

Abstract
Intestinal failure is common in patients with septic shock, with dysfunction of the gut often manifesting as both a cause and consequence of their critical illness. Most studies investigating the pathogenesis of intestinal failure focus on the systemic aspect, although few data examine the inflammatory signaling in the intestinal lumen. Having previously demonstrated apical/luminal chemokine secretion in an in vitro model of intestinal inflammation, we hypothesized that endotoxemia would induce secretion of proinflammatory chemokines into the intestinal lumen. In addition, we examined the contribution of these mediators to intestinal dysmotility. C57/BL6 male mice were injected intraperitoneally with LPS. Serum, intestinal tissue, and intestinal luminal contents were harvested for cytokine analysis. For intestinal motility studies, a transit assay was performed after oral gavage of chemokines. Caco-2 cells grown on Transwell culture inserts were used to examine the role of the intestinal epithelium in chemokine secretion. Monocyte chemoattractant protein 1 (MCP-1/CCL2) and macrophage-derived chemokine (MDC/CCL22) were secreted into the lumen of multiple segments of the gut during endotoxemia in mice. In vitro work showed that the intestinal epithelium participates in monocyte chemoattractant protein 1 and MDC secretion and expresses the CCR2 and CCR4 receptors for these chemokines. Intestinal transit studies show that oral gavage of MDC results in impaired gut motility. This study demonstrates that the intestinal lumen is an active compartment in the gut's inflammatory response. Proinflammatory chemokines are secreted into the intestinal lumen during endotoxemia. These intraluminal chemokines contribute to intestinal dysmotility, complicating intestinal failure.
Keywords: Luminal cytokines, luminal chemokines, MDC, MCP-1, intestinal failure, lumen
INTRODUCTION
Acute intestinal failure is defined as the inability of the gastrointestinal tract to meet the nutritional, hydration, and electrolyte needs of the patient in the setting of critical illness or an acute surgical disease (1). This process is broadly classified, based on the cause and severity of the intestinal dysfunction. Mild disease is often associated with a self-limiting postoperative ileus, where more severe disease is often due to a postoperative bowel obstruction, abdominal contamination, or enterocutaneous fistula. The consequences of acute intestinal failure include the need for parenteral nutrition and invasive central venous access, each of which carries its own risks and complications. Furthermore, there are atrophy of bowel mucosa and impaired mucosal defense due to lack of enteral nutrients, worsening intestinal dysfunction. This process of intestinal failure is too frequently understated in the care of a critically ill patient, whereas emphasis is placed on failure of other organ systems, such as acute respiratory or acute renal failure. This is unfortunate as intestinal dysfunction often manifests as both a cause and consequence of critical illness (2–4).
Adynamic ileus caused by sepsis is a common presentation of intestinal failure. Sepsis induces dysfunction in each of the multiple protective mechanisms of the intestine. There is impaired intestinal motility in the septic patient (5), allowing for bacterial overgrowth (6). Impairment of normal fluid and nutrient absorption is common, resulting in bowel wall and luminal fluid sequestration and thus increasing in intra-abdominal pressure (7). Sepsis is associated with impaired intestinal barrier function, allowing breach of the intestinal lining and opening of the luminal compartment to the systemic circulation (8), which is closely followed by bacterial translocation (9). The intestine participates in this inflammatory response locally, with cytokine and nitric oxide production and leukocyte recruitment, and propagates the systemic response, contributing to distant organ dysfunction (10–12).
Most studies examining the pathogenesis of intestinal dysfunction have focused on the systemic aspect of inflammation. There is relatively little information regarding inflammatory signaling in the lumen of the intestine. Recent studies suggest that loss of the intestinal mucus layer after injury exacerbates the intestinal inflammatory response and that pancreatic proteases in the lumen damage the gut mucosa (13, 14). In addition, it is known that the bacteria in the gut contribute to the inflammatory response, some through complex quorum sensing mechanisms (15). Previously, we have shown that the intestinal epithelium in vitro is capable of responding to apical/luminal tumor necrosis factor α (TNF-α) in a direction fashion, with chemokines secreted into the apical media (16). In addition, we have shown that there are chemokines secreted into the gut lumen after hemorrhagic shock and resuscitation (17).
It is currently unknown if there are proinflammatory mediators secreted into the gut lumen in the setting of sepsis or endotoxemia. In addition, it is unknown if these intraluminal chemokines contribute to intestinal failure. We hypothesized that endotoxemia, a well-studied septic model in mice, results in the secretion of proinflammatory chemokines into the intestinal lumen. In addition, we examined the role of intraluminal chemokines in intestinal dysfunction, as pertains to intestinal dysmotility.
