

Abstract
K-ras mutations promote angiogenesis in lung cancer and contribute to the drug resistance of cancer cells. It is not clear whether K-ras mutated adenocarcinomas are sensitive to anti-angiogenic therapy with monoclonal antibodies (mAbs) that target vascular endothelial growth factor (VEGF). Anti-angiogenic mAbs are usually delivered systemically, but only a small proportion reaches the lung after intravenous injection. We investigated the relevance of a non-invasive pulmonary route for the delivery of anti-VEGF mAbs in the mouse K-rasLA1 model. We found that pulmonary delivery of these mAbs significantly reduced the number of tumor lesions and inhibited malignant progression. The antitumor effect involves the VEGFR2-dependent inhibition of blood vessel growth, which impairs tumor proliferation. Pharmacokinetic analysis of aerosolized anti-VEGF showed its low rate of passage into the bloodstream, suggesting that this delivery route is associated with reduced systemic side effects. Our findings highlight the value of the aerosol route for administration of anti-angiogenic mAbs in pulmonary adenocarcinoma with K-ras activating-mutations.
Keywords: Non-small cell lung cancer, lung adenocarcinoma, anti-VEGF, monoclonal antibody, aerosol, K-ras mutation, anti-angiogenic agent, airways delivery, pharmacokinetics
Introduction
Lung cancer remains the leading cause of cancer-related deaths worldwide, and non-small cell lung cancer (NSCLC) accounts for 85% of lung cancers. Over the past two decades, the treatment of NSCLC has radically changed with the introduction of targeted drugs, e.g., small molecule tyrosine kinase inhibitors and monoclonal antibodies (mAbs), and the selection of patients based on tumor histology and genotyping. This has led to substantial clinical benefits and improved overall survival rates. Notably, adenocarcinoma subtypes with epidermal growth factor receptor mutations and ELM4-ALK translocations have benefited from improvements in the management of advanced NSCLC.1-3 However, new therapeutic options are required for adenocarcinoma with V-Ki-ras2 Kirsten ras sarcoma (K-ras) activating mutations, which are encountered in 25% of adenocarcinomas. Indeed, these cancers are associated with poor survival and rarely respond to chemotherapy and some targeted therapies.4 Clinical trials with MAPK inhibitors in this population are ongoing. It is unclear whether K-ras mutated lung adenocarcinomas are sensitive to anti-angiogenic therapies. Indeed, K-ras mutations promote endothelial cell-dependent tumor angiogenesis, mainly via the upregulation of vascular endothelial growth factor (VEGF) and interleukin (IL)-8 and through the repression of the anti-angiogenic protein thrombospondin-1.5,6 In a mouse model of lung adenocarcinomas mutated for K-ras, the neutralization of CXCR2, the mouse homolog of IL-8 receptor (CXCR2), blocks angiogenesis and impairs the formation of early lung lesions.6
Among the various angiogenesis inhibitors tested recently in NSCLC, bevacizumab (Avastin; Genentech/Roche) is the first molecule to target tumor angiogenesis and is the only mAb approved for use in non-squamous subtype NSCLC. Its addition to chemotherapy regimens results in a significant improvement in overall survival.7Bevacizumab is a recombinant humanized IgG1 directed against soluble human VEGF-A (also called VEGF),8 a crucial regulator of angiogenesis and differentiation of progenitor endothelial cells that interacts with two related tyrosine kinase receptors VEGFR-1 and VEGFR-2.9,10
The airways appear to be to a promising alternative to the intravenous (i.v.) route for the delivery of mAbs into the lungs to treat lung cancer. Although the i.v. route is conventionally used for their administration, the assessment of the partitioning of mAbs from the blood into the lungs shows that only a small portion of injected mAbs reach the lungs.11,12 Thus, the i.v. route may not be optimal to deliver mAbs to the lungs to achieve a therapeutic concentration at this target site. In theory, airways offer several advantages: loco-regional delivery allows the direct administration of the mAbs to the target and limits their passage into the bloodstream, thereby reducing systemic side effects. However, several issues should be considered before administrating mAbs through the airways. We showed previously that the pharmacological properties of mAbs are preserved after aerosolization, and that aerosolized mAbs are well tolerated and result in a therapeutic response in animal models.13 Moreover, passage in the bloodstream was low.14 The aims of this study were to determine whether adenocarcinomas with K-ras mutations benefit from anti-VEGF therapies and whether pulmonary delivery is an effective method of administration.
We treated a mouse model of K-ras-induced lung adenocarcinoma (K-rasLA1)15 with a cross-species anti-VEGF mAb (G6–31) and analyzed the anti-tumor efficacy when administrated systemically or locally into the lungs. We also studied the molecular pathways associated with the antitumor effect and the pharmacokinetics (PK) of the pulmonary and systemic routes to examine the value of the pulmonary route for the local delivery of mAbs.
Results
Systemic administration of anti-VEGF mAb reduces tumor burden in lungs from K-rasLA1 mice
G6–31 is a murine IgG2a mAb that binds with high affinity to both human and murine VEGF-A,5 and high expression of VEGF-A is associated with tumor progression in K-ras mutated NSCLC (Fig. S1). In K-rasLA1 mice, VEGF is expressed in papillary tumor cell subtypes from large adenocarcinomas (> 0.5 mm in diameter), but not in tumor cells from atypical alveolar hyperplasia and adenomas (Fig. S1A-B-C).
We sought to evaluate the efficacy of anti-VEGF mAb against K-ras mutated adenocarcinoma. We treated K-rasLA1 mice intraperitoneally once a week for 4 wk with G6–31 or with an isotype control either at 2.5 or 10 mg/kg (Fig. 1A). Assessment of the total number of visible lung lesions, after the animals were killed, showed that mice treated with G6–31 at 10 mg/kg had significantly fewer lung lesions (P = 0.02) than control mice (Fig. 1B; Fig. S2). Treatment with 2.5 mg/kg of G6–31 did not significantly affect the number of lung lesions. Accordingly, the microscopic count of lesions on lung tissue sections showed significantly fewer lesions (P = 0.02) in mice treated with G6–31 at 10 mg/kg than in control mice (Fig. 1C).
Figure 1.
