

Abstract
Cytoplasmic chloroplast (cp) genomes and nuclear ribosomal DNA (nR) are the primary sequences used to understand plant diversity and evolution. We introduce a high-throughput method to simultaneously obtain complete cp and nR sequences using Illumina platform whole-genome sequence. We applied the method to 30 rice specimens belonging to nine Oryza species. Concurrent phylogenomic analysis using cp and nR of several of specimens of the same Oryza AA genome species provides insight into the evolution and domestication of cultivated rice, clarifying three ambiguous but important issues in the evolution of wild Oryza species. First, cp-based trees clearly classify each lineage but can be biased by inter-subspecies cross-hybridization events during speciation. Second, O. glumaepatula, a South American wild rice, includes two cytoplasm types, one of which is derived from a recent interspecies hybridization with O. longistminata. Third, the Australian O. rufipogan-type rice is a perennial form of O. meridionalis.
Plant cells contain three genomes with different evolutionary origins and history: nuclear, mitochondrial and chloroplastic. Chloroplast (cp) genomes and nuclear ribosomal DNA (nR) units are the primary sequences used to analyze plant genetic diversity as well as evolution1,2 because mitochondrion (mt) genomes show hyper-variable structure in plant genomes3. The cp genomes are 120- to 217-kb circular DNA molecules containing ~30 conserved genes and relatively diverse intergenic spaces (IGSs)4,5,6,7,8,9, and maintained uni-parentally, usually via maternal inheritance10,11. Within plant nuclear genomes, nR is organized into highly abundant tandemly-repeated transcription units12. Due to their conserved roles in ribosome assembly and nucleolus formation, these high-copy nR units have remained homogeneous through concerted genome evolution within species. Four nR gene components usually reside in two independent chromosomal locations, namely the 5S nR (5S) and 45S nR (45S) blocks in higher plants, although some ancient plants such as Ginkgo biloba, moss, and algae maintain the 5S and 45S components in one tandem unit13,14. The 45S blocks include tandemly arrayed copies of the 45S cistron unit, which comprises conserved 18S, 5.8S, and 26S gene clusters, relatively variable internal transcribed spacers (ITS1 and ITS2), and IGSs13,15.
Although next-generation sequencing (NGS) technology has enabled remarkable progress in understanding nuclear genomics, sequencing of cp genomes and nR units has remained in challenging due to their high-copy characteristics. Whereas more than 500 complete cp genome sequences have been reported in GenBank, complete 45S unit sequences are known for only a few species. Most reported cp genome sequences have been obtained by conventional methods16,17 but recently, several studies have utilized NGS platforms to obtain complete cp genome sequences using isolated chloroplast DNA or reference cp-guided mapping, followed by significant efforts to fill gaps using PCR and Sanger sequencing18,19,20,21,22,23. Recently, there has been some improvement of efficiency in obtaining complete cp genome and nR sequences by optimization of reference-guided mapping using several criteria and also a computing program24,25,26,27. Reference-guided mapping is good approach for studies of related species with the known reference sequences. However, if related reference genome sequences are lacking, de novo assembly will be the best way to obtain the complete sequences.
Plant whole-genome shotgun (WGS) sequence data produced by NGS technologies always contains cp sequences to various levels, depending on tissue types and extraction methods used for DNA preparation. Here, we have developed an efficient method, named de novo assembly of low coverage WGS (dnaLCW), to assemble those short NGS reads to simultaneously obtain high-quality complete cp genome and nR units. We provide solutions for gap-filling and error correction in sequence assembly without additional efforts such as PCR and Sanger sequencing. We successfully generated new complete cp genome and nR unit sequences for more than 50 species/cultivars with a range of genome sizes. This method greatly facilitates the use of highly informative plastome and nR data to elucidate the evolution of land plants. We have applied this method to our own NGS sequences as well as to publically available NGS sequences for Oryza species.
Rice is the most important staple crop for human consumption worldwide. Cultivated rice includes three species/subspecies, Oryza sativa ssp. japonica (North Asian rice), O. sativa ssp. indica (South Asian rice), and O. glaberrima (African rice), all of which contain the Oryza AA-type nuclear genome. The Oryza AA genome group consists of eight diploid species distributed worldwide: O. barthii (Africa), O. glaberrima (Africa), O. glumaepatula (South America), O. longistaminata (Africa), O. meridionalis (Australia), O. nivara (Asia), O. rufipogon (Asia and Australia), and O. sativa (Asia and now worldwide)28. The AA genome group is estimated to have diverged 2–3 million years ago (MYA)28,29, and the eight species likely evolved with unique adaptive traits for each native region over time28,30. Although molecular data have improved our understanding of the phylogenetic relationships and evolutionary history of the Oryza AA genome group, the taxonomical classifications and evolutionary relationships for all eight species in the group have not been not fully resolved.
Here, we report cp and nR sequences for 30 Oryza accessions and describe the phylogenomic relationship of wild and cultivated species of the Oryza AA genome. We also discuss the origin of cultivated rice and some ambiguous issues for classification and evolution of wild Oryza AA genome species.
Results
De novo assembly of low coverage WGS
We used rice reference cultivar ‘Nipponbare’ (NP)31 in order to test whether high-copy components such as cp, mt, and nR sequences could be assembled from low-coverage WGS data. In de novo assemblies of rice 1x haploid genome-equivalent WGS data, among the 30 longest assembled contigs were 5, 15, and 1 contigs representing cp, mitochondrial (mt), and nR sequences, respectively, with the remaining 9 contigs representing major repeats, mainly transposable elements (TEs) (Fig. 1a and Supplementary Table S1 online). Importantly, the five cp contigs covered the entire 134,551-bp cp genome with approximately 20-bp overlap between adjacent contigs (Fig. 1b). One 6,889-bp contig covered most of the 45S nR unit (i.e. 86%), while 15 contigs (summing to 130 kb) provided partial coverage of the mt genome (i.e. 26%). Similar results were obtained from de novo assembly of 151.5 Mbp Panax ginseng (ginseng) WGS data (0.05x whole genome coverage) where 3, 12, and 1 contigs represented cp, mt, and nR sequences, respectively, and the remaining 14 contigs were classified as unknown (Fig. 1a, and Supplementary Table S2 online). The complete cp genome was covered by three contigs that overlapped slightly (Fig. 1c) and 10 kb mate-pair read mapping showed that the three contigs were ordered properly (Fig. 1d). One 9,423-bp contig represented the 45S unit and 12 contigs (38 kb) represented the mt genome.
Figure 1. Characterization of the 30 longest contigs derived from de novo genome assembly using 1x and 0.05x haploid genome equivalents of rice and ginseng, respectively.