METHODS
Materials and reagents
Lipopolysaccharide from Escherichia coli 0111:B4 and fluorescein isothiocyanate (FITC)–dextran 70 kd were purchased from Sigma (St Louis, Mo). Whatman 42 filter paper was purchased from GE Healthcare UK Limited (Buckinghamshire, UK). Eagle minimum essential medium (EMEM) with Earle's balanced salt solution and 2 mM l-glutamine were obtained from American Type Culture Collection (Rockville, Md). Sodium pyruvate, nonessential amino acids, penicillin, streptomycin, and fetal bovine serum were purchased from Hyclone Laboratories (Logan, Utah). Recombinant human TNF-α, murine monocyte chemoattractant protein 1 (MCP-1)/CCL2, murine MDC/CCL22, and enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems (Minneapolis, Minn). Rabbit anti–human CCR2 and goat anti–human CCR4 antibodies were purchased from Abcam (San Francisco, Calif). Secondary antibodies used were immunoglobulin G, horse-radish peroxidase–conjugated goat anti–rabbit (Bio-Rad, Hercules, Calif), and donkey anti-goat (Santa Cruz Bio, Santa Cruz, Calif). Culture flasks and Costar Transwells were purchased from Corning, Inc (Corning, NY). EVOM2 Epithelial Voltohmmeter was purchased from World Precision Instruments (Sarasota, Fla). Ninety-six-well plates were purchased from Nunc (Roskilde, Denmark). Custom Multiplex ELISA kits were purchased from Quansys Biosciences (Logan, Utah).
Animals
Male C57/BL6 mice weighing 21 to 29 g were purchased from Charles River Laboratories and fed a standard laboratory diet and water ad libitum. Experiments were performed after acclimation for 2 to 3 weeks in a climate controlled room with a 12-h light-dark cycle and were approved by the Institutional Animal Care and Use committees of the University of Cincinnati.
Endotoxemia model
Endotoxemia was induced as previously described (18). Mice were intraperitoneally injected with LPS (15 mg/kg) dissolved in sterile saline. Control mice received a corresponding volume of sterile saline. All mice then received a 1-mL injection of sterile saline intraperitoneally for resuscitation. Following injection, mice were allowed full access to food and water.
Specimen preparation
At intervals after injection, mice were killed by carbon dioxide inhalation, followed by cervical dislocation. Blood was collected via direct cardiac puncture, allowed to clot, centrifuged at 6,800g to separate the serum and cellular components, and stored at –80°C until analysis.
To examine cytokine production in the intestine and lumen, tissue was harvested as previously described (17). Gut tissue was separated into jejunum, ileum, cecum, and colon segments. Jejunum was defined as a 15-cm segment beginning at the ligament of Treitz, and ileum was defined as a 15-cm segment proximal to the ileocecal valve. Solid and liquid stool was removed from the cecum and colon and placed into 1 mL of extraction buffer (0.1 M Tris-buffered saline with 0.3% bovine serum albumin, 0.01% sodium azide, and 0.002% Tween). The jejunum and ileum were opened longitudinally, and intestinal contents were absorbed with filter paper. Two pieces of 100 × 5-mm filter paper were used on each the jejunum and ileum. The filter paper was placed into 1 mL of extraction buffer. All stool specimens were gently agitated overnight at 4°C then centrifuged at 18,000g for 15 min. Supernatants were transferred to clean containers and stored at –80°C until analysis.
Intestinal tissue was snap frozen in liquid nitrogen. Specimens were homogenized and then sonicated for 10 s in 1 mL phosphate-buffered saline containing complete protease inhibitor cocktail tablets and 2 mM phenylmethylsulfonyl fluoride. Samples were centrifuged at 12,000g at 4°C for 45 min. Supernatant density was determined using BCA Protein Assay Kit. Samples were stored at –80°C until analysis. Serum, tissue, and stool chemokine levels were analyzed using custom multiplex cytokine array.
Cell culture
Caco-2 cells were grown in flasks at 37°C in 5% CO2 in nutrient media consisting of EMEM supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells used for experiments were between passages 5 and 20 and were seeded at a density of 300,000 cells/well onto 24-mm-diameter Transwell permeable inserts with 0.04-μm pores. Cells were grown in supplemented EMEM for 21 days to achieve full differentiation before use. Transepithelial electrical resistance values were measured in all wells, and only cells with transepithelial electrical resistance greater than 500 Ωcm2 were considered fully differentiated and suitable for use. For experiments, cells were placed in serum-free media for 24 h and treated with TNF-α at a concentration of 100 ng/mL into the apical or basolateral compartment. After 24 h of treatment, cell media from the apical and basolateral chamber were harvested. Samples were analyzed by conventional ELISA.