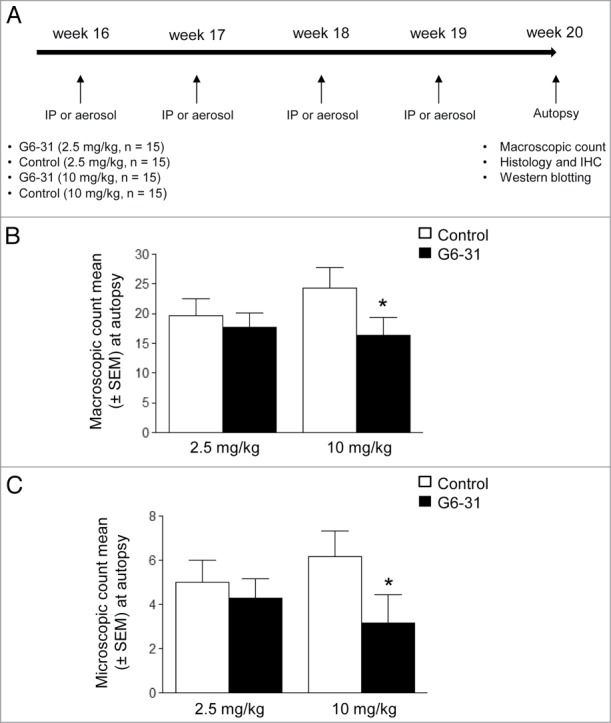
Assessment of G6–31 activity against lung lesions when administrated intraperitoneally. (A) Experimental procedure; Four month-old K-rasLA1 mice received G6–31 or an isotype control, administrated by i.p injection or aerosol, once a week for 4 wk. At the time of death visible lesions were counted on the whole lungs. (B) Quantification of visible nodules per mouse (n = 15 mice per group; 2.5 mg/kg and 10 mg/kg; *P< 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM of nodules. (C) Quantification of lung lesions on H&E stained sections from control and G6–31 treated group (n = 15 mice per group; 2.5 mg/kg and 10 mg/kg; P< 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM of the number of lesions.
Local administration of anti-VEGF mAb into the lungs is well tolerated and reduces tumor burden in the lungs of K-rasLA1 mice
We first confirmed that G6–31 inhibited VEGF-A-mediated VEGFR2 phosphorylation whether it was nebulized or not with MicroSprayer® Aerosolizer (Fig. S3A).
We then determined whether aerosol delivery of G6–31 was well tolerated in 16 wk-old wild-type mice with a genetic background similar to that of K-rasLA1 mice. We treated mice orotracheally with the antibody (10 mg/kg) once a week for 4 wk (Fig. 1A). This treatment was not associated with any secondary effects or changes of behavior and we found no lesions or pulmonary hemorrhage on lungs (Fig. S3B) and no lesions on other organs (kidneys, liver and spleen; data not shown).
The number of lung macroscopic lesions in K-rasLA1 mice that received G6–31 at either dose by pulmonary route was significantly lower than in control mice (2.5 mg/kg dose, P = 0.0010; 10 mg/kg dose, P = 0.0013). Accordingly, the number of lung microscopic lesions in K-rasLA1 mice treated with G6–31 at 10 mg/kg by the pulmonary route was significantly lower than in control mice (P = 0.04) (Fig. 2A-B; Fig. S2). Altogether, these results highlight the effectiveness of the pulmonary route for the delivered of mAb.
Figure 2.

Assessment of aerosolized G6–31 activity against lung lesions. (A) Quantification of visible nodules per mouse (n = 15 mice per group; 2.5 mg/kg and 10 mg/kg; *P < 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM of nodules. (B) Quantification of lung lesions on H&E stained sections from control and G6–31 treated group (n = 15 mice per group; 2.5 mg/kg and 10 mg/kg; P < 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM of the number of lesions.
Anti-VEGF mAb reduces adenocarcinoma lesions
We performed histological analysis to examine the effect of anti-VEGF therapy on malignant progression. Mice treated with G6–31 (10 mg/kg) delivered by intra-peritoneal (i.p.) injection or aerosol had fewer adenocarcinoma lesions than control mice (P = 0.008), whereas the number of atypical alveolar hyperplasia and adenoma was not modified by the treatment (Fig. 3A). These results are in agreement with the restricted expression of VEGF to adenocarcinomas in K-rasLA1 mice that we observed by immunohistochemistry (Fig. S1).
Figure 3.
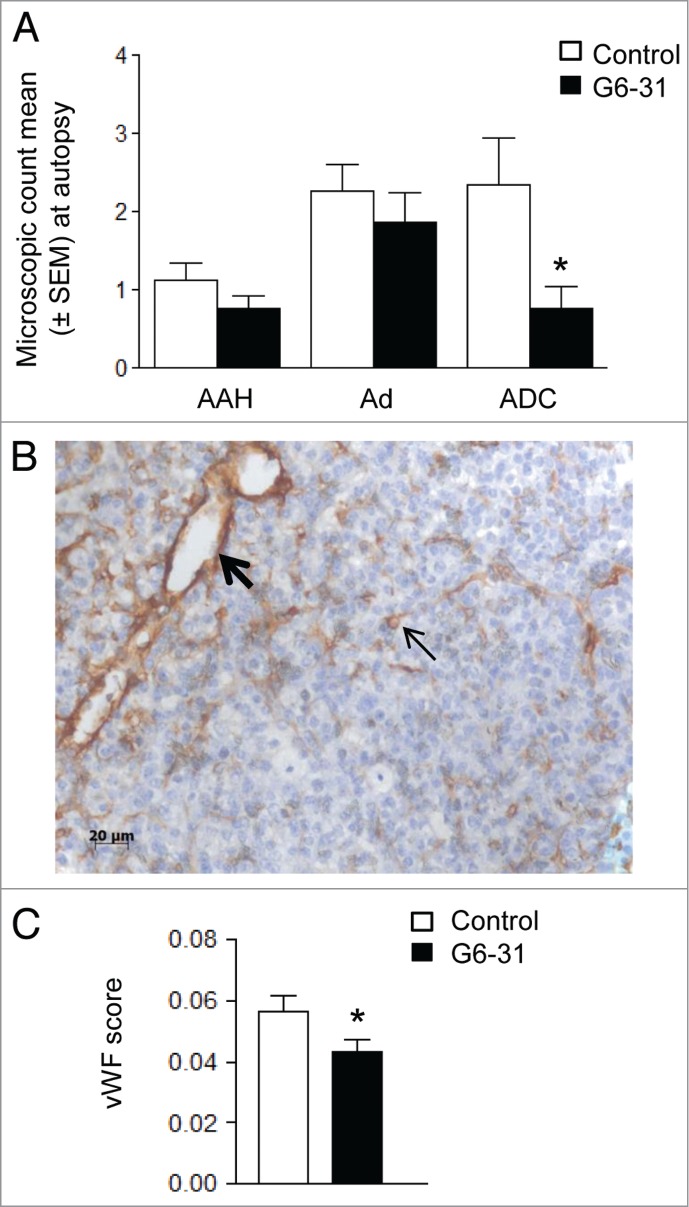
Effect of G6–31 at 10 mg/kg administered by i.p. injection or aerosol on K-rasLA1 lung tumors according to each lesion type and effect on microvascular density. (A) Quantification of AAH (atypical alveolar hyperplasia), Ad (adenoma) and ADC (adenocarcinoma), on H&E stained lung sections from control and G6–31-treated (10 mg/kg dose) mice (n = 30 mice per group; *P < 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM of the number of each lesion type. (B) Representative image of vWFimmunostaining in the lung of K-ras LA1 mice. Bold arrow shows large vessel while standard arrow shows small vessel. (C) Quantification of the microvascular density, from vWFimmunostaining in control and G6–31-treated (10 mg/kg dose) mice (n = 30 mice per group; *P < 0.05 Mann-Whitney test). Results are expressed as the mean ± SEM.
