

Abstract
As the developing zebrafish pancreas matures, hormone-producing endocrine cells differentiate from pancreatic Notch-responsive cells (PNCs) that reside within the ducts. These new endocrine cells form small clusters known as secondary (2°) islets. We use the formation of 2° islets in the pancreatic tail of the larval zebrafish as a model of β-cell neogenesis. Pharmacological inhibition of Notch signaling leads to precocious endocrine differentiation and the early appearance of 2° islets in the tail of the pancreas. Following a chemical screen, we discovered that blocking the retinoic acid (RA)-signaling pathway also leads to the induction of 2° islets. Conversely, the addition of exogenous RA blocks the differentiation caused by Notch inhibition. In this report we characterize the interaction of these two pathways. We first verified that signaling via both RA and Notch ligands act together to regulate pancreatic progenitor differentiation. We produced a transgenic RA reporter, which demonstrated that PNCs directly respond to RA signaling through the canonical transcriptional pathway. Next, using a genetic lineage tracing approach, we demonstrated these progenitors produce endocrine cells following inhibition of RA signaling. Lastly, inhibition of RA signaling using a cell-type specific inducible cre/lox system revealed that RA signaling acts cell-autonomously in PNCs to regulate their differentiation. Importantly, the action of RA inhibition on endocrine formation is evolutionarily conserved, as shown by the differentiation of human embryonic stem cells in a model of human pancreas development. Together, these results revealed a biphasic function for RA in pancreatogenesis. As previously shown by others, RA initially plays an essential role during embryogenesis as it patterns the endoderm and specifies the pancreatic field. We reveal here that later in development RA is involved in negatively regulating the further differentiation of pancreatic progenitors and expands upon the developmental mechanisms by which this occurs.
Keywords: Retinoic acid, pancreas, progenitors, differentiation
Introduction
Type I diabetes is a chronic disease caused by the loss of insulin-expressing β cells, leading to elevated blood glucose levels (hyperglycemia) and tissue damage. Ultimately, type I diabetics require insulin therapy. Long-term insulin therapy can be associated with serious complications such as inadvertent hypoglycemia and insulin resistance (Donga et al., 2013). β-cell replacement therapy is a promising way to cure type I diabetes, but its use is limited by both the paucity of donor islet tissue for transplantation and problems associated with continuous immunosuppression (Robertson, 2004). Human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) possess the ability to differentiate into many derivatives of the three primary germ layers: ectoderm, mesoderm, and endoderm (Murry and Keller, 2008). Progress has been made in generating β cells in vitro from hESCs and iPSCs (Nostro and Keller, 2012); however, because this method is still relatively inefficient and has accompanying safety concerns,, this technique is still some way from becoming a cure. Elucidating mechanisms regulating β-cell development in normal pancreas helps identify crucial signals that improve the efficiency of generating mature β cells in vitro and could potentially point to ways of inducing endogenous pancreatic progenitors to differentiate in diabetic patients.
The development of the zebrafish pancreas has been well studied and is closely conserved with that of the mammalian pancreas (Kinkel and Prince, 2009; Tiso et al., 2009). The first step of pancreatogenesis is the specification of the pancreatic field from nascent foregut endoderm, which in zebrafish occurs in the first day of development. The retinoic acid (RA)-signaling pathway is crucial in specifying the pancreatic field (Kinkel et al., 2009; Stafford and Prince, 2002; Stafford et al., 2006). RA is derived from vitamin A and acts as a ligand for nuclear RA receptors (RARs) that directly regulate the transcription of downstream target genes important for development (Rhinn and Dolle, 2012). The distribution and levels of RA in the embryo are tightly controlled by synthesis enzymes (aldehyde dehydrogenases, Aldhs) and specific degradation enzymes of the cytochrome P450 subfamily (CYP26A1, CYP26B1 and CYP26C1), allowing RA to function like a morphogen to control the differentiation and patterning of different stem and progenitor cell populations (Rhinn and Dolle, 2012). Neckless (nls) mutant fish lack Aldh1a2 activity and, as a consequence, RA production is compromised. In nls mutants there is a dramatic reduction in the number of pancreatic cells formed (Stafford and Prince, 2002). Conversely, increasing RA-signaling activity (either by exogenous RA supplement or the removal of RA-degradation enzymes) leads to an expansion of the pancreatic field (Kinkel et al., 2009; Stafford and Prince, 2002; Stafford et al., 2006).
By 24 hours post fertilization (hpf), dorsal pancreatic endoderm has coalesced at the midline of the zebrafish embryo to form the principal islet. In the majority of fish before 5 days post fertilization (dpf), this islet represents the sole location of the pancreatic endocrine cells (Biemar et al., 2001). These first-transition endocrine cells of the principal islet possess a low proliferative capacity and contribute little to the future adult endocrine system (Hesselson et al., 2009; Wang et al., 2011). Around 32 hpf, ventral endoderm cells start to express the transcription factor Ptf1a (Lin et al., 2004; Zecchin et al., 2004) and migrate in a posterior and dorsal direction to meet and envelop the principal islet and to create a recognizable pancreas. Around 80 hpf a second wave of endocrine differentiation (or secondary transition) occurs as hormone-producing cells differentiate from the extra-pancreatic duct and contribute to the principal islet (Dong et al., 2007; Dong et al., 2008). By 5 dpf, the pancreas is elongated and mostly exocrine tissue derived from the ventral cells, structured with an anterior ‘head’ containing the principal islet and a ‘tail’ containing intrapancreatic ducts. The ducts contain pancreatic Notch-responsive cells (PNCs). These PNCs are larval progenitors that differentiate during later stages of development to form the 2° islets along the duct in the pancreatic tail (Ninov et al., 2012; Wang et al., 2011). The formation of such 2° islets in the larval zebrafish pancreas is analogous to endocrine formation in mammalian pancreas. Both events involve the differentiation of Notch-responsive ductal-associated progenitors. For these reasons studying the formation of endocrine cells of the 2° islets is a promising way to discover new strategies for β-cell recovery in type I diabetics.
Several different signaling pathways are essential for pancreas development (Kimmel and Meyer, 2010; Serup, 2012). For instance, Notch signaling has long been known as central to both mammalian (Apelqvist et al., 1999; Esni et al., 2004; Hald et al., 2003; Jensen et al., 2000; Murtaugh et al., 2003) and zebrafish pancreas development (Esni et al., 2004; Lorent et al., 2004; Ninov et al., 2012; Parsons et al., 2009; Zecchin et al., 2007). Inhibition of Notch signaling leads to precocious differentiation of PNCs and the early appearance of endocrine cell types in the 2° islet position within the pancreatic tail (Parsons et al., 2009). To find pathways other than Notch signaling that regulate 2° islet formation, we performed a chemical screen in larval zebrafish from 3 to 5 dpf. One of the hit compounds we identified was Tetraethylthiuram disulfide (Disulfiram, DSF) and this drug was shown to induce precocious 2° islets by inhibiting the Aldh-dependent production of RA (Rovira et al., 2011). Conversely, increasing levels of RA rescued the effects of Notch inhibition by blocking precocious endocrine differentiation, suggesting RA signaling is also involved in the regulation of endocrine differentiation. Together, these results suggested that RA plays a role in endocrine pancreas differentiation; hence, a pancreatic source of RA should exist. Previously we showed that the developing larval exocrine pancreas (6 dpf) has Aldh enzymatic activity, consistent with RA production (Rovira et al., 2011). As the larval fish mature (from 15 dpf), a second potential RA source appears as Aldh1 positive cells differentiate along the duct from the PNCs. These Aldh1+ cells are rare at first but increase in number with age (Matsuda et al., 2013).