Intestinal transit
To determine intestinal transit, experiments were performed as previously described, with modifications (19). Briefly, mice were given an oral gavage of a solution of MDC (500 ng/mL), MCP-1 (500 ng/mL), or vehicle (0.1% bovine serum albumin in phosphate-buffered saline). Six hours after chemokine gavage, mice were administered a 10 μL intraoral aliquot of FITC-dextran 70-kd solution at a concentration of 10 mg/mL using a micropipette. Exactly 90 min later, animals were killed using CO2 inhalation and cervical dislocation. The gastrointestinal tract was divided into 12 segments: stomach, eight small intestine segments, cecum, and two colon segments. Each segment was placed into a plastic tube containing 1 mL of deionized water. Segments were allowed to elute for 30 min then centrifuged at 12,000g for 5 min to force out luminal contents, including the fluorescent marker. The supernatant was transferred to a 96-well plate, and fluorescence from each gut segment was measured.
Western blot
Caco-2 cytoplasmic extracts were prepared as follows. After scraping from membrane inserts, cells were pelleted by centrifugation at 3,800g for 5 min. Supernatants were removed, and pellets were stored at –80°C until assay. Cytoplasmic fractions were prepared by resuspension of pellets in one packed cell volume of lysis buffer, consisting of 10 mM HEPES, pH 7.9; 10 mM KCL; 0.1 mM EDTA; 1.5 mM MgCl; 0.2% Nonidet P-40 by volume; 1.5 mM dithiothreitol; 0.5 mM phenylmethylsulfonyl fluoride; and 1.5 μM pepstatin. Then cells were incubated on ice for 5 min with intermittent vortexing. Cells were again centrifuged as above. The supernatant was collected and saved as the cytoplasmic fraction. Protein concentrations of the cytoplasmic extracts were determined by the BCA protein assay.
Aliquots of cytoplasmic fractions containing 40 μg of protein were boiled in equal volumes of loading buffer (250 mM Tris, pH 6.8; 10% sodium dodecyl sulfate; 50% glycerol; 0.1% bromophenol blue; and 25% betamercaptoethanol) for 5 min. Proteins were then separated by electrophoresis on a gradient gel with a molecular weight marker. The proteins were transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dried milk in Tris-buffered saline containing 0.05% Tween-20 for 30 min. Membranes incubated overnight with a rabbit monoclonal rabbit anti–human CCR2 antibody and polyclonal goat anti–human CCR4 antibody. After being washed twice in tris-tween buffered saline, the blots were incubated with a peroxidaseconjugated goat anti–rabbit and donkey anti–goat secondary antibodies, respectively, for 1 h. The blots were then washed three times in TTBS for 15 min, followed by incubation in enhanced chemiluminescence reagents and exposure on radiographic film. Where indicated, blots were stripped and reprobed with a commercially available antibody to actin.
Statistical analysis
Statistical analysis was performed using SigmaPlot 11 software (Systat, Chicago, Ill). For in vitro experiments, analysis of variance was used to compare all treatment groups. For in vivo experiments, t test was used to compare vehicle- and LPS-injected animals at each time point. For each, P < 0.05 was considered statistically significant. Data are reported as mean ± SEM.
RESULTS
Increased serum cytokine levels during endotoxemia
Serum samples taken at intervals after LPS versus vehicle injection were analyzed via multiplex ELISA. As is expected during endotoxemia, there was a robust systemic response compared with control groups, with multiple cytokines increased during endotoxemia. Serum MCP-1 and MDC peaked at 6 h after injection, whereas macrophage inflammatory protein (MIP)-1α and MIP-2 peaked early at the 1-h time point, steadily declining afterward. Serum KC and interleukin 6 (IL-6) levels were increased early and were sustained, finally tapering by the 24-h interval (Fig. 1).
Fig. 1.

Serum chemokines during endotoxemia. Animals were injected with vehicle (open bars) or LPS (closed bars) and killed at intervals. *P < 0.05 LPS vs. vehicle by t test. n = 4 animals in 1-h group; n = 7 in 3-, 6-, 12-h groups; n = 3 in 24-h group.
Increased intestinal tissue cytokine levels during endotoxemia
To determine the effect of endotoxemia on cytokine expression in the intestine, whole intestinal tissues were processed and analyzed. We were specifically interested in the chemokines MCP-1 and MDC because of their known contribution to intestinal inflammation, namely, MCP-1's participation in the pathogenesis of ileus and MDC's role in attracting dendritic cells (DCs), a cell known to be present at the luminalmucosal interface. We also measured IL-6 levels given the role of this cytokine in the systemic inflammatory response. In animals receiving vehicle injection, there was little to no MCP-1 production in any intestinal segment. Control animals also had very little MDC production except in the small bowel, where there was a low level of constitutive MDC production, consistent with known data about MDC in other tissues (20). In LPS-injected animals, there was an increase in both MCP-1 and MDC levels in all intestinal segments (Fig. 2). This increase reached statistical significance in the small bowel at 3 and 12 h after injection, with higher levels of MCP-1 found in the cecum and distal colon, whereas MDC levels were higher in the proximal intestinal tissue (Fig. 2). Interleukin 6 levels were also increased in the intestinal tissues at 3 and 12 h after injection in the jejunum, ileum, and colon and at 3 h in the cecum in LPS-injected animals, compared with control groups (data not shown).
