

Abstract
Polyomavirus JC (JCV) is the causative agent of progressive multifocal leukoencephalopathy (PML), a rare and frequently fatal brain disease that afflicts a small fraction of the immune-compromised population, including those affected by AIDS and transplantation recipients on immunosuppressive drug therapy. Currently there is no specific therapy for PML. The major capsid viral protein 1 (VP1) involved in binding to sialic acid cell receptors is believed to be a key player in pathogenesis. PML-specific mutations in JCV VP1 sequences present at the binding pocket of sialic acid cell receptors, such as L55F and S269F, abolish sialic acid recognition and might favor PML onset. Early diagnosis of these PML-specific mutations may help identify patients at high risk of PML, thus reducing the risks associated with immunosuppressive therapy. As a first step in the development of such early diagnostic tools, we report identification and characterization of affinity reagents that specifically recognize PML-specific mutations in VP1 variants using phage display technology. We first identified 2 peptides targeting wild type VP1 with moderate specificity. Fine-tuning via selection of biased libraries designed based on 2 parental peptides yielded peptides with different, yet still moderate, bindinspecificities. In contrast, we had great success in identifying synthetic antibodies that recognize one of the PML-specific mutations (L55F) with high specificity from the phage-displayed libraries. These peptides and synthetic antibodies represent potential candidates for developing tailored immune-based assays for PML risk stratification in addition to complementing affinity reagents currently available for the study of PML and JCV.
Keywords: phage display, synthetic antibody, protein engineering, JC virus, virus-like particle
Abbreviations
- JCV
polyomavirus JC
- PML
progressive multifocal leukoencephalopathy
- VP1
major capsid viral protein 1
- PCR
polymerase chain reaction
- CSF
cerebrospinal fluid
- L55F
Leu to Phe mutation at position 55
- S269F
Ser to Phe mutation at position 269
- VLP
virus-like particle
- WT
type 3 wild type JCV VP1
- D66H
Asp to His mutation at position 66
- ELISA
enzyme linked immunosorbent assay
- CDR
complementarity determining region
- P8
M13 major coat protein
- BSA
bovine serum albumin
- PBS
phosphate-buffered saline
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- HRP
horseradish peroxidase
- TMB
3,3',5,5'-tetramethylbenzidine
- DHFR
dihydrofolate reductase
Introduction
Progressive multifocal leukoencephalopathy (PML) is a rare but frequently fatal brain disease caused by polyomavirus JC (JCV) infection. JCV is a highly prevalent human pathogen that establishes a persistent, mostly asymptomatic infection in a significant fraction of the human population.1 PML onset, however, usually occurs in only a small fraction of the infected population, in particular in those with an impaired immune system, such as HIV patients and patients receiving immunosuppressive drug therapy.2,3 The mechanism leading to PML is believed to include lethal infection of glia in the brain of the afflicted individual leading to severe demyelination and brain necrosis. Concerns regarding PML became more prominent when it was recognized that an increased number of PML cases occur in patients treated for autoimmune diseases with certain immunoregulatory agents, including natalizumab, rituximab, efalizumab and mycophenolate mofetil.4,5 In the absence of immune response restoration, PML is usually fatal within one year of symptom onset. There is currently no specific therapy for PML, and the primary treatment approaches rely on reconstitution of the patient's own immune response.6,7
Due to the limited treatment options, there is substantial clinical interest in developing diagnostic tools that could identify patients at higher risk for the disease to help reduce the risks of PML associated with immunomodulatory therapy.8 Early attempts have been made to measure the presence of JCV DNA in bodily fluids and compartments to stratify patients for PML risk or to monitor PML development. For example, by analyzing the JCV DNA load in patients with or without PML via polymerase chain reaction (PCR), Koralnik et al. demonstrated that the presence of JCV in the cerebrospinal fluid (CSF), but not in the blood or urine, correlated well with PML, suggesting that JCV quantification in CSF may be a possible means to monitor clinical PML treatment trials.9 Likewise, Rudick et al. reported that measuring JCV DNA in blood or urine by quantitative PCR had no clinical utility to predict the risk of PML.10 Gorelik et al. developed a 2-step assay for JCV antibodies in serum as a potential tool for stratifying multiple sclerosis patients for the risk of developing PML.4 Using this method, all 17 of the pre-PML samples collected 16 to 180 months before symptoms developed tested positive, demonstrating the potential usefulness of immuno-based assays in prognosis of PML.
Studies on JCV structure and mechanism of viral entry provide valuable insights into potential PML-specific biomarkers critical for risk stratification of PML development.5,11 As with other members of the polyomaviridae family, a key component of JCV pathogenesis is the major capsid viral protein 1 (VP1). VP1 plays multiple critical roles in JCV infection by mediating cell attachment and viral cell entry via interaction with sialic acid-containing cell receptors.3,11-13 By a comprehensive analysis of JCV VP1 sequences isolated from both PML patients and healthy individuals using statistical method of molecular evolution, Sunyaev et al. demonstrated that at least a subset of PML-specific mutations in VP1 sequences were acquired via adaptive evolution, of which no more than one such mutation was observed in the same JCV isolate.7 Three-dimensional modeling of the VP1 molecular structure further indicated that these substitutions are located at or close to the sialic acid binding site,7 which was later confirmed by the crystal structure of JCV VP1 complexed with a linear sialylated pentasaccharide (Figure S1).5 The most common mutations in PML CSF-derived VP1 sequences are a Leu to Phe mutation at position 55 (L55F) and a Ser to Phe mutation at position 269 (S269F). These residues are part of the sialic acid binding site (Figure S1),5 and these 2 mutations together account for half of the large group of PML patients under study.3,14 The mutations in JCV VP1 caused a loss or dramatic change in its receptor-binding specificity, which might favor JCV dissemination through abrogation of its binding to sialo-containing oligosaccharides expressed on a great majority of peripheral cells while maintaining other entry pathways independent of sialic acid.3 Notably, JCV pseudoviruses with these PML-associated mutations were shown to be noninfectious in cell culture systems.15 However, more recently, an in vivo study demonstrated that these PML-associated mutants, including L55F, are infectious and capable of causing viral spread and disease in a chimeric mouse model.16
Successful identification of PML-specific mutations in JCV VP1 sequences suggested a promising way to stratify patients for high or low risk of PML development. One way to address this is to develop affinity reagents that specifically recognize these PML-specific mutations in VP1 variants. These reagents could potentially be applied in an immunoassay to predict the likelihood of PML development with improved accuracy, and they could also be powerful tools for the study of PML caused by PML-associated mutants. In comparison with traditional hybridoma methods, phage display and other in vitro display technologies have proven advantages for the development of such reagents.17 By selection under controlled conditions, reagents with particular specificities can be obtained. Two recent examples are the identification of antibodies specifically recognizing different conformational states of caspase-1 using phage display18 and identification of antibodies recognizing histone post-translational modifications using yeast and phage display.19 Here, we report the identification and characterization of peptides and antibodies specifically targeting point mutations in different JCV VP1 variants by phage display. These peptides and synthetic antibodies complement current JCV VP1 binding reagents and are potential candidates for developing assays for PML risk stratification.