In this present paper, we further characterize the pancreatic function of the RA-signaling pathway, and show that RA signaling cell-autonomously regulates the differentiation of pancreatic progenitors during the secondary transition of endocrine development. Together with the results of other groups, our results suggest that RA signaling has a biphasic function in pancreatogenesis. RA is both required for the specification of the embryonic pancreas and later in regulating pancreatic progenitor differentiation. Using hESC differentiation protocols to model human pancreas development (Rezania et al., 2012), we demonstrate that the action of RA inhibition on endocrine progenitor differentiation is a conserved phenomenon.
Material and Methods
Transgenic Lines
All transgenic lines used are listed in Supplementary Table S1. All new transgenic lines were generated as described (Wang et al., 2011) using the T2KXIGΔIN backbone and Tol2-mediated transgenesis (Kawakami, 2004). Two cre drivers were used in this report that express 4-hydroxytamoxifen (4OHT) inducible cre (creERT2) and are described in (Wang et al., 2011). Tg(Tp1glob:creERT2)jh12 drives creERT2 in cells undergoing Notch signaling and is called ‘Notch-responsive creERT2 driver’. Tg(β-actin:GFP-F2A-creERT2)jh29 drives creERT2 ubiquitously and is called ‘ubiquitous creERT2 driver’. Two cre-responder lines were utilized: 1) Tg(β-actin:loxP-stop-loxP-hmgb1-mCherry)jh15, which is used in lineage tracing and is referred to as ‘nuclear-red cre responder’ (Wang et al., 2011); and 2) Tg(ubb:loxP-eCFP-loxP-dnRAR-GFP)jh39, a new cre-responder line utilizing the ubiquitous promoter/enhancer element from ubiquitin B (ubb) (Mosimann et al., 2011) and called ‘dnRAR cre responder’. This transgene was generated using a gene cassette encoding a fusion of dominant-negative zebrafish retinoic acid receptor α (dnRAR) fused with GFP (gift from K.Poss (Kikuchi et al., 2011)). Several transgenic lines were used to label specific cell types with fluorescent protein expression. Tg(Tp1bglob:hmgb1-mCherry)jh11 (Notch reporter) labels Notch-responsive cells with nuclear mCherry. Tg(pax6b:GFP)ulg515 (Delporte et al., 2008) labels all endocrine cells; Tg(neuroD:GFP)nl1 labels committed endocrine progenitors (Obholzer et al., 2008). The construct used to label cells responding to RA signaling, Tg(RARE-cFos:QF; QUAS:GFP), here called ‘RA reporter,’, was made using an oligo containing four copies of 5′-ggttca(n5)agttca-3′, each copy separated by 11 nucleotides. This RA-responsive element (RARE) was cloned upstream of the cFos minimal promoter and sequence encoding the QF transcriptional activator. Included in the same transgene were the QF-binding upstream activating sequence (QUAS) and the cFos minimal promoter, which together regulate transcription of gfp (Subedi et al., 2013).
In situ Hybridization and Immunofluorescence
5 dpf larvae were fixed in 4% PFA overnight and in situ hybridization was performed on dissected larval pancreata essentially as in (Xu et al., 2002). The primer sequences used to generate the ribo probes are listed in Supplementary Table S2. Images were recorded using a Nikon AZ100 microscope. Immunofluorescent staining (Costa et al., 2003) was performed on dissected larval pancreata (Parsons et al., 2009). The following antibodies were used: rabbit polyclonal anti-GFP at 1:200 dilution (A11122, Life Technologies), anti-Insulin at 1:400 (A0564, Dako), anti-Glucagon (A0565, Dako) at 1:400, anti-Somatostatin at 1:400 (A0566, Dako), and fluorescently conjugated 2° antibodies at 1:400 dilution (Jackson ImmunoResearch Labs INC).
Confocal Imaging
Dissected larval pancreata were mounted in fluorescent mounting medium (Dako) and imaged with a Nikon A1-si Laser Scanning Confocal Head mounted on a TiE inverted microscope through a Plan Apo 20× VC (N.A. 0.75) (Nikon). Acquisition and analysis was performed using NIS-Elements software (Ver 3.22, build 710). Cell number was quantified by counterstaining nuclei with DAPI (0.2 μg/ml) and counted by using NIS-Elements software (Nikon).
Drug Treatment
Unless otherwise stated in the text, drug treatments were performed as follows. Compounds were made as 10 mM stocks in DMSO and diluted to: 10 μM Disulfiram (DSF; 86720, Sigma), 10 μM Diethylaminobenzaldehyde (DEAB ; D86256, Sigma), 10 μM all-trans retinoic acid (RA; R2625, Sigma), and 0.625 μM RO4929097 (S1575, Selleckchem). 3 dpf embryos were incubated in drug until 5 dpf in the dark at 28 °C. As previously shown, 0.5% DMSO does not significantly change secondary islet formation and is used as a negative control (Rovira et al., 2011). 4-Hydroxytamoxifen (4OHT; T176, Sigma) was dissolved in 100% ethanol to make a 10 mM stock solution. Embryos were incubated in E3 medium (Westerfield, 2000) with 5 μM 4OHT and kept in the dark at 28°C for 24 hours, then washed in fresh E3. After drug treatment we either: 1) quantified numbers of 2° islets following in situ hybridization to detect neuroD expression; or 2) used transgenic markers and confocal microscopy to image and quantify 2° islet cell numbers.
hESC Culture
The H9 hESC line was obtained from the WiCell Research Institute (Madison, WI), and was treated as per the protocol established by the Kieffer lab (Rezania et al., 2012). More details provided in Supplemental Materials and Methods.
Results
RA and Notch signaling act together to regulate secondary islet differentiation
Our lab and others have established the requirement of Notch-signaling to maintain multi-potent progenitor cells in the pancreas (Murtaugh et al., 2003; Parsons et al., 2009). More recently, we demonstrated that the precocious formation of 2° islets can be induced in zebrafish larvae by reducing levels of endogenous RA signaling (Rovira et al., 2011). Furthermore, we found that exogenous RA can prevent the normal formation of 2° islets that form in larval fish (5–8 dpf, Supplemental Fig. S1). This observation is consistent with a reduction in RA levels being required for 2° islet formation during normal larval development. Accordingly, we wanted to determine whether RA and Notch signaling act together in regulating 2° islet formation. We empirically tested a range of concentrations for the Notch inhibitor, RO4929097, and ALDH-1 inhibitor, DEAB, to find concentrations for each compound that led to only low levels of precocious 2° islet formation. To assist in quantifying 2° islet cells we used Tg(neuroD:eGFP)nl1 transgenic larva in which GFP is expressed in all nascent endocrine cells (Obholzer et al., 2008). As is shown in Fig. 1, treatment with 0.625 μM RO4929097 or 12.5 μM DEAB from 3–5 dpf induced only slightly more 2° islet cells than appear in negative controls (2.69±0.32 SE p<0.02, and 2.08±0.29 SE p>0.05, compared to 1.75±0.23 SE). Conversely, a combination of both 0.625 μM RO4929097 and 12.5 μM DEAB gave a highly significant difference (8.49±0.7 SE, p<0.001) when compared to DMSO treatment, and this amount of 2° islet cells is clearly higher than just an additive effect of the two compounds (Fig. 1). This result suggests that the RA and Notch pathways are concordantly acting in the same pathway to regulate the differentiation of 2° islets. To further test whether simultaneous or sequential inhibition of these pathways was most efficacious in 2° islet induction, we carried out the following four different treatments: 1) RO4929097 and DEAB were added together for one day from 3–4dpf; 2) both drugs were added for one day from 4–5dpf; 3) RO4929097 was added for one day (3–4 dpf) before the larvae were washed and incubated with DEAB for the next day (4–5 dpf); and, 4) the order of the drugs was reversed, observing the same time intervals. Induction of 2° islets was quantified for all conditions and compared to negative controls (DMSO). We only observed a significant induction of 2° islet cells if both drugs were simultaneously present (either between 3–4 or 4–5 dpf, p<0.001) (Supplementary Fig. S2). These observations are consistent with RA- and Notch-signaling acting synergistically in the same developmental pathway to regulate 2° islet formation.
