

Abstract
Smoking is a major risk factor in many diseases. Genome wide association studies have linked genes for nicotine dependence and smoking behavior to increased risk of cardiovascular, pulmonary, and malignant diseases. We conducted an epigenome wide association study in peripheral-blood DNA in 464 individuals (22 current smokers and 263 ex-smokers), using the Human Methylation 450 K array. Upon replication in an independent sample of 356 twins (41 current and 104 ex-smokers), we identified 30 probes in 15 distinct loci, all of which reached genome-wide significance in the combined analysis P < 5 × 10−8. All but one probe (cg17024919) remained significant after adjusting for blood cell counts. We replicated all 9 known loci and found an independent signal at CPOX near GPR15. In addition, we found 6 new loci at PRSS23, AVPR1B, PSEN2, LINC00299, RPS6KA2, and KIAA0087. Most of the lead probes (13 out of 15) associated with cigarette smoking, overlapped regions of open chromatin (FAIRE and DNaseI hypersensitive sites) or / and H3K27Ac peaks (ENCODE data set), which mark regulatory elements. The effect of smoking on DNA methylation was partially reversible upon smoking cessation for longer than 3 months. We report the first statistically significant interaction between a SNP (rs2697768) and cigarette smoking on DNA methylation (cg03329539). We provide evidence that the metSNP for cg03329539 regulates expression of the CHRND gene located circa 95 Kb downstream of the methylation site. Our findings suggest the existence of dynamic, reversible site-specific methylation changes in response to cigarette smoking , which may contribute to the extended health risks associated with cigarette smoking.
Keywords: CHRND, CPOX, DNA methylation, epigenome-wide screen, gene network, metQTLs, rs2697768, smoking
Abbreviations
- AHRR
aryl-hydrocarbon receptor repressor
- ALPP
alkaline phosphatase, placental
- AVPR1B
arginine vasopressin
- CHRND
cholinergic nicotinic receptor
- COPD
chronic obstructive pulmonary disease
- CPOX
coproporphyrinogen oxidase
- DNMT
DNA methyltransferase
- EWAS
epigenome wide association study
- FDR
false discovery rate
- GWAS
genome-wide association studies
- metQTL
methylation quantitative trait loci
- PRSS23
serine protease 23
- PSEN2
presenilin-2 gene
- RPS6KA2
ribosomal protein S6 kinase
Introduction
Cigarette smoking is a significant cause of premature death and disease being one of the most important risk factors for cancer, heart disease, stroke, and chronic lung disease. The mechanisms by which tobacco consumption causes harm have not been fully elucidated. Twin studies document substantial heritability to smoking initiation, smoking persistence, and nicotine dependence, suggesting a substantial genetic component in inter-individual differences in the ability to quit smoking.1 There is increasing evidence that epigenetic variation plays an important role in several complex traits,2,3 for example, nicotine exposure has an effect on promoter methylation4 and has also been associated with lung cancer.5 An association of the offspring's DNA methylation with paternal DNA methylation that is strongest if both have never smoked6 has been recently reported while the degree of global hypomethylation was associated with smoking history in squamous cell carcinoma.6,7
Promoter hypermethylation represents an epigenetic hit that inactivates gene expression by extensive methylation of cytosines in CpG dinucleotide-rich islands in the promoter-enhancer region of a gene. Significant associations have been established between smoking and promoter hypermethylation. It has been shown that the frequency of promoter methylation is significantly higher among smokers, compared to never-smokers.8 As DNA hypermethylation is now recognized as an alternative, epigenetic mechanism for gene silencing in lung cancer, several environmental exposures are thought to cause aberrant DNA methylation, including dietary factors and chemotherapeutic agents, among others. Interestingly, as well as gene specific hypermethylation,9 smoking has also been associated with global hypomethylation.10 Genome-wide association studies (GWAS) have established one locus associated with nicotine dependence and smoking quantity, on chromosome 15q25.11 The same locus is also associated with lung cancer, peripheral arterial disease and chronic obstructive pulmonary disease (COPD) and lung function.12 Animal models suggest that epigenetic changes arise in lung tissue following short-term exposure to tobacco smoke condensate and precede histopathological changes.13 Exposure to tobacco smoke is also believed to alter expression of DNA methyltransferase (DNMT) enzymes14 and modulate histone modifications, including acetylation and methylation.15 However, these results do not reveal whether (i) DNA hyper- or hypo-methylation occur early or late in the pathogenesis of tobacco smoke related diseases and; (ii) methylation precedes the pathology of the disease or is a direct consequence of smoking. Early epigenetic changes in carcinogenesis (especially those related to smoking) are hypothesized to occur somewhat diffusely in the lung and may therefore be detectable in noncancerous lung tissue, as well as in any cancers that arise.16,17 Many of the effects of smoking on the lung are thought to result from the direct effects of cigarette smoke on pulmonary epithelium and alveolar macrophages. However, the exact mechanism(s) through which smoking increases the risk for disease in non-pulmonary tissues, such as blood and brain, are unclear. Recently, sets of convergent findings have suggested that a portion of that vulnerability may be driven by differential DNA methylation acquired by smoking.18,19-22 To date, 9 loci have been confirmed at the genome-wide level of significance to show differential methylation between current smokers and non-smokers23-27.
The aim of this study was to investigate the genome-wide methylation status of current and ex-smokers versus non-smokers taking advantage of recent technological advancements that allow interrogation of methylation levels at 485,577 sites in the human genome.28 Those sites are located both at gene promoters and other genomic features (e.g., intra- and inter-genic CpG islands). We hypothesize not only that specific CpG sites are differentially methylated in smokers and non-smokers in a tissue specific manner, but also that specific genes associated with smoking also show altered methylation in peripheral blood.
Results
Epigenome-wide screen
We assessed methylation levels across the genome with the Illumina HumanMethylation450K array in peripheral-blood DNA samples from 464 CARDIOGENICS individuals (Fig. 1). Among this discovery cohort of European descent, 22 were current, 263 were ex-smokers and 179 were never-smokers ranging from 38 to 68 years of age (Table S1). In the discovery phase, we found 53 probes to be associated with smoking at P ≤ 10−6 (Table S2). We then tested these 53 probes for replication in an independent cohort of 356 unselected female twins (41 current, 104 former and 211 non-smokers) from the TwinsUK Registry that were profiled for DNA methylation using the same array platform. In this instance, we applied a linear mixed model to adjust for random effects consisting of family ID and zygosity in this analysis. Of the 53 probes tested, 30 replicated after applying Bonferroni correction (adjusted P < 9.43 × 10−4) and checking that signals were directionally consistent (Table 1) in the 2 cohorts; one probe, cg27537125, was significant but was removed as it showed an opposite effect. We combined P-values from the discovery stage with their respective replication P-values using Fisher's method and found all 30 replicating probes, which represent 15 distinct loci, to exceed the genome-wide threshold of significance (P < 5 × 10−8) (Table 1). We defined independent loci when probes were at least 1 Mb apart.
Figure 1.

EWAS Manhattan plot for smoking status in the CARDIOGENICS cohort and QQ plot (A). The vertical axis indicates (-log10 transformed) observed P-values, and the dotted horizontal line indicates the threshold of significance (P = 10−6) to select markers for replication. Previously reported loci are indicated in blue, new loci and new signals in known loci are marked in red. Panel (B) illustrates a QQ plot of the distribution of the P values.
Table 1.
