5 in any type of the samples analyzed (example shown in supplementary Fig. 1A). RASSF1A showed frequent methylation in breast (21/34, 61.8%), lung (21/40, 52.5%), and melanoma (12/41, 29.3%) metastases. These percentage methylation numbers for breast and melanoma are somewhat lower than the values previously reported for the primary tumors, whereas the percentage for lung tumors is approximately 25% higher.13,14 RASSF2 methylation was observed frequently in breast (10/28, 35.7%) and lung (14/40, 35%) metastases and infrequently in melanoma metastases (3/29, 10.3%). For RASSF6, we found infrequent methylation in metastases from both breast and lung tumors, 5.4% and 10.2%, respectively; however, we identified high methylation frequency in brain metastases from melanomas (28/38, 73.7%). Representative methylation results are shown in Figure 1. Clone sequencing confirmed CoBRA results and demonstrated much lower methylation in non-cancerous brain tissue (between 1 and 13% in the 3 samples examined; representative data is shown in Fig. 2).
Figure 1.
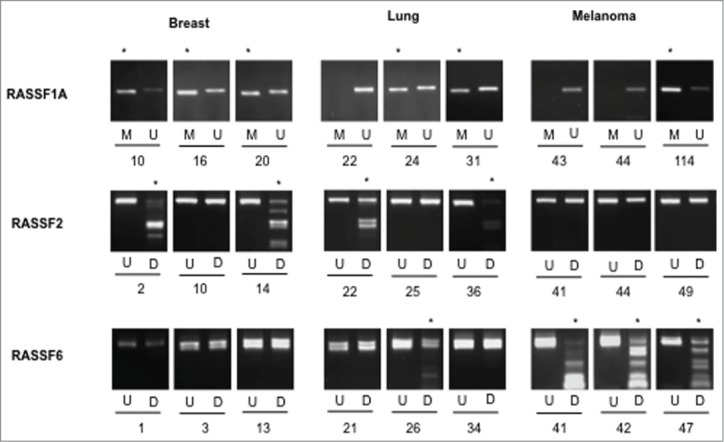
MSP/CoBRA results for RASSF1A, RASSF2 and RASSF6. MSP results are shown for 3 samples each for breast, lung, and melanoma metastases to the brain for RASSF1A, with methylation-specific (MSP) PCR products (M) shown adjacent to unmethylation-specific (USP) PCR products (U). CoBRA results are shown for 3 samples each for breast, lung, and melanoma metastases to the brain for RASSF2, RASSF6. In each case, undigested product (U) is shown next to BstUI digested product (D). * indicates methylation.
Figure 2.

Sequencing results for RASSF6 melanoma metastases to the brain and normal brain samples. The figure shows clone sequencing results for 2 melanoma metastases to the brain (samples 47 and 48) and 2 non-cancerous brain samples (samples non-cancerous brain 1 and non-cancerous brain 3). In each case, a single line represents a single allele and black and white circles represent methylated and unmethylated CpGs respectively. Methylation Indexes (MIs) are given for each sample as a percentage of methylated CpGs out of the total number of CpGs analyzed.
RASSF6 promoter methylation in melanomas
Our results for the RASSF family of genes and results from other studies in cancer metastases suggest that genetic and epigenetic changes are set up in the primary tumor and are maintained in the metastases.15,16 This led us to assess the methylation status of RASSF6 in malignant melanoma primary tumors. Again, we found frequent RASSF6 promoter methylation (7/11, 63.3%). RASSF6 was unmethylated in normal melanocyte controls (Fig. 3).
Figure 3.
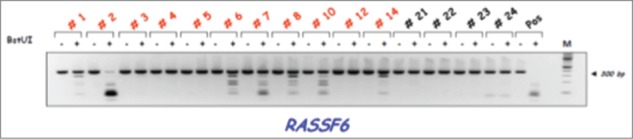
CoBRA results for RASSF6 in primary melanomas. CoBRA results are shown for 11 melanomas (1–14) and 4 normal melanocytes (21–24) with (+) and without (−) BstUI digestion. A positive BstUI control is also shown (Pos).