Fig. 1. Notch and RA signaling regulate the induction of 2° islet cells in a manner consistent with synergy.
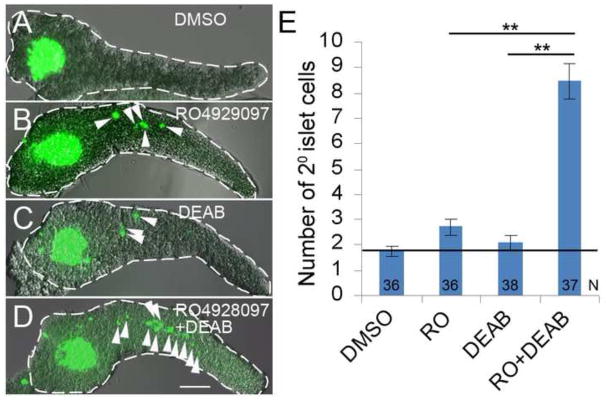
(A–D) Confocal z-stack projections of larval Tg(neuroD:GFP)nl1 pancreata dissected at 5 dpf. Larvae were treated with: A) DMSO, B) RO4929097 (RO) 0.625 μM, C) DEAB 25 μM and D) DEAB 25 μM + RO 0.625 μM, from 3–5 dpf. 2° islet cells (green) in the tail region of pancreata are indicated by arrowheads. Pancreata are outlined in white dashed lines. Scale Bar, 100 μm. (E) Average number of 2° islet cells per 5 dpf larval pancreas. N = number of larval pancreata quantified. Error Bar represents standard error around the mean (SE). Significance **p<0.001
Pancreatic progenitors are RA responsive
We have previously reported ALDH enzymatic activity in larval exocrine tissue, suggesting there is an endogenous source of RA in the larval pancreas (Matsuda et al., 2013; Rovira et al., 2011). To better understand how RA is involved in regulating larval 2° islet formation, we wanted to determine which cells are responding to RA signaling. Several RA-signaling reporter lines have been established (Perz-Edwards et al., 2001; Waxman and Yelon, 2011). Using these RA-signaling reporter lines we could not detect any RA-signaling activity in the larval pancreas (data not shown). We hypothesized that conventional transgenic reporters do not provide a level of sensitivity required to detect RA-dependent transcription in a discrete cell population in the pancreas. We therefore adopted a bipartite transgenic approach by utilizing the QF/QUAS system (Potter et al., 2010; Subedi et al., 2013). Our new construct contains 4 copies of a retinoic acid response element (RARE) found in murine HoxA1 (Langston et al., 1997). This RARE regulates expression of the transactivator QF. Also in the same construct are multimeric binding sites for QF (QUAS) regulating expression of GFP (Fig. 2A). This bipartite system provides the potential for amplification of the reporter signal without the caveats of silencing associated with Gal4/UAS methods (Subedi et al., 2013). The Tg(RARE-cfos:QF;QUAS:GFP) construct was injected into one-cell embryos to generate a stable transgenic line, hereafter referred to as the RA reporter. The ability of the RA reporter line to report on RA signaling was validated by demonstrating sensitivity to RA levels in the whole larvae (Fig. 2B) and by showing an expression pattern in early embryogenesis that is identical to previous reports (Supplementary Fig. S3) (Mandal et al., 2013; Perz-Edwards et al., 2001; Waxman and Yelon, 2011).
Fig. 2. PNCs are RA-responsive.

(A) Schematic of the RA reporter, 4xRARE-cFos:QF; QUAS:GFP. Expression of transcription factor QF (blue arrow) is driven by 4 RA-responsive elements (4xRARE, 4 red ovals). In the opposite orientation, GFP coding sequence (green arrow) is cloned downstream of 5 QF upstream activating sites (5xQUAS, 5 cyan ovals). QF binds to QUAS sequence to activate GFP expression. HS4 Insulator sequence (black rectangle) prevents the interference between the two transcriptional units. Pink rectangles represent cfos minimal promoters. The transgene is flanked by Tol2 arms (grey triangles). (B and C) Fluorescence images of RA reporter larvae. The channel of green fluorescence shows the RA reporter signal. (B) Compared to DMSO control, 3 dpf larvae treated with exogenous RA or DEAB increase or decrease their RA reporter signal, respectively. The larvae were incubated from 1–3 dpf with either 10 μM RA, 25 μM DEAB or DMSO. Insets show images of dissected pancreata from treated larvae at 5dpf, outlined in white dashed lines. (C) RA-responsive cells are Nkx6.1+ in the larval pancreas. A single-plane confocal image of a larval pancreas dissected at 5 dpf and immunostained for Nkx6.1 and Insulin. Arrowheads indicate examples of GFP+ Nkx6.1+ cells. Scale bar, 100 μm.
To investigate which cells of the larval (5 dpf) pancreas are responding to RA signaling we removed and imaged the pancreata from RA reporter fish. GFP expression was localized to the principal islet and intrapancreatic duct regions (Fig. 2C). In keeping with results seen at the level of the whole organism, reporter expression in the pancreas is also sensitive to RA level (Fig. 2B insets). In the absence of exogenous RA, no reporter expression is seen in acinar cells. When exogenous RA is added, however, we do detect variegated GFP expression in acinar cells. There are a few explanations for this variegated expression, including: 1) the acinar population is heterogeneous and only some acinar cells have the capability to respond to RA, albeit at non-physiological RA levels; and 2) as is well reported, the transgenes maybe undergoing stochastic silencing in some cells (Stuart et al., 1990). We next sought to ascertain which ductal cells are RA-responsive. Nkx6.1 is a marker of PNCs (Supplementary Fig. S4). Utilizing our RA reporter, we found RA-responsive GFP positive cells in the intrapancreatic duct region do indeed express Nkx6.1 (Fig. 2C and C′). While some Nkx6.1 positive cells express GFP, not all PNCs are labelled by the transcriptional activity of the RA reporter, demonstrating either heterogeneity within the PNC population or variegated expression from the transgenic RA reporter (Fig. 2C and C′). Taken together, our results demonstrate that a significant sub-population of PNCs is responding to endogenous RA.
Inhibition of RA signaling induces PNC differentiation
PNCs are the larval progenitors of ductal cells, centroacinar cells, and endocrine cells during normal development (Wang et al., 2011). Furthermore, we have shown that a sub-population of PNCs is responding to RA signaling. For these reasons, we wanted to know if the precocious 2° islets induced by inhibiting RA signaling were also formed from differentiating PNCs. To perform genetic inducible fate mapping of larval PNCs we utilized fish transgenic for: 1) our Notch-responsive creERT2 driver; 2) our nuclear-red cre responder (Fig. 3A); and 3) Tg(pax6b:GFP)ulg515, a line in which endocrine cells are labelled with green fluorescence (Delporte et al., 2008). Lineage tracing was performed in these triple transgenic larvae by activating creERT2 with 4OHT incubation from 2–3 dpf (Fig 3B). Next, lineage-labelled larvae were incubated from 3–5 dpf in either DMSO (control, Fig. 3D) or an ALDH-1 inhibitor (either DEAB or DSF). When ALDH-1 activity was inhibited with either DEAB (Fig. 3E) or DSF (Fig. 3F), a proportion of the GFP-positive 2° islet cells were also labeled with nuclear-red fluorescence (Fig. 3E′ and F′). The presence of double-labeled cells indicates that PNCs are contributing to the induced endocrine cell population. While this method of lineage analysis cannot rule out the contribution of other cell types to induced 2° islet cells, we can nevertheless conclude that inhibition of RA signaling directly leads to the differentiation of PNCs into endocrine cells.
