

Abstract
Molecular detection has overcome limitations of microscopic examination by providing greater sensitivity and specificity in Plasmodium species detection. The objective of the present study was to develop a quantitative real-time polymerase chain reaction coupled with high-resolution melting (qRT-PCR-HRM) assay for rapid, accurate and simultaneous detection of all five human Plasmodium spp. A pair of primers targeted the 18S SSU rRNA gene of the Plasmodium spp. was designed for qRT-PCR-HRM assay development. Analytical sensitivity and specificity of the assay were evaluated. Samples collected from 229 malaria suspected patients recruited from Sabah, Malaysia were screened using the assay and results were compared with data obtained using PlasmoNexTM, a hexaplex PCR system. The qRT-PCR-HRM assay was able to detect and discriminate the five Plasmodium spp. with lowest detection limits of 1–100 copy numbers without nonspecific amplifications. The detection of Plasmodium spp. in clinical samples using this assay also achieved 100% concordance with that obtained using PlasmoNexTM. This indicated that the diagnostic sensitivity and specificity of this assay in Plasmodium spp. detection is comparable with those of PlasmoNexTM. The qRT-PCR-HRM assay is simple, produces results in two hours and enables high-throughput screening. Thus, it is an alternative method for rapid and accurate malaria diagnosis.
Symptoms of malaria are nonspecific and similar to other diseases. Without prompt and proper treatment, mild and moderate malaria may also be lethal in children and some adult patients especially for those infected with Plasmodium falciparum. Cases with P. falciparum have higher rates of complications and the symptoms of malaria may suddenly develop, possibly leading to death within 24 hours if treatment is not provided1,2. Hence, a few main factors need to be taken into account when developing a malaria diagnostic kit: rapidity, accuracy, sensitivity, cost-effectiveness, easy handling, and minimal training of personnel3.
Microscopic examination of blood films remains as the most common malaria diagnostic method due to its inexpensiveness and simplicity. It is considered as the “gold standard” for malaria diagnosis4. However, since various limitations which include the requirement of skilled personnel, low sensitivity (100 to 200 parasites/μl of blood), time-consuming and unreliability in species identification especially of P. knowlesi due to its similarities with P. falciparum in the early ring form stage and P. malariae in the latter stages, the microscopic technique needs to be coupled with other alternative diagnostic methods to heighten the accuracy of the identification4,5. Available complementary methods include rapid diagnostic tests (RDTs) and polymerase chain reaction (PCR)-based techniques.
Since the late 1980s, polymerase chain reaction (PCR)-based methods have been applied in malaria diagnosis due to their high sensitivity, rapidity and reproducibility6,7,8. Molecular detection using conventional nested PCR techniques offers detection of parasites at lower concentration of 5 parasites/μl9,10 and mixed infections, as well as discrimination of the Plasmodium spp6,11,12. A number of nested and semi-nested PCR methods have been developed over the years6,7,11,13,14,15,16, which allow for detection of up to four human Plasmodium spp. (P. falciparum, P. vivax, P. malariae and P. ovale), and these methods were replicated in other studies and epidemiological surveillance of malaria cases in numerous countries14,15,17,18,19. When simple dried blood spot sampling on filter papers is coupled with boiling method for DNA extraction, this has increased the robustness of nested PCR Malaria detection system20,21,22. Later on, single round multiplex PCR methods were also developed to overcome the disadvantages of nested and semi-nested PCR methods, such as time-consuming and risk of contamination23,24,25,26. PlasmoNexTM (Reszon Diagnostic International, Selangor, Malaysia) is the commercially available malaria diagnostic kit developed from our previous study which enables detection of the five currently most important human Plasmodium spp. (including P. knowlesi) using hexaplex PCR system26. The emergence of quantitative real-time PCR (qRT-PCR) technology has brought the malaria diagnosis to another level, with greater sensitivity (down to 0.02 parasite/μl)27, easier execution and no post-PCR manipulations compared to the conventional PCR methods. There are two types of qRT-PCR developed for malaria diagnosis: i) fluorescent dye-based qRT-PCR with the use of nonspecific double-stranded DNA (dsDNA) intercalating dyes such as SYBR green28,29,30,31, and ii) probe-based qRT-PCR with the use of specific fluorescent probes such as TaqMan probes32,33,34,35,36,37,38,39. In general, qRT-PCR assay using dsDNA intercalating dye requires lower running cost than that using fluorescent probes. However, intercalating dyes will bind to any dsDNA including non-specific dsDNA sequences and tend to generate false positive results. In 2004, a ready-to-used qRT-PCR system, namely Real Art Malaria LC PCR assay, was introduced and made commercially available in the market. However, Farcas and colleagues35 evaluated this commercial assay on febrile returned travellers and found that it failed to differentiate four Plasmodium spp.
qRT-PCR-SYBR green assay which can detect quantitatively up to five species of Plasmodium has been developed by Oddoux and colleagues31. However, they used a total of nine primers to amplify P. falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi. When the number of primers increases, the risk of unspecific binding between primers also increases. In the present study, we developed a quantitative real-time PCR method for the detection of the five human-infecting Plasmodium spp., but instead of SYBR green dye, high resolution melting (HRM) analysis was incorporated. Similar to qRT-PCR-SYBR green technique, the steps in HRM analysis involve amplification of the region of interest in the presence of a specialized dsDNA binding dye and gradual denaturation of amplicons by increasing the temperature in small increments in order to produce a characteristic melting profile that is called melting analysis. The SYTO® 9 dye used in the HRM analysis produces highly reproducible DNA melting curves over a broader range of dye concentrations than does SYBR Green I40. It is also far less toxic than SYBR Green I and does not appear to selectively detect particular amplicons40. Hence, SYTO® 9 can be used at higher concentration than SYBR Green I for greater saturation of the dsDNA sample in the assay, reducing dye redistribution to non-denatured regions during dissociation of dsDNA41. Owing to this characteristic, fluorescent signals measured in the assay have higher fidelity and provides greater resolution to melting curve analysis. This method has been employed and coupled with nested PCR technique for human malaria diagnostic by Kipanga and colleagues42, with limit of detection of 0.236 parasites/μl. However, it required two rounds of PCR and was able to detect only three species of Plasmodium. Our study aimed to design single-round of qPCR-HRM assay which used lower number of primers to detect all five species of human plasmodium with increased specificity and sensitivity.
Methods
Plasmid DNA preparation
All plasmids were obtained from previous study26. The plasmids containing the conserved region of 18S SSU rRNA gene sequence for each Plasmodium spp. were constructed and served as positive controls to assess the sensitivity and specificity of qRT-PCR-HRM assay.