Known and new loci associated with differential DNA methylation upon cigarette smoking.
| CARDIOGENICS (Unadjusted for Blood Cell Counts) |
EPITWIN (Unadjusted for Blood Cell Counts) |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Difference in median methylation (%) |
Difference in median methylation (%) |
CARDIO GENICSEPITWINAdjusted for Blood Cell Counts* |
metQTL$ |
|||||||||||||
| Probe ID | Ch | Position | Gene Locus (if non overlapping) | Ex-Never | Current-Never | P value | Ex-Never | Current-Never | P value | Fisher's P value | P value | P value | Fisher's | Lead SNP | P value | h2 |
| Known Loci | ||||||||||||||||
| cg06644428 | 2 | 233284112 | (ALPPL2 – CHRND4) | −2.8 | −4.4 | 4.62E-17 | −1.7 | −2.6 | 6.46E-09 | 1.72E-23 | 3.17E-14 | 2.58E-08 | 4.04E-20 | rs909431 | 1.71E-04 | >0.01 |
| cg05951221 | 2 | 233284402 | (ALPPL2 – CHRND4) | −8.4 | −13.2 | 2.35E-32 | −4.8 | −10.9 | 9.62E-25 | 2.92E-54 | 9.80E-30 | 5.83E-22 | 6.67E-49 | rs2697781 | 1.56E-05 | 0.07 |
| cg21566642 | 2 | 233284661 | (ALPPL2 – CHRND4) | −9.9 | −17.6 | 2.80E-31 | −5.1 | −16.2 | 6.89E-25 | 2.45E-53 | 1.10E-25 | 1.93E-21 | 2.26E-44 | 2-232993320 | 1.32E-04 | 0.12 |
| cg01940273 | 2 | 233284934 | (ALPPL2 – CHRND4) | −5.9 | −12.7 | 3.68E-32 | −3.3 | −11.7 | 1.10E-19 | 4.74E-49 | 2.33E-28 | 1.92E-16 | 4.53E-42 | 2-233040600 | 1.17E-04 | >0.01 |
| cg19859270 | 3 | 98251294 | GPR15 | −1.6 | −3.5 | 4.17E-19 | −0.8 | −2.7 | 1.07E-14 | 3.37E-31 | 2.66E-13 | 8.24E-13 | 1.27E-23 | — | — | 0.64 |
| cg05575921 | 5 | 373378 | AHRR | −10.1 | −27.8 | 3.02E-32 | −3 | −19.7 | 1.84E-39 | 9.04E-69 | 8.65E-25 | 6.46E-35 | 7.55E-57 | — | — | 0.41 |
| cg21161138 | 5 | 399360 | AHRR | −3 | −8.7 | 9.29E-22 | −1.2 | −5.6 | 7.00E-18 | 5.78E-37 | 1.04E-14 | 9.44E-16 | 6.64E-28 | — | — | 0.93 |
| cg06126421 | 6 | 30720080 | IER3 | −8.8 | −12.5 | 1.82E-23 | −3.6 | −10.9 | 4.58E-16 | 7.39E-37 | 1.11E-22 | 1.63E-14 | 1.50E-34 | rs12660883 | 2.64E-07 | >0.01 |
| cg03636183 | 19 | 17000585 | F2RL3 | −7.4 | −13.4 | 1.06E-22 | −1.1 | −10.6 | 4.31E-16 | 3.97E-36 | 2.78E-19 | 6.77E-15 | 1.44E-31 | rs2227383 | 1.65E-04 | 0.63 |
| cg27241845# | 2 | 233250370 | (ALPP) | −2.4 | −7 | 1.20E-10 | −1 | −3.1 | 9.21E-10 | 4.93E-18 | 5.48E-06 | 4.28E-09 | 7.60E-13 | rs3748971 | 2.65E-04 | 0.35 |
| cg03991871# | 5 | 368447 | AHRR | −2 | −6.2 | 7.60E-08 | −0.6 | −2.5 | 0.000273 | 5.31E-10 | 2.34E-05 | 2.60E-04 | 1.21E-07 | 5-448883 | 1.46E-12 | >0.01 |
| cg25648203# | 5 | 395444 | AHRR | −2.1 | −6.7 | 6.04E-16 | 0.3 | −3.9 | 5.02E-06 | 1.46E-19 | 4.81E-10 | 2.09E-05 | 3.34E-13 | — | — | >0.01 |
| cg19572487# | 17 | 38476024 | RARA | −3.6 | −7.1 | 3.64E-10 | −3.1 | −3.5 | 1.23E-09 | 1.94E-17 | 6.91E-07 | 3.10E-07 | 6.45E-12 | — | — | 0.83 |
| cg25189904# | 1 | 68299493 | GNG12 / GNG12-AS1 | −5.3 | −7.9 | 4.89E-09 | −3.8 | −6 | 2.71E-09 | 5.28E-16 | 1.97E-05 | 8.40E-09 | 5.04E-12 | rs2419050 | 1.59E-08 | 0.93 |
| cg23480021# | 3 | 22412746 | ZNF385D | 10.8 | 13.8 | 3.23E-11 | −0.3 | 7.4 | 9.36E-06 | 1.11E-14 | 6.86E-09 | 2.40E-04 | 4.63E-11 | — | — | 0.81 |
| cg21611682# | 11 | 68138269 | LRP5 | −1.1 | −4.1 | 2.09E-07 | −0.8 | −3.6 | 3.82E-06 | 2.30E-11 | 4.12E-06 | 5.17E-05 | 4.96E-09 | rs312777 | 3.91E-04 | 0.51 |
| New Signals in Known Loci | ||||||||||||||||
| cg03329539 | 2 | 233283329 | (ALPP) | −2.6 | −5.7 | 7.72E-17 | −2.1 | −2.5 | 1.91E-15 | 1.06E-29 | 6.28E-12 | 2.14E-13 | 7.52E-23 | rs2697794 | 4.41E-06 | 0.53 |
| cg13193840 | 2 | 233285289 | (ALPPL2 – CHRND4) | −1.8 | −2.8 | 1.15E-10 | −0.9 | −2.1 | 1.98E-06 | 8.43E-15 | 5.54E-08 | 4.54E-05 | 6.97E-11 | — | — | >0.01 |
| cg02657160 | 3 | 98311063 | CPOX | −1.3 | −2.6 | 4.90E-10 | −0.8 | −0.8 | 7.79E-05 | 1.22E-12 | 1.23E-06 | 1.31E-04 | 3.80E-09 | — | — | 0.41 |
| cg14817490 | 5 | 392920 | AHRR | −1.9 | −7 | 4.14E-11 | −2 | −4.7 | 4.18E-13 | 9.24E-22 | 2.34E-07 | 1.88E-11 | 1.80E-16 | rs2672725 | 2.13E-04 | >0.01 |
| cg24090911 | 5 | 400732 | AHRR | −1.6 | −6 | 1.39E-09 | −0.7 | −1.8 | 6.72E-05 | 2.90E-12 | 2.48E-07 | 1.04E-04 | 6.55E-10 | rs2671903 | 6.14E-07 | 0.73 |
| cg24859433 | 6 | 30720203 | IER3 | −2.2 | −5 | 7.00E-14 | −0.2 | −2.5 | 2.62E-06 | 8.10E-18 | 6.33E-11 | 2.00E-05 | 4.47E-14 | — | — | 0.00 |
| cg15342087 | 6 | 30720209 | IER3 | −1.9 | −4 | 6.01E-15 | −0.3 | −1.6 | 5.73E-08 | 1.74E-20 | 1.49E-10 | 2.14E-06 | 1.17E-14 | — | — | 0.00 |
| Probes Confounded by Blood Cell Counts | ||||||||||||||||
| cg17024919 | 3 | 21792248 | ZNF385D | −4.6 | −6.3 | 3.83E-07 | −1.8 | −2.4 | 0.00033 | 3.01E-09 | 2.19E-03 | 7.52E-03 | 1.54E-04 | rs7634827 | 1.02E-04 | 0.89 |
| New Loci | ||||||||||||||||
| cg11660018 | 11 | 86510915 | PRSS23 | −2.3 | −5.6 | 3.89E-08 | −2 | −3.9 | 9.10E-08 | 1.21E-13 | 2.66E-07 | 1.21E-07 | 1.04E-12 | — | — | 0.96 |
| cg20295214 | 1 | 206226794 | AVPR1B | −0.2 | −2.4 | 3.11E-07 | −0.1 | −1.9 | 5.97E-06 | 5.20E-11 | 1.69E-06 | 4.12E-05 | 1.69E-09 | — | — | 0.36 |
| cg03547355 | 1 | 227003060 | (PSEN2) | −1.2 | −2.6 | 5.86E-08 | −0.7 | −1.4 | 9.30E-05 | 1.47E-10 | 1.28E-05 | 9.67E-04 | 2.38E-07 | rs61835656 | 4.40E-06 | >0.01 |
| cg23079012 | 2 | 8343710 | LINC00299 | −0.9 | −5.3 | 1.80E-09 | −0.01 | −1.9 | 0.00094 | 4.76E-11 | 4.40E-05 | 6.61E-04 | 5.34E-07 | — | — | >0.01 |
| cg22717080 | 6 | 166959505 | RPS6KA2 | −0.3 | −1.7 | 1.68E-07 | −0.03 | −0.7 | 0.000497 | 2.02E-09 | 6.42E-08 | 8.56E-04 | 1.35E-09 | — | — | 0.13 |
| cg02451831 | 7 | 26578098 | KIAA0087 | −0.8 | −3.2 | 1.32E-07 | −0.1 | −2.8 | 4.67E-06 | 1.79E-11 | 1.34E-05 | 1.26E-07 | 4.75E-11 | — | — | 0.06 |
Adjusted for monocyte, lymphocyte and neutrophil cell counts$ metSNPs passing Bonferroni correction (4.2 × 10−4).