Fig. 3. Inhibition of RA signaling induces PNCs to differentiate into endocrine cells.

(A) Schematic of the nuclear red cre responder, Tg(β-actin:loxP-stop-loxP-hmgb1-mCherry)jh15. The β-actin promoter/enhancer drives ubiquitous expression. Active Cre induces recombination between the two loxP sites (black triangles) removing the STOP signal (yellow box) and allowing production of Hmgb1-mCherry, a nuclear-red fluorescent protein. The transgene is flanked by Tol2 arms (grey triangles). (B) Experimental timeline of 4OHT-dependent fate mapping. 4OHT was applied 2 – 3 dpf. Larvae were then exposed to drugs from 3 – 5 dpf. (C–F) Confocal z-stack projections of dissected pancreata from larvae transgenic for the Notch-responsive creERT2 driver, Tg(Tp1glob:creERT2)jh12, the nuclear red cre responder and the endocrine marker, Tg(pax6b:eGFP)ulg515. These triple transgenic larvae were treated with vehicle (C), 4OHT+DMSO (D), 4OHT+DEAB (E), and 4OHT+DSF (F). The presence of lineage-traced cells (red nuclei) is dependent on 4OHT treatment (D–F). 2° islet cells can be detected in the tail region of larval pancreata after treatment with either DEAB (E) or DSF (F). (E′ and F′) The precocious 2° islet cells are positive for both GFP and mCherry, indicating their PNC origin. Nuclei are counterstained with DAPI (blue). White dashes outline pancreata. Scale Bar, 100 μm.
Generation of dnRAR cre responder transgene to inhibit endogenous RA signaling
In order to inhibit endogenous RA signaling in a cell-specific way, we generated a dnRAR cre responder transgene using sequence encoding a fusion of a dominant-negative form of the zebrafish RA receptor α and GFP (dnRAR-GFP) (Kikuchi et al., 2011). Using this transgene we generated the dnRAR cre responder line of fish, Tg(ubb:loxP-eCFP-loxP-dnRAR-GFP)jh39. The dnRAR cre responder transgene is designed to constitutively express dnRAR-GFP following cre recombination (Supplementary Fig. S5A). To validate this dnRAR cre responder line, we crossed these fish to a ubiquitous creERT2 driver line, Tg(β-actin:GFP-F2A-creERT2)jh29 (Wang et al., 2011). The resulting double-transgenic larvae had a 4OHT-dependant phenotype (Supplementary Fig. S5F) reminiscent of aldh1a2nls/nls mutants, which lack Aldh1a2 activity (Begemann et al., 2001) (Supplementary Fig. S5H). In situ hybridization for myoD and krox20 reveals aberrant anterior-posterior patterning caused by widespread dnRAR-GFP expression (Supplementary Fig. S5G) and is consistent with diminished RA signaling (Supplementary Fig. S5I). We further demonstrated the utility of the dnRAR cre responder line by crossing it with our new RA reporter line. As predicted, widespread expression of dnRAR-GFP dramatically reduced the activity of the RA reporter (Supplementary Fig. S5J–M).
As mentioned previously, RA-signaling is required for pancreas field specification during early development (Kinkel et al., 2009; Stafford and Prince, 2002; Stafford et al., 2006). To further validate our dnRAR cre responder line, we looked at the consequences of inducing early expression of dnRAR-GFP. Again we injected cre mRNA into one-cell dnRAR cre responder embryos to induce widespread expression of dnRAR-GFP (Supplementary Fig. S6 A–B) and examined the formation of pancreatic endocrine and exocrine tissue by in situ hybridization for ins and trypsin, respectively. We found that expression of both ins and trypsin were abrogated following the induction of dnRAR-GFP (Fig. 4C and D), compared to uninjected controls (Fig. 4A and B) or injected controls transgenic for a nuclear-mCherry responder (Supplementary Fig. S6E and F). This observed phenotype is consistent with studies on either aldh1a2nls/nls mutant or ALDH inhibitor-treated fish (Alexa et al., 2009; Stafford and Prince, 2002). Taken together, our results show that over-expression of dnRAR from the dnRAR cre responder transgene can functionally block the endogenous RA-signaling pathway. Hence, we have generated a dnRAR cre responder fish line that allows inhibition of endogenous RA signaling in a Cre-dependent manner.
Fig. 4. Overexpression of dnRAR blocks pancreatic field specification.
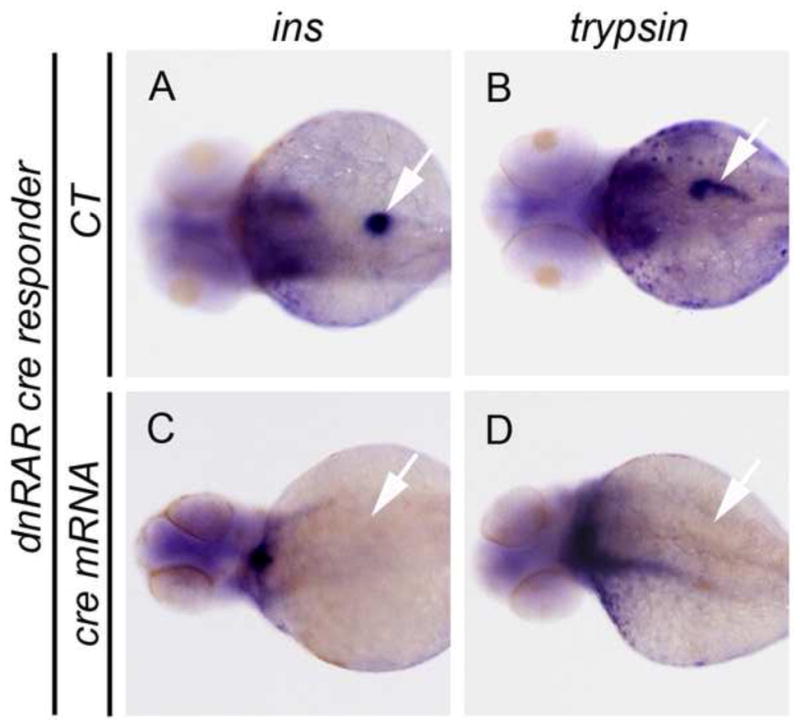
(A–D) The results of whole mount in situ hybridization to detect ins and trypsin expression in, dnRAR cre responder transgenic larvae at 72 hpf. Expression of ins and trypsin demonstrates normal pancreatic field specification. (A and B) Negative control larvae (CT). (C and D) Larvae previously injected with cre mRNA at the one-cell stage and displaying the failure of pancreas specification in presence of cre-dependent dnRAR-GFP. Arrows indicate the location of larval pancreata.