Results
Selection and optimization of peptides binding to JCV virus-like particles
We used phage display to identify peptide ligands for type 3 wild type JCV VP1 (WT) (Genbank: AAQ88264) and 2 point mutation variants (L55F and D66H), using as antigens the corresponding virus-like particles (VLPs), produced by using a baculovirus expression system in insect cells.7 VLPs resemble authentic viruses that present the viral structural proteins assembled in a particle mimicking the coat of authentic viruses, but VLPs are non-infectious because they do not contain any core viral genetic material. Hence, VLPs are often used as delivery vessels for genes or other therapeutics, and as powerful tools for vaccine development.20 D66H contains an Asp to His mutation at position 66, which, like L55F, is located at the sialic acid-binding site on JCV VP1. A highly diverse naïve peptide library X1221 containing over 1010 random dodecameric peptides was utilized to select parental binding clones. Subsequently, we constructed biased libraries based on the peptides identified in initial selections to enhance binding specificities and affinities. The binding specificity of each peptide was tested by use of phage enzyme linked immunosorbent assays (ELISA) with the 3 antigens under study.
Following four rounds of selection for binding to immobilized WT, we obtained 2 unique peptide-phage clones, Pep-1 and Pep-2 (Fig. 1A) that preferentially bound with moderate specificity for WT versus the 2 variants L55F and D66H. As these 2 mutations are located on the surface of VP1 and do not likely change the overall conformation of the protein dramatically, the observed moderate specificity for WT indicates that both peptides likely bind close to the pocket where sialic acid binds to JCV VP1.
Figure 1.

Anti-VLP peptides selected from phage-displayed peptide libraries. (A) The sequences of unique clones, denoted by the single-letter amino acid code, selected against WT, D66H or L55F are listed separately. The parental clones, Pep-1 and Pep-2, isolated against WT, are listed at the top. Cysteine residues are shaded yellow. The relative binding strength of each clone to different VLPs was qualified according to their phage ELISA signals against different VLPs, −: <0.5; +: 0.5–1; ++: 1–1.5; +++: 1.5–2; ++++: 2–2.5; +++++: >2.5. (B) Binding of peptide-Fc fusion proteins against a panel of VLP variants. ELISA signals are presented as mean values ± SD from 2 independent measurements.
We next constructed 2 biased phage-displayed peptide libraries designed to facilitate specificity tuning of the parental anti-VLP peptides. Except for the cysteine residues, which were held constant, we randomized the codons at each position using a 70/10/10/10 nucleotide mix that on average allows for 50% of wild type amino acid and 50% of other amino acids. In addition, we added 2 extra residues at both N- and C-termini of the peptides in order to improve the binding contributions of both terminal regions. These positions were completely randomized with degenerate codons that encode for all 20 amino acids.
Following four rounds of selection for binding to immobilized VLPs, 15 clones from selections against each of the 3 VLPs were sequenced and the clones that showed >1.5-fold binding specificity against any single VLP compared to the other 2 were selected (Fig. 1A). Interestingly, derivatives of Pep-1 and Pep-2 showed distinct binding preferences: Pep-1 derivatives preferably bound to WT and L55F whereas Pep-2 derivatives preferred D66H and L55F.
We expressed these peptides as Fc fusion proteins and tested their binding to a broader panel of VLPs containing single point mutations (including K60E, D66H, N265D, S269F, S269Y and Q271H). As shown in Fig. 1B, the peptide-Fc fusions recognized different VLP variants with different levels of selectivity. Clone A06 preferentially bound to D66H with a >3-fold difference in ELISA signal intensity. In comparison, the other peptide-Fc fusions did not show selectivity for a single point mutant, binding to at least 2 VLP variants to comparable degrees. Moreover, the binding preferences observed using the phage ELISA did not well match those observed using the protein ELISA, the most striking difference being the poor binding of A06 to D66H in phage ELISA vs. the preferential binding of A06 to D66H in protein ELISA. While the cause of this discrepancy is not clear, we reason that it is most likely due to the complexity introduced by phage particles in peptide-displayed phage ELISA as fusion to the large phage particle may change the behavior of the small peptide.
Selection of anti-L55F parental antibody
We first performed selections for antibodies targeting different VLPs (WT, D66H and L55F) using a naïve, phage-displayed synthetic Fab library (library F)22 without performing negative selection with non-cognate VLPs. Using this method, we did not obtain any phage clones that specifically targeted any of the 3 VLPs. We then focused on using more complex selection strategies to develop antibodies specific for one of the VLP variants, L55F.
In the first round of selection, in order to obtain Fabs that specifically bind to L55F, negative selection to deplete cross-reactive antibodies was performed by incubating the phage library in WT-immobilized wells. In successive rounds of selection, competitor WT was added in solution to trap Fab-phage capable of binding to both WT and L55F, keeping L55F-specific clones free for capture by immobilized L55F. Following five rounds of selection, one unique clone, GC058, was identified that preferentially bound to L55F over WT, as evidenced by phage ELISA (Fig. 2).
Figure 2.
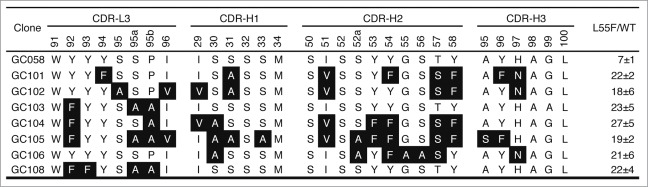
CDR sequences of anti-L55F antibodies. CDR-L3 and the 3 heavy chain CDRs are shown, as CDR-L1 and CDR-L2 were not diversified in the library design. The numbering is according to the nomenclature of Kabat et al.39 Residues in specificity-tuned clones that are different from the parental clone GC058 are shown in white with black background. The signal ratios of Fab-phage bound to L55F vs. WT in phage ELISA (L55/WT) were determined as the mean values from 3 independent measurements.
Specificity tuning of anti-L55F antibodies
Next, we applied a homolog-scan strategy to investigate the functional contributions of individual side chains involved in antigen–antibody interactions in the complementarity-determining regions (CDRs) of GC058. Homolog-scan combinatorial mutagenesis introduces subtle mutations in the CDR sequences of the parental antibody, providing the potential for fine-tuning the existing interactions at the antigen-antibody interface. As a result, antibodies with higher binding affinities or specificities could potentially be obtained under conditions with controlled stringency during selection.
In library F, from which GC058 was isolated, diversity was only introduced into the 3 heavy chain CDRs and light chain CDR3.22 Thus, for the homolog-scan analysis we constructed a single library that targeted a total of 30 residues in these 4 CDRs (Fig. 2). We replaced the wild type codon in each scanned position with a binary codon that encodes only the wild type and a similar amino acid, as described previously.23 The library contained 5.8 × 109 unique members, which exceeded the number of all possible theoretical combinations encoded by the binary degenerate codons (1.1 × 109) by approximately five-fold, thus providing good sampling of the theoretical diversity.