Of the 30 probes corresponding to 15 loci, 6 have not been previously reported at the genome-wide level of significance (Table 1; Box 1 provides a brief description of the genes linked to the associated probes). All 9 loci previously known to be associated with differential methylation upon cigarette smoking18,22-27 replicated in our study reaching genome-wide significance (Table 1; F2RL3, AHRR, GPR15, IER3, ALPP, RARA, GNG12, ZNF385D, and LRP5). The strongest signal was in AHRR (cg05575921; P = 9.04 × 10−69) followed by probe cg05951221 (P = 2.92 × 10−54), which is located near the alkaline phosphatase gene cluster on chromosome 2q3725. In total, 7 probes reached genome wide significance in this locus (Fig. 2A), which harbors 2 CpG islands at 233, 251, 361-233, 253, 414 and 233, 283, 397–233, 285, 959 bp, respectively.28 A single probe, cg27241845, was significant in the first CpG island and was located near the alkaline phosphatase, placental (ALPP) gene. The remaining 6 probes, including cg05951221, are located in the second CpG island nearest to the ALPPL2 gene. Of notice is the presence of a cholinergic nicotinic receptor (CHRND) gene ∼100 Kb downstream of cg05951221. It is also worth noting that in addition to theGPR15 signal (cg19859270, P = 3.37 × 10−31; Fig. 2B), we detected a second one with probe cg02657160 (P = 1.22 × 10−12), which is located 60 Kb away in the intron of the coproporphyrinogen oxidase (CPOX) gene (Fig. 2B).
Box 1.
Literature based functional annotation for candidate genes in the 15 known and new loci associated with DNA methylation in response to cigarette smoking.
| Gene | Function |
|---|---|
| RARA | Retinoic acid receptor, nuclear receptor, steroid hormone receptor |
| PRSS23 | Extracellular region, nucleus, proteolysis, serine-type endopeptidase activity |
| CPOX | Heme biosynthetic process, small molecule metabolic process |
| GNG12 | Energy reserve metabolic process, cerebral cortex development, small molecule metabolic process, phosphate ion binding, G-protein coupled receptor signaling pathway |
| RPS6KA2 | Toll signaling pathway, toll-like receptor 1 signaling pathway, positive regulation of apoptotic process, TRIF-dependent toll-like receptor signaling pathway, cardiac muscle cell apoptotic process |
| AVPR1B | Vasopressin receptor activity, response to stress, positive regulation of blood pressure, positive regulation of heart rate, peptide hormone binding, regulation of systemic arterial blood pressure by vasopressin |
| GPR15 | G-protein coupled receptor signaling pathway, integral to plasma membrane |
| ZNF385D | Nucleic acid binding, zinc ion binding |
| LRP5 | Positive regulation of cell proliferation, cholesterol metabolic process, induction of apoptosis, regulation of blood pressure, regulation of insulin secretion, transcription factor activity, Wnt-activated receptor activity, |
| CHRND | Ion transport, postsynaptic membrane, neuromuscular process, muscle contraction, receptor activity, acetylcholine binding, nicotinic receptor |
| KIAA0087 | lncRNA |
| IER3 | Anti-apoptotic gene involved in cellular stress responses, inflammation and tumorigenesis. May play a role in the ERK signaling pathway by inhibiting the dephosphorylation of ERK by phosphatase PP2A-PPP2R5C holoenzyme |
| AHRR | DNA binding, DNA-dependent, regulation of transcription, negative regulation of transcription from RNA polymerase II promoter, positive regulation of protein sumoylation |
| LRP5 | Wnt receptor signaling pathway |
| LINC00299 | Long intergenic non-protein coding RNA 299 |
| RPS6KA2 | Serine/threonine-protein kinase downstream of ERK, stress-induced activation of transcription factors, May function as tumor suppressor in epithelial ovarian cancer cells |
Figure 2.

Regional plots of known and new loci associated with cigarette smoking. For each locus the association plot (bottom half of each panel plotting all probes and their respective –log10(P) values) is shown in the context of genomic annotation tracks (e.g., CpG islands, RefSeq gene structures, regulatory elements reported by ENCODE) available in the UCSC genome browser (http://genome.ucsc.edu/) – correlated regions are marked by red lines. The location of the 450 K array probes in the UCSC display is shown as gray rectangles. The depicted loci are: (A) the known locus 2q37.1 in which we detected an interaction between rs12996863 and smoking on DNA methylation levels and (B) the known locus GPR15 in which we identified a new, possibly independent, signal near CPOX.
We then examined the location of the most associated probes with respect to regulatory elements from the ENCODE data as well as DNaseI peaks which mark regions of open chromatin.29 Among the 6 new loci (see Fig. S2 for each locus’ regional association plot and genomic context), 2 harbor probes overlapping an H3K27Ac peak. Probe cg03547355 (P = 1.47 × 10−10) is located at 1q42.12 in an intergenic region 54 Kb away from the presenilin-2 gene (PSEN2) and the overlapping H3K27Ac peak was found in 3 cell lines.30 The last significant probe to overlap a strong H3K27Ac peak, cg11660018 (P = 1.21 × 10−13), mapped at the promoter of the serine protease 23 (PRSS23) gene.
A further 2 new loci overlapped open chromatin regions marked by DNaseI and/or FAIRE peaks detected in the ENCODE study.29 Probe cg22717080 (P = 2.02 × 10−09) was located in an intron of the ribosomal protein S6 kinase (RPS6KA2) gene and overlapped a FAIRE peak. Probe cg20295214 (P = 5.20 × 10−11) was located in an intron of the arginine vasopressin (AVPR1B) gene and overlapped a DNaseI hypersensitivity site reported by ENCODE in HepG2 cells (weaker signals were observed in other cell types).
Finally, for 2 of the new loci the associated probe did not overlap any element. Probe cg02451831 (P = 1.79 × 10−11) mapped to the 3’ UTR of the KIAA0087gene andcg23079012 (P = 4.76 × 10−11) in an intron of a long non-coding RNA gene, LINC00299, at chromosome 2q25.1.
Differential DNA methylation is known to be strongly associated with age.3 We found no overlap between the 30 probes significantly associated with smoking and known DMRs associated with age.3 Furthermore, in the discovery cohort we found no overlap between the 30 CpG sites associated with smoking (adjusted for age and gender) and the 1,210 and 11,751 CpG sites associated with age and gender at P < 10−6, respectively. Association results for the 30 probes from the age and gender analyses are given in Table S4. Blood is composed of different cell types and thus signals detected in EWASs may be confounded to DNA methylation changes caused by differences in cell counts of the main blood cell types.30 We compared our EWAS results for exposure to cigarette smoking with EWAS results we generated for lymphocyte, monocyte and neutrophil counts respectively in the discovery cohort. As shown in Figure S3 several probes associated with cigarette smoking in our study, show evidence of association with neutrophil and lymphocyte counts (none with monocyte counts) at P < 10−6. We note that among the significant loci, the probes for RARA and PRSS23 showed very strong evidence for association with lymphocyte (P = 8.1 × 10−14) and neutrophil (P = 1.17 × 10−9) cell counts respectively. To assess confounding effects, we re-analyzed the 30 probes including lymphocyte, monocyte and neutrophil counts as covariates in the model. Table 1 shows that all probes but cg17024919 in ZNF385D remained significant in the adjusted model in both the discovery and replication cohort after Bonferroni correction (P = 1.6 × 10−3). Finally, we cross checked the 30 probes against a recently reported list of 1865 differentially methylated probes in isolated cells from blood31 and found no overlap.
In summary, we identified 6 new loci showing differential DNA methylation upon cigarette smoking of which 4 had the most associated probe overlapping a putative regulatory element. The new loci have effect sizes ranging from 1.7 to 5.6% difference in median methylation between current and never smokers (CARDIOGENICS, Table 1).