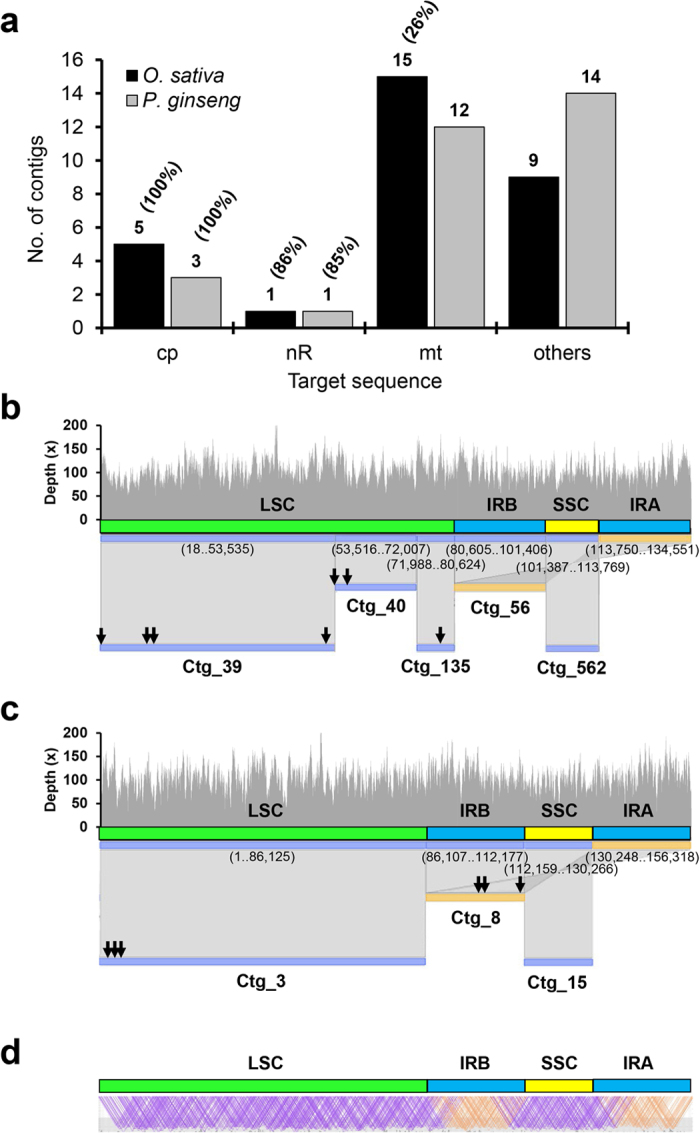
(a) Classification based on best hit (Supplementary Tables S1 and S2 online). Number of contigs and percent coverage of cp, nR, mt and other sequences are presented above the bars. (b,c) Alignment of five and three contigs covering the complete cp genome sequences of rice (b) and ginseng (c), respectively. The contig numbers are indicated under the contigs and hit positions in parentheses are under the reference cp genome sequences for rice (GU592207) and ginseng (NC_006290). Sequence errors identified in the initial contigs are denoted by arrows. The overall structure of the cp genome is denoted with different colored bars: green, blue, and yellow, for LSC, IRs, and SSC, respectively. Mapping of 100x raw reads is shown above alignment. (d) Read mapping of 2x-depth 10-kb mate-pair reads on the assembled sequence. Purple and orange mate information indicates the proper range for 10-kb mate pairs.
Optimization of dnaLCW to obtain complete cp genome sequence
Because we obtained almost complete cp and nR sequences for rice and ginseng with 1x and 0.05x genome equivalent WGS data despite their different genome sizes (430 Mbp and 3,600 Mbp for rice and ginseng, respectively)31,32, we optimized the WGS dataset size needed to obtain complete cp genome assemblies. As NP and ginseng WGS reads included ~1.7 and 6.0% cp genome-derived reads, respectively, we extracted 10 WGS datasets with between 25x and 5,000x coverage of the cp genome for independent assembly (Supplementary Table S3 online).
We used the number of contigs covering the entire cp genome and the number of assembly errors as criteria for assessment of optimal assembly. Datasets 3–6 with 100x to 250x cp coverage, corresponding to 2–10 x haploid genome equivalents, for rice, showed the best assembly performance for cp genomes, whereas assembly errors and contig numbers in rice rapidly increased when NGS reads reached 20x whole genome coverage (~8.6 Gbp WGS sequence) (Supplementary Table S3 online). This suggests that, with higher amounts of rice input data, short NGS reads originating from nuclear or mitochondrial plastid DNAs (NMPTs; cp sequences inserted into the nuclear or mitochondrial genome) were erroneously co-assembled into cp contigs. The different assembly behavior with regard to input data could be attributable to rice having a higher NMPT content compared to ginseng in which number of assembly errors decreased as increase of cp coverage (Supplementary Table S3 online). Therefore, it is important to use the proper amount of data for assembly to minimize erroneous cp contigs caused by NMPTs.
We compared the performance of two popular genome assemblers, SOAPdenovo33 and the CLC de novo assembler (http://www.clcbio.com/products/clc-assembly-cell/), in generating small numbers of longer contigs to cover the entire cp genome using various WGS datasets of rice. The CLC de novo assembler outperformed SOAPdenovo (Supplementary Fig. S1 online).
Identification and correction of de novo assembly errors
We could construct a single circular draft cp genome by joining the initially assembled overlapping cp contigs. However, we identified several types of assembly errors, such as the positions denoted by arrows in Fig. 1b,c, by aligning PE reads onto assembled contigs. The mis-assembled regions were typically characterized by accumulation of discordantly mapped reads or abnormally higher read mapping depth. The identified assembly errors included false gaps, false SNPs, and copy number errors for TR or monopolymers. We developed detailed in silico methods for identification and correction of each type of error (see Materials and Methods, Supplementary Figs S2-S5 online). We obtained a complete 134,551-bp cp genome sequence for NP that was 100% identical to the reference cp sequence of NP (GU592207), using the dnaLCW approach followed by in silico correction of seven errors detected in the initial assembly.
Obtaining complete sequences for major nR units
The dnaLCW assembly also generated contigs representing the 5S and 45S nR units. The initial 5S contigs contained the complete 5S units of 324 bp and 898 bp for NP and ginseng, respectively (Supplementary Figs S6 and S7 online). By contrast, the 45S contig was represented as incomplete contigs longer than 6 kb, including the main 45S transcriptional unit and part of the flanking IGS. We developed a method to extend the IGS sequences based on the highly homogeneous tandemly arrayed nature of the 45S. We generated a two-unit 45S tandem array using the initial contig and manually inserted 100 unknown nucleotides, (N)100, between the two units for the remaining gaps in the IGS (Fig. 2). We then applied iterative gap closing to fill the gaps between the units using Gapcloser with the raw reads. Occasionally, GC-rich regions and sub-repeat elements in IGS made gap-filling ineffective (Fig. 2d,e); however, we successfully obtained representative complete 324-bp 5S and 7,928-bp 45S units from NP that were identical to the 5S and 45S tandem array found in chromosome 11 and 9, respectively (Supplementary Fig. S6 online)34.
Figure 2. Assembly of complete 45S units.