Escherichia coli carrying the desired recombinant plasmid DNA was grown overnight in 10 ml Luria Bertani (LB) broth (BD, NJ, USA) containing 100 μg/ml ampicillin (Merck, Darmstadt, Germany) at 37 °C with vigorous shaking. The bacterial culture was harvested by centrifugation at 6000 × g for 15 minutes at 4 °C. The supernatant were removed and the cell pellets consisting of the recombinant plasmids of the five Plasmodium spp. were isolated and purified with High Yield Plasmid Mini Kit (Yeastern Biotech, Taiwan) according to the manufacturer’s instructions. The purified plasmid DNA samples were quantified spectrophotometrically at 260 nm and then kept at −20 °C.
Clinical samples
A total of 229 clinical DNA samples were collected from Sabah, Malaysia from 2008 to 2010. Prior to sample collection, approval of the ethics application with appropriate experimental protocols was obtained from the Medical Ethics Committee of University of Malaya Medical Centre, Kuala Lumpur, Malaysia. All the protocols used in this study were in accordance with the approved guidelines (Ethics reference no. 709.2) and informed consents were obtained from all patients recruited. Peripheral blood was drawn into EDTA tube (BD) from each patient suspected with malaria but prior to anti-malarial treatment, and an aliquot of each blood sample was subject to routine biological diagnosis. Two hundred μl of the remaining blood sample was extracted according to the manufacturer’s instruction to obtain total human genomic DNA as well as plasmodial DNA using QIAamp DNA mini kit (Qiagen, Hilden, Germany). All the samples were initially assessed using PlasmoNexTM (Reszon Diagnostic International), a hexaplex PCR detection system30. From the results, the clinical samples were found to consist of 92 cases of P. falciparum cases, 1 case of P. ovale, 2 cases of P. malariae cases, 90 cases of P. vivax cases, 36 cases of P. knowlesi cases, 2 cases of mixed infections (P. falciparum and P. vivax), and 6 cases with negative results.
Microscopic examination
Microscopic examination for clinical samples was done in previous study26. The analysis was performed by experienced microscopists in hospital, and the parasitemia level was estimated. Briefly, thin and thick blood smears were prepared and examined under microscopic magnification of ×1000 for the presence of Plasmodium. The parasite counts were estimated with scores ranging from + to ++++, whereby + indicated 4 to 40 parasites/μl, ++ indicated 41 to 400 parasites/μl, +++ indicated 401 to 4000 parasites/μl, and ++++ indicated >4000 parasites/μl of blood43. Parasitemia level was defined as negative if blood films was absence of parasites in 300 microscopic fields.
Quantitative real-time PCR-HRM (qRT-PCR-HRM) assay development
Primer design
Primers were designed from 18S SSU rRNA gene of the five Plasmodium spp. as it contains both highly conserved and variable regions. The 18S SSU rRNA gene sequences of P. falciparum, P. knowlesi, P. ovale, P. malariae and P. vivax were obtained from GenBank43 (NCBI, BethesdaMD, USA) (accession number M19172.1, U83876.1, L48987.1, M54897.1 and X13926.1, respectively) and sequence alignment was carried out using BioEdit Sequence Alignment Editor v7.1.744. Once the specific and conserved regions for each Plasmodium spp. were identified, a pair of common primers was designed for the qRT-PCR-HRM assay using Primer 3 software45, a free online primer design tool (http://simgene.com/Primer3). BLAST analysis (NCBI) was performed on the primer pair to evaluate their specificity. The folding characteristics of these primers and amplicons were evaluated using an internet open access secondary structure profiling software, OligoAnalyzer 3.1 (http://sg.idtdna.com/calc/analyzer) (Integrated DNA Technologies, Coralville, Iowa, USA).
Evaluation of primer efficiency with various annealing temperatures
A series of conventional gradient PCR was carried out to make sure the primer pair was able to amplify the target region of all the five Plasmodium spp. without producing unspecific bands or primer dimers which would interfere with the result interpretation in qRT-PCR-HRM analysis later on. Overall, 20 μl of PCR mixture was prepared, which contained 1× Taq buffer, 1.5 mM of MgCl2, 0.2 mM of dNTPs, 0.5 μM of each primer, 1 U of Taq polymerase and 10 ng of DNA template (plasmid carrying target sequence of each Plasmodium spp.). Conventional gradient PCR was performed using Applied Biosystems Veriti® 96 well thermal cycler (Applied Biosystems, CA, USA). The mixture was heated at 95 °C for 1 minute, followed by 30 cycles of denaturation at 95 °C for 30 seconds, annealing at gradient temperatures from 59 °C to 64 °C for 30 seconds and extension at 72 °C for 30 seconds, and final elongation at 72 °C for 5 minutes. After the gradient conventional PCR, the PCR products were electrophoresed and analysed on 2% (w/v) agarose gel.
Performing qRT-PCR-HRM analysis
qRT-PCR-HRM assay was performed using an Applied Biosystems 7500 Fast Real-Time PCR System. Each reaction was carried out in a total volume of 20 μl reaction mixture containing 10 μl of 2× MeltDoctor® HRM master mix (Applied Biosystems), 0.1 μM of each primer and 1 μl of 10 ng plasmid DNA bearing target sequence of each Plasmodium spp. or 1 μl of each clinical DNA sample. Both MicroAmp® Fast 0.1 ml 8-tube strip with cap (Applied Biosystems) and MicroAmp® Fast Optical 0.1 ml 96-well reaction plate (Applied Biosystems), sealed with MicroAmp® Optical adhesive film (Applied Biosystems), were used to perform qRT-PCR-HRM. The tubes or plates were centrifuged briefly prior to undergoing qRT-PCR-HRM.
The thermal profile comprised initialization step at 95 °C for 10 minutes, followed by 40 cycles of amplification which included denaturation of double stranded DNA at 95 °C for 15 seconds and annealing of DNA strands at 60 °C for 1 minute. The PCR products were then subject to a melt program: denaturation of double stranded DNA at 95 °C for 10 seconds, annealing of double stranded DNA at 60 °C for 1 minute, followed by a gradual temperature increase until 95 °C in 15 seconds and annealing of DNA at 60 °C for 15 seconds. Finally, melting curve plots were generated and analysed using High Resolution Melt software v 3.0.1 (Applied Biosystems) to determine average melting temperature (Tm) for each Plasmodium spp.
Analytical sensitivity and specificity evaluation
The analytical sensitivity or detection limit of qRT-PCR-HRM assay was assessed by using 10-fold serial dilutions of positive control plasmid DNAs, ranging from 100,000 to 1 copy number(s)/μl. The procedure was repeated twice to ensure the reproducibility of the threshold cycle number (Ct) results. Standard curves were also plotted from Ct’s of the 10-fold serial diluted plasmid DNAs using 7500 Software v2.0.1 (Applied Biosystems) to determine the efficiency percentage of each Plasmodium spp. assay.
The specificity of the assay was also evaluated using DNA of other organisms such as human genomic DNA; bacterial DNA of Aeromonas hydrophila, A. punctata, Vibrio parahaemolyticus, V. harveyi; and parasitic DNA of Toxoplasma gondii, Ancylostoma ceylanicum, Haemonchus contortus, Trichostrongylus colubriformis, Nippostrongylus brasiliensis, Giardia duodenalis and Entamoeba histolytica. The assay was performed in duplicates with 10 ng of each DNA.