Phage pools from the homolog-scan library were subjected to 2 parallel selections. Display selection against the anti-FLAG antibody M2 was performed to quantify display biases, since a FLAG tag was fused to the C terminus of the phage-displayed Fab light chain. Statistical analysis of the clones identified from this selection can reveal amino acid preferences at each position for Fab display on phage particles. Following four rounds of selection for binding to anti-FLAG antibody M2, 40 unique binding clones were sequenced. Functional selection was performed to isolate GC058 variants capable of binding to L55F with higher specificity. Following four rounds of selection for binding to L55F, 96 clones from round 3 and 96 clones from round 4 were subjected to phage ELISA to determine the binding specificity of individual clones. Phage ELISAs indicated that 41 clones specifically bound to L55F over WT, with L55F/WT signal ratios >5, and sequencing of these clones revealed 39 unique sequences. The sequences were aligned, and the relative occurrences of the wild type or corresponding mutated residue at each position were calculated for both selections (Fig. 3). Most of the wild type occurrence percentages for the display selection were close to 50%, indicating that the homologous mutations did not significantly affect GC058 display levels (Fig. 3B). At two positions (lW91 and hL100), mutations occurred at a frequency of greater than 70%, indicating that these mutations may increase display. Conversely, wild type residues were preferred at a few positions (lP95b, hS50, hG55 and hG99), indicating that mutations at these positions may decrease display or negatively affect antibody stability.
Figure 3.
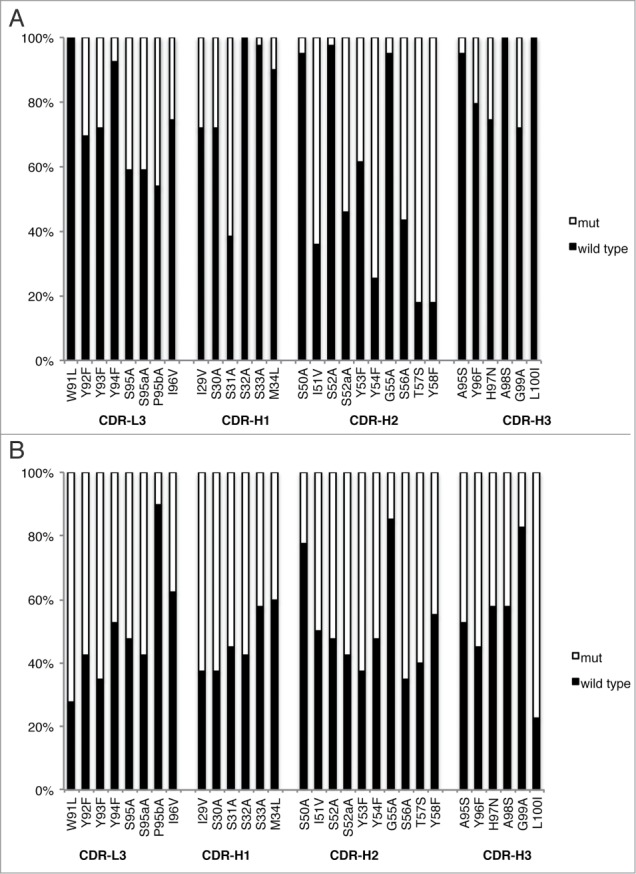
Positional amino acid preferences in Fab variants from homolog scanning of GC058 for functional selection against L55F (A) or display selection against anti-FLAG antibody M2 (B). For each position, the percentage of the population is shown for the wild type or its homolog residue. The wild type GC058 residue is indicated prior to position number, and its homolog residue following the number. The data were compiled from 39 or 40 unique sequences from the functional selection or the display selection, respectively.
As expected, wild type residues were preferred at most positions for the functional selection. At many positions (lW91, lY94, hS32, hS33, hM34, hS50, hS52, hG55, hA95, hA98 and hL100) wild type residues were >95% conserved, suggesting that these residues may be involved in antigen binding or may be critical for maintaining the structural integrity of the antigen-binding site. However, mutations were moderately preferred over wild type at several heavy chain positions (hY54, hT57 and hY58), suggesting that mutations at these sites enhance antigen binding.
In phage ELISAs, most clones exhibited significantly higher L55F/WT binding ratios, indicating that the selection process had been successful in yielding antibodies with enhanced specificity for L55F VLP relative to WT VLP (Fig. 2).
Anti-L55F antibodies are highly specific
The eight most selective clones from the phage ELISAs were expressed as Fab proteins in E. coli, and were purified as described.24 One of the Fab proteins proved to be unstable and was not analyzed further. We tested the direct binding of the purified Fabs to L55F and WT VLPs by ELISA. All Fabs showed high binding specificities for L55F over WT and, as expected, the specificity-tuned Fabs all exhibited improved specificity compared with the parent GC058 (Fig. 4). Fab GC058 and the specificity-tuned Fab GC103 bound to L55F with EC50 values of 26 or 12 nM, respectively. Both bound poorly to WT, with signals only slightly above background even at 200 nM. Fab GC101 bound to L55F with an EC50 value of 26 nM, and the others bound with EC50 values ranging from 50–108 nM; no WT binding was detected up to 200 nM for any of these 6 Fabs.
Figure 4.
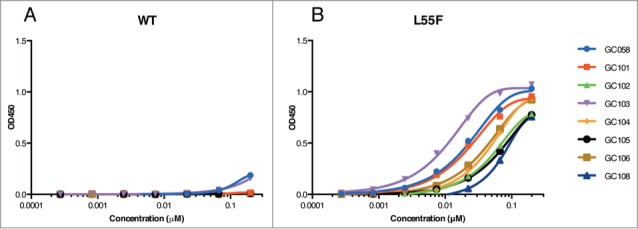
Detection of WT and L55F VLPs by Fabs. ELISAs were performed with serial dilutions of Fab proteins using plates coated with 1 μg/ml of WT VLPs (A) or L55F VLPs (B). Data from L55F-binding ELISAs were fit to a 4-parameter logistic equation.
We then tested the cross-reactivity of the anti-L55F Fabs to a large panel of VLP variants derived from different JCV strains. This panel was composed of mutant VLPs (L55F, K60E, D66H, N265D, S267F and S269F) on the type 3 background, as well as additional variants that belong to different JCV types other than type 3. As shown in Figure 5, some cross-reactivity was observed for GC058 and GC103 toward VLP variants Mad1, 2a, 1b, S267F and S269F, and very slight cross-reactivity was observed with VLP variant Mad1 for GC101, but these Fabs bound to L55F more strongly than to other VLP variants. In contrast, none of the other specificity-tuned Fabs bound to VLP variants other than L55F at the concentrations tested, indicating their high specificity for L55F. Thus, compared with the parent Fab GC058, most specificity-tuned Fabs showed enhanced binding specificity to L55F. Interestingly, the Fabs that showed detectable cross-reactivity to other VLP variants bound to L55F with at least 2-fold higher L55F-binding affinity compared with other Fabs (Fig. 4). This result, combined with the EC50 values shown above, demonstrates that for these specificity-tuned Fabs, increases in L55F-binding specificity came at the cost of decreased antigen binding affinity.
Figure 5.
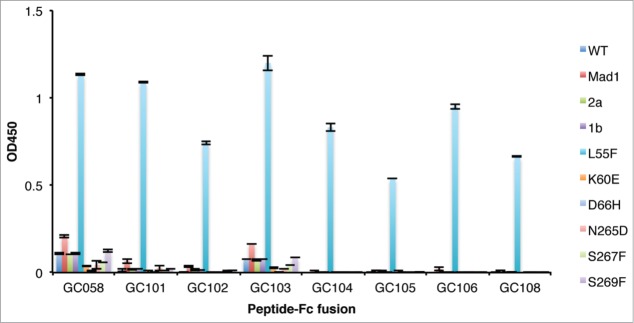
Detection of VLP variants by Fabs. ELISAs were performed with 10 μg/ml of Fab proteins using plates coated with 1 μg/ml of the indicated VLPs. ELISA signals are presented as mean values ± SD from 2 independent measurements.