RASSF6 re-expression in A375 melanoma cells
The promoter methylation results suggested that inactivation of RASSF6 is particularly important for the tumorigenic and metastatic development of melanoma. A375 cells are a metastatic melanoma cell line driven by a V600E activating mutation in B-Raf.17 To assess the RASSF6 inactivation state in A375s, cells were treated with 2 DNA methyltransferase inhibitors, 5-azacitidine and nanaomycin A. RASSF6 re-expression was detected in cells treated with both drugs via immunoprecipitation/Western blot with an antibody directed against RASSF6 (Supplementary Fig. 1B).
RASSF6 inhibits melanoma invasion
We generated a matched pair of A375 cells transfected with a HA-tagged RASSF6 expression plasmid or an empty vector. We confirmed RASSF6 expression via western analysis using antibodies directed against HA (Fig. 5A) and RASSF6 (Supplementary Fig. 1C). The matched pair of cell lines were then assayed for their ability to proliferate and to invade through a collagen matrix. Figure 4A shows that at 5 d after plating the RASSF6-expressing cells showed a ∼20% growth reduction. However, Figure 4B shows that at the same time point, the cells showed a ∼60% inhibition of invasion through collagen. To determine if this result might simply be due to death by anoikis rather than reduced invasion, we assayed the cells for anoikis. Cells were induced to suspension culture by poly-HEMA treatment of the tissue culture dishes. After 5 d, cell viability was assayed by trypan blue staining, revealing little cell death in either cell line (Fig. 4C).
Figure 5.
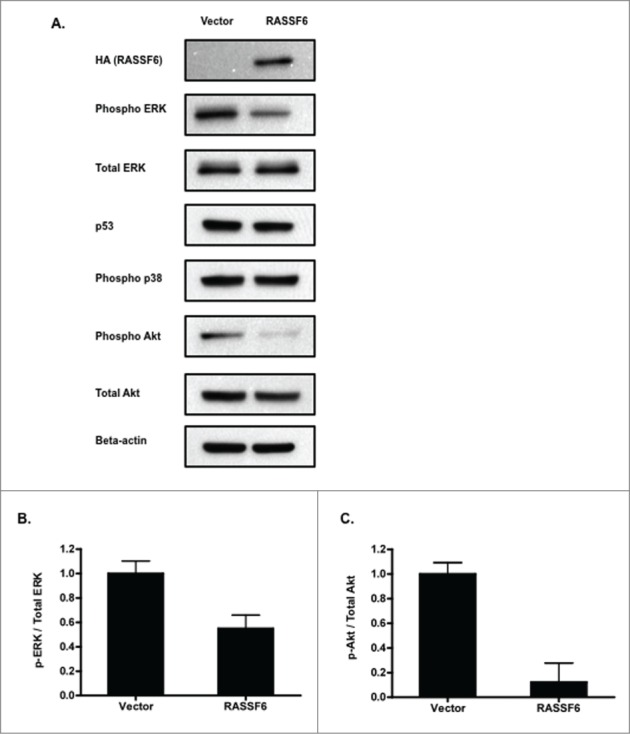
RASSF6 inhibits MAPK and Akt activation. (A). Western analysis followed by densitometry quantification was used to compare the level of MAP Kinase activation in the RASSF6 +/− A375 cells. A representative blot is shown with a bar graph showing the average of 3 assays. The presence of the overexpressed HA-tagged RASSF6 expression construct in the cells was confirmed by western blotting with HA. Additionally, activation of other signaling pathways was examined by western analysis for phospho and total Akt, phospho p38, and p53. Representatives of experiments performed in duplicate are shown. (B). Quantification of densitometric analysis of phospho ERK relative to total ERK. P = 0.0153. (C). Quantification of densitometric analysis of phospho and total Akt. P = 0.0091.
Figure 4.