Anti-VEGF mAb inhibits angiogenesis
We used immunochemistry to analyze microvascular density in the tumor because G6–31 achieves tumor regression by blocking angiogenesis.16-19 Microvascular density, as assessed by Von Willebrand Factor (vWF) immunostaining (Fig. 3B), was lower in G6–31 treated mice than in control mice (Fig. 3C). G6–31 mostly affected the microvascular density of large vascular structures (i.e., those with a diameter > 10 μm20,21) when it was delivered by i.p. injection (Table 1), whereas it mostly affected small vessels (mostly capillaries with a d < 10 μm20,21) when it was administered by aerosol (Table 1).
Table 1.
Effect of G6–31 (10 mg/kg) on microvascular density in K-rasLA1 lung tumors, assessed from vWFimmunostaining. Quantification of the microvascular density (MVD) of small (< 10 μm) and large (> 10 μm) vascular structures (n = 30 mice per group; control and G6–31 10 mg/kg; *P< 0.05 Mann-Whitney test)
| vWF% (± SEM) |
||
|---|---|---|
| I.P. G6–31 / Placebo | Aerosol G6–31 / Placebo | |
| Small vessel (< 10 μm) | + 9.85 (± 20.05) | − 44.21 (± 7.67) |
| P = 0.96 | P = 0.018 | |
| Large vessel (> 10 μm) | − 43.10 (± 12.09) | − 11.50 (± 14.69) |
| P = 0.0096 | P = 0.27 | |
The effect of anti-angiogenic therapies on tumor oxygenation and hypoxia is still unclear and may depend on the tumor model.22-26 Thus, we analyzed the expression of the oxygen-sensitive regulatory subunit hypoxia-inducible factor (HIF)-1α, as a marker of tumor hypoxia, in K-rasLA1 mice treated with G6–31 (Fig. S4; Table S1). Western blotting showed that there was no significant difference in abundance of HIF-1α in whole lung extracts from mice treated with G6–31 (either by i.p. or pulmonary administration) and control mice (Table S1).
Anti-VEGF mAb impairs tumor cell proliferation
We analyzed tumor cell proliferation and apoptosis in the lung of animals treated with G6–31 by i.p. or aerosol, to explore the mechanisms associated with reduced tumor burden. The mean proliferative index measured by proliferating cell nuclear antigen (PCNA) expression was significantly lower in G6–31-treated animals (10 mg/kg) than in control animals, regardless of the route of administration (Fig. 4A; Table 2). However, immunochemistry showed that the abundance of cleaved caspase 3 (CC3), which is a marker of apoptosis, was significantly higher in tumors from mice treated intraperitoneally than in those from control mice. This was not the case for G6–31 administrated via the airways, suggesting different mechanisms of action of G6–31 depending on the delivery route (P< 0.0001; Figure 4B and C).
Figure 4.

(A) Detection of PCNA by western blotting in whole lung protein extracts from control mice (mice 1–4) and G6–31-treated (10 mg/kg) mice (mice 5–9). Pulmonary delivery is on the right and i.p. delivery is on the left. (B) CC-3 immunostaining. Representative images of the control (left) and G6–31 (10 mg/kg dose) delivered either by i.p. (middle) or by pulmonary route (right). Brown cells are positive for CC-3 (arrows show some representative cells). The slices were counterstained with hematoxylin (× 400 magnification). Quantification of apoptosis in tumor lesions of the right lung of K-ras LA1 mice (n = 15 mice per group; *P < 0.05 Mann-Whitney test). Results are expressed as the mean staining scores of CC-3 positive cells relative to the control group ± SEM.
Table 2.
Quantification of the level of PCNA (detected by western blotting) by densitometric analysis in mice treated with G6–31 by i.p. or aerosol (15 mice per group, 10 mg/kg) and in control mice (15 mice per group) (*P < 0.05 for both, Mann-Whitney test). Results are expressed as the mean ratio of PCNA normalized to β-actin and relative to the control group ± SEM
| Densitometry - Mean% (± SEM) |
||
|---|---|---|
| I.P. G6–31 / Placebo | Aerosol G6–31 / Placebo | |
| PCNA/β actin | − 46.80 (± 7.70) | − 31.49 (± 8.97) |
| P = 0.0065 | P = 0.009 | |
VEGF neutralization impairs VEGFR2 activation
VEGFR2 is believed to predominantly contribute to VEGF-mediated tumor angiogenesis; therefore, we analyzed members of its signaling pathway by western blotting in whole lung lysates in response to G6–31 treatment by aerosol or i.p. injection. Neutralization of VEGF by G6–31 impaired VEGFR2 phosphorylation (Y1175), indicating that the VEGFR2 pathway is inhibited in G6–31 treated mice27 (P = 0.0008 for i.p. and P = 0.012 for aerosol; Figure 5; Table 3). Phosphorylation of AKT (Ser 473) was significantly lower in mice treated with G6–31 administrated by aerosol than in control mice (P = 0.0018; Figure 5; Table 3). Phosphorylation of PI-3K (Y458) and ERK (T202/Y204) signaling factors downstream of VEGFR2,27,28 were unaffected by the treatment (Fig. 5; Table 3).
Figure 5.

Blockade of VEGFR2 signaling pathway. Phospho-VEGFR2 Y1175, total VEGFR2, Phospho-PI3K, β-actin, Phospho-AKT, total AKT, Phospho-ERK and total ERK were analyzed by western blotting in whole lung protein extracts from control mice (mice 1–4) and from mice treated with G6–31 at 10 mg/kg (mice 5–9). Pulmonary delivery is on the right and i.p. delivery is on the left.
Table 3.
Quantification of the level of phosphorylated VEGFR2 and total VEGF-2 by densitometric analysis in mice treated with G6–31 by i.p. injection or aerosol (15 mice per group, 10 mg/kg) and in control mice (15 mice per group) (*P< 0.05, Mann-Whitney test). Results are expressed as the mean ratio of phospho-VEGFR2 Y1175 to total VEGFR2 relative to that of the control group ± SEM. The same method of quantification was applied to Phospho-PI3K, β-actin, Phospho-AKT, total AKT, phosphor-ERK and total ERK. Results are expressed as the mean ratio of Phospho-PI3K to β-actin relative to that of the control group ± SEM, of Phospho-AKT to total AKT relative to that of the control group ± SEM and of Phospho-ERK to total ERK relative to that of the control group ± SEM
| Densitometry - Mean% (± SEM) |
||
|---|---|---|
| I.P. G6–31 / Placebo | Aerosol G6–31 / Placebo | |
| Phospho-VEGFR2/ VEGFR2 | − 63.81 (± 5.09) | − 68.80 (± 11.09) |
| P = 0.0008 | P = 0.0127 | |
| Phospho-PI3K/ β actin | − 30.14 (± 13.31) | − 5.13 (± 3.24) |
| P = 0.72 | P = 0.28 | |
| Phospho-AKT / AKT | + 34,23 (± 13.34) | − 44,48 (± 4.19) |
| P = 0.10 | P = 0.0018 | |
| Phospho-ERK / ERK | + 1.29 (± 20.31) | + 3.33 (± 3.40) |
| P = 0.90 | P = 0.28 | |
Altogether, these results show that anti-VEGF mAb delivered by the airways or by i.p. injection reduces tumor burden through similar mechanisms.