Testing for detection of mixed infections
Mock mixed infections were created by spiking plasmid DNA carrying target sequences from two different species at varying ratios [1–10,000 copy number(s)/species] into 50 ng/μl of human DNA sample. Mock samples were subject to qRT-PCR-HRM and the ability of the assay to detect the species present in mock samples was examined. Mock samples with three, four and five mixed Plasmodium species (10,000 copy numbers/species) were also produced to test for the sensitivity of the assay to detect mixed infections.
Statistical analysis
One-way ANOVA test using SPSS statistics software version 22 (IBM, NY, USA) was also performed to examine whether or not there was significant difference in the average Tm values between species. Comparison of sensitivity between microscopy and qRT-PCR-HRM assay (or PlasmoNexTM) in the detection of Plasmodium spp. in clinical samples was examined with Chi-square and Cohen’s kappa coefficient tests using SPSS statistics software (IBM).
Results
Primer design
A pair of common primers was designed to amplify target sequence located on 18S SSU rRNA gene of Plasmodium spp.: forward primer 5′-GRAACTSSSAACGGCTCATT-3′ and reverse primer 5′-ACTCGATTGATACACACTA-3′ (Fig. 1). Seeing that the sequence of forward primer annealing site of P. vivax slightly varied from the other four Plasmodium spp., degenerate bases were incorporated into the forward primer to increase its flexibility in amplifying all the five species. According to the Primer-BLAST analysis, the PCR products might vary between 223 and 229 bp, and they covered 3 parts of species-specific regions allowing species discrimination in the melting curve analysis.
Figure 1. The alignment of consensus sequences of five Plasmodium spp.

The sequences were aligned using BioEdit Sequence Alignment v7.1.7. The common forward and reverse primers flanked three species-specific regions of the five Plasmodium spp.
Primer efficiency
The common primer pairs designed for qRT-PCR-HRM assay demonstrated their ability to amplify all the five Plasmodium spp. using conventional PCR method and gave intense bands from annealing temperatures of 59 °C to 61 °C. No unspecific product was detected in the positive control plasmids as well as the no template controls (NTCs) within the range of annealing temperatures used for PCR optimization. However, the intensity of bands for P. falciparum, P. knowlesi and P. vivax reduced obviously at 61 °C. As for P. ovale, amplifications still occurred at annealing temperatures of 62 and 63 °C. The optimal annealing temperature applied on qRT-PCR-HRM assay was finalized at 60 °C.
qRT-PCR-HRM assay
Melting temperature (Tm) for each Plasmodium spp
The qRT-PCR-HRM assay in this study was found to successfully distinguish all the five Plasmodium spp. The melting curve variance of the five Plasmodium spp. can be plotted in various forms, such as aligned and difference plots, according to the melting behaviour of their amplicons using High Resolution Melt software v 3.0.1 (Applied Biosystems) (Fig. 2). As shown in the plots, the melting curves of Plasmodium spp. can be distinctly separated and Tm for each species can be determined unambiguously. The Tm values were highly reproducible across 11 repeated melt curve runs.
Figure 2. Melting curve variance of the five Plasmodium spp. in (A) aligned, (B) difference and (C) derivative plot analyses.

The melting curve and Tm for each species can be very well discerned.
Table 1 shows the average Tm with its standard deviation (SD) for each Plasmodium spp. identified in plasmids.
Table 1. Average melting curve peak Tm value for each Plasmodium spp. in qRT-PCR-HRM analysis.
| Species | Tm range (°C) |
Average Tm ± SD (°C) | P-value for ANOVA test | |
|---|---|---|---|---|
| Plasmid clones* | Clinical samples | |||
| P. falciparum | 71.67–72.20 | 71.84–72.91 | 72.4 ± 0.5 | |
| P. malariae | 71.52–72.07 | 71.52–71.64† | 71.5 ± 0.1 | |
| P. knowlesi | 73.66–74.26 | 73.65–74.33 | 73.9 ± 0.4 | 0.001 |
| P. vivax | 74.65–75.10 | 74.44–75.13 | 74.7 ± 0.3 | |
| P. ovale | 75.09–75.47 | 75.27‡ | 75.4 ± 0.2 | |
*1 plasmid DNA sample for each species, averaged over 22 runs on separate days.
†2 patients infected with P. malariae.
‡1 patient infected with P. ovale.
Analytical sensitivity of the qRT-PCR-HRM assay
Ten-fold serial dilutions were done on positive control plasmid DNAs bearing target sequence [100,000 to 1 copy number(s)/μl] of each Plasmodium spp. to determine the sensitivity or detection limit of the qRT-PCR-HRM assay. The assay could detect target sequence of P. knowlesi until 1 copy number (Fig. 3). The reproducibility of these results was confirmed by duplicated tests. The lowest detectable limit of the assay for P. vivax was also found to be 1 copy number, whereas for P. ovale the limit of detection (LOD) was 10 copy numbers. Both P. falciparum and P. malariae could be detected down to 100 copy numbers of target sequence by the assay.
Figure 3. Representative amplification plot (A) and derivative melting curves (B) of 10-fold serial diluted P. knowlesi target sequence.
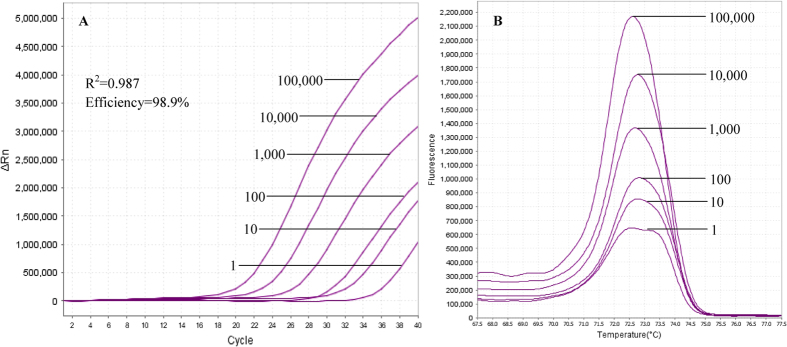
Ct values of six 10-fold serial diluted plasmid DNAs were subject to standard curve analysis using 7500 Software v2.0.1 (Applied Biosystems) and the r2 value and assay efficiency percentage were generated as 0.987 and 98.9%, respectively. The plots also show that the LOD of the assay for P. knowlesi is 1 copy number of target sequence.
Standard curves plotted from the Ct’s of 10-fold serial diluted plasmid DNAs revealed that efficiency percentage achieved were 95.5%, 92.5%, 98.9%, 92.0% and 96.0% for P. falciparum, P. vivax, P. knowlesi, P. malariae and P. ovale, respectively (Table 2). Figure 3 shows the representative amplification plot of 10-fold serial diluted P. knowlesi target sequence [100,000 to 1 copy number(s)], with r2 value of 0.987 for its standard curve, as well as the melting curves.