Based on these results, 5 clones were converted into full-length human IgG1 antibodies and tested for specificity by ELISA. As expected, similar specificity for L55F to the corresponding Fabs was observed for these full-length antibodies (Fig. 6). IgG106 (IgG form of GC106) showed the highest L55F binding response among the 5 IgGs with an EC50 value of 0.27 nM, whereas IgG104 and IgG108 showed the highest L55F selectivity, with no detectable D66H binding and low WT binding slightly above background at 6.7 nM. IgG103 and IgG106 showed weak binding signals to WT and D66H at high concentrations, but their L55F binding reached saturation at a much lower concentration (˜1.33 nM), demonstrating that these antibodies are also specific for L55F. In comparison, the non-specific JCV VLP antibody H0L0, a humanized antibody derived by rabbit immunization against L55F (Biogen Idec, unpublished data), did not differentiate between WT and L55F (Fig. 6). Notably, even the highest affinity discriminatory antibody IgG106 was of lower affinity than the cross-reactive antibody H0L0, demonstrating that the in vitro negative selection process favors clones with desirable binding specificities but lower affinities.
Figure 6.
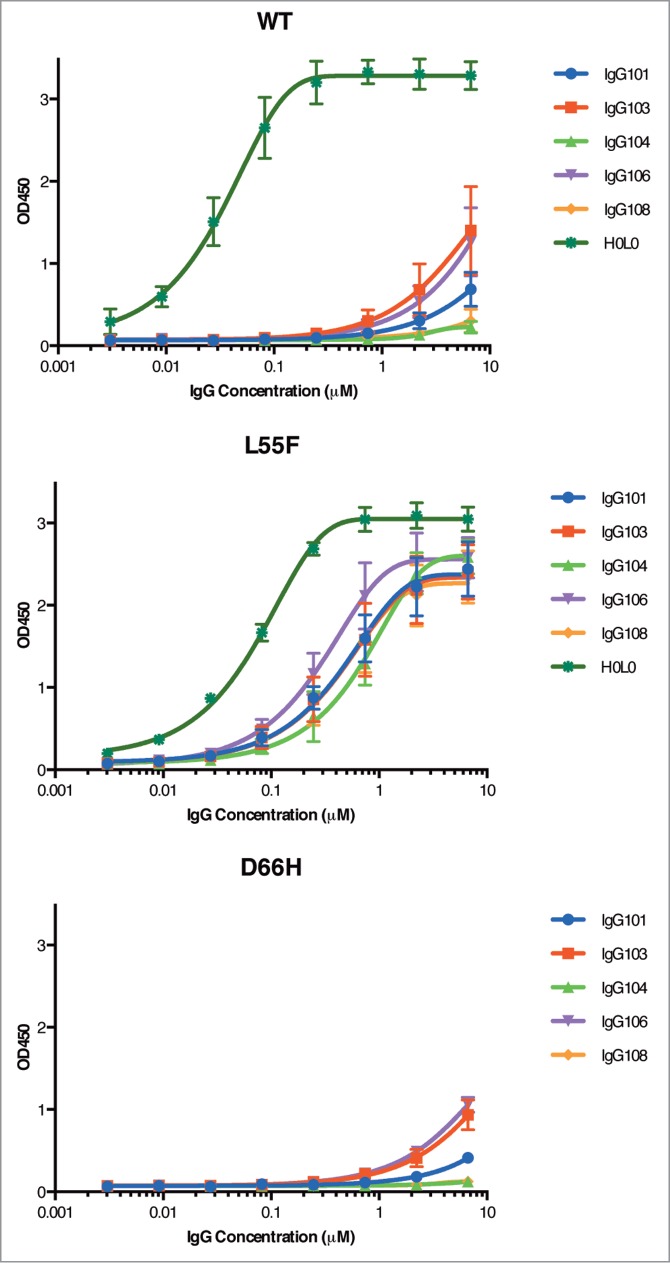
Detection of VLPs by IgGs. ELISAs were performed with serial dilutions of IgG proteins using plates coated with 1 μg/ml of WT VLPs (A), L55F VLPs (B) or D66H VLPs (C). ELISA signals are presented as mean values ± SD from 3 independent measurements.
Discussion
In this study, type 3 JCV VP1 was utilized as part of the continued study based on the research described previously.7 Although type 1 and 2 are more commonly observed in PML patients, whether or not specific JCV genotypes are preferentially associated with PML is still an open question.3,14 Thus, while it would certainly be useful to investigate mutants in the more common genetic backgrounds type 1 or 2, use of the less common type 3 does not affect the validity of our results.
We used phage display to identify and characterize peptides and synthetic antibodies specifically targeting JCV VLP variants. These point mutations occur at positions that are among approximately a dozen residues located at the binding site for putative sialic acid containing receptors and are present exclusively in PML-specific JCV VP1.7 Accurate early detection of these PML-specific mutations using specific reagents might help PML risk stratification of patients receiving immunomodulatory therapies. Phage display is well suited for deriving such highly specific binding reagents, since selections are performed under controlled, in vitro conditions that favor the enrichment of clones with desired properties.
Binding selections with a phage-displayed dodecameric peptide library yielded 2 peptide ligands that recognize WT with moderate specificity. Notably, both clones contain 2 cysteine residues spaced either 6 or 10 residues apart. It is likely that these cysteine residues lead to cyclization of the peptide via intramolecular disulfide bonds, and that the resultant cyclic peptide presents a structure suitable for VLP binding, a common observation for cysteine-containing peptidic ligands.25,26 Specificity tuning of these 2 parental peptides via phage selection yielded 2 or 3 peptides targeting each of the 3 VLPs with moderate specificities. The peptide A06, when expressed as a C-terminal Fc fusion, showed the highest binding specificity for D66H versus a panel of other VLP variants with at least three-fold difference in ELISA signals. This showed that peptide ligands could potentially be utilized to recognize subtle differences in protein structure, such as solvent accessible point mutations in the case herein. Therefore it is reasonable to postulate that this approach might be expanded to generate peptides targeting post-translational modifications and protein allosteric sites, where polyclonal and monoclonal antibodies are currently dominantly applied. Although relatively modest specificity was obtained from selection against VLP variants, the peptides we identified through phage library screenings could potentially be used as lead compounds to develop alternatives to antibodies for differentiating fine structural differences in JCV VP1.
To obtain reagents for detection of VLP variants with higher specificities, we isolated antibodies specifically recognizing individual VLP variants from our highly functional synthetic antibody library Lib F.22 Lib F incorporates improvements upon earlier libraries based on our understanding of antibody structure and function and insights from earlier work on determination of minimal requirements of synthetic antibody libraries.27-30 Conformation-specific antibodies18,31 and antibodies recognizing post-translational modifications (Gang Chen and Sachdev Sidhu, unpublished data) have been successfully identified from these libraries. Initial selections without depletion of undesired cross-reactive antibodies yielded no VLP variant-specific clones. We then focused on isolating L55F-specific antibodies under controlled selection conditions. Either a negative selection or addition of competitor in the solution phase ensured cross-reactive clones were effectively removed, leaving L55F-specific clones captured by immobilized L55F. This procedure yielded the clone GC058, which specifically recognized L55F with an EC50 value of 26 nM but bound poorly to other VLPs.
