

Abstract
To determine the contribution of the somatic point mutations and that of the complementarity-determining region (CDR)3 Arg to DNA binding, we engineered the germ-line VH and Vκ gene revertant and site-mutagenized the CDR3 Arg residues of the mutated and “antigen-selected” mAb 412.67. This anti-DNA autoantibody was derived from B-1 cells of a lupus patient and bore two H-CDR3 Arg, Arg105 and Arg107, encoded by N segment additions, and one κ-CDR3 Arg, Arg97, resulting from a point mutation (Kasaian et al. 1994. J. Immunol. 152: 3137–3151; Kasaian et al. 1995. Ann. N.Y. Acad. Sci. 764: 410–423). The germ-line revertant bound double-stranded (ds) DNA and single-stranded (ss) DNA as effectively as its wild-type counterpart (relative avidity: 6.4×10−7 and 9.9×10−9 vs. 6.7×10−7 and 9.1×10−9 g/μl), raising the possibility that an antigen other than DNA was responsible for the selection of the mAb 412.67 VH and Vκ point mutations. H-CDR3 Arg105 and Arg107 were both required for dsDNA binding, but either Arg105 or Arg107 was sufficient for ssDNA binding. The central role of Arg105 and Arg107 in DNA binding reflected their solvent-exposed orientation at the apex of the H-CDR3 main loop. Consistent with its inward orientation afar from the antigen-binding surface, the κ-CDR3 Arg97 played no role in either dsDNA or ssDNA binding.
Keywords: Autoantibody, B lymphocyte, Complementarity-determining region 3 Arg, DNA, Immunoglobulin gene somatic mutation
1 Introduction
Somatic hypermutation is induced through engagement of the B cell surface receptor for antigen [1, 2], and is thought to play a major role in the generation of high-affinity anti-double-stranded (ds) DNA antibodies. These autoantibodies are characteristic of systemic lupus erythematosus and are responsible for most of the tissue injury in these patients. In the anti-dsDNA autoantibodies in both lupus patients and autoimmune mice, somatic mutations are consistent in distribution and nature with clonal selection by antigen [3–5], as found in T-dependent antibody responses to conjugated haptens and viral antigens [6–9]. They concentrate in the V segment complementarity-determining regions (CDR), and display a preponderance of replacement (R) over silent (S) nucleotide changes (high R: S mutation ratio) [3–5]. However, it remains unclear whether DNA or nucleosomes induce anti-DNA antibodies or whether anti-DNA antibodies are actually generated in response to some other antigens, either self or foreign. If anti-DNA antibodies were induced by DNA, at least some of the point mutations detected in “affinity-mature” anti-DNA auto-antibodies should crucially enhance the DNA binding activity.
The contribution to DNA binding of R point mutations to the basic amino acids Arg, Lys and His, and, perhaps, the uncharged Asn has been emphasized in murine autoantibodies [4]. Arg is by far the most important and versatile amino acid in DNA binding [3, 10]. It can form hydrogen bonds with base-paired guanine, as well as unpaired and base-paired cytosine. It can fit into the major or the minor DNA groove through extensive interactions with the DNA sugar-phosphate backbone, with the flexible side chain facilitating the binding [11]. The pleiotropic activity of Arg in DNA binding is responsible for the binding of both ds and single-stranded (ss) DNA by many murine anti-DNA antibodies. In these autoantibodies, amino acid replacements that correlate with improved DNA binding are located at sites that are predicted to more closely contact the DNA molecule. For instance, in 3H9, a higher affinity for dsDNA was generated by introducing Arg at position 31 of the H-CDR1, positions 56 and 64 of the H-CDR2, and position 76 of the H-FR3 [4]. However, the presence of an Arg in the H-CDR3 may supersede the contribution to anti-dsDNA specificity of the other CDR. In 3H9, reversion of the H-CDR3 Arg97 completely abrogates dsDNA-binding by this mAb [10]. In this and other antibodies with no significant affinity for DNA, the introduction of Arg in the H-CDR3 results in the generation of high-affinity dsDNA binding [4, 10, 12]. In spite of these findings in murine autoantibodies, the role of H-CDR3 Arg in DNA binding in autoantibodies from lupus patients remains to be determined.