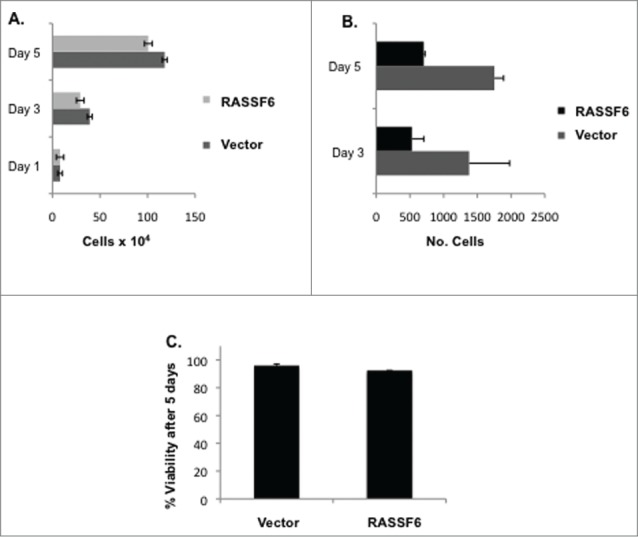
RASSF6 inhibits melanoma invasion. (A). A375 cells transfected with empty vector or RASSF6 expression construct were seeded in 6 well plates and counted over the time course of 3 d. P-values were calculated using an unpaired t test and were as follows: P = 0.2692 for day 3 and P = 0.1186 for day 5. (B). A375 cells transfected with empty vector or RASSF6 expression construct were seeded onto collagen-plugged transwells and incubated for 3 or 5 d. Invasion across the transwell membrane was measured by fixing, staining, and counting cells that had traversed the membrane. Lower axis is number of cells that had invaded to the distal side of each transwell membrane. P = 0.3069 for day 3 and P = 0.0167 for day 5. (C). Cells were suspended in Poly-HEMA treated plates to prevent adhesion and cell viability measured by Trypan blue staining after 5 d. P = 0.0712.
RASSF6 inhibits MAPK activation
Like many melanomas, A375 cells contain an activated B-Raf mutation.17 Activated B-Raf hyperactivates the MAP kinase pathway to drive growth and transformation.18 To investigate the mechanism by which RASSF6 might suppress invasion, we therefore examined the status of the MAPK pathway in the A375 cells. Western blotting with phospho-specific anti-sera showed that the MAPK pathway was approximately 50% less active in the RASSF6-expressing cells than in the vector cells alone. (Fig. 5A).
RASSF6 suppresses Akt phosphorylation
Given the finding that RASSF6 decreases MAPK phosphorylation, several other MAPK markers were examined to see if RASSF6 influenced other signaling pathways. RASSF6 did not appear to influence phosphorylation of p38, but phosphorylated Akt was greatly diminished in cells expressing RASSF6 (Fig. 5A).
RASSF6 has no influence on p53 levels
RASSF6 has been shown to facilitate MDM2 degradation in human colon carcinoma and osteosarcoma cells, leading to p53 stabilization.19 To examine if RASSF6 was able to stabilize p53 in A375 melanoma cells, western analysis was performed for p53 in the RASSF6-expressing and vector cell lines. Surprisingly, p53 protein expression levels were not influenced by the presence of RASSF6 in A375 cells (Fig. 5A).
RASSF1A fails to decrease MAPK activation in A375s
To determine if other RASSF family members were also able to influence MAPK activation in A375 cells, a matched pair of A375 cells transfected with HA-tagged RASSF1A or empty vector were generated. We confirmed RASSF1A expression via western analysis using antibodies directed against HA (Supplementary Fig. 2A). RASSF1A did not decrease phosphorylation of MAPK (Supplementary Fig. 2), showing that RASSF6 has a distinct effect on MAPK activation in A375 cells.
RASSF6 increases mutant B-Raf association with MST1
RASSF1A can negatively modulate the Raf/MAPK pathway by controlling the association of MST2 with c-Raf.20 MST2 does not bind B-raf, but the related MST1 will bind mutant B-Raf.21 In an effort to determine a mechanism by which RASSF6 might suppress MAPK activation in A375 cells, we examined the interaction of RASSF6, MST1, and mutant B-RafV600E. We performed co-immunoprecipitation studies using a GFP-tagged B-RafV600E, Myc-tagged MST1, and Flag-tagged RASSF6. While RASSF6 does not bind B-RafV600E directly, it does appear to increase the association of B-RafV600E and MST1 (Fig. 6).