The RA-signaling pathway has cell-autonomous effects on PNC differentiation in the formation of 2° islets
From our earlier work we knew that PNCs could be induced to differentiate to endocrine cells by RA inhibition. To determine whether RA signaling acts on PNC differentiation in a cell-autonomous manner we again used the cre/lox system. As previously reported, in the pancreata of Notch-responsive creERT2 driver larvae up to 75% of PNCs will undergo cre-dependent recombination following 4OHT treatment (Wang et al., 2011). Larvae transgenic for both this driver and the dnRAR cre responder were treated with 4OHT from 2–5 dpf to induce PNC expression of the dnRAR-GFP fusion protein. Use of this fusion precludes the use of other GFP reporters; consequently, the formation of 2° islets was assessed at 5 dpf by the following methods: 1) in situ hybridization to detect the expression of the pan-endocrine marker neuroD (Fig. 5A–C); and 2) immunofluorescent detection of all α, β and δ cells (Fig. 5D–G). As in situ hybridization affords a lower level of resolution, with this method we were limited to counting the number of 2° islets rather than individual cells. We found that the average number of 2° islets/pancreas in 4OHT-treated transgenic larvae (0.95±0.12 SE) was significantly increased compared to vehicle-treated larvae (0.48±0.11 SE) (p<0.001) (Fig. 5A–C). Using immunofluorescent detection of hormone-expressing cells (with antibodies to Insulin, Glucagon and Somatostatin to respectively detect α, β and δ cells), we quantified the number of individually induced endocrine cells in 4OHT-treated transgenic larvae (Fig. 5D–G). Again we found that the effects of cre-dependent dnRAR expression significantly increased the number of 2° endocrine cells when compared to controls (1.29±0.17 SE 2° islet cells/pancreas vs 0.58±0.14 SE, p<0.001). Using immunofluorescent detection, we saw similar numbers of induced 2° islet cells in 4OHT treated transgenic larvae (where RA signaling is reduced in just PNCs), as compared to sibling fish treated with DEAB (1.11±0.21 SE) (where RA production is compromised throughout the fish) (Fig. 5F–G). Additionally, use of a ubiquitous creERT2 driver allowing widespread expression of dnRAR did not induce a significantly different number of 2° islets than when dnRAR expression was limited to PNCs (data not shown). As cre-activity is not uniform in all PNCs it is difficult to make direct comparisons between the effects of pharmacological and transgenic RA inhibition. Nevertheless it is clear that inhibiting RA signal transduction in just PNCs is sufficient to obtain precocious 2° islet cells, further supporting the hypothesis that the RA-signaling pathway has a cell-autonomous effect in controlling PNC differentiation.
Fig. 5. PNC differentiation is regulated by endogenous RA signaling.

(A and B) in situ hybridization for neuroD expression to detect 2° islets in 5 dpf pancreata from larvae transgenic for both the Notch-responsive creERT2 driver, Tg(Tp1glob:creERT2)jh12, and the dnRAR cre responder, Tg(ubb:loxP-eCFP-loxP-dnRAR-GFP)jh39. These larvae had been incubated at 2 dpf with either vehicle (A) or 4OHT (B). (C) Average number of 2° islets/pancreas in vehicle or 4OHT treated larvae calculated by counting neuroD expressing islets. (D–G) Fluorescence immunostaining to detect hormone-expressing cells (insulin+/glucagons+/somatostatin+), facilitating quantification of 2° islet cells in larvae that were treated from 2–5 dpf with either vehicle (D), or 4OHT (E), or vehicle + DEAB (F). (E″) enlarged image of (E′). (G) Average number of 2° islet cells/pancreas in vehicle, 4OHT, and vehicle + DEAB treated larvae calculated by counting for the number of hormone-expressing cells. **p<0.001. White dashes outline pancreata. 2° islets in the tail region of the pancreas are indicated by arrowheads. N = number of larval pancreata quantified. Error Bar = SE. Scale Bar, 100 μm.
Inhibition of RA signaling in hESC-derived pancreatic endoderm promotes production of endocrine cells
To test if the action of RA inhibition is conserved from zebrafish to human we took advantage of the protocol from the Kieffer laboratory (Rezania et al., 2012). This method allows the differentiation of pluripotent hESCs to pancreatic cell types by recapitulating in culture the developmental signaling events that occur during embryonic pancreas formation. This protocol is divided into 4 stages. Each stage has a treatment regimen that mimics a different stage of pancreas development during embryogenesis and leads to the formation of hESC-derived cells with the following characteristics: stage 1, definitive endoderm; stage 2, primitive gut tube; stage 3, posterior foregut; and, stage 4, pancreatic endoderm and endocrine precursors (Rezania et al., 2012) (see Supplemental Materials and Methods for details). In this protocol, RA is required for the specification of pancreatic lineages in stage 3, but is removed by stage 4. We used this established hESC differentiation protocol to assess if the suppression of RA signaling to promote endocrine formation was conserved in humans.
Obviously, for RA inhibition to have any effect on endocrine differentiation a local source of RA synthesis in stage 4 cultured hESCs needs to be present. Previous reports have demonstrated that pancreatic multi-potent progenitors express Aldh1 in E12.5 mouse embryos (Rovira et al., 2010). Therefore, we hypothesized that stage 4 cultured hESCs also produce their own RA. To test this hypothesis we detected the percentage of cells with ALDH-enzymatic activity in stage 4 cultured hESCs using the Aldefluor Kit (StemCell Technologies). This kit allows the detection of endogenous ALDH activity by fluorescently labelling the ALDH active cells, which can then be quantified by flow cytometry (see Supplemental Materials and Methods for details). A negative control of cells co-treated with DEAB to inhibit ALDH activity is used to obtain the level of background fluorescence. As shown in Fig. 6A, 70.1% of stage 4 cells have ALDH activity, suggesting that a potential source of endogenous RA indeed exists. Maintaining levels of exogenous RA through stage 4 resulted in a lower proportion of cells with ALDH activity (56.2%, Fig. 6B), consistent with a negative feedback mechanism regulating ALDH1 positive cell numbers.
Fig. 6. Endogenous RA impedes endocrine cell differentiation from hESC-derived pancreas cells.

(A) Flow cytometric analysis of endogenous ALDH activity in stage 4 hESCs. Cells were incubated with Aldefluor reagent in the absence (black line) or presence (grey line) of the ALDH inhibitor (DEAB), then washed before being analyzed for fluorescence and quantified by flow cytometry. Graph shows distribution of cells (% of Max) against relative fluorescent intensity. % of Max is calculated from (the number of cells in an event)/(the number of cells in the maximum event). (B) Flow cytometric analysis of ALDH activity in stage 4 hESCs with additional exogenous RA. (C) Quantitative RT-PCR to detect expression of hormone genes. During stage 4 of hESC differentiation, 1 μM DEAB was added to inhibit the endogenous synthesis of RA, while non-treated cells were used as a control (labeled as “Stage 4”). DEAB-treated samples (black bars) were normalized to the controls (grey bar). (D) Quantification of the number of hormone-producing endocrine cells (GCG+, or INS+ cells). Shown are numbers of cells in DEAB-treated samples normalized to the controls. *p<0.05, ***p<0.005. (E) Confocal images of the differentiated hESCs after immunostaining for GCG and INS. Scale Bar, 50 μm.
Next, we wanted to ask what the effects of inhibiting RA synthesis are on the production of hormone-positive endocrine cells. We compared samples of stage 4 cells cultured following the reported protocol (Rezania et al., 2012) (used as control) to samples in which 1 μM DEAB was added during stage 4. This experiment was repeated 4 times with 3 biological replicates for each condition (n=12). By using quantitative RT-PCR (see Supplemental Materials and Methods for details), we demonstrated that after DEAB treatment the levels of mRNAs encoding for endocrine hormones (such as insulin (INS), glucagon (GCG) and somatostatin (STT)) are significantly increased compared to controls, with fold increases of 6.27±0.69 SE, p<0.001; 6.16±0.79 SE, p<0.001 and 2.67±0.3 SE, p<0.05 respectively (Fig. 6C). We then quantified hESC-derived cells expressing INS or GCG cultured with or without 1 μM DEAB at the end of stage 4 to ascertain if the increase in transcripts corresponded to an increase in endocrine cell numbers. We observed a 1.86±0.12 SE fold increase (p<0.005) in the number of INS positive cells following DEAB treatment compared to cells cultured following the normal stage 4 protocol (Fig. 6D and E). We also quantified the number of polyhormone-producing cells in the presence and absence of DEAB, but did not detect a significant difference (data not shown). These results demonstrate that inhibition of endogenous RA signaling during stage 4 of hESC differentiation significantly increases the number of endocrine cells, (especially INS positive cells). In conclusion, our results with differentiating hESCs are consistent with our findings in the larval zebrafish pancreas, and demonstrate a novel and conserved function for RA signaling in regulating endocrine differentiation.