Table 2. R2 values and efficiency percentages of qRT-PCR-HRM assays for Plasmodium spp.
| Species | R2 value | Assay efficiency percentage |
|---|---|---|
| P. falciparum | 0.988 | 95.5% |
| P. vivax | 0.989 | 92.5% |
| P. knowlesi | 0.987 | 98.9% |
| P. malariae | 0.995 | 92.0% |
| P. ovale | 0.993 | 96.0% |
Analytical specificity of the real-time PCR coupled with HRM
The specificity of qRT-PCR-HRM assay was evaluated by testing DNA from other organisms (as mentioned in the methods). As seen in Fig. 4, no amplification and thus no melting curve was detected from these organisms. These results showed that this assay had high specificity to the target sequence of the five Plasmodium spp.
Figure 4. Raw data melt curve analysis of positive control plasmid DNAs and DNAs of organisms other than Plasmodium spp.
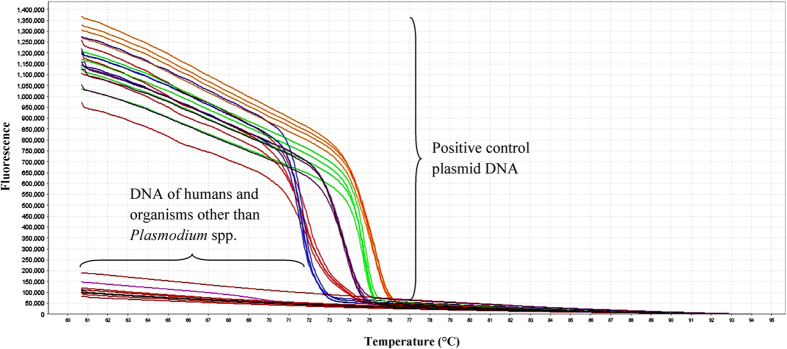
No amplification occurred in DNA samples of non-Plasmodium organisms and thus no melting curve was observed.
Ability to detect mixed infections
Among the mock double species mixed infections, only the combinations of P. falciparum/P. vivax and P. malariae/P. vivax could be accurately distinguished by the qRT-PCR-HRM assay (Fig. 5). Two melting curve peaks contributed by the two component species could be clearly seen on the derivative melting curve plot of samples with these two combinations. However, the ratios between each species within the combinations that could give rise to this pattern were unable to be determined as the results were found to be inconsistent.
Figure 5. Evaluation for detection of mixed infection.
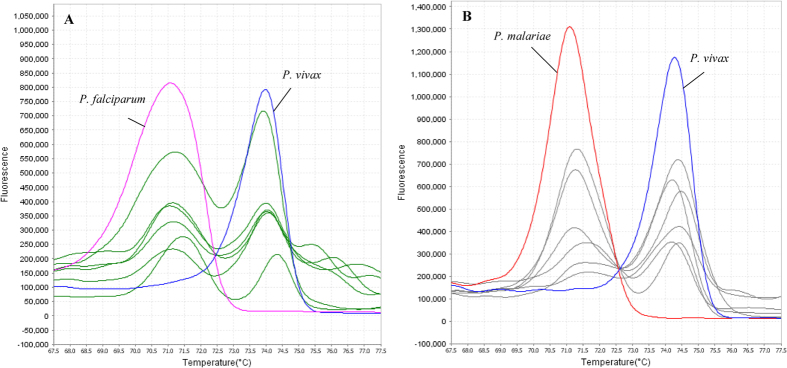
Mock mixed infections with different combinations of any two Plasmodium spp. were created and detection ability of the assay was evaluated. The figures show the two combinations of mixed infections that could be detected by the assay, i.e. (A) P. falciparum/P. vivax and (B) P. malariae/P. vivax, whereby the derivative melting curve peaks corresponding to Tm’s of each respective species can be clearly seen on the plots for each sample. Melting curves in (A) green and (B) gray were produced by samples with mock mixed infections.
While for the other double species combinations, only one peak was observed on the derivative melting curve plot for each combination and species ratio, and the Tm’s of the peaks were ambiguous. Thus, the assay was unsuitable to be used for detection of mixed infections other than the two above mentioned combinations.
Apart from that, the qRT-PCR-HRM assay in this study also failed to detect mixed infections with triple, quadruple and five Plasmodium spp. (10,000 copy numbers/species) as only one derivative melting peak with unspecific Tm was produced each time due to template interference.
Screening of clinical samples
A total of 229 clinical DNA samples collected were screened using qRT-PCR-HRM assay in this study. The identity of Plasmodium spp. present in all the clinical samples was initially determined using PlasmoNexTM. Based on that, the melting curves produced by the clinical samples were evaluated and it was observed that the Plasmodium spp. in the clinical samples showed consistency in their corresponding Tm values without much shift from those generated by positive control plasmid DNAs (Fig. 6). In the end, the melting curves resulted from the clinical samples could be clustered into five groups using auto-calling mode of High Resolution Melt software v 3.0.1 (Applied Biosystems), and the identification of the Plasmodium spp. present in the clinical samples could be done by referring their Tm values to those of plasmids. The confidence interval for auto-called results ranged between 80–100%.
Figure 6. Derivative melt curve analysis for the positive control plasmid DNAs of each Plasmodium spp. and clinical samples.
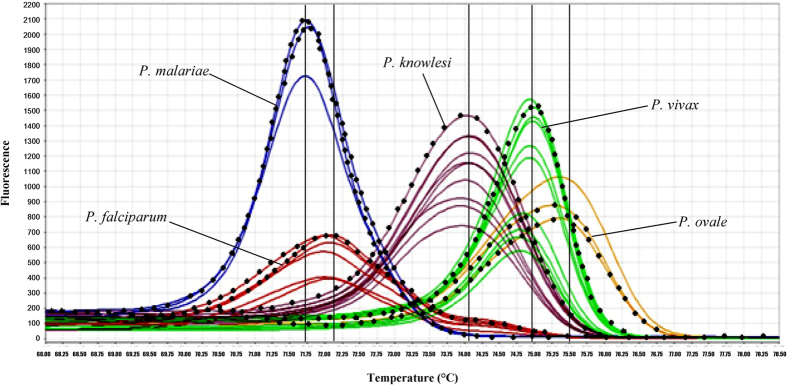
Vertical lines indicate Tm values for each Plasmodium spp. cloned in plasmids, and their melting curves are plotted with solid dotted lines while those of the clinical samples are plotted with solid colour lines. In this study, the melting curves of the clinical samples can be clustered into 5 groups and the identity of Plasmodium spp. present in the clinical samples can be clearly distinguished by referring their Tm values to those provided by the positive control plasmids.