Figure 6.
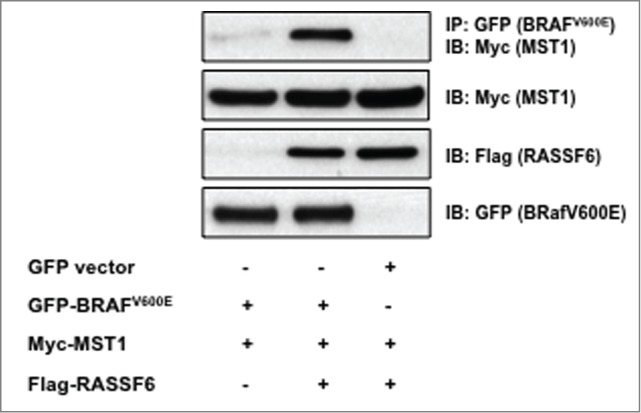
RASSF6 increases the association of BRAFV600E and MST1. HEK 293 cells were transfected with constructs for mutant B-Raf (BRAFV600E), MST1, and RASSF6. Cell lysates were immunoprecipitated (IP) with GFP-Trap® Beads (Allele Biotechnology, ACT-CM-GFA0050) and immunoblotted (IB) for the presence of MST1 (top panel). Cell lysates were also immunoblotted for total levels of MST1, RASSF6, and BRAFV600E. A representative blot is shown.
Discussion
Certain members of the RASSF family of tumor suppressors are inactivated by promoter methylation at high frequency in a broad range of cancers.22 However, RASSF6 has only previously been reported to be epigenetically inactivated at high frequency in neuroblastomas23 and leukemias.24 Here we show that RASSF6 has an extraordinarily high frequency of promoter methylation in melanomas and melanoma brain metastases, but not in other solid tumors. Indeed, the frequency of methylation in the brain metastases far exceeds that of RASS1A or RASSF2. Previously, this methylation has been shown to correlate well with loss of expression of RASSF6.24 This suggests that the tumor suppressor role of RASSF6 may be particularly pertinent to melanoma development and metastasis. Thus, RASSF6 methylation status could potentially act as a marker for predisposition to melanoma brain metastasis. We also assayed methylation for multiple RASSF family members in addition to RASSF6. We were unable to discern any particular pattern or trend of co-methylation in the tumors in this relatively small sample size.
These data implicate RASSF6 downregulation as a component of metastasis and invasion. The collagen invasion assays we performed support this concept, as RASSF6 was an effective suppressor of invasion. The reduced invasion observed in the presence of RASSF6 was not simply due to reduced growth or anoikis upon suspension, as these properties were little affected by the addition of RASSF6. The best-characterized tumor suppressor pathway modulated by the RASSF family is the Hippo pathway. Classically, RASSF1A binds the MST kinases and promotes their activation to initiate a kinase cascade leading to the regulation of the transcriptional regulator YAP.11 RASSF6 also binds the MST kinases but curiously appears to inhibit their activation.10 When we examined the effects of RASSF6 on Hippo signaling in the A375 cells, we observed no change in the status of phosphorylated YAP, the classic assay for Hippo activity (Supplementary Figs. 1D and 1E). Therefore, RASSF6 must be acting via some other pathway. As mutant B-Raf is a key driver of melanoma and constitutively activates the MAP Kinase pathway, we also assayed the effects of RASSF6 on the steady state levels of MAPK phosphorylation. A significant suppression of MAPK signaling was observed in the RASSF6-transfected A375 cells. This suppression was not observed in A375 cells expressing RASSF1A, suggesting RASSF6 has a distinct ability to modulate MAPK signaling in A375 melanoma cells.