Discussion
Retinoic acid signaling is necessary for the formation of pancreatic endoderm, as has been demonstrated by others in both animal models (Chen et al., 2004; Kinkel et al., 2009; Martin et al., 2005; Ostrom et al., 2008; Penny and Kramer, 2000; Stafford et al., 2004; Stafford and Prince, 2002; Stafford et al., 2006; Tulachan et al., 2003) and in hESC in vitro differentiation (D’Amour et al., 2006; Jiang et al., 2007; Johannesson et al., 2009; Kroon et al., 2008; Rezania et al., 2012; Rezania et al., 2013; Zhang et al., 2009). In zebrafish embryos, RA is synthesized in the anterior trunk mesoderm (8–13 hpf) and patterns the overlaying RAR-expressing endoderm to become the pancreatic anlagen (Stafford and Prince, 2002). As in other models, RA promotes the formation of pancreas in early development and thus ultimately the generation of insulin producing β cells (Kinkel and Prince, 2009). Conversely, impeding RA signaling early in development, either by pharmacological inhibition (DEAB) (Alexa et al., 2009; Martin et al., 2005), mutations in aldh1a2 (Alexa et al., 2009; Martin et al., 2005), or by overexpression of dnRAR, results in the loss of all pancreatic markers. All these previous studies were focused on early embryonic events (around 12 hpf) during stages when the pancreatic field is specified from nascent endoderm. In this report, we have presented new evidence that later on in development (3–5dpf) RA signaling negatively regulates the differentiation of PNCs during organogenesis and expands upon the developmental mechanisms by which this function is realized. Using our new QF/QUAS based RA reporter, we were able to detect RA signaling in PNCs. We also showed that RA signaling opposes PNC differentiation and endocrine cell formation in a cell-autonomous manner. We conclude, therefore, that RA has a biphasic function in regulating the formation of endocrine cells in the larval pancreas (Fig. 7): 1) RA is required for pancreatic field specification during the early stages of pancreas development; and 2) RA negatively regulates PNC differentiation at the secondary transition of larval pancreas.
Fig. 7. Proposed model of the biphasic function of RA signaling in the development of the pancreas.
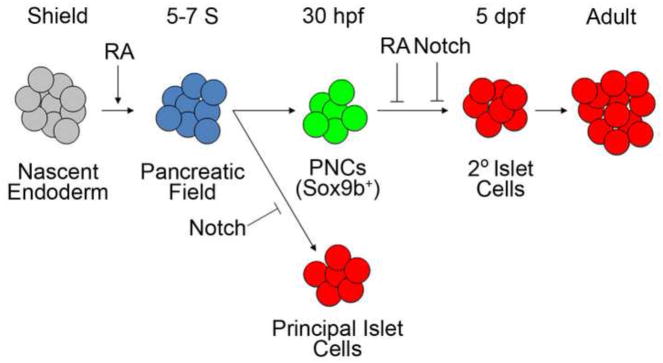
The pancreatic field is specified from the nascent endoderm by RA at very early stages of development (5–7 Somites (S)). Exogenous RA treatment expands the pancreatic field. In the dorsal bud, Notch signaling inhibits the formation of endocrine cells in the primary transition to form the principal islet. The ducts contain pancreatic Notch-responsive cells (PNCs), which express Sox9b. PNCs are larval progenitors contributing to the formation of 2° islets, which form the majority of endocrine tissue in adult pancreas. A subpopulation of these pancreatic progenitors also respond to RA signaling. Inhibition of RA signaling leads to precocious formation of the 2° islets. RA and Notch signaling synergistically regulate the differentiation of pancreatic progenitors.
Besides RA signaling, several other pathways have been shown to be essential for pancreas development (Kimmel and Meyer, 2010; Serup, 2012). Notch signaling is one such pathway, and it is known to negatively regulate the differentiation of pancreatic progenitors. In this present study, we demonstrated that the Notch- and RA-signaling pathways act in a manner consistent with synergy and regulate PNC differentiation, suggesting that there is a common downstream target shared by both cascades. This interaction needs further investigation; however, some clues to potential mechanisms can be inferred from the published research of others. Müller et al were interested in how RA blocked proliferation in a breast cancer cell line, and their discoveries demonstrated that RA up-regulated expression of the transcription factor SOX9 (Muller et al., 2010). It is known that Notch signaling directly regulates Sox9 expression in pancreatic progenitors, both in mouse (Sox9) (Shih et al., 2012) and in zebrafish (sox9b) (Delous et al., 2012). Furthermore, Sox9 not only marks pancreatic progenitors, but also is essential in regulating progenitor differentiation in both mice and zebrafish (Lynn et al., 2007; Seymour et al., 2007). Thus, Sox9 represents a promising candidate as a downstream target of both Notch- and RA-signaling pathways in pancreatic progenitors. Future work will test whether the action of RA signaling regulates PNC differentiation in a Sox9b dependent manner.
Following from our zebrafish research, we wanted to determine if the function of RA in the later stages of pancreas development was conserved in humans. To test the effects of RA signaling on human pancreas development, we utilized the in vitro differentiation of hESCs to pancreatic endocrine hormone-producing cells. These pluripotent stem cells can be induced to differentiate along different development programs and produce a wide variety of cell types (Thomson et al., 1998). We selected the Kieffer laboratory protocol for its efficiency and reproducibility on different commercially available hESC lines (Rezania et al., 2012). In this protocol, as with specification of the pancreatic anlage in the embryo, RA is required in stage 3 to drive differentiation of hESC-derived cells with characteristics of gut-tube endoderm to cells with characteristics reminiscent of posterior foregut (D’Amour et al., 2006). In the last step of this protocol during the differentiation of cells with pancreas progenitor characteristics to endocrine cell fates (stage 4), exogenous RA is not added to the culture media. Instead, we showed that during stage 4, hESC derived cells are capable of synthesizing RA themselves. The majority of stage 4 cells possess ALDH enzymatic activity and can utilize the RA synthesis substrate retinol (vitamin A) found in the B-27 serum supplement used in the media.
In this present study, we did not address if cells reminiscent of PNCs exist during the hESC differentiation protocol or if human equivalent cells give rise in vitro to hormone-positive endocrine cells. However, others have clearly shown that as hESCs differentiate, numerous cells express the endocrine precursor and PNC marker, NKX6.1. These NKX6.1 positive cells form before the appearance of hormone-expressing endocrine cells (D’Amour et al., 2006; Rezania et al., 2013). This observation suggests a common differentiation progression in the developing pancreatic endoderm of both zebrafish larvae and differentiating hESCs. Of potential therapeutic importance, we showed that inhibition of RA synthesis by DEAB treatment during stage 4 of hESC differentiation significantly enhances hormone-producing cell production. Our results with this in vitro model of human pancreas development demonstrate that, as in larval zebrafish, inhibition of RA signaling promotes the differentiation of endocrine cells from pancreatic progenitors.
When stage 3 levels of exogenous RA were maintained at stage 4 of hESC differentiation, the proportion of cells with ALDH activity was significantly reduced (Fig. 6B). The fact that elevated RA levels lead to fewer cells capable of making RA implies that a feedback mechanism is operating to regulate the production of ALDH expressing cells. This observation is reminiscent of Aldh1-expressing cells in the juvenile zebrafish pancreas. As the larval pancreas matures (after 15 dpf), Aldh1-expressing cells start to form from ductal epithelial cells (Matsuda, et al, 2013). These cells are a likely new source of RA. Inhibition of Aldh1 enzymatic activity with DEAB increases the number of these Aldh1 positive cells (Matsuda, et al, 2013). This result also supports the hypothesis that RA levels regulate numbers of Aldh positive cells in the pancreas.