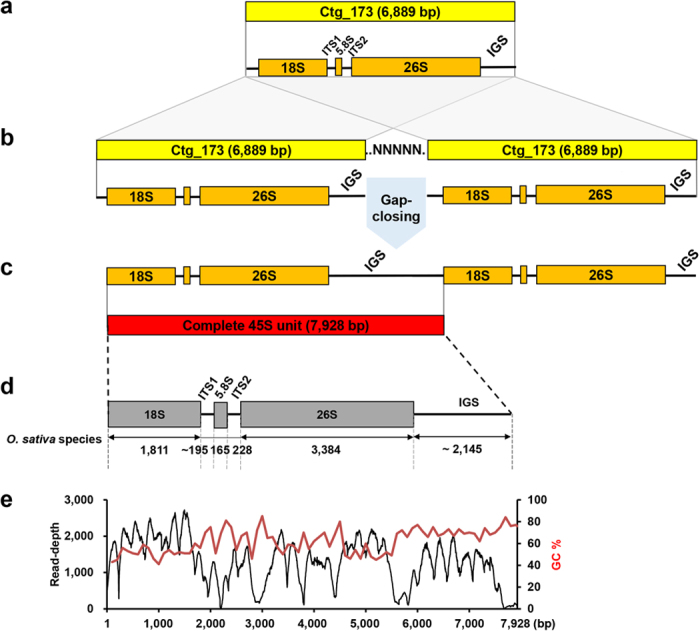
(a–c) Schematic diagram of the method used to obtain a complete 45S unit. (a) A draft single contig included the 45S transcription unit and occasionally part of the IGS. In this example, Ctg_173 assembled using a rice dataset contained a partial IGS. (b) To obtain the full-length IGS, a hypothetical tandem array was constructed using two copies of the contig and intervening Ns. Through a gap-closing process, the Ns were filled in by nucleotide sequences originating from IGS regions. (c) If the IGS remains partial, adjustment of the intervening N length and repeated gap-closing will be necessary. Ultimately, a complete 45S unit with the full-length IGS can be obtained. (d) Structure of the complete 45S unit of Oryza species. (e) Status of read mapping on the assembled 45S units. The Os5 dataset was mapped again to assembled single contigs covering the entire 45S unit sequence (black line). Red line indicates GC content per 100-bp unit length.
Complete cp and nR sequences of 30 Oryza species
We next applied our method to generate complete cp genome and nR sequences for additional 29 Oryza accessions, including five O. sativa cultivars (one japonica cultivar, two indica cultivars, and two cultivars derived from an indica x japonica hybrid) and 24 Oryza wild relatives using WGS data produced by four independent groups in Korea, USA, Australia, and China (Table 1, and Supplementary Figs S8 and S9 online)30,35,36,37. The cp genomes varied from 134,296 [O. glumaepatula (IRGC88793)] to 134,678 bp [O. barthii (WAB0028903, WAB0028952)] among the Oryza species and representative InDels between species are shown in Supplementary Fig. S10 online. The complete 5S units varied from 302 to 499 bp due to sequence divergence in the IGS, although the coding sequence was highly conserved (Table 1 and Supplementary Fig. S7 online). The 45S units were 7,745–8,190 bp and sequence variations were more frequent in the IGS region (Table 1 and Supplementary Fig. S11 online).
Table 1. Statistics for assembly of cp and nR sequences from 30 Oryza species.
| Species | Genome size (Mbp) | WGS reads for cp assembly |
Complete sequence (bp) |
Estimated copy numberb |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Amount (Mbp) | Coverage (x) |
|||||||||
| Genome | Cp | Cp | 45Sa | 5Sa | 45S | 5S | ||||
| 1 | O. sativa J. NP | 430 | 860 (SRR1178954c) | 2.0` | 99 | 134,551 (KM088016c) | 7,928 (KM036282c) | 324 (KM036298c) | 390 | 593 |
| 2 | O. sativa J. Yukara | 430 | 303 (SRR1182447) | 0.7 | 227 | 134,551 (KM088017) | 7,929 (KM036283) | 324 (KM036299) | 186 | 458 |
| 3 | O. sativa JxI. M23 | 460 | 303 (SRR1182807) | 0.7 | 147 | 134,551 (KM103382) | 8,160 (KM036287) | 322 (KM036303) | 131 | 971 |
| 4 | O. sativa IxJ. Tongil | 460 | 253 (SRR1182443) | 0.5 | 69 | 134,502 (KM103369) | 8,160 (KM036286) | 322 (KM036302) | 255 | 1,593 |
| 5 | O. sativa I. IR8 | 460 | 327 (SRR921498) | 0.7 | 251 | 134,502 (KM103367) | 8,166 (KM036284) | 322 (KM036300) | 140 | 746 |
| 6 | O. sativa I. TN1 | 460 | 303 (SRR921505) | 0.7 | 206 | 134,502 (KM103368) | 8,164 (KM036285) | 322 (KM036301) | 210 | 894 |
| 7 | O. rufipogon (W1943)36 | 439 | 500 (ERX096841) | 1.1 | 128 | 134,510 (KM103372) | 8,004 (KM117266) | 303 (KM036304) | 275 | 495 |
| 8 | O. rufipogon (AC11-1008369)37 | 38034 | 1,442 (SRX480817) | 3.8 | 245 | 134,586 (Dataset S1) | 6,090d (Dataset S2) | 322 (Dataset S3) | 793 | 2,718 |
| 9 | O. rufipogon (AC01-1002323)37 | 38034 | 712 (SRX480820) | 1.9 | 685 | 134,572 (Dataset S1) | 5,816d (Dataset S2) | 499 (Dataset S3) | 310 | 354 |
| 10 | O. nivara (IRGC88812)30 | 39535 | 571 (SRX809784) | 1.5 | 203 | 134,483 (Dataset S1) | 5,823d (Dataset S2) | 322 (Dataset S3) | 439 | 5,561 |
| 11 | O. nivara (IRGC100897) | 448 | 775 (SRR1264534) | 1.4 | 112 | 134,516 (KM088022) | 7,904 (KM036288) | 322 (KM036305) | 441 | 1,129 |
| 12 | O. barthii (WAB0028976) | 411 | 159 (SRX502175) | 0.4 | 91 | 134,585 (KM103378) | 7,835 (KM117256) | 325 (KM117247) | 667 | 4,418 |
| 13 | O. barthii (WAB0028903) | 411 | 437 (SRX502171) | 1.1 | 257 | 134,678 (KM103379) | 7,845 (KM117257) | 325 (KM117248) | 748 | 2,380 |
| 14 | O. barthii (WAB0028952) | 411 | 441 (SRX502173) | 1.1 | 319 | 134,678 (KM103380) | 7,845 (KM117258) | 325 (KM117249) | 771 | 1,291 |
| 15 | O. barthii (WAB0028987) | 411 | 163 (SRX502178) | 0.4 | 97 | 134,613 (KM103381) | 7,836 (KM117252) | 325 (KM117250) | 368 | 1,989 |
| 16 | O. barthii (IRGC101252)30 | 37635 | 1,113 (SRX809864) | 3.0 | 1,277 | 134,598 (Dataset S1) | 5,888d (Dataset S2) | 325 (Dataset S3) | 3,769 | 5,984 |
| 17 | O. barthii (IRGC100934) | 411 | 343 (SRX502164) | 0.8 | 113 | 134,598 (KM103371) | 7,836 (KM117253) | 325 (KM117251) | 630 | 4,414 |
| 18 | O. barthii (W1588) | 411 | 375 (SRR1264535) | 0.9 | 60 | 134,590 (KM088023) | 7,836 (KM036290) | 325 (KM036307) | 617 | 6,045 |
| 19 | O. glaberrima (IRGC104574) | 357 | 330 (SRX502311) | 0.9 | 343 | 134,586 (KM103377) | 7,836 (KM117254) | 325 (KM117246) | 198 | 1,820 |
| 20 | O. glaberrima (IRGC96717) | 357 | 188 (SRR1181643) | 0.5 | 50 | 134,598 (KM088021) | 7,836 (KM036289) | 325 (KM036306) | 455 | 3,670 |
| 21 | O. glaberrima (IRGC103486)30 | 37035 | 647 (SRX809780) | 1.7 | 218 | 134,614 (Dataset S1) | 5,899d (Dataset S2) | 325 (Dataset S3) | 773 | 6,447 |
| 22 | O. glaberrima (IRGC103937) | 357 | 268 (SRX502309) | 0.8 | 257 | 134,598 (KM103370) | 7,836 (KM117255) | 325 (KM117245) | 358 | 2,374 |
| 23 | O. glumaepatula (W1187) | 400 | 1,536 (SRR1997915) | 3.9 | 1,338 | 134,606 (KR364802) | 6,427d (KR364804) | 440 (KR364807) | 1,461 | 528 |
| 24 | O. glumaepatula (IRGC88793)30 | 36635 | 397 (SRX809892) | 1.1 | 91 | 134,296 (Dataset S1) | 5,841d (Dataset S2) | 440 (Dataset S3) | 554 | 278 |
| 25 | O. glumaepatula (W2201) | 400 | 1,728 (SRR1997912) | 4.3 | 1,446 | 134,575 (KR364803) | 5,830d (KR364805) | 440 (KR364806) | 2,292 | 1,107 |
| 26 | O. glumaepatula (GEN1233) | 464 | 253 (SRR1264537) | 0.5 | 78 | 134,575 (KM103374) | 8,074 (KM036292) | 460 (KM036309) | 598 | 191 |
| 27 | O. meridionalis (IRGC105298)30 | 38835 | 760 (SRX809898) | 2.0 | 270 | 134,555 (Dataset S1) | 5,839d (Dataset S2) | 499 (Dataset S3) | 1,367 | 731 |