MAPK activation can promote invasion,25 and B-Raf-induced MAPK activation has been reported to play a key role in extravasation.6 Thus, the loss of an inhibitor of B-Raf/MAPK should promote invasion and metastasis.
The mechanism by which RASSF6 acts to suppress the B-Raf/MAPK pathway is still unclear but may involve a similar mechanism to that reported for RASSF1A and c-Raf. RASSF1A modulates the binding of MST2 with c-Raf, a negative regulatory interaction.20 MST2 does not bind B-Raf, but MST1 does bind with mutant B-RafV600E.21 Our finding that RASSF6 strongly enhances the association of MST1 with mutant B-RafV600E provides a plausible potential novel mechanism for the action of RASSF6 on B-Raf signaling.
We also found that RASSF6 can suppress AKT activation in the B-Raf-transformed melanoma cells. AKT activity has been linked to invasion and metastasis,26 suggesting that this may also contribute to the action of RASSF6. How RASSF6 suppresses AKT is not known; however, MST2 can interact with AKT27 and RASSF6 binds MST2, suggesting there are ample further opportunities for cross regulation.
Thus, we identified a potential role for RASSF6 loss of function in the development of melanoma and melanoma brain metastasis. Interestingly, RASSF10 is also methylated in malignant melanomas.12 RASSF10 is an N-terminal RASSF family member, while RASSF1–6 are C-terminal members, and both RASSF6 and RASSF10 exhibit aberrant promoter methylation in childhood leukemias .24 Future research may involve the analysis of RASSF10 overexpression in malignant melanoma cells to determine if this N-terminal RASSF member has similar effects on invasion and MAPK activation as RASSF6, and if the proteins co-operate. The high frequency of RASSF6 promoter methylation in melanoma may serve as a novel diagnostic tool to monitor melanoma development. The suppression of invasion by RASSF6 in a melanoma cell line supports the concept of epigenetic therapy aimed at reactivating RASSF6 to inhibit melanoma invasion and metastasis.
Materials and Methods
Patient samples
A selection of 40 lung, 40 breast, and 43 melanoma metastases to the brain, 11 non-cancerous brain samples, 10 primary melanomas and 4 normal melanocyte samples were used for this study. Ethical guidelines of the University Hospital Carl Gustav Carus Dresden were followed for sample collection and institutional review board approval was obtained for the study from the University of Birmingham.
Methylation analysis
All DNA for this was study was bisulphite modified by using the Qiagen EpiTect kit (Qiagen, Rockville MD) according to the manufacturer's instructions. Combined bisulphite restriction analysis (CoBRA) was used to analyze the methylation status of RASSF2-RASSF6 using the primer sequences shown in Supplementary Table 1, Essentially as described in.24,28-31 All CoBRA PCR products were digested with BstUI (Fermentas, Pittsburgh, PA) overnight at 37°C prior to visualization on a 2% agarose gel. The methylation status of RASSF1A was assessed using methylation -specific PCR (MSP) using primers listed in Supplementary Table 1. Selected CoBRA PCR products were cloned into the pGEM-T Easy vector system (Promega, A1360) according to the manufacturer's instructions. Single colony PCR was performed using the forward primer: 5′-TAATACGACTCACTATAGGG-3; and reverse primer: 5′-ACACTATAGAATACTCAAGC-3′. Sequencing was then carried out on up to 12 colonies using a 3730 DNA Analyzer (Applied Biosystems, 3730S).