Aerosolized anti-VEGF mAb passes slowly and poorly into the systemic circulation
We developed a quantitative ELISA to analyze the passage of anti-VEGF mAb into the circulation after pulmonary delivery in K-rasLA1 mice treated with a unique dose of antibody (10 mg/kg) (Fig. S5). Only a small amount of G6–31 enters the bloodstream after pulmonary delivery (Fig. S6). The maximum serum concentration was observed about 6 h after G6–31 aerosolization and was about 100-fold lower than that after systemic delivery (mean of 1.25 μg/mL for the highest values of the pulmonary route vs. 150 μg/mL for the systemic delivery) (Fig. 6A). We first used a non-compartmental analysis to describe the PK parameters of aerosolized G6–31. However, this approach was invalidated because the MRT was higher in the i.v. group than in the pulmonary group, and consequently the mean absorption time was negative, which is impossible given that pulmonary administration should lead to a longer persistence of the drug in the organism (Table S2). This bias can be most likely due to the nonlinear profile of G6–31 PK as observed in Figure 6A. Thus, we used a compartmental analysis as presented in Figure 6B. This model was able to describe the concentration-time profiles for G6–31 following i.v. and pulmonary administration with a good fit between the observed and predicted values (Fig. 6A). The estimated PK parameter values of the compartmental approach are summarized in Table 4. The estimated bio-available fraction was 5.1%. This value is consistent with the bio-available fraction already estimated for other mAbs.14 We used a Michaelis–Menten equation to quantify the saturable component of the elimination of G6–31; the maximum elimination rate Vmax was 12.8 d−1 and a Michaelis constant kM was 0.45 μg·mL−1 (Fig. S7).
Figure 6.
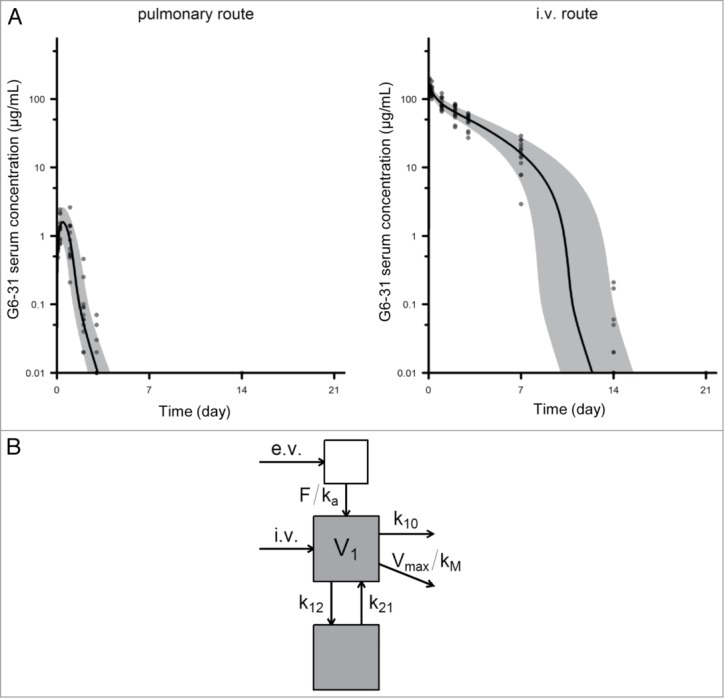
Pharmacokinetics of G6–31. (A) Serum concentration of G6–31 mAb was measured by ELISA in the serum collected at different time points from mice that received a single dose of the mAb (10 mg/kg) either by the pulmonary route (left, n = 13) or intravenously (right, n = 15). Gray area and solid line show the 5th-95th and 50th percentiles of the model-predicted time-concentration profiles, respectively. (B) Schematic representation of the pharmacokinetic compartmental model of G6–31.
Table 4.
Estimated values of compartmental pharmacokinetic parameters. Abbreviations: F, bio available fraction; ka, absorption rate; V1, central volume of distribution; k10 systemic elimination and k12, k21 distribution rates. Vmax maximum elimination rate; and kM, Michaelis constant
| Parameter | Value (CV%) |
|---|---|
| F (%) | 5.1 (−) |
| ka (day−1) | 1.59 (18) |
| k10 (day−1) | 0.2 (13) |
| V1 (mL) | 2 (13) |
| k12 (day−1) | 0.46 (−) |
| k21 (day−1) | 0.94 (60) |
| Vmax (day−1) | 12.8 (23) |
| kM (μg/mL) | 0.46 (13) |
| a | 0.026 |
| b | 0.15 |
Discussion
The first aim of this study was to evaluate whether K-ras mutated lung tumors benefit from an anti-angiogenic drug given as a monotherapy. We showed that VEGF neutralization with a mAb (G6–31, IgG2a,k) markedly inhibits lung tumorigenesis. It reduced the number of K-ras mutated adenocarcinomatous lesions in a mouse model of lung adenocarcinomas in which VEGF is highly expressed.29 We found that the anti-VEGF mAb mediated its antitumor activity partly through the alteration of VEGF-dependent angiogenic mechanisms, as shown by the inhibition of VEGFR2 signaling, and the regression of blood vessels. The VEGF/VEGFR2 pathway plays a central role in angiogenesis and regulates endothelial cell migration, proliferation, permeability and survival.27,30,31 Both VEGF and VEGFR2 are commonly expressed in NSCLC32 and are important components that promote tumor growth, progression and metastasis.33 In agreement with these findings, we found that VEGF neutralization by G6–31 impairs cell proliferation in the lungs of K-rasLA1 mice. But the antitumor efficacy of G6–31 in K-rasLA1 mice may not depend only on the angiogenic properties of the VEGF/VEGFR pathway since autocrine or paracrine VEGF promotes cancer cell survival.31
As described herein, we found an induction of apoptosis after anti-VEGF mAb treatment. The reason why CC3, a marker of apoptosis, was increased only in the tumor lesions after a systemic treatment with G6–31 has not been clarified. This discrepancy may be due to a concentration-dependent effect of the anti-VEGF mAb, which is undoubtedly more concentrated within the lungs following pulmonary delivery than following systemic delivery.34,35 Similar differential apoptotic effects were previously reported for Taxol.36
Intratumoral hypoxia regulates VEGF levels and is often encountered in NSCLC.37 We analyzed HIF-1 α as a molecular marker of tumor hypoxia and did not find any change to HIF-1 α expression by western blotting with whole lung protein extract, or by IHC on tumor area sections. This suggests that the intrinsic tumor cell characteristics of K-ras mutated NSCLC do not lead to tumor adaptation or evasion of anti-angiogenic agents through therapy-induced hypoxia.38-40 These findings support the rationale for targeting angiogenesis alone in NSCLC and particularly in K-ras mutated adenocarcinomas. Moreover, neutralizing VEGF may be doubly relevant in NSCLC with K-ras mutations, allowing the blockade of both VEGF-VEGFR1 and -VEGFR2 molecular pathways. Recently, VEGF-VEGFR1 signaling pathway has been shown to contribute to tumor survival and metastasis.31,41
We also showed here that the local delivery of G6–31 through the pulmonary route achieved an antitumor response in K-rasLA1 mice. The pulmonary delivery of G6–31 was well tolerated and significantly reduced the number of tumor lesions. Neutralization of VEGF by aerosolized G6–31 inhibited tumor proliferation, angiogenesis and blocked VEGF/VEGFR2 molecular pathways. Although the systemic delivery of G6–31 gave similar results, the effects on blood vessels differed according to the route of administration. G6–31 delivered through the pulmonary route correlated with a significant reduction in the number of small vessels, whereas systemic injection mostly affected large vessels. Moreover, downstream mediators of VEGF/VEGFR2 (PI3K/AKT), and apoptosis were also differently modulated according to the delivery route, suggesting the involvement of several signaling pathways in the pharmacodynamics of G6–31 dependent on the route of administration.