To further improve antibody specificity, we applied a homolog-scanning combinatorial mutagenesis strategy to GC058 to analyze the functional contributions of individual side chains of selected CDR loops to specific antigen recognition. Homolog-scanning mutagenesis is extremely helpful in inferring energetic consequences of mutations introduced at protein binding interfaces in a high throughput fashion, especially in the case where structural information about the complex is unavailable.23,32 The information obtained from this analysis can then be applied to design next generation libraries for further affinity maturation or specificity optimization. Alternatively, homolog-scanning libraries may directly yield antibodies with improved properties by introducing favorable interactions or removing unfavorable interactions at the antibody-antigen interface with subtle mutations in CDR sequences. For instance, a similar strategy applied in the selection of human antibodies targeting Staphylococcus aureus enterotoxin SEB yielded antibodies with higher affinities and better toxin neutralization activities.24
Following specificity tuning, we found that most clones were more specific for L55F recognition compared to the parent clone GC058. Among seven Fabs selected for detailed analysis, cross-reactivity with VLPs other than L55F was observed for only one Fab (GC103). However, there seems to be a trade-off between affinity and specificity, since clones with higher specificity for L55F generally bound to L55F with lower affinity. Similar binding profiles were observed with monovalent Fabs and bivalent IgGs.
The methodical approach to in vitro specificity optimization used in this study resulted in discovery of highly specific antibodies capable of recognizing single point mutation differences in JCV VLPs. These peptides and synthetic antibodies could be useful in JCV and PML-related basic and preclinical research, and have the potential to complement currently available JCV VP1 affinity reagents. In particular, the L55F-specific antibodies we developed should be valuable when applied to future research aimed at understanding the infection biology of the virus harboring the L55F mutation. Moreover, these peptides and synthetic antibodies represent potential starting points for developing tailored immune-based assays for PML risk stratification.
Materials and Methods
Oligonucleotides
Oligonucleotides were purchased from Integrated DNA Technologies. Two mutagenic oligonucleotides Pep-1AM and Pep-2AM (Table 1) were used for construction of phage-displayed peptide libraries for fine-tuning binding specificity of the parental clones. The mutagenic oligonucleotides HS-L3, −H1, −H2 and −H3 (Table 1) were used for construction of phage-displayed antibody libraries to homolog scan residues in CDR3 of the light chain and all 3 CDRs of the heavy chain in clone GC058, respectively. The mutagenic oligonucleotides used for phage-displayed antibody library construction were designed to simultaneously randomize predefined codon positions while ensuring equimolar starting frequency of wild type and a codon for a homologous residue in the homolog-scanning libraries. Degenerate codons are shown in bold text and the amino acid types coded by these codons have been described previously.23
Table 1.
Mutagenic oligonucleotides used for peptide and antibody library construction*
| Oligonucleotide | Sequence (5'–3') |
|---|---|
| Pep-1AM | GCA GCC TCT TCA TCT GGC NNK NNK aaa cgc TGT ctc cgc tcc ggt tat TGC tac gtc atg NNK NNK GGT GGA GGA TCC GGA |
| Pep-2AM | GCA GCC TCT TCA TCT GGC NNK NNK ctc TGT tgg cgc gtc ggt cgc gac gcc tgg gtc TGC NNK NNK GGT GGA GGA TCC GGA |
| HS-L3 | ACT TAT TAC TGT CAG CAA TKG TWC TWC TWC KCC KCC SCA RTT ACG TTC GGA CAG GGT AC |
| HS-H1 | GCA GCT TCT GGC TTC AAC RTT KCC KCC KCC KCC MTG CAC TGG GTG CGT CAG |
| HS-H2 | GGC CTG GAA TGG GTT GCA KCC RTT KCC KCC TWC TWC GST KCC ASC TWC TAT GCC GAT AGC GTC AAG |
| HS-H3 | GTC TAT TAT TGT GCT CGC KCT TWC MAC KCT GST MTC GAC TAC TGG GGT CAA GG |
Degenerate codons are shown in bold text. Equimolar DNA degeneracies are represented in the IUB code (K = G/T, M = A/C, R = A/G, S = G/C, W = A/T, Y = C/T, N = A/T/G/C). The positions targeted for non-equimolar DNA degeneracies are shown in lower case. These codons are soft randomized using a 70/10/10/10 nucleotide mix that on average allows for 50% of the wild type amino acid and 50% of other amino acids.
Production of JCV VP1 virus-like particles
The information on sequences of VP1 proteins from different JCV strains was described previously.7 VLPs were expressed and purified as described previously.7 Briefly, JCV VLPs were expressed in insect cell line SF9 and isolated from whole cell extract using OptiPrep™ Density Gradient Medium (Sigma-Aldrich, #D1556), followed by buffer exchange and concentrating in an Amicon stirred cell with a 300,000 MWCO membrane and protein quantitation by BCA Protein Assay Kit (Thermo Scientific, # PI-23227).
Construction of phage-displayed peptide libraries
The naïve phage-displayed dodecapeptide library (X12, where X is any amino acid) was reported previously.21 The library was displayed in a polyvalent format on the M13 major coat protein (P8) with a phagemid containing isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible Ptac promoter. Two biased peptide-phage libraries for binding specificity tuning were constructed in a similar manner using the 2 oligonucleotides listed in Table 1. These two libraries were constructed using a “stop template” M13 phagemid pRSTOP4, a vector modified from Stop4 33 by introducing a spacer linker between the stII secretion signal sequence and the region containing 4 stop codons, for polyvalent display of peptides. A Ptac promoter is utilized to drive the expression of open-reading frames encoding the peptide-P8 fusion proteins in the following form: the stII secretion signal sequence followed by a random peptide flanked with spacer linkers at both N- and C-termini, and the C-terminal spacer links the peptide to the N-terminus of the major coat protein P8. Each constructed library contained >1010 unique members. The two libraries were pooled and a combined diversity of 5 × 1010 independent sequences was obtained.
Construction of phage-displayed antibody libraries
Construction of the naïve phage-displayed synthetic antibody library F was described previously.22 Library F was constructed on a single human anti-maltose binding protein antibody framework as previously reported Library D,29 but with greater diversity allowed in the third hypervariable loops of the light and heavy chains. This framework used for library construction contains variable domains from VH3 and Vκ1 subgroups that are highly prevalent in natural human antibodies and have favorable characteristics such as high expression, stability and tolerance to mutations.29,34,35 The library for specificity tuning was constructed using optimized site-directed mutagenesis methodology as previously described36,37 with the appropriately designed “stop template” version of pGC058. The stop template contained TAA stop codons in CDR-L3 and CDR-H3 whereas CDR-H1 and CDR-H2 to be mutated were kept as wild type sequences. Mutagenic oligonucleotides listed in Table 1, designed to simultaneously repair the stop codons and introduce mutations at the desired sites in the stop template, were used in the mutagenesis. Mutations were introduced simultaneously into CDR-L3 and CDR-H3, and possibly CDR-H1 or CDR-H2. The library for affinity maturation contained 5 × 109 unique clones (theoretical diversity 1.1 × 109).