From the screening results, Plasmodium spp. identified in the clinical samples were: P. falciparum (n = 92), P. ovale (n = 1), P. malariae (n = 2), P. vivax (n = 90), P. knowlesi (n = 36), mixed infection of P. falciparum and P. vivax (n = 2) (Table 3). There was no amplification in 6 clinical samples from the non-malaria patients with similar symptoms to malaria. All melting curve peak Tm for each Plasmodium spp. are illustrated in box plot (Fig. 7) and the average Tm with SD for each species was shown in Table 1. The ANOVA test also showed that Tm values between species could be significantly differentiated by having p-value of 0.001. Overall, the results produced by the qRT-PCR-HRM assay in this study were 100% in agreement with the previous results of PlasmoNexTM.
Table 3. Comparison of Plasmodium spp. identification results for 229 clinical samples between qRT-PCR-HRM assay and PlasmoNexTM.
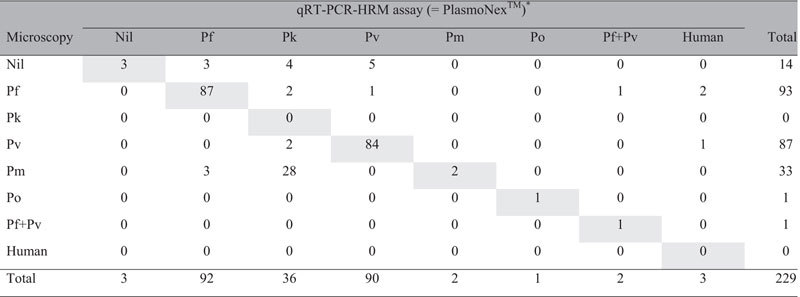
Chi-square value between microscopy and qRT-PCR-HRM assay = 756.710 (p < 0.001). Cohen’s kappa coefficient = 0.675 (p < 0.001). Note: Pf-P. falciparum; Pk-P. knowlesi; Pv-P. vivax; Pm-P. malariae; Po-P. ovale. *100% concordance in results obtained from both qRT-PCR-HRM and PlasmoNexTM assays. Concordant results by all three methods are shaded in grey.
Figure 7. Box plot showing melting curve temperatures (Tm) for each Plasmodium spp.
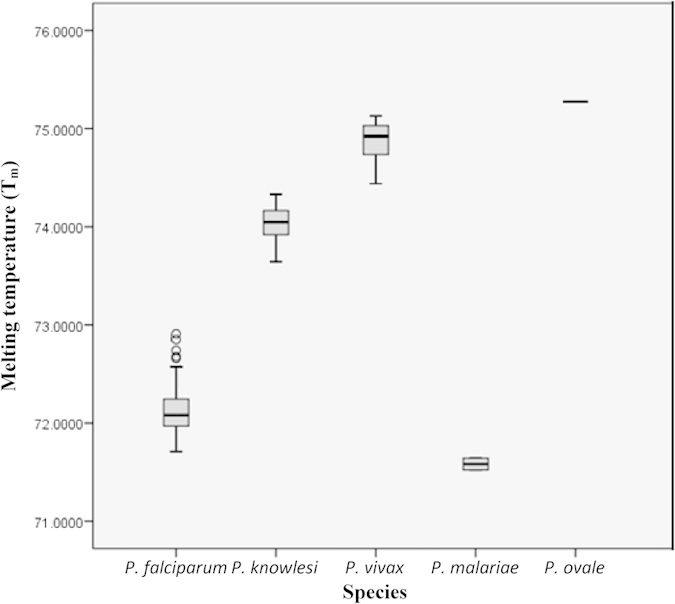
Note that P. malariae and P. ovale had only 2 and 1 sample(s), respectively. Four outliers are noticed for P. falciparum. The ANOVA test showed significant difference of average Tm values between species (p = 0.001).
Among the clinical samples, 3 of them previously identified by microscopic examination as two P. falciparum and one P. vivax-infected cases were otherwise identified as negative by both PlasmoNexTM and qRT-PCR-HRM assay (Table 3). As for the 14 clinical samples from patients with presumptive clinical diagnosis of malaria but were microscopically negative for Plasmodium spp., only 3 of them showed negative results in qRT-PCR-HRM assay while the other 2 were identified as P. falciparum-infected specimens, 4 were identified as P. knowlesi, and the remaining 5 were P. vivax-infected specimens. The same results were also obtained by using PlasmoNexTM.
The other two clinical samples which were previously diagnosed by PlasmoNexTM as mixed infections of P. falciparum and P. vivax were also detected as mixed infections using qRT-PCR-HRM assay in this study (Table 3). In this case, two melting curve peaks close to the corresponding Tm values of P. falciparum and P. vivax were observed concurrently in the derivative melt curve plot (Fig. 8).
Figure 8. Derivative melt curve analysis for the positive control plasmids cloned with P. falciparum and P. vivax, and a clinical sample with mixed infection.

The melting curve of this clinical sample shows two significant peaks with Tm values close to those of the two plasmid controls (vertical lines). This denotes that the clinical sample contains mixed infection of P. falciparum and P. vivax.
None of the clinical sample containing P. knowlesi could be identified by microscopic method. Among the 36 clinical samples identified with P. knowlesi by PlasmoNexTM and qRT-PCR-HRM assay, 28 (78%) were misdiagnosed by microscopy as P. malariae, 2 (6%) as P. falciparum, 2 (6%) as P. vivax, and 4 (10%) as negative (Table 3).
In addition, another 3 cases of previously microscopically identified P. malariae were detected as P. falciparum by both qRT-PCR-HRM assay and PlasmoNexTM, whilst 4 cases of P. knowlesi were mistaken for P. falciparum and P. vivax and 1 case of P. vivax was initially distinguished as P. falciparum by microscopic method (Table 3). Microscopy also failed to identify a case mix-infection with P. falciparum and P. vivax by reporting only P. falciparum.
Chi-square test showed that sensitivity in detecting Plasmodium spp. in clinical samples between microscopy and qRT-PCR-HRM assay was significantly independent between each other (p < 0.001) with Cohen’s kappa coefficient value of 0.675 (p < 0.001) (Table 3).
Discussion
Plasmodium 18S SSU rRNA gene was selected in this study because it contains both highly conserved and variable regions for Plasmodium spp. It has multiple copies scattered throughout the Plasmodium genome, which provides greater sensitivity during amplification than those single-copy genes46,47. Besides that, the RNA content in Plasmodium spp. is 1.5 times the amount of DNA and the rRNA subunit represents 85% to 95% of the total cellular RNA. This in turn causes the rRNA probes to have greater potential than DNA probes in molecular studies of malaria48. Hence, positive control clones containing partial sequence of 18S SSU rRNA gene was used to permit easy access to individual species-specific controls throughout the study.