Fig. 2.
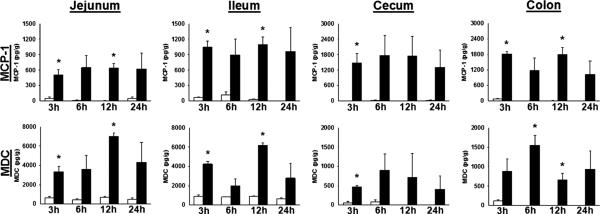
Intestinal tissue chemokines during endotoxemia. Animals were injected with vehicle (open bars) or LPS (closed bars) and killed at intervals. *P < 0.05 LPS vs. vehicle by t test. n = 3 at each time point.
Increased luminal chemokine levels during endotoxemia
Intestinal contents were harvested from each intestinal segment for cytokine analysis. Monocyte chemoattractant protein 1 was significantly increased in the ileal lumen at 3, 6, and 12 h after LPS injection compared with controls (Fig. 3). There was a trend toward higher levels in the proximal intestine early after injection and increased levels in the cecum and distal colon at later times during endotoxemia. Macrophage-derived chemokine was present at a level of approximately 150 to 300 pg/mL in the lumen of control animals at nearly all time points studied (Fig. 3). In LPS-injected animals, MDC was increased in the lumen of the jejunum and ileum at 3 and 12 h into endotoxemia. In the cecum, luminal MDC was increased at 6 and 24 h after endotoxin injection. In the lumen of the colon, there was no significant difference between vehicle and experimental animals except for a trend toward increased MDC at 12 and 24 h after injection in the experimental group. Interleukin 6 levels were measured in the lumen of each intestinal segment with no significant increase in luminal IL-6 except for one 3-h time point in the ileum, suggesting that increased MCP-1 and MDC levels were not simply a part of a generalized response (data not shown).
Fig. 3.
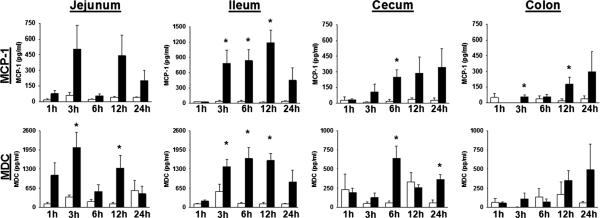
Intestinal lumen chemokines during endotoxemia. Animals were injected with vehicle (open bars) or LPS (closed bars) and killed at intervals. *P < 0.05 LPS vs. vehicle by t test. n = 4 animals in 1-h group; n = 7 in 3-, 6-, 12-h groups; n = 3 in 24-h group.
Intestinal epithelial cells secrete MCP-1 and MDC
We hypothesized that the intestinal epithelium participated in the secretion of MCP-1 and MDC into the lumen intestinal lumen. To test this hypothesis, we used our in vitro model of intestinal inflammation (16). Caco-2 cells grown on Transwell membranes were treated with TNF-α either apically (luminally) or basolaterally (systemically). Control cells received treatment with serum-free media alone. Control groups produced no MCP-1 at baseline, but after apical treatment with TNF-α, the cells produced MCP-1 into the apical chamber. With basolateral TNF-α treatment, the Caco-2 cells secreted MCP-1 into the apical and basolateral chamber (Fig. 4). The cells secreted a small amount of MDC into the apical media at baseline and, when stimulated apically, increased their apical MDC production. Cells stimulated with basolateral TNF-α produced MDC into the apical and basolateral direction (Fig. 4).
Fig. 4.
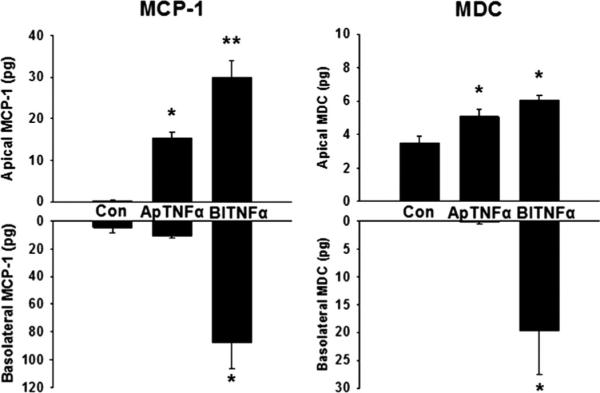
Intestinal epithelial cell chemokine production. Caco-2 cells were treated with apical or basolateral TNF-α or serum-free media. Monocyte chemoattractant protein 1 and MDC were measured in the apical and basolateral compartments by ELISA. *P < 0.05 vs. control, **P < 0.05 vs. control and apical treatment. n = 6.