Selection of JCV VLP variant-specific peptides
Library biopanning for different JCV VLP variant-binding peptides was performed separately with WT and its 2 point mutation variants L55F and D66H by following the published protocol37 with minor modifications. Maxisorp immunoplates (Thermo Scientific, #442404) were coated with 5 μg/ml of WT or its mutant and incubated overnight with gentle shaking. The antigen-immobilized immunoplates were blocked with bovine serum albumin (BSA) (Bioshop, #ALB001) for 1.5 h before naïve peptide-displayed phage library (1012–1013 phage particles/ml) for initial screening or the biased library for specificity tuning was added. Following 1 h incubation to allow for phage binding, the plates were washed 10 times with phosphate-buffered saline (PBS) supplemented with 0.05% Tween 20 (PT buffer). Bound phage particles were eluted with 0.1 M HCl for 5 min and the eluant was neutralized with 1.0 M Tris base (pH 11). Eluted phage particles were amplified by super-infecting mid-log phase E. coli XL1-blue with the addition of M13KO7 helper phage (New England Biolabs, #N0315S). After overnight growth at 37°C in 2YT medium supplemented with 100 μg/ml carbenicillin, 25 μg/ml kanamycin and 100 uM IPTG, phage particles were concentrated by precipitation with 20% PEG/2.5M NaCl, resuspended in PBS supplemented with 0.5% (w/v) BSA and 0.05% Tween 20 (PBT buffer), and used for additional rounds of binding selection. Four rounds of selection were performed for each protein. Individual clones from rounds 3 and 4 of binding selections were grown overnight in 96-well format in 1 ml of 2YT medium supplemented with 100 μg/ml carbenicillin and M13KO7 helper phage to a final concentration of 1010 cfu/ml. The culture supernatants were used directly in phage ELISAs to detect phage-displayed polypeptides that specifically bound to antigen. The positive clones were subject to DNA sequencing. In addition, phage ELISAs were used to compare the relative binding strength of these positive clones against WT and 2 variants: L55F and D66H.
Selection of anti-L55F Fabs
Library sorting was performed by following the protocol similar to that of peptide selection described above with modifications tailored to prioritize specificity selection. Briefly, in the first round of selection, in addition to one Maxisorp immunoplate coated with 5 μg/ml of L55F, a depletion Maxisorp immunoplate was coated with 5 μg/ml of WT and incubated overnight with gentle shaking. Both immunoplates were blocked with BSA for 1.5 h. Antibody-displayed phage library (1012–1013 phage particles/ml) was pre-incubated in the WT-coated immunoplate for 1 h to deplete WT-specific antibodies before transfer into L55F-coated immunoplate. Following 1 h incubation to allow for phage binding, the plate was then washed 10 times with PT buffer. Bound phage particles were eluted with 0.1 M HCl for 5 min and the eluant was neutralized with 1.0 M Tris base (pH 11). Eluted phage particles were amplified by super-infecting mid-log phase E. coli XL1-blue with the addition of M13KO7 helper phage. After overnight growth at 37°C in 2YT medium supplemented with 100 μg/ml carbenicillin and 25 μg/ml kanamycin, phage particles were concentrated by precipitation with 20% PEG/2.5M NaCl and resuspended in PBT buffer, and were used for additional rounds of binding selection. From rounds 2–5 of selection, phage solution was pre-incubated with 10 μg/ml of WT in solution for 1 h before being added to L55F-immobilized immunoplate. The concentration of WT used for pre-incubation was increased to 50 μg/ml in the specificity tuning selection. Individual clones from rounds 4 and 5 of binding selection were grown overnight in 96-well format in 1 ml of 2YT medium supplemented with 100 μg/ml carbenicillin and M13KO7 helper phage to a final concentration of 1010 cfu/ml. The culture supernatants were used directly in phage ELISAs to detect phage-displayed antibodies specifically targeting L55F over WT. For biopanning against anti-FLAG antibody with the homolog-scan library, pre-depletion treatment, as described above for L55F selection, was omitted.
Phage ELISAs for identification of anti-VLP peptides and Fabs
A phage ELISA was performed to determine the binding specificity of phage-displayed anti-VLP peptides or antibodies, as described previously.25 Briefly, phage ELISAs were carried out on plates coated with WT or the variants D66H and L55F. Phage particles displaying peptides or Fabs were diluted in PBT buffer and were transferred to wells coated with different antigens and blocked with BSA. The plates were incubated for 30 min, washed with PT buffer, and incubated for 30 min with horseradish peroxidase (HRP)/anti-M13 antibody conjugate (GE Healthcare, #27942101). The plates were washed with PT buffer, developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (KPL, #50-76-00), quenched with 1.0 M H3PO4, and read spectrophotometrically at 450 nm. The cross-reactivity of the anti-VLP peptides or antibodies was quantitated as the signal ratios of the OD450 value obtained from the peptide- or antibody-displaying phage particles binding to the immobilized cognate antigen divided by the value from the same phage particles binding to other antigen.
Expression and purification of peptide-Fc fusion proteins
Stop template version of the Fc fusion expression vector pGC130 was used as the template in site-directed mutagenesis to construct peptide-Fc fusion expression plasmids designed to express fusion proteins consisting of individual peptide sequence fused to N terminus of the human Fc domain. The template pGC130 was constructed by replacing the scFv sequence in a Ptac driven scFv display vector 1.10scFv (Nicolas Economopoulos and Sachdev Sidhu; unpublished data) designed for phage display of a human scFv fused to the C-terminal domain of the M13 gene-3 minor coat protein with a human Fc region and inserting an amber codon at the C-terminus of Fc domain. Standard molecular cloning techniques were applied to construct plasmid pGC130. The linker between the peptide and Fc domain in the resultant peptide-Fc fusion is GGGSGGG; the flanking sequence at the N terminus of the peptide is ASSSG.
E. coli BL21(DE3)-pLysS (Promega, #L1191) harboring the appropriate expression plasmid were grown in LB broth overnight at 37°C. The culture was used to inoculate 500 ml of LB/carb broth in a 2-L baffled flask at 37°C and induced for fusion protein expression at 16°C with 100 uM IPTG once the OD600 of the culture reached 0.6–0.8. Cells were harvested after 24 h of growth and the pellet was resuspended in lysis buffer (50 mM Tris, 150 mM NaCl, pH 8.0, 0.5 mg/ml lysozyme (Bioshop, #LYS702), 0.2 U/ml benzonase (Sigma-Aldrich, #E1014), 10 mM MgCl2) and incubated on ice for 1 h. The crude lysate was spun down and the supernatant was applied to an rProtein A affinity column (GE Healthcare, #17-1279-01). The loaded column was then washed with 10 column volumes of PBS followed by elution of Fab protein with elution buffer (50 mM NaH2PO4, 100 mM H3PO4, 140 mM NaCl, pH 2.0). The eulate was immediately neutralized with neutralization buffer (1 M Na2HPO4, 140 mM NaCl, pH 8.6). Protein concentrations were determined by Bio-Rad Protein Assay (Bio-Rad, #500-0001) using bovine gamma globulin as standard and protein purity was verified by SDS-PAGE.