The PK study of anti-VEGF mAb revealed significant differences between the local and systemic routes. Non-compartmental and compartmental analyses estimated a low value of bio-availability of G6–31 in serum after pulmonary delivery, in agreement with previous results.14 This poor passage into the bloodstream after pulmonary delivery is advantageous because of the pulmonary localization of the multiple aberrant foci. We found that the non-compartmental analysis was not relevant for estimating the PK parameters, and we used a compartmental approach to take into account the nonlinear elimination of G6–31. Passage into the circulation may be related to FcRn-mediated active transport and lymphatic drainage, as described previously.14,42,43 A linear elimination of mAbs targeting soluble antigens is generally observed,44 whereas those targeting membrane-bound antigen often display nonlinear kinetics mainly due to target-mediated drug disposition. We found that the kinetics of G6–31 elimination were nonlinear, and its estimated parameters were similar to mAbs with target mediated elimination. This may be explained by the high affinity of G6–31 for its target antigen VEGF-A.18 By reducing systemic exposure, the pulmonary route may reduce systemic adverse side effects. However, the low passage of the drug into the bloodstream may also limit the therapeutic effect of anti-VEGF mAb on extra-pulmonary metastasis.
In conclusion, our results show the antitumor efficacy of G6–31 in K-rasLA1 visualized by a blood vessel regression and a decrease in tumor growth rate. This antitumor effect occurs through inhibition of angiogenic and non-angiogenic mechanisms related to the VEGF/VEGFR pathway.31 Our findings highlight the clinical value of anti-VEGF inhibitors for patients with K-ras mutated lung adenocarcinoma. They also emphasize the relevance of using local delivery of mAbs through the pulmonary route to treat lung adenocarcinomas.
Material and Methods
Antibodies
Murine anti-human/mouse VEGF-A G6–31 (batch PUR21821, 8 mg/mL), was kindly provided by Genentech (MTA OR-209 778). IgG2a,k from murine myeloma that was used as a control isotype antibody, was purchased from Sigma Aldrich (St Quentin Fallavier, France). For immunohistochemistry, we purchased rabbit polyclonal anti human/mouse CC3 (Asp175) (Cell Signaling Technology, Ozyme, St Quentin en Yvelines, France), anti-human/mouse vWF (DakoCytomation, Trappes, France), anti-human/mouse VEGF (SC-507, Santa Cruz, Tebu-Bio SA, Le Perray-en-Yvelines, France), rat mAbs against F4/80 (Interchim, Montluçon, France) and mouse anti-HIF1α [H1alpha67] - ChIP Grade (ab1) (Abcam, Paris, France).
For western blotting, we used the following primary antibodies: rabbit monoclonal anti-human/mouse phospho-VEGF receptor 2 (Tyr 1175), anti-human/mouse VEGF receptor 2, anti-human/mouse phospho-AKT (Ser 473), rabbit polyclonal anti-human/mouse AKT, rabbit polyclonal anti-mouse phospho-ERK1/2 (T202/Y204), rabbit polyclonal anti-human/mouse ERK1/2 and anti-mouse phospho-PI3K (Tyr 458)/p55 (Tyr 199) (Cell Signaling Technology), rabbit polyclonal anti-human/mouse active CC3 and rabbit monoclonal anti-human/mouse HIF-1α from Novus Biologicals (Interchim, France), rabbit polyclonal anti-human/mouse PCNA (proliferating cell nuclear antigen) and rabbit polyclonal anti-human/mouse β-actin from Sigma Aldrich. Secondary antibodies for western blotting were peroxidase-conjugated Affini-Pure goat anti-rabbit IgG (H+L) and peroxidase-conjugated Affini-Pure goat anti-mouse IgG (H+L) from Jackson ImmunoResearch (Interchim).
Cell culture
Human umbilical vein endothelial cells (HUVEC) were grown according to the manufacturer's instructions (PromoCell GmbH, Heidelberg, Germany). HUVECs were seeded in 6-well plates and maintained up to 80% confluence. Native or aerosolized G6–31 at 1, 5 or 10 nM in 1X PBS was pre-incubated with 0.5 nM of human VEGF-A (Cell Signaling) for 60 min, and then added to the cells. After 7 min incubation at 37 °C, cells were lysed in RIPA buffer and VEGFR 2 phosphorylation (Phospho-VEGFR 2 Tyr 1175 and total VEGFR Ab from Cell Signaling) was analyzed by western blotting.
Animal experiments
Animals
K-rasLA1 or wild-type mice with the same genetic background (C57BL/6N) were housed and handled according to the guidelines from the European Animal Care and Use Committee (agreement #2010/11), and body weight was measured weekly. K-rasLA1 is a murine model of lung adenocarcinoma with K-ras mutations.15 The K-rasLA1 mice were provided by the NCI Mouse Models of Human Cancers Consortium (MMHCC) (NCI Mouse Repository/NIH, Rockville, MD). These mice carry a latent K-ras allele with two copies of exon 1: one wild-type copy and one copy with the G12D mutation. The latent allele is stochastically activated in cells through homologous recombination, which results in the deletion of the wild-type copy of exon 1 and the expression of an oncogenic form of the K-ras gene. Substantial epithelial changes were observed at 4–8 wk, and included atypical adenomatous hyperplasia and small adenomas, followed by adenomas with papillary or atypical features, and finally well-differentiated adenocarcinomas at 6–8 mo.6,15 All animals were censored for survival based either on mortality or pre-determined morbidity criteria for euthanasia. Mice were monitored three times a week by an investigator who was blind to the data and were euthanized if they showed signs of respiratory distress which included hunching, ruffled fur, belabored breathing, low body temperature, lack of mobility, lethargy, > 20% decrease in body weight, or abdominal distension, according to the standards of the Institutional Animal Care and Use Committee.