Correlation of DNA methylation with gene expression levels
DNA methylation is known to play an important role in transcriptional regulation. We set to explore whether the observed changes in DNA methylation levels upon exposure to cigarette smoking were correlated to changes in gene expression levels of the corresponding gene(s). To do so we analyzed whole blood RNA-Seq data from 322 female Twins with available smoking history which overlapped by 129 samples with the EPITWIN replication cohort; 77 never, 39 former and 13 current smokers (data were scaled to 10 million reads per sample filtering out any exons with multiple missing values). In this data set, 8 of the 16 genes linked to the top associated probes showed evidence of expression, PSEN2, PRSS23, RARA, F2RL3, GPR15, CPOX, AHRR, and RPS6KA2. Only GPR15 showed a clear trend of increased gene expression in smokers compared to non-smokers (Fig. S4) suggesting that the reduction in methylation levels we observed in smokers leads to increased transcription (the reverse cannot be excluded). We then tested whether there is any correlation between gene expression and DNA methylation (accounting for age and chip batch id) levels for these genes and found a strong negative correlation for GPR15 (β = −713.01, P = 1.05 × 10−7) and weaker correlations for CPOX (β = 31.31, P = 7.1 × 10−3) and AHRR (β = −45.5, P = 2.19 × 10−2) (Table S5). To assess the effect of smoking and DNA methylation on gene expression we analyzed the 77 never and 13 current smokers with a linear regression model (see Methods). We found a strong positive effect of cigarette smoke exposure on GPR15 expression (β = 0.489, P = 2.52 × 10−9) and weaker, nominally significant, signals for CPOX and RARA (Table S5). In contrast, we found no statistically significant effects of DNA methylation on gene expression after correcting for cigarette smoke exposure (Table S5). Taken together, our data suggests that exposure to cigarette smoking leads to differential DNA methylation in PSEN2, PRSS23, RARA, F2RL3, GPR15, CPOX, AHRR, and RPS6KA2, which is not linked to differential gene expression. However, exposure to cigarette smoking is associated with differential gene expression in GPR15 and possibly CPOX and AHRR.
DNA methylation patterns upon smoking cessation
All but one (cg23480021) of the 30 probes showed a clear trend that smokers have lower methylation levels than non-smokers. Interestingly, we observed that for all 30 probes methylation levels were, at least partially, restored in former-smokers (Fig. 3A and data not shown). However, for none of the probes is DNA methylation completely reversed to non-smoking levels. This suggests that the impact of cigarette exposure on DNA methylation is partially reversible after smoking cessation at least for our 30 most significant probes. We were able to assess, though in a crude way, the timing of this reversal effect in the CARDIOGENICS study where it was recorded whether participants ceased smoking more or less than 12 weeks prior to the recruitment day. We divided all ex-smokers (n = 263) in 2 groups (A) those who had ceased smoking for more than 12 weeks (n = 251) and (B) those who were still ‘active smokers’ up to 12 weeks prior to the recruitment day (n = 12) and compared the 2 groups to the never- and current-smokers. Figure S5 shows the results for the lead probe of each locus associated with cigarette smoking in this study, in most cases (12 out of 15 loci) group (A) had DNA methylation levels almost identical to the never-smokers and group (B) had DNA methylation levels similar to those of the current-smokers (results were consistent for loci with multiple probes, data not shown).
Figure 3.

Genetic interaction at the 2q37.1 locus. The first 2 panels show the effect of (A) smoking on methylation levels at probe cg03329539 in current-, ex- and never- smokers and (B) of SNP rs2697768 on DNA methylation levels of probe cg03329539. (C) shows the allele effect of rs2697768 on methylation levels in current, former and never smokers. The rs2697768-C allele is associated with lower methylation levels in the never-smokers but appears to stay stable in the current smokers where most of the observed reduction in methylation levels is driven by the major allele, rs2697768-A.
cis metQTL analysis
To assess whether any of the probes associated with cigarette smoking (Table 1) is under genetic control in cis we undertook methylation quantitative trait loci (metQTL) analysis in a subset of the CARDIOGENICS samples corresponding to healthy controls (n = 247). We considered all SNPs (1000 Genomes imputed GWAS data) within a 200 Kb window centered on the probe position. In total, we found 61,951 out of the 355,628 (17.42%) CpG probes to have at least one significant metSNP at an FDR of 1%; we note that probes overlapping known SNPs had been removed during QC (Methods).
We then assessed whether probes associated with smoking exposure are more likely to be under genetic control. Looking at the discovery EWAS, we found 329 of the 1,172 probes with a suggestive association (P ≤ 1.00 × 10−3) to have a metSNP (28.07%). We observed a further enrichment among the 31 probes that replicated in our study (Table 1); 45.16% (15 probes in 8 loci) had a metSNP.
Table 1 lists the lead metSNP of each of the 15 probes associated with smoking exposure (see Table S6 for a full list). Taking advantage of the twin design in the EPITWIN study we also estimated narrow-sense heritability (h2) for all significant probes in Table 1. We found 15 probes (10 loci) being heritable (h 2 0.3) of which 7 have metQTLs (Table 1). Highly heritable probes (h2 > 0.5) were more likely to have a metQTL (55%).
In summary, we found wide spread genetic regulation of DNA methylation in 8 of the loci associated with cigarette smoking. Methylation was heritable in 6 out of these 8 loci.
Interaction analysis in loci associated with cigarette smoking and harboring metSNPs
We then tested whether there is an interaction between genetic effects (metSNPs) and smoking on DNA methylation levels in the 8 loci, which were both associated with smoking status and harbored metQTLs. Of the 495 metSNPs, we considered for further analysis 119 LD pruned metSNPs (removed SNPs with r2 0.9 with a lead metSNP) present in both cohorts (CARDIOGENICS and EPITWIN; SNPs are listed in Table S6). Interaction analysis was carried out in PLINK and association results were meta-analyzed using GWAMA to increase power. Table 2 lists the top 17 SNPxSmoking interactions significant at P < 10−3. After applying a Bonferroni correction for the 119 tests (P < 4.2 × 10−4), we found 2 probe-metSNP pairs, cg03329539-rs2697768 and cg03329539-rs55781386, both at the chromosome 2q37.1 locus (Table 2) showing evidence of interaction. Methylation at cg03329539 is highly heritable (h2 = 0.53). The 2 SNPs are in LD r2 = 0.826 and as expected were not independent upon conditional analysis (P = 0.27 for rs2697768 conditioned on rs55781386). Therefore, we considered only rs2697768 for further analyses. Figure 3A shows that current smokers have reduced methylation levels (5.7% in CARDIOGENICS and 2.5% in the EPITWIN cohort) compared to the never-smokers for the top SNP. The minor allele (rs2697768-C) is associated with lower methylation levels in the never-smokers (Fig. 3B) but appears to have a ‘protective effect’ in the current smokers (Fig. 3C) where most of the observed reduction in methylation levels is driven by the major allele, rs2697768-A. The SNP rs2697768 (2:233,270,916) maps between 2 ENCODE DNaseI hypersensitive site clusters at 233,270,707–233,270,898 and 233,271,004–233,275,292, 18bp and 88bp away respectively. Only the latter site was found in a blood related cell line (lymphoblastoid).
Table 2.