In this study, qRT-PCR-HRM assay had been successfully developed for rapid and accurate detection of five human-infecting Plasmodium spp. compared to the qRT-PCR green assay developed by Oddoux and colleagues which used nine primers31. In contrast, a pair of primers was already adequate for our assay31. Our assay was found to be highly specific as none other organism than the five Plasmodium spp. could be amplified, and no fluorescence signal was detected in NTCs as well. The average difference in Tm values of 0.5 to 1 °C was sufficient enough to enable discrimination between the five Plasmodium spp. during the melting curve analysis. The confidence interval for auto-called results of 80–100% also indicated that the species discrimination was rather convincing. Albeit the Tm produced by target sequence of P. falciparum and P. malariae were very near to each other, they could still be significantly distinguishable given that p-value obtained from T-test between Tm’s of both species was 0.001. Furthermore, both species could also be discerned by the shape of their melting curves, especially in the difference melting curve plot, whereby the melting curve peak of P. malariae pointed downwards while that of P. falciparum was very short and pointed upwards instead (Fig. 2). P. falciparum also tended to display a very small shoulder peak after the main peak in the derivative melting curve plot (Fig. 2). The melting curve profiles between species in clinical samples could be easily discriminated when positive controls were included in each run of qRT-PCR-HRM.
The performance the qRT-PCR-HRM assay in the present study was evaluated and compared with PlasmoNexTM which served as the primary reference standard, given its established sensitivity and specificity over microscopic examination in malaria diagnosis26. It achieved 100% concordance with the results obtained using PlasmoNexTM in the detection of Plasmodium spp. in clinical samples. The lower detection limits of PlasmoNexTM was reported as 0.025, 0.25, 0.027, 0.27 and 0.15 parasites/μl for P. vivax, P. knowlesi, P. ovale, P. malariae and P. falciparum, respectively26. This implies that the qRT-PCR-HRM assay could have at least the same level of diagnostic sensitivity and specificity for Plasmodium spp. detection compared to PlasmoNexTM.
The sensitivity of the qRT-PCR-HRM assay was again justified when, likewise in PlasmoNexTM, it was able to detect parasitemia and identify Plasmodium spp. in the 11 out of 14 previously microscopically negative clinical samples (Table 3). Errors in microscopy are always reported in differentiating between P. vivax and P. ovale as well as between P. vivax and P. falciparum49,50. Besides that, the morphology of P. knowlesi is similar to that of P. malariae and correct microscopic identification between the two species is often an issue5. The above mentioned scenarios were exactly what occurred in 28 cases of P. knowlesi in this study which had been misdiagnosed as P. malariae by microscopy, as well as the other 8 cases. In this situation, molecular detection assays, such as qRT-PCR-HRM assay and PlasmoNexTM, should be brought into play to overcome these shortcomings of microscopic method. Low parasitemia levels could have also brought about the misdiagnosis of these samples during microscopic examination.
However, the presence of Plasmodium in the three clinical samples which was previously determined by microscopic examination, failed to be detected in qRT-PCR-HRM assay. This was in agreement with the results obtained using PlasmoNexTM. This may be due to the extremely low parasitemia in these samples which in turn led to the failure in or inadequate plasmodial DNA extraction from the samples, given that only 200 μl of blood was extracted for DNA sample. False positive microscopic detection of malaria can also occur when poor blood film preparation creates artefacts commonly mistaken for malaria parasites, including bacteria, fungi, stain precipitation, and dirt and cell debris50,51. Hence, although the PCR-based diagnostic methods are relatively sensitive compared to microscopic method, the plasmodial DNA extraction is the key factor that dictate the successfulness and accuracy of the diagnosis.
Malaria patients may harbour multiple species of Plasmodium. The qRT-PCR-HRM assay in this study was able to detect mixed infections. Yet, limitations were reported when the assay could only afford to differentiate two combinations of mixed infections, i.e. P. falciparum/P. vivax and P. malariae/P. vivax, and moreover, with unknown ratios between the species. Co-infection with P. falciparum and P. vivax is believed to be a prevalent type of mixed infection, and in Thailand, over one-third of patients manifesting acute malaria with P. falciparum were found to be actually infected with P. vivax simultaneously52. It was suggested that P. falciparum tends to suppress P. vivax and treatment for P. falciparum malaria would cause the subsequent relapse of P. vivax53. Hence, the detection for P. falciparum/P. vivax mixed infection is important for proper medical management and the assay developed in this study fits the need. This combination of mixed infection also seems common in Malaysia, as observed in clinical samples in this study and reviewed by William and Menon54.
As it was a quantitative real-time PCR, quantitation of parasitemia level in a clinical sample was feasible. The threshold cycle (Ct) value reflects the amount of starting material subject to qRT-PCR. The quantitation of the parasites in the clinical samples can be done by performing a comparative Ct experiment, using the standard curve constructed with plasmid control to determine the quantity of target in a sample relative to that in a reference sample38. However, quantitation of parasites in clinical sample was not performed in this study as there was no statistical record of parasitemia level from microscopic examination as well as PlasmoNexTM available for comparison. This was also one of the advantages of qRT-PCR-HRM assay against PlasmoNexTM.
Similar diagnosis of malaria using nested PCR coupled with qRT-HRM approach had been reported by Kipanga and colleagues42. Although the method had tremendous sensitivity (limit of detection of 0.236 parasites/μl) for detecting low parasitemia in patient samples, it was very time-consuming as it involved two round of PCR and one round of melting phase. This may not be very practical to use this method of diagnosis if the case is urgent, especially in the case of P. falciparum infection which can lead to death within 24 hours after symptoms developed1,2. Furthermore, the method could only detect three species, i.e. P. falciparum, P. malariae and P. ovale, as shown in the data of this study. Thus, diagnostic method developed in this study that enables detection of all five species, including P. knowlesi and P. vivax, can be applied everywhere without geographical concern of Plasmodium spp. distribution. These two species together with P. falciparum are commonly found in the Southeast Asian region. Since copy numbers of target DNA was used as measured unit in this study, the sensitivity of assay in this study could not be compared with that of assay developed by Kipanga and colleagues, where parasites/μl was used.
Besides the aforementioned study, other similar malaria diagnostic methods using qRT-PCR approaches include those developed by Fabre et al.55, Mangold et al.28 and Oddoux et al.31 , but SYBR green was employed instead. Similar to this study, method by Mangold and team also used a pair of primers to amplify Plasmodium spp., yet only 4 species (excluding P. knowlesi)28 were detected. However, their method only took 1 hour to complete, which is an advantage over the method developed in this study. The average Tm value difference across all species ranged from 1.1–4.8 °C, and similar to this study, the Tm values between P. falciparum and P. malariae could be as minute as 0.3 °C. Oddoux and colleagues managed to diagnose all 5 species of human plasmodium but 9 primers were used31. Although qRT-PCR diagnostic methods using TaqMan chemistry, which is relatively specific, sensitive and rapid, were also designed by several groups33,34,39, thus far only up to 4 human Plasmodium spp. were on the detection list and the methods were comparatively expensive compared to HRM approach. Again, since the prevalence of P. knowlesi is high in the Southeast Asian region, inclusion of this species for detection is essentially important when developing a malaria diagnostic assay.
qRT-PCR-HRM technique has also been employed to develop rapid detection of various clinically important microorganisms, such as nosocomial bacteria56, as well as those with drug-resistance, such as rifampicin-resistant Mycobacterium tuberculosis57 and erythromycin-resistant Bordetella pertussis58. Researchers opt for qRT-PCR-based methods over conventional PCR for the development of detection assay because of their high sensitivity, although this might not be always true as sensitivity relies on many factors such as primer design59. However, when speaking of having wider dynamic range, rapidness, needless of post-PCR or laborious processing, and rendering quantitation analysis, qRT-PCR-HRM technique is one of the methods to be considered as it has all these advantages over conventional PCR methods. Moreover, it is also relatively cost-effective than the probe-based qRT-PCR methods.