Intestinal epithelial cells express CCR2 and CCR4 receptors
We hypothesized that the intestinal epithelium had the capacity to respond to intraluminal MCP-1 and MDC and therefore expressed the associated chemokine receptors CCR2 (for MCP-1) and CCR4 (for MDC). To test this, untreated Caco-2 cells were scraped from the Transwell membrane and lysed. Western blotting was performed on this cell fraction to determine if the cells expressed the CCR2 and CCR4 receptors. Differentiated Caco-2 cells on Transwells express the CCR2 and CCR4 receptor proteins (Fig. 5).
Fig. 5.
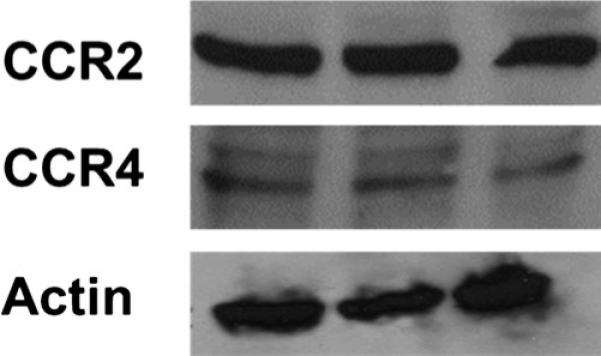
Intestinal epithelial cell chemokine receptor expression. Caco-2 cell cytoplasmic/membrane fractions were analyzed with Western blot for CCR2 and CCR4 receptors. n = 3.
Intraluminal cytokines induce intestinal dysmotility
One potential role for increased chemokines in the intestinal lumen after endotoxemia is in the pathophysiology of adynamic ileus. To determine this, we performed an intestinal transit assay after giving MCP-1 or MDC by oral gavage. As a positive control, the transit assay was performed on endotoxemic animals as well. After intraperitoneal LPS injection, approximately 95% of the FITC marker was retained in the small intestine (data not shown). Animals receiving vehicle had normal gastrointestinal motility, with approximately 95% of the FITC-dextran 70 kd reaching the cecum or distal colon at 90 min (Fig. 6, A, D, and E). Macrophage-derived chemokine gavage resulted in a significant increase in the amount of fluorescent marker retained in the small bowel and failing to reach the colon (Fig. 6, CYE). There was a nonsignificant trend toward increased dysmotility in MCP-1 gavaged mice (Fig. 6, B, D, and E). When used in combination, MDC and MCP-1 resulted in a significant increase in FITC marker being retained in the small bowel compared with vehicle but did not differ from MDC used alone (data not shown).
Fig. 6.

Gavage with chemokines induces intestinal dysmotility. Mice were treated with oral gavage of vehicle, MCP-1, or MDC, followed 6 h later by oral 70-kd FITC-dextran marker to determine intestinal transit. *P < 0.05 compared with vehicle. n = 4, 7, and 7 in vehicle, MCP-1, and MDC groups, respectively. Stom indicates stomach; SI, small intestine; ce, cecum; co, colon.
DISCUSSION
In the present study, we have demonstrated secretion of MCP-1 and MDC into the intestinal lumen in multiple segments of the gut during endotoxemia in mice. These intraluminal chemokines appear to be secreted with directional selectivity into the lumen as increased luminal concentrations were not in concordance with increased tissue levels at all time points. In addition, the cytokine IL-6 was increased systemically in serum and intestinal tissue but was found to be increased in the lumen at only one time point in the ileum, suggesting that luminal secretion of MCP-1 and MDC is unlikely to be a non-specific response to systemic inflammation.
Our data demonstrate that intestinal epithelial cells have the ability to secrete MCP-1 and MDC chemokines in the luminal direction in a dual-chamber, in vitro model of intestinal inflammation. Directional secretion of MDC and MCP-1 was observed, with apical TNF-α treatment inducing only apical secretion, but basolateral stimulation resulting in apical and basolateral secretion in approximately a 1:3 ratio in both MDC and MCP-1. The intestinal epithelial cells also possess the cognate receptor for these chemokines, as demonstrated by CCR2 and CCR4 expression on Western blot of Caco-2 cell lysates. Taken together with our in vivo data, these data support the concept that the intestine is able to specifically secrete and respond to inflammatory mediators via the intestinal lumen. Additional experiments will be needed to determine if the chemokines act either by increasing inflammation in the mucosa or by directly affecting the mucosa and gut function. We feel that it is possible that both are occurring, but further experiments will be needed to increase our understanding of this process.