Interaction analysis of probes associated with differential methylation upon cigarette smoking and which have a metQTL
| CpG-metSNP | Locus | Alleles |
MAF | BETA | SE | P-value | Q P-value | i2 | Number of samples | Direction of Effect | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | A2 | ||||||||||
| cg03329539-rs2697768 | chr2:233283397-233285959 | C | A | 0.263973 | 0.015895 | 0.004231 | 1.73E-04 | 7.10E-01 | 0.00E+00 | 510 | ++ |
| cg03329539-rs55781386 | chr2:233283397-233285959 | C | G | 0.24386 | 0.015623 | 0.004357 | 3.38E-04 | 6.47E-01 | 0.00E+00 | 510 | ++ |
| cg03329539-rs2697766 | chr2:233283397-233285959 | T | C | 0.352653 | 0.013271 | 0.0039 | 6.72E-04 | 2.33E-01 | 2.98E-01 | 510 | ++ |
| cg03329539-rs2853377 | chr2:233283397-233285959 | A | G | 0.343313 | 0.013237 | 0.003917 | 7.31E-04 | 4.34E-01 | 0.00E+00 | 510 | ++ |
| cg03329539-rs1048988 | chr2:233283397-233285959 | C | G | 0.247151 | 0.01498 | 0.004572 | 1.06E-03 | 3.90E-01 | 0.00E+00 | 410 | ++ |
| cg03329539-rs13017092 | chr2:233283397-233285959 | T | C | 0.294833 | 0.012711 | 0.003926 | 1.21E-03 | 4.19E-01 | 0.00E+00 | 510 | ++ |
| cg14817490-rs7712269 | AHRR | G | C | 0.088767 | -0.024011 | 0.00749 | 1.35E-03 | 6.70E-01 | 0.00E+00 | 510 | – |
| cg03329539-rs62192181 | chr2:233283397-233285959 | T | A | 0.287669 | 0.012454 | 0.003888 | 0.001367 | 0.588166 | 0 | 509 | ++ |
| cg03329539-rs13016319 | chr2:233283397-233285959 | A | T | 0.279882 | 0.012476 | 0.003958 | 0.001626 | 0.211756 | 0.35872 | 509 | ++ |
| cg03329539-rs2344360 | chr2:233283397-233285959 | C | T | 0.473067 | 0.011004 | 0.003598 | 0.002237 | 0.404628 | 0 | 510 | ++ |
| cg03329539-rs13023370 | chr2:233283397-233285959 | G | T | 0.264173 | 0.011811 | 0.004162 | 0.004558 | 0.396488 | 0 | 510 | ++ |
| cg03329539-rs35849718 | chr2:233283397-233285959 | T | C | 0.2939 | 0.011091 | 0.00396 | 0.005112 | 0.627957 | 0 | 510 | ++ |
| cg03329539-rs790040 | chr2:233283397-233285959 | G | A | 0.4826 | 0.009923 | 0.003616 | 0.006081 | 0.772092 | 0 | 510 | ++ |
| cg03329539-rs790039 | chr2:233283397-233285959 | G | C | 0.488293 | 0.009864 | 0.003599 | 0.006152 | 0.780298 | 0 | 510 | ++ |
| cg03329539-rs3762524 | chr2:233283397-233285959 | T | C | 0.46995 | 0.009619 | 0.003609 | 0.007715 | 0.595701 | 0 | 509 | ++ |
| cg03991871-rs2672723 | AHRR | G | A | 0.182547 | 0.012535 | 0.004808 | 0.00915 | 0.013642 | 0.835633 | 510 | −+ |
| cg03991871-rs2671903 | AHRR | C | T | 0.19964 | 0.012203 | 0.004718 | 0.009713 | 0.023106 | 0.806222 | 510 | −+ |
Is the interaction SNP rs2697768 affecting expression levels of a nearby gene?
We undertook expression QTL analysis of rs2697768 (included all 1000 Genomes proxies at an r2 > 0.6) in 7 different cell types (monocytes, macrophages, lymphoblastoid cell lines (LCLs), T-cells, fibroblasts, fat and skin) from published data sets.32-35 Analysis was performed in a 2 Mb window centered on the SNP position. The only association detected at an FDR of 5% was between rs12996863 (r2 of 0.66 with rs2697768) and CHRND expression in LCLs (P = 9.9 × 10−4).35 Interestingly, rs12996863 which is the lead eSNP for CHRND, is in LD (r2 = 0.73) with rs2697794, which is the lead metSNP for the interaction probe cg03329539 (Fig. 4).
Figure 4.
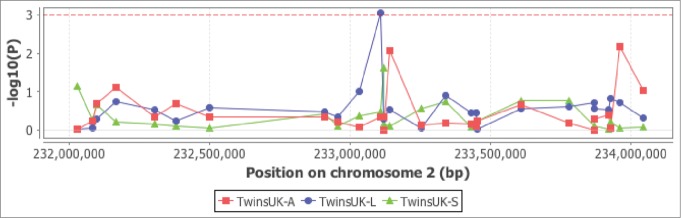
eQTL SNP (rs12996863) of the CHRND gene is associated with regulation of DNA methylation levels at cg03329539. A regional plot of rs12996863 showing a weak eQTL effect (blue line) on CHRND expression in lymphoblastoid cell lines from the MuTHER study [27. No eQTL effect was detected for rs12996863 in adipose (red) or skin (green) tissue from the same MuTHER individuals.
Based on Illumina's human BodyMap v2.0 data (this data was recently added to Ensembl release 62 and is presented as an optional track), expression levels of CHRND in whole blood are very low (< 10−4 fragments per Kb of exon per million fragments mapped). We also assessed CHRND expression levels based in our whole blood RNA-seq data. Given the very low expression of CHRND in whole blood and the available RNA sequence depth in the 322 twins (129 overlap with the EPITWIN cohort) we cannot extract firm conclusions but we did observe that current smokers have a higher median RNA read count than never smokers (Table 3; scaled read count ranged from 0 to 12). The data, although not confirming, point to the assumption that reduced DNA methylation at cg03329539 caused by smoking may induce CHRND expression.
Table 3.
Whole blood RNA-Seq data of CHRND in female twins
| Smoking Status | Available No of Subjects | Median (scl)1 |
|---|---|---|
| Non smoker | 89 | 1.50173 |
| Ex-smoker | 50 | 1.62522 |
| Current smoker | 10 | 2.10951 |
1 Scaled data: number of reads / library depth *10.000.000.
Network analysis
To assess whether genes in the loci showing differential DNA methylation in response to cigarette smoke fall in to specific biological pathways, we performed network analysis with the 17 genes (GNG12, AVPR1B, LINC00299, ALPPL2-CHRND, PSEN2, ZNF385D, GPR15-CPOX, AHRR, LRP5, PRSS23, RPS6KA2, KIAA0087, RARA, F2RL3, and IER3) in the 15 distinct loci significantly associated with smoking. Genes were selected on the basis of overlap or proximity with one of the 30 associated probes except CHRND, which was included because of a common metQTL/eQTL. Including neighboring genes, Ingenuity generated 2 networks, A and B, which included 10 (AHRR, AVPR1B, CHRND, CPOX, F2RL3, GNG12, LRP5, PRSS23, RPS6KA2, and PSEN2) and 2 (IER3 and RARA) of the 17 genes, respectively (Fig. S6). The 2 networks were enriched for genes known to be involved in cardiovascular disease (P = 6.95 × 10−4), cancer (P = 9.77 × 10−4), connective tissue and developmental disorders (P = 9.77 × 10−4), and cell death, cell survival and cell-cell interactions (P = 9.77 × 10−4). We then assessed how genes in the networks overlap with canonical pathways in the Ingenuity database. The top canonical pathways associated with the 2 networks were the heme biosynthetic pathways III and II (P = 3.9 × 10−3) and the Aryl Hydrocarbon receptor signaling (P = 7.69 × 10−3).
Discussion
We undertook an EWAS for cigarette smoking in 464 individuals of European descent and identified 30 probes that replicated in an independent sample of 356 individuals after Bonferroni correction. We replicated all 9 known loci associated with differential DNA methylation upon cigarette smoking in adults. Two of these loci AHRR and F2LR3 have been recently associated with DNA methylation changes in newborns whose mothers smoked during pregnancy.24 The same study reported GFI1, which did not reach significance in our study although several probes showed evidence for association (cg12876356 had P = 1.65 × 10−4 in CARDIOGENICS and P = 9.81 × 10−4 in EPITWIN). GFI1 encodes a nuclear zinc finger protein that functions as a transcriptional repressor involved in diverse developmental contexts, including hematopoiesis and oncogenesis. Therefore it is possible that GFI1 methylation status is affected by exposure to cigarette smoking in newborns as found by Joubert et al. 24 but this effect is much weaker in adults (this study). We note that probes in CYP1A1 and MYO1G showed strong evidence for association at the discovery stage in both our study and that by Joubert et al but did not replicate at the Bonferroni threshold of significance (Table S2, this study). Finally, the study by Sun et al.,27 analyzed an African American sample and reported 5 significant loci of which 3, HNRPUL1, LIM2, and AKT3, have not been reported by any study in Caucasians (including this study in which none of the reported probes showed any trend of association in the discovery cohort P > 0.1). This may be due to differences in smoking habits and / or diversity of the genetic background. Furthermore, the significance of the genetic background has been highlighted by a recent report that showed differences in DNA methylation patterns in loci 6p21.23 and GNG12 in Europeans and East Asians.36
Beside the 9 known loci, 6 other loci had probes that reached genome-wide significance (P < 5 × 10−8) for the first time (Table 1). Several of the lead probes in both known and new loci showed strong association with neutrophil and lymphocyte cell counts but their association to cigarette smoke exposure remained significant (pass Bonferroni correction and reach genome-wide significance in the combined analysis) after adjustment for blood cell counts. Whether blood cell counts change in response to differential DNA methylation triggered by cigarette smoking in some loci e.g RARA and PRSS23 cannot be resolved with the current study design and will require further experiments. The new loci that remained highly significant, after adjusting for blood cell counts, brought the total number of independent CpG sites to 17 (15 distinct loci) showing differential methylation in response to cigarette smoking. Based on the ENCODE annotation, we found that most of the lead probes (13 out of 15) showing differential methylation with smoking status are located in putative regulatory regions in blood related cell types. Most of the putative regulatory elements appear to be distal to the TSS as they were marked by H3K27Ac (7 out of 11), which is associated with active enhancers. For the known loci, we saw the same trend for effect sizes in our study compared to other published reports23-26 with AHRR and ALPPL2-CHRND being the strongest, followed by IER3 and GPR15 but absolute values fluctuated significantly between all studies compared. The new loci (6) had small effect sizes, with the EPITWIN sizes (replication) being consistently lower than in CARDIOGENICS and although this may in part be attributable to the winner's curse, over-adjusting for family ID in EPITWIN is also very likely to contribute to this effect.