| 28 | O. meridionalis (W2112) | 435 | 1,000 (SRR1264536) | 2.3 | 109 | 134,556 (KM103373) | 8,190 (KM036291) | 499 (KM036308) | 461 | 2,525 |
| 29 | O. longistaminata (IRGC110404) | 352 | 563 (SRR1264538) | 1.6 | 187 | 134,558 (KM088024) | 7,844 (KM036293) | 302 (KM036310) | 200 | 69 |
| 30 | O. punctata (IRGC105690) | 423 | 250 (SRR1264539) | 0.6 | 60 | 134,604 (KM103375) | 7,745 (KM036294) | 326 (KM036311) | 307 | 1,317 |
a The lengths of the most redundant and longest representative nR units are given for each species. The 45S transcription units were 5,769–5,783 bp long for Oryza species. We cannot rule out the presence of other nR units in each species because there is some variance in the length of the IGS.
bCopy numbers of 45S and 5S are based on the average depth of raw reads mapping to each sequence and were calculated based on the 1x haploid genome equivalent of raw reads.
cSRA and accession numbers of reads and assembled sequences deposited in GenBank.
dLength of 45S transcription units.
Phylogenomic analysis of cultivated rice including indica-japonica hybrid cultivars and their ancestors
We obtained cp and nR sequences for 30 accessions that belong to nine Oryza species. The nine Oryza species included all eight species belonging to the Oryza AA genome group, as well as O. punctata, the best outgroup Oryza BB-genome species for phylogenomic analysis of AA-genome species28,29.
The phylogenomic analyses based on the cp genomes and on 45S sequences agreed with each other for the most part, with the exception of the placement of japonica-indica hybrid M23 and one O. rufipogon accession (Fig. 3a,b). The cp genome-based tree clearly distinguished O. sativa subspecies japonica (NP, Yukara) and indica (IR8, TN1) from each other. Cultivars Tongil and M23, bred by crossing O. sativa ssp. japonica and indica, had cp genomes identical to the indica and japonica types, respectively, in accordance with their last maternal parent even though both show indica-like plant architecture and nuclear genome sequence (Fig. 3c,d, and Supplementary Figs. S8 and S9 online)38,39. In the 45S-based phylogenetic analysis, M23, the cultivar derived from japonica x indica hybridization, belonged to the indica group, in accord with its phenotype even though its cp genome was identical to those of japonica cultivars due to maternal inheritance36. Conversely, the O. rufipogon used in this study was grouped with japonica based on 45S, but with indica based on the cp genome (Fig. 3a,b).
Figure 3. Phylogenomic tree of cultivated rice in Asia and Africa with their putative ancestor species.

(a,b) Phylogenetic trees were built based on the complete cp genome (a) and 45S cistron sequences (b). O. sativa ssp. japonica and indica groups are denoted as J and I, respectively. Two cultivars, M23 (no. 3, red thick line) and Tongil (no. 4, blue thick line), derived from japonica x indica hybridization and vice versa are denoted as JxI and IxJ, respectively. Different species/subspecies are indicated with different colored labels. Lines connect the positions of each accession/cultivar in the two trees. Numbers in colored circles represent accessions labeled in Table 1. The phylogenetic tree was generated using Poisson correction and the neighbor-joining (NJ) method in MEGA6. Bootstrap values calculated for 1000 replicates are shown on the branches; the values less than 50% are not shown. (c,d) Pedigree of two cultivars, M23 (c) and Tongil (d), bred by crossing between O. sativa ssp japonica and indica38. Red and blue thick lines indicate final maternal genotype backgrounds for M23 and Tongil, respectively.
The African cultivated rice O. glaberrima and its wild relative O. barthi were grouped together by both cp and nR-based trees (Fig. 3a,b). It was previously reported that O. barthii accessions are divided into five independent subgroups (OB-I to OB-V) and O. glaberrima was domesticated from OB-V40. Our cp-based phylogeny showed that all the O. glaberrima accessions are grouped with the O. barthi OB-V group, and the 45S-based phylogeny is in accordance with the cp-based tree although the OB-IV and OB-V groups are not distinguished (Fig. 3b).
Phylogenomic analysis of wild Oryza AA genome species
Our concurrent analysis using cp and 45S-based phylogenomic analysis revealed that the cp-based trees can be biased by one event of inter-subspecies or inter-species cross-hybridization during speciation, which was shown by one artificially bred cultivar, M23 developed by hybridization between japonica and indica rice (Fig. 3). Accordingly, we next included several different accessions as representative of each species in analysis to clarify the phylogenomic relationships using cp as well as nR sequences. The topology of the cp-based tree was well in accordance with the 45S-based tree as well as the previous reports with some exceptions (Fig. 4). In addition, phylogenetic analysis based on the maximum likelihood (ML) method (Supplementary Fig. S12 online) produced a similar topology as that in Fig. 4. All accessions belonging to same species were placed into the same clade, with two exceptions. Three O. rufipogon specimens were positioned independently by both cp and 45S trees. Accession no. 7 (Chinese rufipogan) was intermingled with Asian cultivated rice species. Accession no. 8 (Vietnamese rufipogan) was intermediated between Asian and African cultivated rice species. Accession no. 9 (Australian rufipogan) was grouped with Australian wild rice, O. meridionalis. We also analyzed four different O. glumaepatula accessions, and the four South American wild rice accessions were grouped as two independent groups by the cp-based tree. Among four O. glumaepatula accessions, nos. 23 and 24 were placed between O. meridionalis (Australian AA) and O. barthi (African AA), however, accessions nos. 25 and 26 were grouped with the basal AA genome species O. longistaminata in the cp-based tree. However, all four accessions were grouped together and independently from O. longistaminata by the 45S tree.
Figure 4. Phylogenomic tree of Oryza species.