Molecular cloning
GFP-BRAFV600E was generated by PCR amplification of human BRAFV600E, obtained from Addgene. The PCR product was TOPO cloned into pGEM-T-Easy (Promega, Madison WI) and subcloned into pEGFP-C1 (Clontech, Mountain View CA) using SalI and XmaI restriction enzymes. Myc-MST1 was a generous gift from Jonathan Chernoff (Fox Chase Cancer Center, PA). Flag-RASSF6 has been previously described.32
Tissue culture and transfections
A375 melanoma cells (ATCC, CRL-1619TM) were grown in DMEM/10% fetal bovine serum and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) before selecting in 500 μg/mL G418. HA-tagged RASSF6 expression plasmids have been described previously.32 HA-tagged RASSF1A was cloned into the pBABE expression construct.33 Growth curve analysis was performed by plating 50,000 cells per well in 6 well plates. Cells were allowed 24-hour growth after plating, and then one well was trypsinized and counted using a hemocytometer every 24 hours for 6 d. All experiments were repeated in triplicate for each cell line. Collagen invasion assays were performed by preparing 30% collagen plugs from Rat tail collagen I (BD sciences, San Jose CA) in 8 μM pore transwell membrane inserts. Collagen (200 μl)was used for each 24 well transwell insert. Cells (5,000) were reconstituted in 500 μL of serum-free DMEM and added to the top of each collagen-filled insert. Complete medium (2 mL) was placed in each well. Cells were allowed to invade for 3 and 5 d before membranes were isolated, fixed and stained for cells using a Diff-Quik Stain Set (Fisher, Pittsburgh, PA). Each assay was performed in duplicate. Anoikis assays were performed by plating cells in Poly-HEMA (Sigma, St. Louis MS)) coated dishes and measuring cell death by trypan blue staining after 5 d.
Drug treatments and immunoprecipitation studies
A375 cells were treated with 0.2 μM 5-azacitidine (Sigma) or 0.5 μM nanaomycin A (obtained from the National Cancer Institute/Developmental Therapeutics Program Open Chemical Repository) for 48 hours. Control cells were treated with DMSO. Cells were lysed in RIPA buffer as previously described.31 Cell lysates (1 mg) were immunoprecipitated using RASSF6 antibody (Proteintech, Chicago, IL) and TrueBlot® Anti-Rabbit Ig IP beads (eBioscience). Experiments were performed in duplicate.
For co-immunoprecipitation studies, GFP-tagged BRAFV600E, Myc-tagged MST1, and Flag-tagged RASSF6 were transfected into HEK-293 cells via jetPRIME® transfection reagent (Polyplus transfection, Illkirch Fr). After 48 hours, cells were lysed in modified RIPA buffer (50 mM Tris-HCl, ph 7.4, 200 mM NaCl, 1% NP-40). Lysates were incubated with GFP-Trap® beads (Allele Biotechnology, San Diego, CA), and washed with lysis buffer. Co-immunoprecipitation experiments were performed in triplicate.
Western blotting
Cells were lysed in RIPA buffer as previously described.32 Proteins were run on a 4–15% Tris-Glycine gel (Bio-Rad, Hercules, CA) and transferred to 0.2 μM nitrocellulose membranes (Bio-Rad). Antibodies used were: phospho-p44/p42 MAP Kinase (Thr 202/Thr 204), p44/p42 MAP Kinase, phospho p38 MAPK (Thr 180/Tyr 182), YAP and phospho YAP (S127), Akt and Phospho Akt (S473) (Cell Signaling Boston MA), RASSF6 (Proteintech, Chicago, IL), β-actin (Sigma), p53 (Do-1) (Santa Cruz, Santa Cruz, CA) and HA (Covance, MMS-101P). Secondary antibodies were goat anti-mouse and goat anti-rabbit from KPL (54–12–50) and TrueBlot® anti-rabbit IgG HRP (Rockland, Rockville, MD). Western Blots were developed using an enhanced chemiluminescence kit from West Pico (Thermo Scientific, Pittsburgh, PA) or West Femto Enchance ECL (Pierce, Pittsburgh, PA).
Statistical analysis
P values were calculated via unpaired t-tests for all indicated samples. Microsoft Excel software was used for all statistical analyses.
Supplementary Material
Author Contributions
JM performed tissue culture assays, immunological studies and contributed to manuscript preparation. VH, ST and T.S. performed methylation analysis, MLS performed cloning and molecular biology assays, DK and GS performed tissue acquisition, GPP generated primary melanoma data, GJC and FL conceived and drafted the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The work was funded in part by the Brown Cancer Center MD/PhD program, University of Louisville, the Department of Neurosurgery, University Hospital Dresden, Germany, and King Abdulaziz University, Saudi Arabia.