Eight of the 17 CpG sites associated with exposure to cigarette smoking, PSEN2, PRSS23, RARA, F2RL3, GPR15, CPOX, AHRR, and RPS6KA2, are linked to genes showing expression in whole blood (RNA-Seq data from 322 individuals). For the remaining sites which are linked to genes showing no detectable levels of expression in blood, we know that they are overall hyper-methylated suggesting that the reduction in methylation levels we observed in the current smokers may have an effect on gene expression but at levels not detectable in our data set. However, although we found that exposure to cigarette smoking is associated with differential gene expression in GPR15 and possibly CPOX and AHRR, we did not observe an association with DNA methylation after correcting for cigarette smoke exposure. We cannot exclude that methylation at other untested CpG sites in these loci is associated with differential gene expression.
In 10 of the loci associated with differential methylation upon exposure to cigarette smoke we found heritable probes (15 probes; h2 > 0.3). Several of the heritable probes had metQTLs (7 out of 15). In total, 8 of the 15 distinct loci (53.3%) had a metSNP, confirming the previously reported abundance of genetic regulation of DNA methylation levels. Environmental variables such as smoking may interact with genetic factors. Despite the small size of our sample (n = 820), we found a significant interaction (Bonferroni correction) between rs2697768 and smoking status altering methylation levels of cg03329539 in the 2q37.1 locus. A second SNP (rs55781386) was also significant for the same probe but the signal was not independent upon conditional analysis. SNP rs2697768 has not been previously associated with smoking or any other related phenotypes. To the best of our knowledge this is the first report of a SNP x smoking interaction on DNA methylation levels. It has previously been suggested that in gene environment interaction studies, proximal variants rather than those with a main-effect (GWAS) are more likely to show a significant association because the interaction tends to weaken the statistical significance of the main effect.37 It is therefore possible that it is the lead metSNP, rs62192178 or a proxy, which interacts with smoking to affect methylation levels at cg03329539. We found that the lead metSNP, rs62192178, was in LD with the lead eSNP of a weak eQTL (5% FDR) we detected for the CHRND gene in LCLs. CHRND which encodes the delta subunit of the acetylcholine receptor of muscle, is located circa 95 Kb downstream the methylation probe cg03329539. The very low expression of CHRND in whole blood did not allow eQTL analysis. Nonetheless, our RNA-seq data showed a trend of higher median expression among current smokers when compared to never smokers. We found no evidence of rs2697768 regulating expression of its adjacent gene alkaline phosphatase, placental-like 2 (ALPPL2), which is known to be transcriptionally stimulated by heme oxygenase-1 (HMOX1).38 We note that the signal (cg02657160) in CPOX, although it did not meet our definition of an independent locus as it is located 70 Kb away from GPR15, implicates another gene of the heme biosynthetic pathway. Heme biosynthesis was the top canonical pathway mapping to the main network A, we obtained using the Ingenuity database.
In all loci (29 out of 30 probes) associated with smoking status we observed a partially reversible pattern of DNA methylation upon smoking cessation. Loci in which methylation patterns are partially reversible may be associated with milder effects of smoking such as inflammation, high blood pressure and vasoconstriction. Under this scenario, multiple genes may interact to form a complex biological web of the symptoms and diseases associated with smoking. On the other hand, such loci may be related to pathways associated with the systemic effect of smoking leading to more severe phenotypes such as COPD and cancer, 2 major consequences of smoking. Studies comparing methylation levels between long term heavy smokers and non-smokers have reported genes associated with cancer such as the p16 promoter17 and RARbeta2.39 The 2 networks, A and B, we constructed and which harbor 12 of the 17 genes associated with differential methylation in response to cigarette smoking in our study, were enriched for genes linked to cancer and cardiovascular disease for which smoking is a known risk factor. It should be noted, however, that one important caveat is that the 450 K array design is biased toward cancer loci. In terms of molecular functions, the 2 networks were enriched in cell cycle regulation, cell death and survival, as well as cellular development and inflammation. It is evident, therefore, that gene interactions may be influenced by changes in DNA methylation in response to smoking. Further analysis to correlate methylation and expression levels in disease relevant tissues, e.g., lung biopsies and samples such as sputum, is required to aid in unraveling the level of significance and specificity of interactions between those loci.
In contrast to studies showing the long-term effects of smoking and the timely process of reversing the effect of smoking after quitting, our data suggests that in specific loci methylation changes toward non-smoking status can be detected within 12 weeks of smoking cessation. Larger samples with more precise information on the timing since cessation are needed to corroborate and fully dissect these initial observations. Furthermore, a new study should include comprehensive information on smoking behavior, which will allow assessing important aspects, such as duration of smoking, smoking burden, and age of smoking initiation. Using a single blood cell type, for example monocytes from peripheral blood, can overcome some of the limitations paused by EWAS studies in blood samples. Nonetheless, expanding the list of biomarkers for cigarette exposure in blood is scalable due to sample accessibility and can lead to comprehensive monitoring of long-term and short-term epigenetic changes in response to smoking.
Materials and Methods
Ethics statement
Both EPITWIN and CARDIOGENICS cohorts recruited under ethically-approved protocols. The protocol was approved by the local Ethical Board at each of the recruitment sites and all subjects provided written informed consent.
Subjects/cohorts
The discovery cohort consisted of subjects participating in the CARDIOGENICS Consortium, a study that recruited coronary artery disease (CAD) and healthy individuals between the ages of 38–67 (Average age: 55.39 ± 6.6). Samples were collected in 3 centers (Paris, Leicester, and Cambridge) and subjects were asked to complete a questionnaire recording their smoking history, all were of self-reported Caucasian ancestry. At the discovery phase 464 subjects were analyzed, of which 238 were CAD cases. The replication cohort consisted of 356 subjects recruited in the EPITWIN study,40 which comprises female twins between the ages of 34–84 (average age: 60.14 ± 8.8). All subjects completed a questionnaire documenting their smoking history and habits (cigarettes smoked per day) and had whole blood drawn for DNA analysis (Supplementary Table 1).
DNA extraction and methylation profiling
For both cohorts, DNA from whole blood was extracted using the DNeasy kit (Qiagen, Inc.). Bisulfite modification of 750 ng of DNA was performed using the 96 well EZ DNA Methylation kit (Zymo Research) according to manufacturer's instructions. DNA methylation levels were assessed using the Infinium HumanMethylation450 K BeadChip (Illumina), which assays 485,577 cytosine positions in the human genome (mainly CpG sites, but also non-CpG sites and 65 random SNPs). The intensities of the images were extracted using the GenomeStudio (2010.3) Methylation module (1.8.5) software. We excluded samples with more than 5% missing probes at detection P < 0.01 (GenomeStudio P-values of detection of signal above background) before normalization. The signal intensities for the methylated and unmethylated state were then quantile normalized for each probe type separately. Beta values which are the ratio of the normalized intensity of the methylated bead type to the combined normalized locus intensity and range from 0 (hypomethylated) to 1 (hypermethylated), were calculated using R 2.12 (Team 2010). Principal component analysis (PCA) of the β values was then performed to assess the impact of known technical factors to the variation in β values as well as to detect any potential outliers. Figure S1 summarizes QC steps and exclusion of extreme outliers by PCA. This procedure was performed iteratively, re-normalizing the data matrix after removing technical outliers at each step. Recruitment site, Beadchip, and BS-treated DNA input were shown to contribute significantly to the variation in β levels and thus together with age and gender were included as covariates in subsequent analyses. Probes included in the analysis were restricted to 459,433 (out of 485,577) whose sequences were uniquely mapped to the GRCh37 genome with up to 2 mismatches using MAQ. Probes overlapping a SNP (MAF > 1%) or CNV reported in the CEU population in the 1000 Genome Project (version 3) were further excluded, resulting in a total of 357,700 probes. Of those, 355,628 were kept for further analysis after removing probes with missing values.