We found that MCP-1 and MDC secretion into the intestinal lumen was increased during endotoxemia. The mechanism of luminal directional secretion in vivo is not known, and there is no previously existing literature describing the function of these chemokines in the luminal compartment of the intestine. Their systemic functions have been previously described. Monocyte chemoattractant protein 1 (CCL2) is the prototypical member of the CC family of chemokines and is produced by many cell types including macrophages, endothelial cells, and epithelial cells (21). Through the CCR2 receptor, MCP-1 signaling is important in the systemic inflammatory response during sepsis. Although the primary known function of MCP-1 is as a potent chemoattractant for recruiting monocytes and macrophages to diseased tissues from the serum and bone marrow, MCP-1 signaling is also vital to the inflammatory response of intestinal tissues. Monocyte chemoattractant protein 1 mRNA is upregulated in surgical specimens from patients with Crohn disease and ulcerative colitis (22). Monocyte chemoattractant protein 1 also inhibits the differentiation of monocytes into tolerant intestinal-type macrophages, resulting in the recruitment of a more inflammatory phenotype of macrophage (23). In experimental colitis, treatment with bindarit, an oral MCP-1 inhibitor, reduced severity of the colitis (24). In a coculture in vitro model, MCP-1 induces transmigration of macrophages into a monolayer of intestinal epithelial cells (25). Monocyte chemoattractant protein 1 may be an important mediator in endotoxemic and postoperative ileus, with the intestinal epithelium and resident macrophages secreting MCP-1 into the lamina propria, leading to influx of circulating monocytes into the muscularis layer of the intestine, where they cause local tissue damage and intestinal dysmotility (26, 27).
Macrophage-derived chemokine (CCL22) is also a member of the CC chemokines family. Macrophage-derived chemokine was first described as being produced by macrophages but is now known to be produced by many tissues, including constitutive expression in the thymus, spleen, and small intestine (20). Signaling through its receptor CCR4, MDC serves as a potent chemoattractant of T cells and monocytes but is approximately 100 times more potent in recruiting immature DCs of monocyte origin than monocytes and macrophages (28). Dendritic cells are antigen-presenting cells of specific immunity. In the intestine, they are located between epithelial cells and sample the microenvironment and report back to the local lymphoid tissues. This increased reactivity of DCs to MDC implies that a lower concentration gradient would be required to recruit DCs to the site of inflammation. In the gut, MDC is constitutively produced, and production is increased in response to TNF-α, IL-1β, and interferon γ (29). In the intestine of patients with inflammatory bowel disease, MDC transcription is increased in specimens exhibiting grossly inflamed mucosa but not in the noninflamed tissue (30).
In our study, we found that MDC was constitutively produced, both in the tissue and into the intestinal lumen, at low but readily detectible levels. Both luminal and tissue levels were increased during endotoxemia. This is in agreement with previous work (29) but represents a novel finding, in that neither constitutive nor activated MDC production into the gut lumen has been demonstrated previously. Our data suggest a potential novel role for MDC in the intestine during endotoxemia.
Having demonstrated that the intestinal epithelium was exposed to these luminal chemokines during endotoxemia in vivo and that intestinal epithelial cells in vitro were capable of secreting these chemokines, MCP-1 and MDC, into the apical or luminal compartment and expressed their associated receptors, CCR2 and CCR4, respectively, we sought to test whether these luminal chemokines were capable of generating a functional response in vivo. One of the manifestations of intestinal failure in sepsis is intestinal dysmotility, and our intestinal transit experiment was designed to elucidate the role of MCP-1 and MDC in this process. We found a significant decrease in intestinal motility after gavage of MDC. Monocyte chemoattractant protein 1 was associated with a trend toward dysfunctional motility but without statistical significance. This is the first time that an intraluminal chemokine has been demonstrated to be associated with intestinal dysfunction.
Intestinal motility is a complex process. Alterations in intestinal motility potentially involve many different cell types and mediators. The current experiments suggest that intestinal mucosa may secrete chemokines, which could, in turn, alter motility. Our finding that gavaged chemokines were biologically active suggests that endogenously secreted chemokines would be active as well. These data are not without limitations and should not be overinterpreted. Although intriguing, the current experiments do not provide data directly linking MCP-1 and MDC to intestinal dysfunction. Additional experiments, ideally with the use of intestinal mucosa–specific MCP-1 or MDC knockout mice, will be needed to more directly address this question. In addition, we do not currently know the mechanism of this process, but signaling may be mediated through intestinal epithelial-derived mediators such as nitric oxide (31) or through signaling by the resident antigen-presenting cells (32, 33). Although currently unknown, we are interested in exploring the half-life of these chemokines in the intestinal lumen, how much they are absorbed, and how far distal they transit before being degraded by intestinal proteases. Furthermore, the intestinal mucosa is composed of a variety of individual cell types. Although our in vitro data suggest that intestinal epithelial cells are capable of producing chemokines in a vectorial fashion, the cell type(s) involved in luminal chemokine production in vivo in the present study are unknown.
In conclusion, our data demonstrate that there is directional and selective secretion of proinflammatory chemokines into the intestinal lumen during endotoxemia in mice. Based on our in vitro data, these chemokines are in part produced by intestinal epithelial cells, which in turn express the CCR2 and CCR4 receptors. Using a gain-of-function approach, we demonstrate that enteral MDC and, to some extent, MCP-1 provoke dysmotility of the intestine and likely contribute to the intestinal failure of sepsis. In total, our data suggest a potentially novel role for chemokines in the development of intestinal dys-function during endotoxemia.