Expression and purification of Fab and IgG proteins
The expression and purification of Fab proteins was performed as described.24 Briefly, the Fab expression vector was constructed using appropriate display phagemid by site-directed mutagenesis to insert an amber stop codon upstream of gene III. The resultant expression vector was transformed into amber non-suppressor strain E. coli 55244 and the transformants were used directly to inoculate low phosphate C.R.A.P. medium. The culture was incubated at 30°C with shaking at 200 rpm for 24 h before harvest. The pellet was stored at −80°C before proceeding with the protein A purification protocol described above in the section for peptide-Fc fusion protein purification.
For production of full-length IgG antibodies, DNA sequences of variable domains were subcloned into proprietary mammalian expression vectors for heavy chain and light chain expression. The heavy chain expression cassette is under transcriptional control of the human cytomegalovirus promoter and the human growth hormone polyadenylation sequence. A separate expression cassette on the plasmid encodes the murine dihydrofolate reductase (DHFR) gene for selection in DHFR-deficient cells. The light chain expression cassette is under the same transcription control elements as the heavy chain and these plasmids encode a separate expression cassette for neomycin phosphotransferase providing for selection with G418. Vectors were co-transfected into CHO-DG44i cells using the FuGENE® 6 Transfection Reagent (Promega, #E2691), according to the manufacturer's instructions. Stable cell lines were generated by adding DHFR and neomycin selection reagents to cell culture media 2–3 d following transfection, and grown for 14 d in selection media followed by growth expansion in 1-L shake flasks. After incubation, the cultures were spun down and filtered. The supernatant was applied to an rProtein A affinity column. IgG proteins were eluted with 25 mM H3PO4, pH 2.8, 100 mM NaCl and neutralized with 0.5 M Na3PO4 pH 8.6. Eluted fractions of interest were combined, concentrated and dialyzed into PBS, pH 7.4.
ELISA for detection of WT, D66H and L55F by peptide-Fc fusion proteins
Immulon® 4HBX immunoplates (Thermo Scientific, #3855) wells were coated with 0.1 ml of 10 μg/ml of peptide-Fc fusion proteins in PBS and incubated overnight at 4°C. Plates were washed once with 0.3 ml/well of PBS and blocked with 0.3 ml/well of PBS containing 1 mM CaCl2 and 0.5 mM of MgCl2, supplemented with 1% BSA and 0.1% Tween-20 (block buffer) for 1 h. VLPs were prepared at 30 μg/ml in block buffer and 0.1 ml VLP was added to each well. Following 2 h incubation, plates were washed 3 times with 0.3 ml/well of PBS supplemented with 0.1% Tween-20. The VP1-specific murine antibody, PAB597 (courtesy of Edward Harlow, Harvard Medical School), prepared at 2 μg/ml in block buffer, was added at 0.1 ml/well and incubated for 1 h.38 Plates were washed and 0.1 ml HRP labeled anti-mouse IgG1 (γ1-specifc) (Jackson ImmunoResearch, #115-035-205) diluted 1:5000 in block buffer was added to each well. After 45 min incubation, plates were washed, developed with TMB substrate (SurModics, #TMBK-0200-2C), quenched with 2.0 M H3PO4, and read spectrophotometrically at 450 nm.
ELISAs for detection of JCV VP1 WT and variants
Immulon® 4HBX immunoplates (Thermo Scientific, #3855) wells were coated with 100 μl of VLP (1 μg/ml) diluted in 50 mM NaHCO3, pH 9.2 and incubated overnight at 4°C. Coating material was decanted and the plates were incubated for 1 h at room temperature with 200 μl per well of Blocking Buffer (Tris-buffered saline, 0.05% Tween 20, 1x Casein solution (Vector Laboratories, #SP-5020)). Plates were washed 4 times with 350 μl of PBS, 0.05% Tween 20 on a plate washer. Serial dilutions of Fabs or IgGs in Blocking Buffer were incubated in the plate for 1 h at room temperature. The plate was washed as described above and 100 μl per well of secondary antibody, diluted in Blocking Buffer, was incubated in the plate for 40 min at room temperature. The plate was washed as described above followed by detection with either 1:5000 dilution of HRP-tagged anti-FLAG antibody (Sigma Aldrich, #A8592) or 1:10000 dilution of anti-human antibody (Jackson ImmunoResearch Laboratories, #209-035-098). The plate was washed as described above and 100 μl per well of TMB substrate was added to the plate and incubated 2–5 min. The reaction was stopped with 100 μl per well of 1 M H2SO4 and the absorbance was read at 450 nm.
For single point analysis of Fab binding to JCV VP1 mutant protein, the assay was performed as described above using a single Fab concentration of 10 μg/ml.
Disclosure of Potential Conflicts of Interest
AC, MB and PHW are employees and shareholders of Biogen Idec, Inc. All authors declare no conflicts of interest with the subject matter or materials discussed in the manuscript.
Acknowledgments
The authors would like to thank Dr Raffi Tonikian and Mrs Danielle Carranza for help and advice with phage work.
Funding
This work was supported by Biogen Idec Inc.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
References
- 1.Knowles WA. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV). Polyomaviruses Hum Dis 2006; 577:19-45; PMID:16626025; http://dx.doi.org/ 10.1007/0-387-32957-9_2 [DOI] [PubMed] [Google Scholar]
- 2.Pietropaolo V, Videtta M, Fioriti D, Mischitelli M, Arancio A, Orsi N, Degener AM. Rearrangement patterns of JC virus noncoding control region from different biological samples. J Neurovirol 2003; 9:603-11; PMID:14602573; http://dx.doi.org/ 10.1080/714044482 [DOI] [PubMed] [Google Scholar]
- 3.Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, et al.. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change itsreceptor specificity. J Infect Dis 2011; 204:103-14; PMID:21628664; http://dx.doi.org/ 10.1093/infdis/jir198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorelik L, Lerner M, Bixler S, Crossman M, Schlain B, Simon K, Pace A, Cheung A, Chen LL, Berman M, et al.. Anti-JC virus antibodies: Implications for PML risk stratification. Annal Neurol 2010; 68:295-303; PMID:20737510; http://dx.doi.org/ 10.1002/ana.22128 [DOI] [PubMed] [Google Scholar]
- 5.Neu U, Maginnis MS, Palma AS, Stroeh LJ, Nelson CDS, Feizi T, Atwood WJ, Stehle T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host & Microbe 2010; 8:309-19; PMID:20951965; http://dx.doi.org/ 10.1016/j.chom.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kishida S. Progressive multifocal leukoencephalopathy: epidemiology, clinical pictures, diagnosis and therapy. Brain and Nerve (Tokyo) 2007; 59:125-37; PMID:17380777 [PubMed] [Google Scholar]
- 7.Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). Plos Genetics 2009; 5:e1000368; PMID:19197354; http://dx.doi.org/ 10.1371/journal.pgen.1000368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyler KL. Progressive multifocal leukoencephalopathy: can we reduce risk in patients receiving biological immunomodulatory therapies? Annal Neurol 2010; 68:271-4; PMID:20818784; http://dx.doi.org/ 10.1002/ana.22185 [DOI] [PubMed] [Google Scholar]