In conclusion, the qRT-PCR-HRM assay developed in this study appears to be a promising tool for simultaneous detection and discrimination of the five human-infecting Plasmodium spp. by using only a single pair of primers. Despite the cost of instrument, each reaction of qRT-PCR-HRM assay costs USD 1.50–4.50, depending on the DNA extraction method used. Albeit the cost is slightly higher and reaction time is longer than those of RDT (USD 0.55–1.50) and microscopy (USD 0.12–0.40)50, it compensates for the sensitivity and limited species (P. falciparum, non-P. falciparum and P. vivax) detection issues of RDT, as well as tendency of false results and requirement of skilled microscopist for microscopic method. Owing to the advantages mentioned earlier, this assay may be a practical and potential alternative for rapid and accurate diagnosis of malaria infections. Direct loading of blood samples to the assay could be one of the improvements to be done in the future to facilitate the diagnosis especially in the remote areas.
Additional Information
How to cite this article: Chua, K. H. et al. Development of High Resolution Melting Analysis for the Diagnosis of Human Malaria. Sci. Rep. 5, 15671; doi: 10.1038/srep15671 (2015).
Acknowledgments
This research was supported by High Impact Research MoE Grant UM.C/625/1/HIR/MoE/E000044-20001.
Footnotes
Author Contributions K.H.C., C.C.N., P.C.L., Y.A.L.L., T.P.L. and H.C.C. contributed in conception and design of the study. S.C.L. and H.C.C. carried out the study and performed literature search. S.C.L., T.P.L. and H.C.C. conducted data acquisition and statistical analyses. All authors were involved in the manuscript preparation and revision, approval of the final version of the manuscript, and agreed for all aspects of the work.
References
- Perlmann P. & Troye-Blomberg M. Malaria blood-stage infection and its control by the immune system. Folia Biol (Praha) 46, 210–218 (2000). [DOI] [PubMed] [Google Scholar]
- Trampuz A., Jereb M., Muzlovic I. & Prabhu R. M. Clinical review: Severe malaria. Crit Care 7, 315–323 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev 15, 66–78 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouatcho J. C. & Goldring J. P. Malaria rapid diagnostic tests: challenges and prospects. J Med Microbiol 62, 1491–1505 (2013). [DOI] [PubMed] [Google Scholar]
- Singh B. et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 363, 1017–1024 (2004). [DOI] [PubMed] [Google Scholar]
- Snounou G., Viriyakosol S., Jarra W., Thaithong S. & Brown K. N. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol 58, 283–292 (1993). [DOI] [PubMed] [Google Scholar]
- Snounou G. et al. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol 61, 315–320 (1993). [DOI] [PubMed] [Google Scholar]
- Johnston S. P. et al. PCR as a confirmatory technique for laboratory diagnosis of malaria. J Clin Microbiol 44, 1087–1089 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers D., Ryan S., Harnett G. & Chidlow G. Diagnosis of malaria aided by polymerase chain reaction in two cases with low‐level parasitaemia. Intern Med J 33, 613–615 (2003). [DOI] [PubMed] [Google Scholar]
- Farcas G. A., Zhong K. J., Lovegrove F. E., Graham C. M. & Kain K. C. Evaluation of the Binax NOW® ICT test versus polymerase chain reaction and microscopy for the detection of malaria in returned travelers. Am J Trop Med Hyg 69, 589–592 (2003). [PubMed] [Google Scholar]
- Singh B., Bohogare A., Cox-Singh J. & Snounou G. A genus-and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am J Trop Med Hyg 60, 687–692 (1999). [DOI] [PubMed] [Google Scholar]
- Brown A. E., Kain K. C., Pipithkul J. & Webster H. K. Demonstration by the polymerase chain reaction of mixed Plasmodium falciparum and P. vivax infections undetected by conventional microscopy. Trans R Soc Trop Med Hyg 86, 609–612 (1992). [DOI] [PubMed] [Google Scholar]
- Kimura M. et al. Identification of the four species of human malaria parasites by nested PCR that targets variant sequences in the small subunit rRNA gene. Parasitol Int 46, 91–95 (1997). [Google Scholar]
- Rubio J. et al. Usefulness of seminested multiplex PCR in surveillance of imported malaria in Spain. J Clin Microbiol 37, 3260–3264 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio J. et al. Alternative polymerase chain reaction method to identify Plasmodium species in human blood samples: the semi-nested multiplex malaria PCR (SnM-PCR). Trans R Soc Trop Med Hyg 96, S199–S204 (2002). [DOI] [PubMed] [Google Scholar]
- Montenegro L. M. et al. Development of a single tube hemi-nested PCR for genus-specific detection of Plasmodium in oligoparasitemic patients. Trans R Soc Trop Med Hyg 98, 619–625 (2004). [DOI] [PubMed] [Google Scholar]
- Ndao M. et al. Comparison of blood smear, antigen detection, and nested-PCR methods for screening refugees from regions where malaria is endemic after a malaria outbreak in Quebec, Canada. J Clin Microbiol 42, 2694–2700 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper C. et al. Detection of very low level Plasmodium falciparum infections using the nested polymerase chain reaction and a reassessment of the epidemiology of unstable malaria in Sudan. Am J Trop Med Hyg 54, 325–331 (1996). [DOI] [PubMed] [Google Scholar]
- Zakeri S., Najafabadi S., Zare A. & Djadid N. Detection of malaria parasites by nested PCR in south-eastern, Iran: evidence of highly mixed infections in Chahbahar district. Malar J 1, 2 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B. et al. Detection of malaria in Malaysia by nested polymerase chain reaction amplification of dried blood spots on filter papers. Trans R Soc Trop Med Hyg 90, 519–521 (1996). [DOI] [PubMed] [Google Scholar]
- Cox-Singh J., Mahayet S., Abdullah M. S. & Singh B. Increased sensitivity of malaria detection by nested polymerase chain reaction using simple sampling and DNA extraction. Int J Parasitol 27, 1575–1577 (1997). [DOI] [PubMed] [Google Scholar]