(a,b) Phylogenetic trees were built based on the complete cp genome (a) and 45S cistron sequences (b). O. sativa ssp. japonica and indica groups are denoted as J and I, respectively. Different species/subspecies are indicated with different colored labels. Numbers in colored circles represent accessions labeled in Table 1. Dashed lines connect the positions of each accession/cultivar in the two trees; red highlights major differences between trees. The tree was generated based on Bayesian Inference analysis using BEAST (version 1.8.1) as mentioned in Materials and Methods. Posterior probability (pp) above 0.5 are shown on the branches. Divergence time was calculated based on 9 million years ago (MYA) when Oryza species with AA and BB genome were estimated to be speciation29.
Discussion
The dnaLCW workflow for simultaneous determination of complete cp and nR sequences
Currently, most leading NGS-read assembly programs use a computational algorithm known as a de Bruijn graph, and 15 assemblers have been developed to improve genome assembly41. Repeat sequences generally hinder genome assembly; accordingly, many efforts have focused on removing repeat sequences to avoid the noise they create42,43,44,45. Conversely, we have developed an efficient workflow to obtain complete cp and nR sequences simultaneously by taking advantage of the high copy genomic elements and using subsequent in silico solutions for error correction (see Materials and Methods). We used standard procedures for DNA preparation, PE library construction and Illumina sequencing. Small amounts of NGS data from WGS reads sufficed to assemble complete cp and nR sequences using our approach.
Origin of cultivated rice species revealed by concurrent cp and 45S-based phylogenomics
Oryza species are self-pollinating. The maternally inherited cp genome often most accurately represents the lineage, whereas nuclear rDNA could be intermingled by chance cross-hybridization between evolving sub-groups. However, our results demonstrated that cross hybridization between diverging groups can also produce altered genotype results in cp genome-based phylogenies, as exemplified by the biased positioning of one japonica-indica hybrid cultivar, M23 (no. 3 in Figs 3 and 4).
During the last four decades, there have been many breeding efforts involving inter-subspecies hybridization between indica and japonica. Two famous rice cultivars, Tong-il and M23, were bred by inter-subspecies hybridization and subsequent inbreeding and selection in Korea (Fig. 3c,d)38. Both cultivars have an indica-type appearance even though a little portion of each genotype is derived from japonica-type rice39. Our cp-based phylogenetic analysis revealed that the Tongil cp genome is identical with indica rice accessions. By contrast, M23 cp genome is identical to those of the japonica accessions rather than those of the indica group, a finding that is contradictory to the 45S-based tree as well as to the phenotype and genotypes (Fig. 3a,b)39. However, this finding is consistent with the breeding history: Tongil likely contains the indica-type cp genome and M23 the japonica-type cp genome because they were maternally inherited from the inter-subspecies hybridization (Fig. 3c,d). Based on our results, we conclude that the cp genome reports genetic diversity well for most plant species; however, the cp-based phylogeny can be sometimes be biased if there was an inter-species cross hybridization event such as in the case of M23. Similar results are observed from some accessions that were estimated to be derived from natural inter-species pollination, one O. rufipogon accession (no. 7), and two O. glumaepatula accessions (nos. 23 and 24) (red lines in Fig. 4). Our data illustrate that phylogenomic analysis based on simultaneous use of both cp and nR sequences will further promote elucidation of the relationships among closely related species over approaches using only one type of sequence.
Genome-wide analysis of large collections of O. sativa relatives has revealed that Asian O. rufipogon genomes are the most diverse and are classified into three major groups, of which Or-I/II and Or-III are thought to be ancestors of the indica and japonica types of rice, respectively, and O. rufipogon W1943 accession (no. 7) is classified as Or-III ecotype36, which coincides with our phylogenetic analysis with 45S. However, our cp-based phylogeny indicated that O. rufipogon W1943 is close to the indica type.
Although the 45S-based phylogeny did not classify the OB-IV&-V group, our cp-based phylogeny clearly distinguished the five O. barthii subgroups and demonstrated that the African cultivated rice was domesticated from the OB-V group, in agreement with a previous report (Fig. 3a,b)40.
Phylogenetic relationships of Oryza AA genomes
Comparison of genome sequences of Oryza sativa and related five AA genome species revealed the rapid diversification of each species30. Phylogenetic analyses based on 53 conserved nuclear genes had O. meridionalis as the basal species with the AA genome29,30. By contrast, our analysis showed that O. longistaminata is the basal AA genome species, based on the cp genome as well as nR sequence (Fig. 4). Although there is contradictory positioning of O. longistaminata and O. meridionalis between these two analyses (Our cp-based tree vs. ref. 44), the overall topology is in agreement with other previous reports46,47,48,49,50,51. The cp-based phylogeny also showed very rapid divergence of AA genome species during the last 2.3 million years, in agreement with data based on the nuclear genome (Fig. 4)29,30.
In this work, we analyzed several different specimens as representatives of the same species, an approach that differs from the other previous studies and promotes better understanding of the divergence of wild species. Our analysis revealed that there were very recent cp genome exchanges between diverging species such as two O. glumaepatula accessions (nos. 23 and 24) which have O. longistaminata-like cp genomes even though the rDNA and phenotypes are similar to two other distinct O. glumaepatula accessions (nos. 25 and 26) (Fig. 4). We assume that there was a cross pollination event with some ancestor of O. glumaepatula (such as accessions nos. 23 and 24) as the male and O. longistaminata as the female between 0.5–0.3 million years ago (MYA) (Fig. 4). As described above, we identified and confirmed a similar phenomenon in one inter-subspecies hybridization-derived cultivar, M23, which was bred by cross-hybridization between O. sativa ssp. japonica x O. sativa ssp. indica according to the breeding history (Fig. 3c).
Three O. rufipogon specimens were placed in different groups. The Chinese O. rufipogon specimen (W1943, no. 7) was grouped with O. nivara and O. sativa. A wild specimen from Vietnam (AC11-1008369, no. 8) was placed independently as basal group of O. sativa indicating that it is a wild ancestor of O. sativa, in agreement with previous reports (Fig. 4a)36,52,53. However, another wild O. rufipogon specimen from Australia (AC01-1002323, no. 9) was grouped with O. meridionalis, indicating that the naming of the species should be reconsidered, consistent with a previous suggestion that the Australian O. rufipogon may be a perennial form of O. meridionalis52.
Advantage of complete cp genome information for barcoding within species
The diversity of the cp genome within the genus or species level is an advantage to analyze domestication of crop plants and for development of barcoding markers for certain cultivars. The origin of domesticated apples has been clarified based on 47 cp genome sequence of Malus species23. The cp genomes of ginseng and American ginseng (P. quinquefolius) showed 138 SNPs and 40 InDels relative to one another (Supplementary Figs. S13 and S14 online). Although there is abundant polymorphism between different Panax species, previous PCR surveys did not detect polymorphism among P. ginseng cultivars in the cp intergenic regions54,55. In this study, we could identify one SNP and two InDels between the P. ginseng cultivars ChP and YP (Supplementary Figs. S13 and S14 online), and one SNP and three InDels between ChP and three reported P. ginseng specimens from China (GenBank Accession nos. KC686331, KC686332, KC686333). We further identified a total of six SNPs and six InDels by comparison of nine more complete cp genomes of P. ginseng cultivars or landraces56. By contrast, when compared with the previously reported wild P. ginseng cp genome (NC_006290)57, our P. ginseng cp genome sequences showed 117 SNPs and 51 InDels, likely due to differences in plant material and/or sequencing errors (Supplementary Fig. S13 online). We also applied this approach to complete cp genomes of tartary buckwheat and three onion accessions to identify useful barcoding markers to classify adjacent species58 and different onion cytotypes59.