Epigenome-wide association analysis
Multivariate linear regression was used to model the relationship between DNA methylation levels and smoking status. Smoking status was grouped into 3 categories of cigarette exposure (never-, former-, and current-smokers) for both cohorts and coded as a factor (0, 1, and 2) in our model. To regress out known confounders we included in the model age, gender, disease status (coronary artery disease cases and controls), recruitment site, chip batch effect, and BS-treated DNA input. The above model was also run with the inclusion of monocyte, neutrophil, and lymphocyte counts to assess confounding effects (see also below). A mixed-effects model was used to account for relatedness in the EPITWIN analyses, by including random effects for family ID and zygosity in addition to the fixed-effect factors (age, chip, and BS-treated DNA input) applying to this study (all female, non-diseased subjects, and single recruitment site). Monozygotic and dizygotic twins were treated in the same manner in the model.
To assess the statistical significance of the association between methylation levels and smoking status we fitted 2 linear models (a full model with the predictor and a null model without) and compared the 2 by Analysis of Variance (ANOVA). We used Bonferroni adjusted P values to test for replication in the EPITWIN study. Meta-analysis of the discovery and replication results was performed using Fisher's combined P-values method to cover for differences in effect sizes. The data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). The accession number is GSE50660.
Blood phenotype association
We performed EWAS for lymphocyte, monocyte and neutrophil cell counts with the models described above in 419 CARDIOGENICS and 351 EPITWIN samples for which blood counts information was available.
RNA-Seq analysis
RNA-Seq analysis was performed in 322 female Twins with available smoking history of which 129 (77 never, 39 former and 13 current smokers) overlapped with the EPITWIN replication cohort. Samples were prepared for sequencing with the TruSeq sample preparation kit (Illumina) as indicated by the manufacturer's instructions. Libraries were sequenced in sets of 12 samples per lane using Hi-Seq. The 49 bp sequenced paired-end reads were mapped to the reference genome GRCh37 with BWA v0.5.95. Using SAMtools, reads mapping uniquely to the genome were kept, with MAPQ > = 10 and properly paired. In order to quantify exons in a non-redundant way, we created a set of merged exons from the GENCODE v10 annotation. In more detail, all transcripts were merged with any overlapping exons into new exon units. The number of reads mapping to each exon unit were counted. Technical outliers having less than 5 M exonic reads were removed from the study. The raw exon counts were normalized by scaling all libraries to 10 million reads.
Methylation QTL and heritability analysis
Methylation QTL (metQTL) analysis was performed in the CARDIOGENICS control (non-CAD) samples (n = 247), which were recruited in a single site, using PLINK.41 Like in the EWAS, we adjusted for age, gender and chip batch effect. The latter was input to PLINK as residuals after regressing out on chip batch effect (as a factor) given that PLINK cannot handle categorical covariates by default.42 Analysis was limited to SNPs located within a 200 Kb window centered on the probe position, resulting in an average of 402 SNPs per probe. We considered all signals at P < 10−3 and applied a Bonferroni correction (based on the number of tests per probe window) to report significant metQTLs.
Based on the classical twin design we assessed the similarity of mono- (MZ) and di-zygotic (DZ) twins using the ACE model, which partitions the variance into additive genetic (A), common environment (variance due to environmental effects shared within twin pairs; C) and unique environment (environmental effects not shared within twin pairs; E). Twins visited the clinic in pairs and as MZ twins share 100% of their genes, any differences arising between them in these circumstances are unique (E). A standard linear mixed model was used to estimate these variance components, as previously described.43
Correlation of DNA methylation with gene expression levels
To test the correlation between DNA methylation and gene expression levels we used a linear regression model with gene expression level as the outcome and residualized (for age and chip batch ID) DNA methylation levels as the predictor. To assess the effect of smoking and DNA methylation on gene expression we analyzed the 77 never and 13 current smokers with a linear regression model. In this case, smoking was added as a categorical variable in the above linear regression model (gene expression ∼ residualized DNA methylation + smoking).
Interaction analysis and combination of evidence
Linear regression models were applied study-wise to estimate the magnitude of the SNP interactions (assuming an additive model) with smoking on DNA methylation levels (DNA methylation levels ∼ SNP + SNP × Smoking + Smoking + Covariates). For CARDIOGENICS, we added as covariates in to the model age, sex and chip batch effect (input to PLINK as residuals after regressing out on chip batch effect as a factor), see also above. For EPITWIN, covariates were age and the first 4 components (adjusting for zygosity, family ID, and chip batch effect) of the Multi Dimensional Scaling (MDS) function of PLINK. Interactions analyses were implemented in PLINK.41 We used an inverse-variance weighted approach to conduct fixed-effect meta-analysis on cohort summary statistics (GWAMA software) for SNPs and smoking interaction effects. Conditional analysis was performed using the GCTA software.44
eQTL analysis
eQTL association analyses from published studies33-35 were assessed with the Genevar user interface.45 A SNP-centric approach was taken to investigate SNP-gene associations within a 2-Mb window centered on the SNP followed by a gene-centric approach to identify the lead eSNP of an associated expression probe.
Network analysis
Network analysis was performed using the Ingenuity Pathway Analysis software tool (IPA, Ingenuity Systems; http://www.ingenuity.com/science/platform). We considered molecules and/or relationships available in The IPA Knowledge Base for human or mouse and set the confidence filter to experimentally observed or high (predicted). Networks were generated with a maximum size of 35 genes, allowing up to 25 networks. Molecules in the query set with recorded interactions were 'eligible' for network construction using the IPA algorithm (http://www.ingenuity.com/wp-content/themes/ingenuitytheme/pdf/ipa/IPA-netgen-algorithm-whitepaper.pdf). Networks were ranked according to their degree of relevance to the eligible molecules in the query data set. The score takes into account the number of eligible molecules in the network and its size, as well as the total number of eligible molecules analyzed and the total number of molecules in the Ingenuity Knowledge Base that could potentially be included in the networks.46 The Network Score is based on the hypergeometric distribution and is calculated with the right-tailed Fisher's Exact Test. The significance P- value associated with enrichment of functional processes is calculated using the right-tailed Fisher's Exact Test by considering the number of query molecules that participate in that function and the total number of molecules that are known to be associated with that function in the Ingenuity Knowledge Base.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Ingebrigtsen TS, Thomsen SF, van der Sluis S, Miller M, Christensen K, Sigsgaard T, Backer V. Genetic influences on pulmonary function: a large sample twin study. Lung 2011; 189(4): 323-30; PMID:21660583; http://dx.doi.org/ 10.1007/s00408-011-9306-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, Widschwendter M. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med 2012; 4(3): 24; PMID:22453031; http://dx.doi.org/ 10.1186/gm323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. . Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 2012; 8(4): e1002629; PMID:22532803; http://dx.doi.org/ 10.1371/journal.pgen.1002629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soma T, Kaganoi J, Kawabe A, Kondo K, Imamura M, Shimada Y. Nicotine induces the fragile histidine triad methylation in human esophageal squamous epithelial cells. Int J Cancer 2006; 119(5): 1023-27; PMID:16570269; http://dx.doi.org/ 10.1002/ijc.21948 [DOI] [PubMed] [Google Scholar]
- 5.Dammann R, Strunnikova M, Schagdarsurengin U, Rastetter M, Papritz M, Hattenhorst UE, Hofmann HS, Silber RE, Burdach S, Hansen G. CpG island methylation and expression of tumor-associated genes in lung carcinoma. Eur J Cancer 2005; 41(8): 1223-36; PMID:15911247; http://dx.doi.org/ 10.1016/j.ejca.2005.02.020 [DOI] [PubMed] [Google Scholar]