Tolerance of aerosolized G6–31 in normal mice
Sixteen week-old wild-type mice were anesthetized with anesthetic gases and received, either G6–31 (10 mg/kg) or NaCl 0.9%, by the pulmonary route (aerosol) administered with a MicroSprayer® Aerosolizer - Model IA-1C - connected to a FMJ-250 High Pressure Syringe (Penn Century, Philadelphia, PA). The mice received this treatment once a week for 4 wk (n = 15 per group). At the end of the experiment, mice were killed and lungs, liver, spleen and kidneys were collected for histological and biochemical studies.
Antitumor efficacy of repeated doses of G6–31 when administrated by the intra-peritoneal or aerosol route
Sixteen week-old K-rasLA1 mice received either G6–31 (2.5 or 10 mg/ kg) or a control isotype mouse IgG2a,k (2.5 or 10 mg/kg), once a week for 4 wk (n = 15 per group). G6–31 or IgG2a,k were administrated by i.p. injection or pulmonary route. Visible surface lung lesions were counted during autopsy by investigators who were blind to the treatment group. After perfusion with NaCl 0.9%, one lung was snap-frozen to extract protein and the other one was fixed in 4% formaldehyde and embedded in paraffin for histological and immunohistochemistry studies as described previously.6
Collection of blood for pharmacokinetics from mice that received G6–31 by the intravenous or aerosol route
A PK study was conducted in 16-wk-old K-rasLA1mice. At this age, multiple aberrant lung foci have already developed.6,15 A unique dose of G6–31 (10 mg/kg) was administrated by either the i.v. or the pulmonary route (n = 15 per group). Blood samples (100–200 μL) were collected submandibularly, at different times after G6–31 delivery: 0, 2, 6, 24, 48, 72 h, 7, 14, 21 and 28 d. Concentrations of G6–31 in sera were measured by quantitative ELISA and PK of G6–31 was studied by non-compartmental and compartmental approaches.
Histological analysis
All lesions were identified by histological analysis on hematoxylin and eosin (H&E) staining. According to the histological criteria established by Johnson et al.,15 two investigators blind to the treatment group counted atypical adenomatous hyperplasias, adenomas and adenocarcinomas from one H&E section per lung for each mouse at × 20 magnification. Results were given as microscopic lesion count (mean+/−SEM) for all lesions and for each histological subtype.
Immunohistochemical analysis
4-μm sections were deparaffinized, rehydrated, and washed with PBS pH7.4. The VEGF, CC3, F4/80 and HIF1α antigens were retrieved with 0.01 M citrate buffer (pH 6) (DakoCytomation) for 30 min in a steamer. Slides were exposed for 10 min to protease 1 (Ventana, Roche Diagnostics, Meylan, France) to detect vWF. Samples were blocked for endogenous activity in 3% hydrogen peroxide-PBS, avidin/biotin blocking kit (Invitrogen, Life Technologies, Saint-Aubin, France), and Protein Block serum-free (Dakocytomation), and were then incubated with the primary antibody overnight at 4 °C. Standard avidin–biotin immunoperoxidase methods, with diaminobenzine as the chromogen, were used for detection. The primary antibody was omitted as a negative control.
We used a quantitative approach to score CC3 and F4/80 staining. We counted positive intra-lesional cells for the whole tumor section and expressed this number as the number of positive cells per tumor-section area. Tissues were visualized at × 20 magnificationto score all antigens, except for CC3, which was scored from tissue visualized at × 40 magnification.
For the quantification of microvascular density, we evaluated the total number of vWF positive sections in the whole tumor section and discriminated small (< 10 μm) from large (> 10 μm) vessels.20,21 Microvascular density was expressed as the percentage of vessels per tumor-section area.
Western blot analysis
Lysates from lung tissue samples were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride nitrocellulose membrane (Invitrogen). After saturation, membranes were immunoblotted, overnight at 4 °C or for 2 h at room temperature, with primary antibodies in TBS containing 5% nonfat dry milk or 5% bovine serum albumin (BSA) for Cell Signaling antibodies. The membranes were washed and then incubated with secondary antibody for 1 h. Antibody binding was detected with an enhanced chemiluminescence kit according to the manufacturer's instructions (Super Signal West Pico ChemiluminescentSubstrat, Pierce, Illkirch, France). The films were subsequently scanned, and band intensity was quantified with densitometry software (Image J, NIH).
Measurement of G6–31 concentrations by a quantitative ELISA
The concentration of G6–31 antibodies in sera was determined by a quantitative ELISA, especially developed for this study. Microplates (NuncMaxisorpimmunoplate, Thermo Scientific, Courtaboeuf, France) were coated with 7.5 ng of recombinant human VEGF-A (Ozyme, St Quentin Yvelines, France), diluted in 100 μL of 1 M carbonate buffer pH 9.6 and were incubated overnight at 4 °C. After three washes with PBS pH 7.4 containing 0.05% Tween 20, the plates were incubated for 90 min at 37 °C with 1% of BSA) (Sigma, St Quentin Fallavier, France) prepared in PBS to block unsaturated binding sites (200 μL/well). After one wash, serum samples and standards (dilutions of G6–31) were distributed (100 μL/well) in the wells and incubated for 60 min at 37 °C. Serum samples were initially diluted from 1:25 to 1:32000 in PBS containing 1% BSA (PBS-BSA). Standard dilutions of G6–31 were serially diluted from 125 to 0.25 ng/mL in PBS-BSA. After five washes, a peroxidase-conjugated rat anti-mouse IgG2a polyclonal antibody (BD Biosciences, Le Pont de Claix, France) was added (final dilution 1:10000, 100 μL/well) and incubated for 60 min at 37 °C. Peroxidase activity was detected with 100 μL of freshly prepared SigmaFastTM OPD (Sigma, St Quentin Fallavier, France) substrate added to every well. After a 10 min incubation at room temperature, the reaction was stopped by the addition of 50 μL/well of 4 N H2SO4. After incubation for 30 min at room temperature, absorbance was measured at 490 nm with an automated microplate reader (Versa Max, Molecular Devices, SaintGregoire, France).
Serum G6–31 concentration was calculated from the linear part of the standard curve. The lower limit of quantification (LLOQ) was 0.114 ng/mL and the lower limit of detection (LLD) was 0.069 ng/mL. Correction was made for serum dilution and G6–31 concentration was expressed in μg/mL of serum. For each microtiter plate, the assay included antigen, sera and enzyme conjugate controls.
Pharmacokinetic analysis
The non-compartmental approach describes the PK profiles in terms of area under the concentration-time curve (AUC0→∞ in mg·L−1·day), area under the first moment concentration-time curve (AUMC in mg·L−1·day2), mean residence time (MRT in day), elimination half-life (t1/2 in day), mean absorption time (MAT in day) and bio-available fraction (F in%). This analysis was performed using R (version 3.0.1, Vienna, Austria.45
The compartmental approach consisted of population PK modeling, with Monolix (version 4.2.2), LixsoftOrsay, France.46 This method describes the concentrations measured after both routes of administration with the same model, optimizes the estimation of common parameters, and allows the calculation of the fraction absorbed after pulmonary delivery (F). Analysis of the data revealed that G6–31 elimination is accelerated at low concentrations. One- and two-compartment models with first-order or saturable elimination from the central compartment were tested to describe this nonlinearity. A Michaelis–Menten equation with maximum elimination rate (Vmax) and the Michaelis–Menten constant (kM) was used to describe the saturable component of the elimination of G6–31. The other PK parameters were the central volume of distribution (V1) and systemic elimination (k10) and distribution (k12, k21) rates. A first-order absorption rate (ka) was also used for the pulmonary route.