Evolution of nR DNA
The copy number of nR unit varies in different plant genomes. The copy numbers of each nR was estimated based on average depth coverage of 1x genome-equivalent WGS reads (Table 1) and the value was roughly in accord with, but slightly lower than, estimates derived from FISH signals, likely because FISH signals amplify the hybridization signal by rendering a two dimensional signal from the three dimensional chromosome structure (Table 1 and Supplementary Fig. S15 and Table S4 online). We found a remarkable range of estimated copy numbers for 5S and 45S. The 5S copy and 45S copy number estimates ranged from 69–6,045 and 131–2,292, respectively, among Oryza species (Table 1). This raises the question of whether there is any association of rDNA diversity and copy number variation with biological function.
The 5S and 45S units are found in independent chromosomal regions in rice (Supplementary Figs. S6 and S15 online) and in most genomes of higher plants. By contrast, the 45S and 5S units co-exist as one tandem repeat unit in some ancient plants13,14, and it is assumed that the single unit was divided into separate 45S and 5S units in higher plants during evolution. However, it is not clear how and when the units evolved because of the lack of complete nR unit sequences from diverse plants. Our high throughput approach can thus contribute to revealing how nR evolved in the plant kingdom.
Methods
Preparation of whole-genome NGS reads
Leaf samples were harvested from plants of rice and ginseng grown in a farm of Seoul National University, Suwon, Korea, and high-quality genomic DNA was extracted using a modified CTAB method60. A paired-end (PE) library with 500-bp insert size was constructed using the Illumina PE DNA library kit according to the manufacturer’s instructions and sequenced using an Illumina Hiseq2000 by the National Instrumentation Center and Environmental Management (NICEM, http://nicem.snu.ac.kr/, Korea) and Macrogen (http://dna.macrogen.com/, Korea) and Illumina MiSeq or NextSeq500 by LabGenomics (www.labgenomics.co.kr, Korea). Illumina Hiseq2000 reads of six O. sativa and eight related Oryza species were provided by Prof. Hee-Jong Koh (Seoul National University, Korea) and the Arizona Genomics Institute (AGI, http://www.genome.arizona.edu/, USA), respectively. WGS sequence data of additional Oryza species were downloaded from the SRA database and used for assembly of cp genomes and nR sequences (Table 1).
WGS assembly and building of complete cp genome and nR sequences
Raw reads with Phred scores of 20 or less were removed from among the total NGS PE reads using the CLC-quality trim tool (quality_trim software included in CLC ASSEMBLY CELL package ver. 4.06 beta. 67189, http://www.clcbio.com/products/clc-assembly-cell/). In assemblies of WGS reads representing more than 70x genome coverage in rice and ginseng, we identified no proper long, unique cp contigs. We then tested assembly of cp genome and nR using low-coverage WGS sequences. Sub-datasets with various levels of cp genome coverage were extracted from trimmed NP and ChP WGS reads and assembled using the CLC de novo assembler included in the CLC ASSEMBLY CELL package or SOAPdenovo included in the SOAP package (ver. 1.12) with default parameters. Sequence gaps were filled by Gapcloser included in the SOAP package (ver. 1.12). Representative contigs for the cp genome or nRs were retrieved from the total assembled contigs using Nucmer61 with reference sequences. Extracted contigs were ordered and oriented based on built-in BLASTZ analysis (http://nature.snu.ac.kr/tools/blastz_v3.php)62 with the cp sequence of related genome and then connected into single draft sequence by joining overlapping terminal sequences.
In silico finishing: Identification and correction of errors in dnaLCW assembly
Tentative error sites were identified by mapping raw reads to draft sequences using the CLC mapping tool (clc_ref_assemble in the CLC ASSEMBLY CELL package) and visualized using CLC viewer (clc_assembly_viewer in the CLC ASSEMBLY CELL package). The mis-assembled error sequences were characterized by accumulation of discordantly mapped reads or abnormally higher read mapping depth. The identified assembly errors were classified as false gaps, false SNPs, or copy number errors for tandem repeats (TR) or monopolymers. Each type of errors was corrected by following in silico manual curation and validated by PCR amplification and Sanger sequencing.
False gaps: This type of error occurs in the regions where ambiguous “N” nucleotides present in draft assembly contigs. The left and right sequence flanking an “N” are duplicated, leading to accumulation of commonly mis-mapped reads at the flanking regions (Supplementary Fig. S2 online). Such errors can be corrected by merging the common duplicated sequences flanking the “N”, and the correction validated by re-mapping reads on the edited sequence. If the edited sequence is correct, read mapping will show clear matches on the sequence.
False SNPs: DNA fragments homologous to those of the cp genome are ubiquitous in mitochondrial and nuclear genomes of rice63,64 and can interfere with cp genome assembly44, leading to create false SNPs (Supplementary Fig. S3a online). Each false SNP could be corrected by assigning the consensus nucleotide sequence to the false SNP location based on the reads showing the highest depth in the paired read mapping, because ~8–100-fold more reads originate from the cp genome than from the nuclear or mitochondrial genome. For example, the assembly of the Os5 dataset, which provides 4x and 200x coverage of the nuclear and cp genomes, respectively, showed two false SNPs, G/T at 51,940 nt and T/A at 51,944 nt (Supplementary Fig. S3b online). The 212 reads mapped to the region revealed clear patterns of origin, in which 186 reads (from the cp) contained T and A nucleotides at those positions, 24 reads (from the mt) contained G and T, and 2 (from the nucleus) contained T and T. Overall, false SNPs in the initial contigs can be easily corrected using read mapping followed by assigning the consensus nucleotide with the highest depth.
Tandem repeat copy number error: There are many chances for copy number error to arise during de novo assembly using short reads42,43,45. Our data show that 18-bp TR units were mis-assembled into 2 copies by default assembly options, whereas four complete copies of 18-bp TRs were correctly assembled with using a k-mer length of 64 (Supplementary Fig. S4a online). When repeats are shorter than the read length, increasing the k-mer value above the TR unit length can reduce mis-assembly. Copy number errors in the assembly can be identified by comparing read-depth at the TR and the flanking region. If raw reads map to a region incorrectly assembled with too few copies of a TR, mis-mapped reads will be abundant and abnormal high read-depth can be found at the collapsed regions (Supplementary Fig. S4b,c online). Most TR units found in cp genomes are simple and less than 100 bp, unlike those in the nuclear genome. Therefore, most errors derived from copy number variance of TRs can be fixed.
Monopolymer copy number error: A total of 95 and 91 regions contained monopolymer tracts of more than 8 nt in the cp genomes of NP and ChP, respectively. Most monopolymers were poly A or T (Supplementary Table S5 online). Monopolymer regions in the cp genome are susceptible to sequencing errors due to polymerase slippage and mis-assembly caused by interruption of homologous mitochondrial or nuclear sequences containing monopolymers of different lengths. Such monopolymer assembly error was detected at the poly T tract region at 78,424 bp in the NP cp genome (Supplementary Fig. S5a online). Similar sequences with different poly T tracts (7, 8, 9, 10, 11, 12, 15 and 17 nt long) were found in 10 chromosomal regions of the NP nuclear genome (Supplementary Fig. S5b online). The initial assembly of the Os3 dataset generated a (T)8 monopolymer tract assembly error caused by interruption with T monopolymers derived from sequences of rice chromosome 5, 6, 7, and 9 (Supplementary Fig. S5b online). This error could be corrected by selection of T monopolymer tracts showing the highest read-depth after raw-read mapping on hypothetical T monopolymer sequences with 100% identity. The draft sequence with the correct (T)17 monopolymer among the eight putative sequences showed the highest mapping depth of 33.14, as expected (Supplementary Fig. S5c online).