Footnotes
This work was presented at the Shock Society's 34th Annual Conference on Shock in Norfolk, Virginia, on June 11–14, 2011.
REFERENCES
- 1.Carlson GL, Dark P. Acute intestinal failure. Curr Opin Crit Care. 2010;16(4):347–352. doi: 10.1097/MCC.0b013e328339fabe. [DOI] [PubMed] [Google Scholar]
- 2.Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “Bmotor” of critical illness. Shock. 2007;28(4):384–393. doi: 10.1097/shk.0b013e31805569df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reintam A, Parm P, Kitus R, Kern H, Starkopf J. Gastrointestinal symptoms in intensive care patients. Acta Anaesthesiol Scand. 2009;53(3):318–324. doi: 10.1111/j.1399-6576.2008.01860.x. [DOI] [PubMed] [Google Scholar]
- 4.Ukleja A. Altered GI motility in critically Ill patients: current understanding of pathophysiology, clinical impact, and diagnostic approach. Nutr Clin Pract. 2010;25(1):16–25. doi: 10.1177/0884533609357568. [DOI] [PubMed] [Google Scholar]
- 5.Bauer AJ, Schwarz NT, Moore BA, Turler A, Kalff JC. Ileus in critical illness: mechanisms and management. Curr Opin Crit Care. 2002;8(2):152–157. doi: 10.1097/00075198-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Jones MP, Bratten JR. Small intestinal motility. Curr Opin Gastroenterol. 2008;24(2):164–172. doi: 10.1097/MOG.0b013e3282f33f5c. [DOI] [PubMed] [Google Scholar]
- 7.Madl C, Druml W. Gastrointestinal disorders of the critically ill. Systemic consequences of ileus. Best Pract Res Clin Gastroenterol. 2003;17(3):445–456. doi: 10.1016/s1521-6918(03)00022-2. [DOI] [PubMed] [Google Scholar]
- 8.MacFie J, O'Boyle C, Mitchell CJ, Buckley PM, Johnstone D, Sudworth P. Gut origin of sepsis: a prospective study investigating associations between bacterial translocation, gastric microflora, and septic morbidity. Gut. 1999;45(2):223–228. doi: 10.1136/gut.45.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexander JW, Boyce ST, Babcock GF, Gianotti L, Peck MD, Dunn DL, Pyles T, Childress CP, Ash SK. The process of microbial translocation. Ann Surg. 1990;212(4):496–510. doi: 10.1097/00000658-199010000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care. 2001;7(2):92–98. doi: 10.1097/00075198-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Alican I, Kubes P. A critical role for nitric oxide in intestinal barrier function and dysfunction. Am J Physiol. 1996;270(2 Pt 1):G225YG237. doi: 10.1152/ajpgi.1996.270.2.G225. [DOI] [PubMed] [Google Scholar]
- 12.Mainous MR, Ertel W, Chaudry IH, Deitch EA. The gut: a cytokine-generating organ in systemic inflammation? Shock. 1995;4(3):193–199. [PubMed] [Google Scholar]
- 13.Caputo FJ, Rupani B, Watkins AC, Barlos D, Vega D, Senthil M, Deitch EA. Pancreatic duct ligation abrogates the trauma hemorrhage-induced gut barrier failure and the subsequent production of biologically active intestinal lymph. Shock. 2007;28(4):441–446. doi: 10.1097/shk.0b013e31804858f2. [DOI] [PubMed] [Google Scholar]
- 14.Sheth SU, Lu Q, Twelker K, Sharpe SM, Qin X, Reino DC, Lee MA, Xu DZ, Deitch EA. Intestinal mucus layer preservation in female rats attenuates gut injury after trauma-hemorrhagic shock. J Trauma. 2010;68(2):279–288. doi: 10.1097/TA.0b013e3181caa6bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L, Estrada O, Zaborina O, Bains M, Shen L, Kohler JE, Patel N, Musch MW, Chang EB, Fu YX, et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science. 2005;309(5735):774–777. doi: 10.1126/science.1112422. [DOI] [PubMed] [Google Scholar]
- 16.Sonnier DI, Bailey SR, Schuster RM, Lentsch AB, Pritts TA. TNF-alpha induces vectorial secretion of IL-8 in Caco-2 cells. J Gastrointest Surg. 2010;14(10):1592–1599. doi: 10.1007/s11605-010-1321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sonnier DI, Makley AT, Friend LA, Bailey SR, Lentsch AB, Pritts TA. Hemorrhagic shock induces a proinflammatory milieu in the gut lumen. J Surg Res. 2011;170(2):272–279. doi: 10.1016/j.jss.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritts TA, Moon MR, Wang Q, Hungness ES, Salzman AL, Fischer JE, Hasselgren PO. Activation of NF-kappaB varies in different regions of the gastrointestinal tract during endotoxemia. Shock. 2000;14(2):118–122. doi: 10.1097/00024382-200014020-00007. [DOI] [PubMed] [Google Scholar]