To date there is no in vitro protocol that can produce mature, glucose-sensitive β cells in a manner pure enough to be safe for transplantation into diabetic humans. Experiments in mice, however, have demonstrated that mature, glucose-responsive β cells can be formed in vivo using transplantation of hESC-derived cells consisting of endocrine precursors and poly-hormonal endocrine cells (Kroon et al., 2008). Such transplantations into diabetic mice can even alleviate hyperglycemia, but such in vivo differentiation requires a significant period of time before normoglycemia is reached (Bruin et al., 2013; Rezania et al., 2012). Any additional modifications to current protocols that ultimately enrich the yield of β cells or reduce heterogeneity of the final cell population would be advantageous. Methods to differentiate hESCs to a pancreatic fate were developed by understanding signaling events during embryogenesis. These protocols can be further improved by applying what can be learned from later signaling events during organogenesis, such as those described here and elsewhere.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Retinoic acid and Notch cooperatively regulate pancreatic Notch-responsive cells.
These cells directly produce new endocrine cells upon inhibition of signaling.
Retinoic acid signaling works cell-autonomously in PNCs.
The role of retinoic acid signaling in endocrine progenitors is conserved.
These results reveal new developmental mechanisms for retinoic acid in the pancreas.
Acknowledgments
We thank Dr. V. Prince (University of Chicago, IL) for Neckless (nls) mutant fish and Dr. Ken Poss (Duke University, NC) for plasmids. We are grateful to SueJeanne Koh and Dr. Steven D. Leach for critical reading of this manuscript. This work was supported by the Juvenile Diabetes Research Foundation (17-2012-408), the JHU-UMD Diabetes Research Center, and the NIH (R01DK080730, R01HD058530). F.D. was supported by an MSCRF 2013 Postdoctoral Fellowship. R. L. B. was supported by an NIH Ruth L. Kirschstein NRSA (1F32DK101289). This work is dedicated to the memory of Larysa H. Pevny.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexa K, Choe SK, Hirsch N, Etheridge L, Laver E, Sagerstrom CG. Maternal and zygotic aldh1a2 activity is required for pancreas development in zebrafish. PloS one. 2009;4:e8261. doi: 10.1371/journal.pone.0008261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- Begemann G, Schilling TF, Rauch GJ, Geisler R, Ingham PW. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development. 2001;128:3081–3094. doi: 10.1242/dev.128.16.3081. [DOI] [PubMed] [Google Scholar]
- Biemar F, Argenton F, Schmidtke R, Epperlein S, Peers B, Driever W. Pancreas development in zebrafish: early dispersed appearance of endocrine hormone expressing cells and their convergence to form the definitive islet. Dev Biol. 2001;230:189–203. doi: 10.1006/dbio.2000.0103. [DOI] [PubMed] [Google Scholar]
- Bruin JE, Rezania A, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia. 2013;56:1987–1998. doi: 10.1007/s00125-013-2955-4. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pan FC, Brandes N, Afelik S, Solter M, Pieler T. Retinoic acid signaling is essential for pancreas development and promotes endocrine at the expense of exocrine cell differentiation in Xenopus. Dev Biol. 2004;271:144–160. doi: 10.1016/j.ydbio.2004.03.030. [DOI] [PubMed] [Google Scholar]
- Costa ML, Escaleira R, Manasfi M, de Souza LF, Mermelstein CS. Cytoskeletal and cellular adhesion proteins in zebrafish (Danio rerio) myogenesis. Braz J Med Biol Res. 2003;36:1117–1120. doi: 10.1590/s0100-879x2003000800019. [DOI] [PubMed] [Google Scholar]
- D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nature biotechnology. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Delous M, Yin C, Shin D, Ninov N, Debrito Carten J, Pan L, Ma TP, Farber SA, Moens CB, Stainier DY. Sox9b is a key regulator of pancreaticobiliary ductal system development. PLoS genetics. 2012;8:e1002754. doi: 10.1371/journal.pgen.1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delporte FM, Pasque V, Devos N, Manfroid I, Voz ML, Motte P, Biemar F, Martial JA, Peers B. Expression of zebrafish pax6b in pancreas is regulated by two enhancers containing highly conserved cis-elements bound by PDX1, PBX and PREP factors. BMC Dev Biol. 2008;8:53. doi: 10.1186/1471-213X-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong PD, Munson CA, Norton W, Crosnier C, Pan X, Gong Z, Neumann CJ, Stainier DY. Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nat Genet. 2007;39:397–402. doi: 10.1038/ng1961. [DOI] [PubMed] [Google Scholar]
- Dong PD, Provost E, Leach SD, Stainier DY. Graded levels of Ptf1a differentially regulate endocrine and exocrine fates in the developing pancreas. Genes Dev. 2008;22:1445–1450. doi: 10.1101/gad.1663208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donga E, van Dijk M, Hoogma RP, Corssmit EP, Romijn JA. Insulin resistance in multiple tissues in patients with type 1 diabetes mellitus on long-term continuous subcutaneous insulin infusion therapy. Diabetes/metabolism research and reviews. 2013;29:33–38. doi: 10.1002/dmrr.2343. [DOI] [PubMed] [Google Scholar]
- Esni F, Ghosh B, Biankin AV, Lin JW, Albert MA, Yu X, MacDonald RJ, Civin CI, Real FX, Pack MA, Ball DW, Leach SD. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131:4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- Hald J, Hjorth JP, German MS, Madsen OD, Serup P, Jensen J. Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev Biol. 2003;260:426–437. doi: 10.1016/s0012-1606(03)00326-9. [DOI] [PubMed] [Google Scholar]
- Hesselson D, Anderson RM, Beinat M, Stainier DY. Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc Natl Acad Sci U S A. 2009;106:14896–14901. doi: 10.1073/pnas.0906348106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J, Heller RS, Funder-Nielsen T, Pedersen EE, Lindsell C, Weinmaster G, Madsen OD, Serup P. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49:163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- Jiang W, Shi Y, Zhao D, Chen S, Yong J, Zhang J, Qing T, Sun X, Zhang P, Ding M, Li D, Deng H. In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell research. 2007;17:333–344. doi: 10.1038/cr.2007.28. [DOI] [PubMed] [Google Scholar]
- Johannesson M, Stahlberg A, Ameri J, Sand FW, Norrman K, Semb H. FGF4 and retinoic acid direct differentiation of hESCs into PDX1-expressing foregut endoderm in a time- and concentration-dependent manner. PloS one. 2009;4:e4794. doi: 10.1371/journal.pone.0004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol. 2004;77:201–222. doi: 10.1016/s0091-679x(04)77011-9. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 2011;20:397–404. doi: 10.1016/j.devcel.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel RA, Meyer D. Molecular regulation of pancreas development in zebrafish. Methods Cell Biol. 2010;100:261–280. doi: 10.1016/B978-0-12-384892-5.00010-4. [DOI] [PubMed] [Google Scholar]
- Kinkel MD, Prince VE. On the diabetic menu: zebrafish as a model for pancreas development and function. BioEssays: news and reviews in molecular, cellular and developmental biology. 2009;31:139–152. doi: 10.1002/bies.200800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkel MD, Sefton EM, Kikuchi Y, Mizoguchi T, Ward AB, Prince VE. Cyp26 enzymes function in endoderm to regulate pancreatic field size. Proc Natl Acad Sci U S A. 2009;106:7864–7869. doi: 10.1073/pnas.0813108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, Agulnick AD, D’Amour KA, Carpenter MK, Baetge EE. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Langston AW, Thompson JR, Gudas LJ. Retinoic acid-responsive enhancers located 3’ of the Hox A and Hox B homeobox gene clusters. Functional analysis. J Biol Chem. 1997;272:2167–2175. doi: 10.1074/jbc.272.4.2167. [DOI] [PubMed] [Google Scholar]