Annotation and comparative analysis of cp and nR sequence
The cp genome sequence was annotated using the DOGMA program (http://dogma.ccbb.utexas.edu/)65 and BLAST searches. Circular and comparative maps of the cp genome were generated using OGDRAW (http://ogdraw.mpimp-golm.mpg.de/)66 and mVISTA (http://genome.lbl.gov/vista/mvista/submit.shtml)67, respectively. The gene structure of rRNAs, ITS, and IGS in assembled 45S sequences were determined by comparison with reported sequences and BLAST searches. Phylogenetic tree construction and the reliability assessment of internal branches were conducted by the neighbor-joining method with 1,000 bootstrap replicates using MEGA668 (Fig. 3). The phylogenetic tree and molecular clock dating was generated based on Bayesian Inference analysis using BEAST (version 1.8.1)69 (Fig. 4). We performed 10 million generation of MCMC and sampled every 1000 generations, effective sample size of 592 with parameter as an uncorrelated lognormal relaxed-clock model, with Yule prior on the tree, general time reversible (GTR + I + Γ) as a substitution model and the default priors for generated random starting tree. The BEAST runs were obtained using Tracer (version 1.6) after discarding as burn-in of 10% of generations and the remaining was used to estimate the posterior probability. Numbers on each branches indicate posterior probability (pp) (>0.5). The divergence times were calculated using TreeAnnotator (version 1.8.1) and constrained to be 9 MYA as root age based on recently reported divergence time between Oryza AA genome and BB genome (O. punctata)29.
Validation of polymorphic regions in cp genome sequences
Specific primers were designed from conserved sequences flanking polymorphic regions such as SNPs and InDels found among cp genomes (Supplementary Table S6 online). Genomic DNA was used as template for PCR amplification using Ex-Taq polymerase (Takara, Japan) and the amplified fragments were analyzed using a Fragment Analyzer (Advanced Analytical Technologies Inc., USA), according to manufacturer’s instructions. DNA fragments amplified using dCAPS primers were digested with appropriate restriction enzyme and then separated by a Fragment Analyzer (Advanced Analytical Technologies Inc., USA).
Additional Information
Accession codes: Whole-genome NGS reads used in this study have been deposited in the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) and final assembled cp genome and nR unit sequences from this study were deposited in the GenBank database (for accession number, see Table 1).http://www.nature.com/srep
How to cite this article: Kim, K. et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5, 15655; doi: 10.1038/srep15655 (2015).
Supplementary Material
Acknowledgments
We thank all members of the Laboratory of Functional Crop Genomics and Biotechnology, Seoul National University, Korea and Phyzen (http://www.phyzen.com/), for their technical assistance. This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01103001, PJ01100801)”, Rural Development Administration, Republic of Korea and partially supported by a grant entitled “The Genetic Evaluation of Important Biological Resources 2012, 2013” from the National Institute of Biological Resources under the Ministry of Environment, Republic of Korea. Preparation of the NGS data sets of wild Oryza species was supported by the US NSF Plant Genome Program’s Oryza Genome Evolution project #1026200 to R.A.W. & Y.Y.
Footnotes
Author Contributions T.J.Y. designed the research and organized the manuscript. K.K., Jun.L., K.Y., B.S.C., H.I.C., H.S.P., Jon.L., H.O.L., H.J.J., H.J.L., J.Y.P., S.P., M.J. and Y.D.K. participated in assembly of cp genomes and rDNAs, bioinformatic analysis, annotation, and validation. H.J.K., Y.Y., N.H.K., W.J., Y.S.L., B.K., Su.K. and K.B.L. conducted sampling, genomic DNA isolation and sequencing. N.E.W conducted FISH analysis. K.K., S.C.L., Jun.L., Y.Y., K.Y., H.J.K., D.C., So.K., Jun.L., R.A.W., B.S.P., K.S.C. and T.J.Y. wrote and revised the manuscript. All authors approved the final manuscript.
References
- Qiu Y.-L. et al. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature 402, 404–407 (1999). [DOI] [PubMed] [Google Scholar]
- Soltis P. S., Soltis D. E. & Chase M. W. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402, 402–404 (1999). [DOI] [PubMed] [Google Scholar]
- Park J. Y. et al. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor. Appl. Genet. 126, 1763–1774 (2013). [DOI] [PubMed] [Google Scholar]
- Palmer J. D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354 (1985). [DOI] [PubMed] [Google Scholar]
- Harris S. A. & Ingram R. Chloroplast DNA and biosystematics: The effects of intraspecific diversity and plastid transmission. Taxon 40, 393–412 (1991). [Google Scholar]
- Wolfe A. D. & Randle C. P. Recombination, heteroplasmy, haplotype polymorphism, and paralogy in plastid genes: Implications for plant molecular systematics. Systematic Botany 29, 1011–1020 (2004). [Google Scholar]
- Shaver J. M., Oldenburg D. J. & Bendich A. J. Changes in chloroplast DNA during development in tobacco, Medicago truncatula, pea, and maize. Planta 224, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- Rivarola M. et al. Castor bean organelle genome sequencing and worldwide genetic diversity analysis. PLoS ONE 6, e21743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. & Messing J. High-throughput sequencing of three Lemnoideae (Duckweeds) chloroplast genomes from total DNA. PLoS ONE 6, e24670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe K. H., Gouy M., Yang Y. W., Sharp P. M. & Li W. H. Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proc. Natl. Acad. Sci. USA 86, 6201–6205 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboud X. & Zeyl C. Organelle inheritance in plants. Heredity 72, 132–140 (1994). [Google Scholar]
- Rogers S. O. & Bendich A. J. Heritability and variability in ribosomal RNA genes of Vicia faba. Genetics 117, 285–295 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Costa A., Muñoz J. & Dietmar Q. Restless 5S: The re-arrangement(s) and evolution of the nuclear ribosomal DNA in land plants. Mol. Phyl. Evol. 61, 321–332 (2011). [DOI] [PubMed] [Google Scholar]
- Galián J. A., Rosato M. & Rosselló J. A. Early evolutionary colocalization of the nuclear ribosomal 5S and 45S gene families in seed plants: evidence from the living fossil gymnosperm Ginkgo biloba. Heredity 108, 640–646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez I. & Wendel J. F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434 (2003). [DOI] [PubMed] [Google Scholar]
- Golenberg E. M. et al. Chloroplast DNA sequence from a miocene Magnolia species. Nature 344, 656–658 (1990). [DOI] [PubMed] [Google Scholar]
- Sang T., Crawford D. & Stuessy T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 84, 1120 (1997). [PubMed] [Google Scholar]
- Burger G., Lavrov D. V., Forget L. & Lang B. F. Sequencing complete mitochondrial and plastid genomes. Nat. Protoc. 2, 603–614 (2007). [DOI] [PubMed] [Google Scholar]
- Nock C. J. et al. Chloroplast genome sequences from total DNA for plant identification. Plant Biotech. J. 9, 328–333 (2010). [DOI] [PubMed] [Google Scholar]
- Zhang Y. J., Ma P. F. & Li D. Z. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6, e20596 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Forrest L. L., Bainard J. D., Budke J. M. & Goffinet B. Organellar genome, nuclear ribosomal DNA repeat unit, and microsatellites isolated from a small-scale of 454 GS FLX sequencing on two mosses. Mol. Phylogenet. Evol. 66, 1089–1094 (2013). [DOI] [PubMed] [Google Scholar]