- 19.Stoffels B, Turler A, Schmidt J, Nazir A, Tsukamoto T, Moore BA, Schnurr C, Kalff JC, Bauer AJ. Anti-inflammatory role of glycine in reducing rodent postoperative inflammatory ileus. Neurogastroenterol Motil. 2011;23(1):76–87. e78. doi: 10.1111/j.1365-2982.2010.01603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantovani A, Gray PA, Van Damme J, Sozzani S. Macrophage-derived chemokine (MDC). J Leukoc Biol. 2000;68(3):400–404. [PubMed] [Google Scholar]
- 21.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411(21–22):1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Grimm MC, Elsbury SK, Pavli P, Doe WF. Enhanced expression and production of monocyte chemoattractant protein-1 in inflammatory bowel disease mucosa. J Leukoc Biol. 1996;59(6):804–812. doi: 10.1002/jlb.59.6.804. [DOI] [PubMed] [Google Scholar]
- 23.Spoettl T, Hausmann M, Herlyn M, Gunckel M, Dirmeier A, Falk W, Herfarth H, Schoelmerich J, Rogler G. Monocyte chemoattractant protein-1 (MCP-1) in hibits the intestinal-like differentiation of monocytes. Clin Exp Immunol. 2006;145(1):190–199. doi: 10.1111/j.1365-2249.2006.03113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatia M, Landolfi C, Basta F, Bovi G, Ramnath RD, de Joannon AC, Guglielmotti A. Treatment with bindarit, an inhibitor of MCP-1 synthesis, protects mice against trinitrobenzene sulfonic acid-induced colitis. Inflamm Res. 2008;57(10):464–471. doi: 10.1007/s00011-008-7210-y. [DOI] [PubMed] [Google Scholar]
- 25.Mori A, Satsu H, Shimizu M. New model for studying the migration of immune cells into intestinal epithelial cell monolayers. Cytotechnology. 2003;43(1–3):57–64. doi: 10.1023/B:CYTO.0000039910.30540.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turler A, Schwarz NT, Turler E, Kalff JC, Bauer AJ. MCP-1 causes leukocyte recruitment and subsequently endotoxemic ileus in rat. Am J Physiol Gastrointest Liver Physiol. 2002;282(1):G145YG155. doi: 10.1152/ajpgi.00263.2001. [DOI] [PubMed] [Google Scholar]
- 27.Wehner S, Behrendt FF, Lyutenski BN, Lysson M, Bauer AJ, Hirner A, Kalff JC. Inhibition of macrophage function prevents intestinal inflammation and postoperative ileus in rodents. Gut. 2007;56(2):176–185. doi: 10.1136/gut.2005.089615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, Mantovani A, Gray PW. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. 1997;185(9):1595–1604. doi: 10.1084/jem.185.9.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berin MC, Dwinell MB, Eckmann L, Kagnoff MF. Production of MDC/CCL22 by human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2001;280(6):G1217YG1226. doi: 10.1152/ajpgi.2001.280.6.G1217. [DOI] [PubMed] [Google Scholar]
- 30.Jugde F, Alizadeh M, Boissier C, Chantry D, Siproudhis L, Corbinais S, Quelvennec E, Dyard F, Campion JP, Gosselin M, et al. Quantitation of chemokines (MDC, TARC) expression in mucosa from Crohn's disease and ulcerative colitis. Eur Cytokine Netw. 2001;12(3):468–477. [PubMed] [Google Scholar]
- 31.Gan HT, Chen JD. Roles of nitric oxide and prostaglandins in pathogenesis of delayed colonic transit after burn injury in rats. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1316YR1324. doi: 10.1152/ajpregu.00733.2004. [DOI] [PubMed] [Google Scholar]
- 32.Schaefer N, Tahara K, Pech T, Websky MV, Fujishiro J, Pantelis D, Abu-Elmagd K, Kalff JC, Hirner A, Turler A. Inducible nitric oxide synthase expression in the intestinal muscularis mediates severe smooth muscle dysfunction during acute rejection in allogenic rodent small bowel transplantation. J Surg Res. 2008;150(2):159–168. doi: 10.1016/j.jss.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Turler A, Kalff JC, Moore BA, Hoffman RA, Billiar TR, Simmons RL, Bauer AJ. Leukocyte-derived inducible nitric oxide synthase mediates murine postoperative ileus. Ann Surg. 2006;244(2):220–229. doi: 10.1097/01.sla.0000229963.37544.59. [DOI] [PMC free article] [PubMed] [Google Scholar]