Statistical analysis
The differences between groups (mean ± SEM) were compared with the non-parametric Mann-Whitney test. P values < 0.05 (*) were considered statistically significant (GraphPad Prism 5.0 software).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank PST Animaleries (Université de Tours, France) and Caroline Martin and Jessy Renciot from INSERM U702 Tenon Hospital Animal Facility (France) for technical assistance and advice. We also thank M. Julien Taurelle and JérômeAmiaud (INSERM UMR957, Nantes, France) for their technical advices and kind assistance during immunohistochemistry analyses.
Funding
This work was supported by various sources of funding. NHV received financial support from the French Higher Education and Research Ministry under the program “Investissementsd’avenir,” Grant agreement: LabExMAbImprove ANR-10-LABX-53, from ARAIR (Tours, France), Canceropôle Grand Ouest and Région Centre (Grant Stabiomed). MW received Grants from the Legs Poix 2011 Chancellerie des Universités de Paris. MD was supported by Le Fonds de Dotation “Recherche en Santé Respiratoire.”
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Cadranel J, Mauguen A, Faller M, Zalcman G, Buisine MP, Westeel V, Longchampt E, Wislez M, Coudert B, Daniel C, et al. . Impact of systematic EGFR and KRAS mutation evaluation on progression-free survival and overall survival in patients with advanced non-small-cell lung cancer treated by erlotinib in a French prospective cohort (ERMETIC project–part 2). J ThoracOncol 2012; 7:1490-502; PMID:22982650; http://dx.doi.org/ 10.1097/JTO.0b013e318265b2b5 [DOI] [PubMed] [Google Scholar]
- 2.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. . Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947-57; PMID:19692680; http://dx.doi.org/ 10.1056/NEJMoa0810699 [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. . Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368:2385-94; PMID:23724913; http://dx.doi.org/ 10.1056/NEJMoa1214886 [DOI] [PubMed] [Google Scholar]
- 4.Binder D, Hegenbarth K. Emerging options for the management of non-small cell lung cancer. Clin Med Insights Oncol 2013; 7:221-34; PMID:24179413; http://dx.doi.org/ 10.4137/CMO.S10269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konishi T, Huang CL, Adachi M, Taki T, Inufusa H, Kodama K, Kohno N, Miyake M. The K-ras gene regulates vascular endothelial growth factor gene expression in non-small cell lung cancers. Int J Oncol 2000; 16:501-11; PMID:10675482 [DOI] [PubMed] [Google Scholar]
- 6.Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, Tang H, Burdick MD, Sato M, Minna JD, et al. . High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res 2006; 66:4198-207; PMID:16618742; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3842 [DOI] [PubMed] [Google Scholar]
- 7.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542-50; PMID:17167137; http://dx.doi.org/ 10.1056/NEJMoa061884 [DOI] [PubMed] [Google Scholar]
- 8.Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res 1997; 57:4593-9; PMID:9377574 [PubMed] [Google Scholar]
- 9.Ferrara N, Gerber HP, Le Couter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669-76; PMID:12778165; http://dx.doi.org/ 10.1038/nm0603-669 [DOI] [PubMed] [Google Scholar]
- 10.Takahashi S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull 2011; 34:1785-8; PMID:22130231; http://dx.doi.org/ 10.1248/bpb.34.1785 [DOI] [PubMed] [Google Scholar]
- 11.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J BiolChem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 12.Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, Maleeff BE, Eustis S, Schwartz LW, Tsui P, Appelbaum ER, et al. . Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Allergy ClinImmunol 2001; 108:250-7; PMID:11496242; http://dx.doi.org/ 10.1067/mai.2001.116576 [DOI] [PubMed] [Google Scholar]
- 13.Maillet A, Congy-Jolivet N, Le Guellec S, Vecellio L, Hamard S, Courty Y, Courtois A, Gauthier F, Diot P, Thibault G, et al. . Aerodynamical, immunological and pharmacological properties of the anticancer antibody cetuximab following nebulization. PharmRes 2008; 25:1318-26; PMID:18030605; http://dx.doi.org/ 10.1007/s11095-007-9481-3 [DOI] [PubMed] [Google Scholar]
- 14.Maillet A, Guilleminault L, Lemarié E, Lerondel S, Azzopardi N, Montharu J, Congy-Jolivet N, Reverdiau P, Legrain B, Parent C, et al. . The airways, a novel route for delivering monoclonal antibodies to treat lung tumors. Pharm Res 2011; 28:2147-56; PMID:21491145; http://dx.doi.org/ 10.1007/s11095-011-0442-5 [DOI] [PubMed] [Google Scholar]
- 15.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001; 410:1111-6; PMID:11323676; http://dx.doi.org/ 10.1038/35074129 [DOI] [PubMed] [Google Scholar]
- 16.Gerber HP, Wu X, Yu L, Wiesmann C, Liang XH, Lee CV, Fuh G, Olsson C, Damico L, Xie D, et al. . Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. ProcNatlAcadSci U S A 2007; 104:3478-83; PMID:17360669; http://dx.doi.org/ 10.1073/pnas.0611492104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korsisaari N, Ross J, Wu X, Kowanetz M, Pal N, Hall L, Eastham-Anderson J, Forrest WF, Van Bruggen N, Peale FV, et al. . Blocking vascular endothelial growth factor-A inhibits the growth of pituitary adenomas and lowers serum prolactin level in a mouse model of multiple endocrine neoplasia type 1. Clin Cancer Res 2008; 14:249-58; PMID:18172277; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1552 [DOI] [PubMed] [Google Scholar]
- 18.Liang WC, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J, Fu L, Malik AK, Gerber HP, Ferrara N, et al. . Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J BiolChem 2006; 281:951-61; PMID:16278208; http://dx.doi.org/ 10.1074/jbc.M508199200 [DOI] [PubMed] [Google Scholar]
- 19.Yu L, Wu X, Cheng Z, Lee CV, Le Couter J, Campa C, Fuh G, Lowman H, Ferrara N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci 2008; 49:522-7; PMID:18234994; http://dx.doi.org/ 10.1167/iovs.07-1175 [DOI] [PubMed] [Google Scholar]
- 20.Glaw JT, Skalak TC, Peirce SM. Inhibition of canonical Wnt signaling increases microvascular hemorrhaging and venular remodeling in adult rats. Microcirculation 2010; 17:348-57; PMID:20618692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko HC, Milthorpe BK, Mc Farland CD. Engineering thick tissues–the vascularisation problem. Eur Cell Mater 2007; 14:1-18, discussion 18-9; PMID:17654452 [DOI] [PubMed] [Google Scholar]