References
- 1. Regad T. Molecular and cellular pathogenesis of melanoma initiation and progression. Cell Mol Life Sci 2013; 70:4055-65; PMID:23532409; http://dx.doi.org/ 10.1007/s00018-013-1324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Finn L, Markovic SN, Joseph RW. Therapy for metastatic melanoma: the past, present, and future. BMC Med 2012; 10:23; PMID:22385436; http://dx.doi.org/ 10.1186/1741-7015-10-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goulart CR, Mattei TA, Ramina R. Cerebral melanoma metastases: a critical review on diagnostic methods and therapeutic options. ISRN Surg 2011; 2011:276908; PMID:22084751; http://dx.doi.org/ 10.5402/2011/276908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949-54; PMID:12068308; http://dx.doi.org/ 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 5. Osborne JK, Zaganjor E, Cobb MH. Signal control through Raf: in sickness and in health. Cell Res 2012; 22:14-22; PMID:22143568; http://dx.doi.org/ 10.1038/cr.2011.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liang S, Sharma A, Peng HH, Robertson G, Dong C. Targeting mutant (V600E) B-Raf in melanoma interrupts immunoediting of leukocyte functions and melanoma extravasation. Cancer Res 2007; 67:5814-20; PMID:17575149; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer 2013; 49:1297-304; PMID:23290787; http://dx.doi.org/ 10.1016/j.ejca.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 8. van der Weyden L., Adams DJ. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta 2007; 1776:58-85; PMID:17692468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avruch J, Praskova M, Ortiz-Vega S, Liu M, Zhang XF. Nore1 and RASSF1 regulation of cell proliferation and of the MST12 kinases. Methods Enzymol 2005; 407:290-310; PMID:16757333; http://dx.doi.org/ 10.1016/S0076-6879(05)07025-4 [DOI] [PubMed] [Google Scholar]
- 10. Ikeda M, Kawata A, Nishikawa M, Tateishi Y, Yamaguchi M, Nakagawa K, Hirabayashi S, Bao Y, Hidaka S, Hirata Y, et al. Hippo pathway-dependent and -independent roles of RASSF6. Sci Signal 2009; 2:ra59; PMID:19797269; http://dx.doi.org/ 10.1126/scisignal.2000300 [DOI] [PubMed] [Google Scholar]
- 11. Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev 2013; 27:355-71; PMID:23431053; http://dx.doi.org/ 10.1101/gad.210773.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helmbold P, Richter AM, Walesch S, Skorokhod A, Marsch WC, Enk A, Dammann RH. RASSF10 promoter hypermethylation is frequent in malignant melanoma of the skin but uncommon in nevus cell nevi. J Invest Dermatol 2012; 132:687-94; PMID:22113481; http://dx.doi.org/ 10.1038/jid.2011.380 [DOI] [PubMed] [Google Scholar]
- 13. Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci 2007; 120:3163-72; PMID:17878233; http://dx.doi.org/ 10.1242/jcs.010389 [DOI] [PubMed] [Google Scholar]
- 14. Furuta J, Umebayashi Y, Miyamoto K, Kikuchi K, Otsuka F, Sugimura T, Ushijima T. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci 2004; 95:962-8; PMID:15596045; http://dx.doi.org/ 10.1111/j.1349-7006.2004.tb03184.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004; 64:1975-86; PMID:15026333; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3972 [DOI] [PubMed] [Google Scholar]
- 16. Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, Van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A 2003; 100:15901-5; PMID:14665696; http://dx.doi.org/ 10.1073/pnas.2634067100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carnahan J, Beltran PJ, Babij C, Le Q, Rose MJ, Vonderfecht S, Kim JL, Smith AL, Nagapudi K, Broome MA, et al. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther 2010; 9:2399-410; PMID:20663930; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0181 [DOI] [PubMed] [Google Scholar]