- 6.Hillemacher T, Frieling H, Moskau S, Muschler MA, Semmler A, Kornhuber J, Klockgether T, Bleich S, Linnebank M. Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 2008; 18(4): 295-98; PMID:18242065; http://dx.doi.org/ 10.1016/j.euroneuro.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 7.Smith IM, Mydlarz WK, Mithani SK, Califano JA. DNA global hypomethylation in squamous cell head and neck cancer associated with smoking, alcohol consumption and stage. Int J Cancer 2007; 121(8): 1724-28; PMID:17582607; http://dx.doi.org/ 10.1002/ijc.22889 [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Lan Q, Siegfried JM, Luketich JD, Keohavong P. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia 2006; 8: 46-51; PMID:16533425; http://dx.doi.org/ 10.1593/neo.05586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur J, Demokan S, Tripathi SC, Macha MA, Begum S, Califano JA, Ralhan R. Promoter hypermethylation in Indian primary oral squamous cell carcinoma. Int J Cancer 2010; 127(10): 2367-73; PMID:20473870; http://dx.doi.org/ 10.1002/ijc.25377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, Kelsey KT. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2007; 16(1): 108-14; PMID:17220338; http://dx.doi.org/ 10.1158/1055-9965.EPI-06-0636 [DOI] [PubMed] [Google Scholar]
- 11.Broms U, Wedenoja J, Largeau MR, Korhonen T, Pitkäniemi J, Keskitalo-Vuokko K, Häppölä A, Heikkilä KH, Heikkilä K, Ripatti S, et al. . Analysis of detailed phenotype profiles reveals CHRNA5-CHRNA3-CHRNB4 gene cluster association with several nicotine dependence traits. Nicotine Tob Res 2012; 14(6): 720-33; PMID:22241830; http://dx.doi.org/ 10.1093/ntr/ntr283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, et al. . Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 2007; 16: 36-49; PMID:17135278; http://dx.doi.org/ 10.1093/hmg/ddl438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Port JL, Yamaguchi K, Du B, De Lorenzo M, Chang M, Heerdt PM, Kopelovich L, Marcus CB, Altorki NK, Subbaramaiah K, Dannenberg AJ. Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesis 2004; 25(11): 2275-81; PMID:15297370; http://dx.doi.org/ 10.1093/carcin/bgh243 [DOI] [PubMed] [Google Scholar]
- 14.Tang M, Xu W, Wang Q, Xiao W, Xu R. Potential of DNMT and its epigenetic regulation for lung cancer therapy. Curr Genomics 2009; 10(5): 336-52; PMID:20119531; http://dx.doi.org/ 10.2174/138920209788920994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol 2010; 126(3): 453-65; PMID:20816181; http://dx.doi.org/ 10.1016/j.jaci.2010.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerr KM, Galler JS, Hagen JA, Laird PW, Laird-Offringa IA. The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 2007; 23(1-2): 5-30; PMID:17325423; http://dx.doi.org/ 10.1155/2007/985474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgiou E, Valeri R, Tzimagiorgis G, Anzel J, Krikelis D, Tsilikas C, Sarikos G, Destouni C, Dimitriadou A, Kouidou S. Aberrant p16 promoter methylation among Greek lung cancer patients and smokers: correlation with smoking. Eur J Cancer Prev 2007; 16(5): 396-402; PMID:17923809; http://dx.doi.org/ 10.1097/01.cej.0000236260.26265.d6 [DOI] [PubMed] [Google Scholar]
- 18.Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, Philibert RA. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet 2012; 159B(2): 141-51; PMID:22232023; http://dx.doi.org/ 10.1002/ajmg.b.32021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang HW, Ling GS, Wei WI, Yuen AP. Smoking and drinking can induce p15 methylation in the upper aerodigestive tract of healthy individuals and patients with head and neck squamous cell carcinoma. Cancer 2004; 101(1): 125-32; PMID:15221997; http://dx.doi.org/ 10.1002/cncr.20323 [DOI] [PubMed] [Google Scholar]
- 20.Suga Y, Miyajima K, Oikawa T, Maeda J, Usuda J, Kajiwara N, Ohira T, Uchida O, Tsuboi M, Hirano T, et al. . Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncol Rep 2008; 20(5):1137-42; PMID:18949413 [PubMed] [Google Scholar]
- 21.Philibert RA, Beach SR, Gunter TD, Brody GH, Madan A, Gerrard M. The effect of smoking on MAOA promoter methylation in DNA prepared from lymphoblasts and whole blood. Am J Med Genet B Neuropsychiatr Genet 2010; 153B(2): 619-28; PMID:19777560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88(4): 450-57; PMID:21457905; http://dx.doi.org/ 10.1016/j.ajhg.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, Agusti A, Anderson W, Lomas DA, Demeo DL. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet 2012; 21(13): 3073-82; PMID:22492999; http://dx.doi.org/ 10.1093/hmg/dds135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun Ø, Cupul-Uicab LA, et al. . 450K Epigenome-Wide Scan Identifies Differential DNA Methylation in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect 2012; 120(10): 1425-31; PMID:22851337; http://dx.doi.org/ 10.1289/ehp.1205412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 2013; 22(5): 843-51; PMID:23175441; http://dx.doi.org/ 10.1093/hmg/dds488 [DOI] [PubMed] [Google Scholar]
- 26.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. . Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 2013; 8(5): e63812; PMID:23691101; http://dx.doi.org/ 10.1371/journal.pone.0063812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, Smith JA, Almli LM, Binder EB, Klengel T, et al. . Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet 2013; 132: 1027-37; PMID:23657504; http://dx.doi.org/ 10.1007/s00439-013-1311-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. . High density DNA methylation array with single CpG site resolution. Genomics 2011; 98: 288-95; PMID:21839163; http://dx.doi.org/ 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- 29.The Encode Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57-74; PMID:22955616; http://dx.doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, Smith AV, Gudnason V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS ONE 2012; 7(10): e46705; PMID:23071618; http://dx.doi.org/ 10.1371/journal.pone.0046705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahle´n S-E, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified hHuman blood cells: implications for cell lineage and studies on disease usceptibility. PLoS One 2012; 7(7): e41361; PMID:22848472; http://dx.doi.org/ 10.1371/journal.pone.0041361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garnier S, Truong V, Brocheton J, Zeller T, Rovital M, Wild PS, Ziegler A; Cardiogenics Consortium, Munzel T, Tiret L, et al. . Genome-wide haplotype analysis of cis expression quantitative trait Loci in monocytes. PLoS Genet 2013; 9(1): e1003240; PMID:23382694; http://dx.doi.org/ 10.1371/journal.pgen.1003240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. . Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 2009; 325: 1246-50; PMID:19644074; http://dx.doi.org/ 10.1126/science.1174148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. . Patterns of cis regulatory variation in diverse human populations. PLoS Genet 2012; 8(4): e1002639; http://dx.doi.org/ 10.1371/journal.pgen.1002639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. . Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 2012; 44(10): 1084-89; PMID:22941192; http://dx.doi.org/ 10.1038/ng.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, Davey Smith G, Hughes AD, Chaturvedi N, Relton CL. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics 2014; 6(1):4; PMID:24485148; http://dx.doi.org/ 10.1186/1868-7083-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murcray CE, Lewinger JP, Gauderman WJ. Gene-environment interaction in genome-wide association studies. Am J Epidemiol 2009; 169: 219-26; PMID:19022827; http://dx.doi.org/ 10.1093/aje/kwn353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tauber S, Jais A, Jeitler M, Haider S, Husa J, Lindroos J, Knöfler M, Mayerhofer M, Pehamberger H, Wagner O, Bilban M. Transcriptome analysis of human cancer reveals a functional role of heme oxygenase-1 in tumor cell adhesion. Mol Cancer 2010; 9: 200; PMID:20667089; http://dx.doi.org/ 10.1186/1476-4598-9-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zöchbauer-Müller S, Lam S, Toyooka S, Virmani AK, Toyooka KO, Seidl S, Minna JD, Gazdar AF. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer 2003; 107(4): 612-16; PMID:14520700; http://dx.doi.org/ 10.1002/ijc.11458 [DOI] [PubMed] [Google Scholar]
- 40.Bell JT, Spector TD. DNA methylation studies using twins: what are they telling us? Genome Biol 2013; 13(10): 172; http://dx.doi.org/ 10.1186/gb-2012-13-10-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J of Hum Genet 2007; 81: 559-75; http://dx.doi.org/ 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaffney DJ, Veyrieras J-B, Degner JF, Pique-Regi R, Pai AA, Crawford GE, Stephens M, Gilad Y, Pritchard JK. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol 2012; 13:R7; PMID:22293038; http://dx.doi.org/ 10.1186/gb-2012-13-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Visscher PM, Benyamin B, White I. The use of linear mixed models to estimate variance components from data on twin pairs by maximum likelihood. Twin Res 2004; 7(6): 670-74; PMID:15607018; http://dx.doi.org/ 10.1375/1369052042663742 [DOI] [PubMed] [Google Scholar]
- 44.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ANthropometric Traits (GIANT) Consortium; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Madden PA, Heath AC, Martin NG, et al. Conditional and joint multiple SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2011; 44: 369-75; http://dx.doi.org/ 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez-Arcelus M, Stranger BE, Deloukas P, Dermitzakis ET. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 2010; 26(19): 2474-76; PMID:20702402; http://dx.doi.org/ 10.1093/bioinformatics/btq452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, et al. . Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013; 45: 25-33; PMID:23202125; http://dx.doi.org/ 10.1038/ng.2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.