- 22.Ban HS, Uno M, Nakamura H. Suppression of hypoxia-induced HIF-1alpha accumulation by VEGFR inhibitors: Different profiles of AAL993 versus SU5416 and KRN633. Cancer Lett 2010; 296:17-26; PMID:20378243; http://dx.doi.org/ 10.1016/j.canlet.2010.03.010 [DOI] [PubMed] [Google Scholar]
- 23.De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev ClinOncol 2011; 8:393-404; PMID:21629216; http://dx.doi.org/ 10.1038/nrclinonc.2011.83 [DOI] [PubMed] [Google Scholar]
- 24.Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM, Brown AS, Hicklin DJ, Foster FS, Kerbel RS. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res 2006; 66:3639-48; PMID:16585189; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3295 [DOI] [PubMed] [Google Scholar]
- 25.Rapisarda A, Shoemaker RH, Melillo G. Antiangiogenic agents and HIF-1 inhibitors meet at the crossroads. Cell Cycle 2009; 8:4040-3; PMID:19923892; http://dx.doi.org/ 10.4161/cc.8.24.10145 [DOI] [PubMed] [Google Scholar]
- 26.Wu JM, Staton CA. Anti-angiogenic drug discovery: lessons from the past and thoughts for the future. Expert Opin Drug Discov 2012; 7:723-43; PMID:22716277; http://dx.doi.org/ 10.1517/17460441.2012.695774 [DOI] [PubMed] [Google Scholar]
- 27.Rapisarda A, Melillo G. Role of the VEGF/VEGFR axis in cancer biology and therapy. Adv Cancer Res 2012; 114:237-67; PMID:22588059; http://dx.doi.org/ 10.1016/B978-0-12-386503-8.00006-5 [DOI] [PubMed] [Google Scholar]
- 28.Jiang T, Zhuang J, Duan H, Luo Y, Zeng Q, Fan K, Yan H, Lu D, Ye Z, Hao J, et al. . CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 2012; 120:2330-9; PMID:22718841; http://dx.doi.org/ 10.1182/blood-2012-01-406108 [DOI] [PubMed] [Google Scholar]
- 29.Belmont L, Rabbe N, Antoine M, Cathelin D, Guignabert C, Kurie J, Cadranel J, Wislez M. Expression of TLR9 in tumor-infiltrating mononuclear cells enhances angiogenesis and is associated with a worse survival in lung cancer. Int J Cancer 2014; 134:765-77; PMID:23913633; http://dx.doi.org/ 10.1002/ijc.28413 [DOI] [PubMed] [Google Scholar]
- 30.Jackson AL, Zhou B, Kim WY. HIF, hypoxia and the role of angiogenesis in non-small cell lung cancer. Expert OpinTher Targets 2010; 14:1047-57; PMID:20854179; http://dx.doi.org/ 10.1517/14728222.2010.511617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Epstein RJ. VEGF signaling inhibitors: more pro-apoptotic than anti-angiogenic. Cancer Metastasis Rev 2007; 26:443-52; PMID:17786538; http://dx.doi.org/ 10.1007/s10555-007-9071-1 [DOI] [PubMed] [Google Scholar]
- 32.Bonnesen B, Pappot H, Holmstav J, Skov BG. Vascular endothelial growth factor A and vascular endothelial growth factor receptor 2 expression in non-small cell lung cancer patients: relation to prognosis. Lung Cancer 2009; 66:314-8; PMID:19324448; http://dx.doi.org/ 10.1016/j.lungcan.2009.02.013 [DOI] [PubMed] [Google Scholar]
- 33.Kerbel RS. Tumor angiogenesis. N Engl J Med 2008; 358:2039-49; PMID:18463380; http://dx.doi.org/ 10.1056/NEJMra0706596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casulli S, Topçu S, Fattoum L, von Gunten S, Simon HU, Teillaud JL, Bayry J, Kaveri SV, Elbim C. A differential concentration-dependent effect of IVIg on neutrophil functions: relevance for anti-microbial and anti-inflammatory mechanisms. PLoS One 2011; 6:e26469; PMID:22065996; http://dx.doi.org/ 10.1371/journal.pone.0026469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Baumgarten L, Brucker D, Tirniceru A, Kienast Y, Grau S, Burgold S, Herms J, Winkler F. Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin Cancer Res 2011; 17:6192-205; PMID:21788357; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1868 [DOI] [PubMed] [Google Scholar]
- 36.Pushkarev VM, Starenki DV, Saenko VA, Pushkarev VV, Kovzun OI, Tronko MD, Popadiuk ID, Yamashita S. Differential effects of low and high doses of Taxol in anaplastic thyroid cancer cells: possible implication of the Pin1 prolyl isomerase. ExpOncol 2008; 30:190-4; PMID:18806740 [PubMed] [Google Scholar]
- 37.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, et al. . Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 2012; 104:228-39; PMID:22247021; http://dx.doi.org/ 10.1093/jnci/djr523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592-603; PMID:18650835; http://dx.doi.org/ 10.1038/nrc2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. ProcNatlAcadSci U S A 2007; 104:17069-74; PMID:17942672; http://dx.doi.org/ 10.1073/pnas.0708148104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moserle L, Casanovas O. Exploiting pleiotropic activities of semaphorins as multi-target therapies for cancer. EMBO Mol Med 2012; 4:168-70; PMID:22323445; http://dx.doi.org/ 10.1002/emmm.201200206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Xu J, Wang M, Wang Q, Bi Y, Han M. Tumor-derived vascular endothelial growth factor (VEGF)-a facilitates tumor metastasis through the VEGF-VEGFR1 signaling pathway. Int J Oncol 2011; 39:1213-20; PMID:21785819 [DOI] [PubMed] [Google Scholar]
- 42.Bitonti AJ, Dumont JA. Pulmonary administration of therapeutic proteins using an immunoglobulin transport pathway. Adv Drug Deliv Rev 2006; 58:1106-18; PMID:16997417; http://dx.doi.org/ 10.1016/j.addr.2006.07.015 [DOI] [PubMed] [Google Scholar]
- 43.Dumont JA, Bitonti AJ, Clark D, Evans S, Pickford M, Newman SP. Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway. J Aerosol Med 2005; 18:294-303; PMID:16181004; http://dx.doi.org/ 10.1089/jam.2005.18.294 [DOI] [PubMed] [Google Scholar]
- 44.Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. ClinPharmacokinet 2013; 52:83-124; PMID:23299465; http://dx.doi.org/ 10.1007/s40262-012-0027-4 [DOI] [PubMed] [Google Scholar]
- 45.RCoreTeam Computational Many-Particle Physics. In: Fehske H, Schneider R, Weiße A, eds.: Springer Berlin Heidelberg, 2008. [Google Scholar]
- 46.Bauer RJ, Guzy S, Ng C. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J 2007; 9:E60-83; PMID:17408237; http://dx.doi.org/ 10.1208/aapsj0901007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.