- 18. Cantwell-Dorris ER, O’Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther 2011; 10:385-94; PMID:21388974; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0799 [DOI] [PubMed] [Google Scholar]
- 19. Iwasa H, Kudo T, Maimaiti S, Ikeda M, Maruyama J, Nakagawa K, Hata Y. The RASSF6 tumor suppressor protein regulates apoptosis and the cell cycle via MDM2 protein and p53 protein. J Biol Chem 2013; 288:30320-9; PMID:24003224; http://dx.doi.org/ 10.1074/jbc.M113.507384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. Protein interaction switches coordinate Raf-1 and MST2Hippo signalling. Nat Cell Biol 2014; 16:673-84; PMID:24929361; http://dx.doi.org/ 10.1038/ncb2986 [DOI] [PubMed] [Google Scholar]
- 21. Lee SJ, Lee MH, Kim DW, Lee S, Huang S, Ryu MJ, Kim YK, Kim SJ, Kim SJ, Hwang JH, et al. Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1 pathway in papillary thyroid carcinoma. PLoS One 2011; 6:e16180; PMID:21249150; http://dx.doi.org/ 10.1371/journal.pone.0016180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta 2009; 1796:114-28; PMID:19344752 [DOI] [PubMed] [Google Scholar]
- 23. Djos A, Martinsson T, Kogner P, Caren H. The RASSF gene family members RASSF5, RASSF6 and RASSF7 show frequent DNA methylation in neuroblastoma. Mol Cancer 2012; 11:40; PMID:22695170; http://dx.doi.org/ 10.1186/1476-4598-11-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hesson LB, Dunwell TL, Cooper WN, Catchpoole D, Brini AT, Chiaramonte R, Griffiths M, Chalmers AD, Maher ER, Latif F. The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol Cancer 2009; 8:42; PMID:19570220; http://dx.doi.org/ 10.1186/1476-4598-8-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev 2003; 22:395-403; PMID:12884914; http://dx.doi.org/ 10.1023/A:1023781114568 [DOI] [PubMed] [Google Scholar]
- 26. Li C, Li F, Zhao K, Yao J, Cheng Y, Zhao L, Li Z, Lu N, Guo Q. LFG-500 inhibits the invasion of cancer cells via down-regulation of PI3KAKTNF-kappaB signaling pathway. PLoS One 2014; 9:e91332; PMID:24618693; http://dx.doi.org/ 10.1371/journal.pone.0091332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res 2010; 70:1195-203; PMID:20086174; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hesson LB, Wilson R, Morton D, Adams C, Walker M, Maher ER, Latif F. CpG island promoter hypermethylation of a novel Ras-effector gene RASSF2A is an early event in colon carcinogenesis and correlates inversely with K-ras mutations. Oncogene 2005; 24:3987-94; PMID:15806169; http://dx.doi.org/ 10.1038/sj.onc.1208566 [DOI] [PubMed] [Google Scholar]
- 29. Hesson L, Dallol A, Minna JD, Maher ER, Latif F. NORE1A, a homologue of RASSF1A tumour suppressor gene is inactivated in human cancers. Oncogene 2003; 22:947-54; PMID:12584574; http://dx.doi.org/ 10.1038/sj.onc.1206191 [DOI] [PubMed] [Google Scholar]
- 30. Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, Fullwood P, Chauhan A, Walker R, Shaw JA, et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001; 20:1509-18; PMID:11313894; http://dx.doi.org/ 10.1038/sj.onc.1204175 [DOI] [PubMed] [Google Scholar]
- 31. Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4AD037 is a potential ras effectortumor suppressor of the RASSF family. Cancer Res 2004; 64:8688-93; PMID:15574778; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2065 [DOI] [PubMed] [Google Scholar]
- 32. Allen NP, Donninger H, Vos MD, Eckfeld K, Hesson L, Gordon L, Birrer MJ, Latif F, Clark GJ. RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene 2007; 26:6203-11; PMID:17404571 [DOI] [PubMed] [Google Scholar]
- 33. Morgenstern JP, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res 1990; 18:1068; PMID:2156225; http://dx.doi.org/ 10.1093/nar/18.4.1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.