- Fuehrer H.-P. et al. Novel nested direct PCR technique for malaria diagnosis using filter paper samples. J Clin Microbiol 49, 1628–1630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixson-Hayden T., Lucchi N. & Udhayakumar V. Evaluation of three PCR-based diagnostic assays for detecting mixed Plasmodium infection. BMC Res Notes 3, 88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonma P., Christensen P. R., Suwanarusk R., Price R. N. & Russell B. Comparison of three molecular methods for the detection and speciation of Plasmodium vivax and Plasmodium falciparum. Malar J 6, 124 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haanshuus C. G. et al. A novel, single-amplification PCR targeting mitochondrial genome highly sensitive and specific in diagnosing malaria among returned travellers in Bergen, Norway. Malar J 12, 26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew C. H., Lim Y. A., Lee P. C., Mahmud R. & Chua K. H. Hexaplex PCR detection system for identification of five human Plasmodium species with an internal control. J Clin Microbiol 50, 4012–4019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mens P. F., Schoone G. J., Kager P. A. & Schallig H. D. Detection and identification of human Plasmodium species with real-time quantitative nucleic acid sequence-based amplification. Malar J 5, 80 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold K. A. et al. Real-time PCR for detection and identification of Plasmodium spp. J Clin Microbiol 43, 2435–2440 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor B. J. et al. Real-time PCR detection of Plasmodium directly from whole blood and filter paper samples. Malar J 10, 244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Monbrison F., Angei C., Staal A., Kaiser K. & Picot S. Simultaneous identification of the four human Plasmodium species and quantification of Plasmodium DNA load in human blood by real-time polymerase chain reaction. Trans R Soc Trop Med Hyg 97, 387–390 (2003). [DOI] [PubMed] [Google Scholar]
- Oddoux O. et al. Identification of the five human Plasmodium species including P. knowlesi by real-time polymerase chain reaction. Eur J Clin Microbiol Infect Dis 30, 597–601 (2011). [DOI] [PubMed] [Google Scholar]
- Divis P. C., Shokoples S. E., Singh B. & Yanow S. K. A TaqMan real-time PCR assay for the detection and quantitation of Plasmodium knowlesi. Malar J 9, 344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemont M. et al. Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J Clin Microbiol 42, 5636–5643 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perandin F. et al. Development of a Real-Time PCR Assay for Detection of Plasmodium falciparum, Plasmodium vivax, and Plasmodium ovale for Routine Clinical Diagnosis. J Clin Microbiol 42, 1214–1219 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farcas G. A., Zhong K. J., Mazzulli T. & Kain K. C. Evaluation of the RealArt Malaria LC real-time PCR assay for malaria diagnosis. J Clin Microbiol 42, 636–638 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokoples S. E., Ndao M., Kowalewska-Grochowska K. & Yanow S. K. Multiplexed real-time PCR assay for discrimination of Plasmodium species with improved sensitivity for mixed infections. J Clin Microbiol 47, 975–980 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass C. et al. PCR-based detection of Plasmodium in Anopheles mosquitoes: a comparison of a new high-throughput assay with existing methods. Malar J 7, 177 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama B. E. et al. Real-time PCR versus conventional PCR for malaria parasite detection in low-grade parasitemia. Exp Parasitol 116, 427–432 (2007). [DOI] [PubMed] [Google Scholar]
- Lee M.-A. et al. Real-time fluorescence-based PCR for detection of malaria parasites. J Clin Microbiol 40, 4343–4345 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monis P. T., Giglio S. & Saint C. P. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal Biochem 340, 24–34 (2005). [DOI] [PubMed] [Google Scholar]
- Reed G. H., Kent J. O. & Wittwer C. T. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics 8, 597–608 (2007). [DOI] [PubMed] [Google Scholar]
- Kipanga P. N. et al. High-resolution melting analysis reveals low Plasmodium parasitaemia infections among microscopically negative febrile patients in western Kenya. Malar J 13, 429 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A. et al. GenBank. Nucleic Acids Res 41, D36–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41, 95–98 (1999). [Google Scholar]
- Rozen S. & Skaletsky H. J. in Bioinformatics Methods and Protocols: Methods in Molecular Biology (eds Krawetz S. & Misener S.) 365–386 (Human Press, 2000). [DOI] [PubMed] [Google Scholar]
- Escalante A. A. & Ayala F. J. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci USA 91, 11373–11377 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharavanij S. New developments in malaria diagnostic techniques. Southeast Asian J Trop Med Public Health 21, 3–16 (1990). [PubMed] [Google Scholar]
- Milne L. M., Kyi M. S., Chiodini P. L. & Warhurst D. C. Accuracy of routine laboratory diagnosis of malaria in the United Kingdom. J Clin Pathol 47, 740–742 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongsrichanalai C., Barcus M. J., Muth S., Sutamihardja A. & Wernsdorfer W. H. A review of malaria diagnostic tools: microscopy and rapid diagnostic test (RDT). Am J Trop Med Hyg 77, 119–127 (2007). [PubMed] [Google Scholar]
- Houwen B. Blood film preparation and staining procedures. Clin Lab Med 22, 1–14, v (2002). [DOI] [PubMed] [Google Scholar]
- Mayxay M., Pukrittayakamee S., Newton P. N. & White N. J. Mixed-species malaria infections in humans. Trends Parasitol 20, 233–240 (2004). [DOI] [PubMed] [Google Scholar]
- Looareesuwan S., White N. J., Chittamas S., Bunnag D. & Harinasuta T. High rate of Plasmodium vivax relapse following treatment of falciparum malaria in Thailand. Lancet 2, 1052–1055 (1987). [DOI] [PubMed] [Google Scholar]
- William T. & Menon J. A review of malaria research in malaysia. Med J Malaysia 69 Suppl A, 82–87 (2014). [PubMed] [Google Scholar]
- Fabre R., Berry A., Morassin B. & Magnaval J. F. Comparative assessment of conventional PCR with multiplex real-time PCR using SYBR Green I detection for the molecular diagnosis of imported malaria. Parasitology 128, 15–21 (2004). [DOI] [PubMed] [Google Scholar]
- Wong Y. P., Chua K. H. & Thong K. L. One-step species-specific high resolution melting analysis for nosocomial bacteria detection. J Microbiol Methods 107, 133–137 (2014). [DOI] [PubMed] [Google Scholar]
- Yin X. et al. High-resolution melting curve analysis for rapid detection of rifampin resistance in Mycobacterium tuberculosis: a meta-analysis. J Clin Microbiol 51, 3294–3299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Li M., Wang L., Xin T. & He Q. High-resolution melting analysis for the detection of two erythromycin-resistant Bordetella pertussis strains carried by healthy schoolchildren in China. Clin Microbiol Infect 19, E260–262 (2013). [DOI] [PubMed] [Google Scholar]
- Bastien P., Procop G. W. & Reischl U. Quantitative real-time PCR is not more sensitive than “conventional” PCR. J Clin Microbiol 46, 1897–1900 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]