- 9.Koralnik IJ, Boden D, Mai VX, Lord CI, Letvin NL. JC virus DNA load in patients with and without progressive multifocal leukoencephalopathy. Neurology 1999; 52:253-60; PMID:9932940; http://dx.doi.org/ 10.1212/WNL.52.2.253 [DOI] [PubMed] [Google Scholar]
- 10.Rudick RA, O'Connor PW, Polman CH, Goodman AD, Ray SS, Griffith NM, Jurgensen SA, Gorelik L, Forrestal F, Sandrock AW, et al.. Assessment of JC virus DNA in blood and urine from natalizumab-treated patients. Annal Neurol 2010; 68:304-10; PMID:20737514; http://dx.doi.org/ 10.1002/ana.22107 [DOI] [PubMed] [Google Scholar]
- 11.Neu U, Stehle T, Atwood WJ. The Polyomaviridae: contributions of virus structure to our understanding of virus receptors and infectious entry. Virology 2009; 384:389-99; PMID:19157478; http://dx.doi.org/ 10.1016/j.virol.2008.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu CK, Wei G, Atwood WJ. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal alpha(2-6)-linked sialic acids. J Virol 1998; 72:4643-9; PMID:9573227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen BJ, Atwood WJ. Construction of a novel JCV/SV40 hybrid virus (JCSV) reveals a role for the JCV capsid in viral tropism. Virology 2002; 300:282-90; PMID:12350358; http://dx.doi.org/ 10.1006/viro.2002.1522 [DOI] [PubMed] [Google Scholar]
- 14.Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, McAuliffe M, Tonkin C, Simon K, Goelz S, et al.. Sequencing and analysis of JC virus DNA from Natalizumab-treated PML patients. J Infect Dis 2011; 204:237-44; PMID:21673034; http://dx.doi.org/ 10.1093/infdis/jir256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maginnis MS, Ströh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 2013; 4:e00247-13; PMID:23760462; http://dx.doi.org/ 10.1128/mBio.00247-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, et al.. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest 2014; 124:5323-36; PMID:25401469; http://dx.doi.org/ 10.1172/JCI76629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011; 29:245-54; PMID:21390033; http://dx.doi.org/ 10.1038/nbt.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Sidhu SS, Wells JA. Two-state selection of conformation-specific antibodies. Proc Natl Acad Sci U S A 2009; 106:3071-6; PMID:19208804; http://dx.doi.org/ 10.1073/pnas.0812952106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattori T, Taft JM, Swist KM, Luo H, Witt H, Slattery M, Koide A, Ruthenburg AJ, Krajewski K, Strahl BD, et al.. Recombinant antibodies to histone post-translational modifications. Nat Methods 2013; 10:992-5; PMID:23955773; http://dx.doi.org/ 10.1038/nmeth.2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lua LHL, Connors NK, Sainsbury F, Chuan YP, Wibowo N, Middelberg APJ.. Bioengineering virus-like particles as vaccines. Biotech Bioeng 2014; 111:425-40; PMID:24347238; http://dx.doi.org/ 10.1002/bit.25159 [DOI] [PubMed] [Google Scholar]
- 21.Xin X, Gfeller D, Cheng J, Tonikian R, Sun L, Guo A, Lopez L, Pavlenco A, Akintobi A, Zhang Y, et al.. SH3 interactome conserves general function over specific form. Mol Sys Biol 2013; 9; PMID:23549480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Persson H, Ye W, Wernimont A, Adams JJ, Koide A, Koide S, Lam R, Sidhu SS. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol 2013; 425:803-11; PMID:23219464; http://dx.doi.org/ 10.1016/j.jmb.2012.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vajdos FF, Adams CW, Breece TN, Presta LG, De Vos AM, Sidhu SS. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J Mol Biol 2002; 320:415-28; PMID:12079396; http://dx.doi.org/ 10.1016/S0022-2836(02)00264-4 [DOI] [PubMed] [Google Scholar]
- 24.Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, Devi VS, Stavale E, Warfield KL, Zeitlin L, et al.. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J Biol Chem 2012; 287:25203-15; PMID:22645125; http://dx.doi.org/ 10.1074/jbc.M112.364075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Applications of Chimeric Genes and Hybrid Proteins, Pt C, 2000:333-63; PMID:11075354; http://dx.doi.org/ 10.1016/S0076-6879(00)28406-1 [DOI] [PubMed] [Google Scholar]
- 26.Sidhu SS. Phage display in pharmaceutical biotechnology. Curr Opin Biotech 2000; 11:610-6; PMID:11102798; http://dx.doi.org/ 10.1016/S0958-1669(00)00152-X [DOI] [PubMed] [Google Scholar]
- 27.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol 2004; 338:299-310; PMID:15066433; http://dx.doi.org/ 10.1016/j.jmb.2004.02.050 [DOI] [PubMed] [Google Scholar]
- 28.Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol 2004; 340:1073-93; PMID:15236968; http://dx.doi.org/ 10.1016/j.jmb.2004.05.051 [DOI] [PubMed] [Google Scholar]
- 29.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al.. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 2007; 373:924-40; PMID:17825836; http://dx.doi.org/ 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 30.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nat Chem Biol 2006; 2:682-8; PMID:17108986; http://dx.doi.org/ 10.1038/nchembio843 [DOI] [PubMed] [Google Scholar]
- 31.Paduch M, Koide A, Uysal S, Rizk SS, Koide S, Kossiakoff AA. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 2013; 60:3-14; PMID:23280336; http://dx.doi.org/ 10.1016/j.ymeth.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pál G, Fong SY, Kossiakoff AA, Sidhu SS. Alternative views of functional protein binding epitopes obtained by combinatorial shotgun scanning mutagenesis. Protein Sci 2005; 14:2405-13; PMID:16131663; http://dx.doi.org/ 10.1110/ps.051519805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murase K, Morrison KL, Tam PY, Stafford RL, Jurnak F, Weiss GA. EF-Tu binding peptides identified, dissected, and affinity optimized by phage display. Chem Biol 2003; 10:161-8; PMID:12618188; http://dx.doi.org/ 10.1016/S1074-5521(03)00025-5 [DOI] [PubMed] [Google Scholar]
- 34.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol 2005; 348:1153-62; PMID:15854651; http://dx.doi.org/ 10.1016/j.jmb.2005.03.041 [DOI] [PubMed] [Google Scholar]
- 35.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A 2004; 101:12467-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Method Enzymol 1987; 154:367-82; PMID:3323813; http://dx.doi.org/ 10.1016/0076-6879(87)54085-X [DOI] [PubMed] [Google Scholar]
- 37.Fellouse FA, Sidhu SS. Making antibodies in bacteria In: Howard GC, Kaser MR, eds. Making and using antibodies: a practical handbook. Boca Raton, FL: CRC Press; 2006, 157-80. [Google Scholar]
- 38.Atwood WJ, Wang L, Durham LC, Amemiya K, Traub RG, Major EO. Evaluation of the role of cytokine activation in the multiplication of JC virus (JCV) in human fetal glial-cells. J Neurovirol 1995; 1:40-9; PMID:9222341; http://dx.doi.org/ 10.3109/13550289509111009 [DOI] [PubMed] [Google Scholar]
- 39.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest Bethesda, MD: National Institutes of Health; 1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.