- McPherson H. et al. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 13, 8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforova S. V., Cavalieri D., Velasco R. & Goremykin V. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol. Biol. Evol. 30, 1751–1760 (2013). [DOI] [PubMed] [Google Scholar]
- Straub S. C. et al. Building a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genomics 4, 12–211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub S. C. et al. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364 (2012). [DOI] [PubMed] [Google Scholar]
- Wysocki W. P. et al. A multi-step comparison of short-read full plastome sequence assembly methods in grasses. Taxon 63, 899–910 (2014). [Google Scholar]
- Weitemier K., Straub S. C., Fishbein M. & Liston A. Intragenomic polymorphisms among high-copy loci: a genus-wide study of nuclear ribosomal DNA in Asclepias (Apocynaceae). PeerJ. 3, e718 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan D. A., Lu B. R. & Tomooka N. The evolving story of rice evolution. Plant Science 174, 394–408 (2008). [Google Scholar]
- Zhu T. et al. Phylogenetic relationships and genome divergence among the AA- genome species of the genus Oryza as revealed by 53 nuclear genes and 16 intergenic regions. Mol. Phylogenet. Evol. 70, 348–361 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang Q. J. et al. Rapid diversification of five Oryza AA genomes associated with rice adaptation. Proc. Natl. Acad. Sci. USA 111, E4954–4962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005). [DOI] [PubMed] [Google Scholar]
- Choi H. I. et al. Major repeat components covering one third of the ginseng (Panax ginseng C.A. Meyer) genome and evidence for allotetraploidy. Plant J. 77, 906–916 (2014). [DOI] [PubMed] [Google Scholar]
- Li R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido R., Sano Y. & Fukui K. Ribosomal DNAs: an exception to the conservation of gene order in rice genomes. Mol. Gen. Genet. 263, 586–591 (2000). [DOI] [PubMed] [Google Scholar]
- Ammiraju J. S. S. et al. The Oryza BAC resource: a genus-wide and genome scale tool for exploring rice genome evolution and leveraging useful genetic diversity from wild relatives. Breeding Science 60, 536–543 (2010). [Google Scholar]
- Huang X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S. G., Waters D. L. & Henry R. J. Australian wild rice reveals pre-domestication origin of polymorphism deserts in rice genome. PLoS ONE. 9, e98843 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung G. S. & Heu M. H. Improvement of Tongil-type rice cultivars from indica/japonica hybridization in Korea. In Biotechnology in Agriculture and Forestry 14, 105–112 (Springer-Verlag, 1991). [Google Scholar]
- Kim B. et al. Defining the genome structure of “Tongil Rice”, a cultivar for Korean green revolution, derived from indica x japonica hybridization. Rice 7, 22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 46, 982–988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W. & Li R. Test driving genome assemblers. Nat. Biotechnol. 30, 330–331 (2012). [DOI] [PubMed] [Google Scholar]
- Phillippy A. M., Schatz M. C. & Pop M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biol. 9, R55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C., Coe B. P. & Eichler E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeau P. E., Pevzner P. A. & Tesler G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 29, 987–991 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen T. J. & Salzberg S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Nagashima H., Nagamine T., Higo H. & Higo K. P-SINE1-like intron of the CatA catalase homologs and phylogenetic relationships among AA-genome Oryza and related species. Theor. Appl. Genet. 98, 853–861 (1999). [Google Scholar]
- Cheng C., Tsuchimoto S., Ohtsubo H. & Ohtsubo E. Evolutionary relationships among rice species with AA genome based on SINE insertion analysis. Genes. Genet. Syst. 77, 323–334 (2002). [DOI] [PubMed] [Google Scholar]
- Park K. et al. Genetic variations of AA genome Oryza species measured by MITE-AFLP. Theor. Appl. Genet. 107, 203–209 (2003). [DOI] [PubMed] [Google Scholar]
- Ren F., Lu B. R., Li S., Huang J. & Zhu Y. A comparative study of genetic relationships among the AA-genome Oryza species using RAPD and SSR markers. Theor. Appl. Genet. 108, 113–120 (2003). [DOI] [PubMed] [Google Scholar]
- Zhu Q. & Ge S. Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol. 167, 249–265 (2005). [DOI] [PubMed] [Google Scholar]
- Duan S. et al. Phylogenetic analysis of AA-genome Oryza species (Poaceae) based on chloroplast, mitochondrial, and nuclear DNA sequences. Biochem. Genet. 45, 113–129 (2007). [DOI] [PubMed] [Google Scholar]
- Waters D. L., Nock C. J., Ishikawa R., Rice N. & Henry R. J. Chloroplast genome sequence confirms distinctness of Australian and Asian wild rice. Ecol. Evol. 2, 211–217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. High occurrence of functional new chimeric genes in survey of rice chromosome 3 short arm genome sequences. Genome Biol. Evol. 5, 1038–1048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H. et al. Diversity and evolution of major Panax species revealed by scanning the entire chloroplast intergenic spacer sequences. Gent. Resour. Crop Evol. 60, 413–425 (2013). [Google Scholar]
- Jung J., Kim K. H., Yang K., Bang K. H. & Yang T. J. Practical application of DNA markers for high-throuput authentication of Panax ginseng and Panax quinquifolius from commercial ginseng products. J. Ginseng Res. 38, 123–129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. et al. Comprehensive survey of genetic diversity in chloroplast genomes and 45S rDNAs within Panax ginseng species. PLoS ONE 10, e0117159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. J. & Lee H. L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261 (2004). [DOI] [PubMed] [Google Scholar]
- Cho K. S. et al. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 10, e0125332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Park J. Y. & Yang T. J. Comparative analysis of complete chloroplast genome sequences of a normal male-fertile cytoplasm and two different cytoplasms conferring cytoplasmic male sterility in onion (Allium cepa L.). The Journal of Horticultural Science & Biotechnology 90, 459–468 (2015). [Google Scholar]
- Allen G. C., Flores-Vergara M. A., Krasynanski S., Kumar S. & Thompson W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325 (2006). [DOI] [PubMed] [Google Scholar]
- Kurtz S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. et al. Human-mouse alignments with BLASTZ. Genome Res. 13, 103–107 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan M. et al. Analysis of 1.9Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391, 485–488 (1998). [DOI] [PubMed] [Google Scholar]
- Matsuo M., Ito Y., Yamauchi R. & Obokata J. The rice nuclear genome continuously integrates, shuffles, and eliminates the chloroplast genome to cause chloroplast-nuclear DNA flux. Plant Cell 17, 665–675 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman S. K., Jansen R. K. & Boore J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20, 3252–3255 (2004). [DOI] [PubMed] [Google Scholar]
- Lohse M., Drechsel O., Kahlau S. & Bock R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K. A., Pachter L., Poliakov A., Rubin E. M. & Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Suchard M. A., Xie D. & Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.