- Lin JW, Biankin AV, Horb ME, Ghosh B, Prasad NB, Yee NS, Pack MA, Leach SD. Differential requirement for ptf1a in endocrine and exocrine lineages of developing zebrafish pancreas. Dev Biol. 2004;274:491–503. doi: 10.1016/j.ydbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Lorent K, Yeo SY, Oda T, Chandrasekharappa S, Chitnis A, Matthews RP, Pack M. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131:5753–5766. doi: 10.1242/dev.01411. [DOI] [PubMed] [Google Scholar]
- Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci U S A. 2007;104:10500–10505. doi: 10.1073/pnas.0704054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Rydeen A, Anderson J, Sorrell MR, Zygmunt T, Torres-Vazquez J, Waxman JS. Transgenic retinoic acid sensor lines in zebrafish indicate regions of available embryonic retinoic acid. Developmental dynamics: an official publication of the American Association of Anatomists. 2013;242:989–1000. doi: 10.1002/dvdy.23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P, Dolle P, Gradwohl G. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol. 2005;284:399–411. doi: 10.1016/j.ydbio.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Matsuda H, Parsons MJ, Leach SD. Aldh1-expressing endocrine progenitor cells regulate secondary islet formation in larval zebrafish pancreas. PloS one. 2013;8:e74350. doi: 10.1371/journal.pone.0074350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development. 2011;138:169–177. doi: 10.1242/dev.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Crofts JD, Newman BS, Bridgewater LC, Lin CY, Gustafsson JA, Strom A. SOX9 mediates the retinoic acid-induced HES-1 gene expression in human breast cancer cells. Breast Cancer Res Treat. 2010;120:317–326. doi: 10.1007/s10549-009-0381-6. [DOI] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninov N, Borius M, Stainier DY. Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors. Development. 2012;139:1557–1567. doi: 10.1242/dev.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro MC, Keller G. Generation of beta cells from human pluripotent stem cells: Potential for regenerative medicine. Semin Cell Dev Biol. 2012;23:701–710. doi: 10.1016/j.semcdb.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obholzer N, Wolfson S, Trapani JG, Mo W, Nechiporuk A, Busch-Nentwich E, Seiler C, Sidi S, Sollner C, Duncan RN, Boehland A, Nicolson T. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci. 2008;28:2110–2118. doi: 10.1523/JNEUROSCI.5230-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom M, Loffler KA, Edfalk S, Selander L, Dahl U, Ricordi C, Jeon J, Correa-Medina M, Diez J, Edlund H. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PloS one. 2008;3:e2841. doi: 10.1371/journal.pone.0002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, Leach SD. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev. 2009;126:898–912. doi: 10.1016/j.mod.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny C, Kramer B. The effect of retinoic acid on the proportion of insulin cells in the developing chick pancreas. In Vitro Cell Dev Biol Anim. 2000;36:14–18. doi: 10.1290/1071-2690(2000)036<0014:TEORAO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Perz-Edwards A, Hardison NL, Linney E. Retinoic acid-mediated gene expression in transgenic reporter zebrafish. Dev Biol. 2001;229:89–101. doi: 10.1006/dbio.2000.9979. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Tasic B, Russler EV, Liang L, Luo L. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 2010;141:536–548. doi: 10.1016/j.cell.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, Gauvin R, Narayan K, Karanu F, O’Neil JJ, Ao Z, Warnock GL, Kieffer TJ. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Xu J, Narayan K, Fox JK, O’Neil JJ, Kieffer TJ. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem cells. 2013;31:2432–2442. doi: 10.1002/stem.1489. [DOI] [PubMed] [Google Scholar]
- Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139:843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- Robertson RP. Islet transplantation as a treatment for diabetes - a work in progress. The New England journal of medicine. 2004;350:694–705. doi: 10.1056/NEJMra032425. [DOI] [PubMed] [Google Scholar]
- Rovira M, Huang W, Yusuff S, Shim JS, Ferrante AA, Liu JO, Parsons MJ. Chemical screen identifies FDA-approved drugs and target pathways that induce precocious pancreatic endocrine differentiation. Proc Natl Acad Sci U S A. 2011;108:19264–19269. doi: 10.1073/pnas.1113081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serup P. Signaling pathways regulating murine pancreatic development. Semin Cell Dev Biol. 2012;23:663–672. doi: 10.1016/j.semcdb.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Kopp JL, Sandhu M, Dubois CL, Seymour PA, Grapin-Botton A, Sander M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford D, Hornbruch A, Mueller PR, Prince VE. A conserved role for retinoid signaling in vertebrate pancreas development. Dev Genes Evol. 2004;214:432–441. doi: 10.1007/s00427-004-0420-6. [DOI] [PubMed] [Google Scholar]
- Stafford D, Prince VE. Retinoic acid signaling is required for a critical early step in zebrafish pancreatic development. Curr Biol. 2002;12:1215–1220. doi: 10.1016/s0960-9822(02)00929-6. [DOI] [PubMed] [Google Scholar]
- Stafford D, White RJ, Kinkel MD, Linville A, Schilling TF, Prince VE. Retinoids signal directly to zebrafish endoderm to specify insulin-expressing beta-cells. Development. 2006;133:949–956. doi: 10.1242/dev.02263. [DOI] [PubMed] [Google Scholar]
- Stuart GW, Vielkind JR, McMurray JV, Westerfield M. Stable lines of transgenic zebrafish exhibit reproducible patterns of transgene expression. Development. 1990;109:577–584. doi: 10.1242/dev.109.3.577. [DOI] [PubMed] [Google Scholar]
- Subedi A, Macurak M, Gee ST, Monge E, Goll MG, Potter CJ, Parsons MJ, Halpern ME. Adoption of the Q transcriptional regulatory system for zebrafish transgenesis. Methods. 2013 doi: 10.1016/j.ymeth.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tiso N, Moro E, Argenton F. Zebrafish pancreas development. Molecular and cellular endocrinology. 2009;312:24–30. doi: 10.1016/j.mce.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Tulachan SS, Doi R, Kawaguchi Y, Tsuji S, Nakajima S, Masui T, Koizumi M, Toyoda E, Mori T, Ito D, Kami K, Fujimoto K, Imamura M. All-trans retinoic acid induces differentiation of ducts and endocrine cells by mesenchymal/epithelial interactions in embryonic pancreas. Diabetes. 2003;52:76–84. doi: 10.2337/diabetes.52.1.76. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rovira M, Yusuff S, Parsons MJ. Genetic inducible fate mapping in larval zebrafish reveals origins of adult insulin-producing beta-cells. Development. 2011;138:609–617. doi: 10.1242/dev.059097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman JS, Yelon D. Zebrafish retinoic acid receptors function as context-dependent transcriptional activators. Dev Biol. 2011;352:128–140. doi: 10.1016/j.ydbio.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. A guide for the laboratory use of zebrafish (Danio rerio) 4. University of Oregon Press; Eugene: 2000. The zebrafish book. [Google Scholar]
- Xu X, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, Fishman MC. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet. 2002;30:205–209. doi: 10.1038/ng816. [DOI] [PubMed] [Google Scholar]
- Zecchin E, Filippi A, Biemar F, Tiso N, Pauls S, Ellertsdottir E, Gnugge L, Bortolussi M, Driever W, Argenton F. Distinct delta and jagged genes control sequential segregation of pancreatic cell types from precursor pools in zebrafish. Dev Biol. 2007;301:192–204. doi: 10.1016/j.ydbio.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Zecchin E, Mavropoulos A, Devos N, Filippi A, Tiso N, Meyer D, Peers B, Bortolussi M, Argenton F. Evolutionary conserved role of ptf1a in the specification of exocrine pancreatic fates. Dev Biol. 2004;268:174–184. doi: 10.1016/j.ydbio.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell research. 2009;19:429–438. doi: 10.1038/cr.2009.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.