Here we report experiments showing that R mutations which accumulated in the VH and Vκ segment CDR did not confer any significantly higher DNA binding to the somatically mutated anti-DNA mAb 412.67, which was derived from B-1 cells of a lupus patient [5, 13]. Rather, the whole dsDNA and ssDNA binding activity was mediated by the two H-CDR3 Arg105 and Arg107, which protrude from the apex of the H-CDR3 loop. The implications of these findings with respect to the generation of anti-dsDNA autoantibodies are discussed.
2 Results
2.1 Point mutations in the anti-dsDNA mAb 412.67 V(D)J gene segments
The VHDJH gene segment of the B-1 cell-derived anti-dsDNA mAb 412.67 accumulated 11 somatic point mutations, of which three were in CDR1, three in CDR2, one in CDR3, and four in framework region 3 (FR3) (Figs. 1 and 2, Table 1). The seven CDR mutations resulted in six (five actual and one potential due to two adjacent CDR1 nucleotide changes) amino acid substitutions, yielding a significantly higher number of R mutations in the CDR than that expected to occur by chance alone, and an R:S mutation ratio of 6:1. The four FR3 mutations resulted in two amino acid substitutions, yielding a significantly lower number of R mutations in the FR than that expected by chance alone and an R:S mutation ratio of 1.0. The mAb 412.67 VκJκ gene sequence accumulated five somatic point mutations (four in Vκ and one in the 5′ portion of Jκ1), of which two were in CDR1, one in CDR3, one in FR2, and one in FR3 (Figs. 1 and 2, Table 1). The three CDR mutations resulted in three (two actual and one potential due to two adjacent CDR1 nucleotide changes) amino acid substitutions, yielding a significantly higher number of R mutations in the CDR than that expected by chance alone, and an R:S mutation ratio of 3:0. The two nucleotide mutations in the FR resulted in one amino acid substitution, yielding a significantly lower number of R mutations in the FR than that expected by chance alone and an R:S mutation ratio of 1.0. All the H-CDR1 and H-CDR2 mutations and the κ-CDR1 mutations targeted RGYW motifs which are known as mutational hot spots. Thus, the distribution of the R mutations in mAb 412.67 was consistent with a process of selection driven by antigen [14].
Fig. 1.

Nucleotide sequences of the wild-type, variously substituted, and germ-line (top row) mAb 412.67 VHDJH and VκJκ gene segments. 412.67.gl.5a is the autologous germ-line gene sequence. Dashes indicate identities. Solid lines on top of each cluster depict CDR. Lowercase letters denote vector sequences. The sequences or reverse complementary sequences encompassed by the H-Ov1, H-Ov2, H-FR3, 67cFR3, H-FR4, 67-CDR3 A, and 67-CDR3 B or κ-Ov1, κ-Ov2, κIII-FR3 A, κIII-FR3 B, and κ-FR4 mutagenized primers are underlined.
Fig. 2.
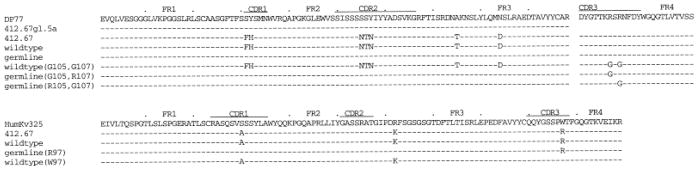
Deduced amino acid sequences of the wild-type, variously substituted, and germ-line (top row) mAb 412.67 VHDJH and VκJκ gene segments. 412.67.gl.5a is the autologous germ-line sequence. Dashes indicate identities. Solid lines on top of each cluster depict CDR.
Table 1.
Nature and distribution of the somatic mutations in the mAb 412.67 V(D)J genes
| Mutations | VHDJH | VκJκ | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| CDR | FR | CDR | FR | |||||
|
| ||||||||
| R | S | R | S | R | S | R | S | |
| Observed | 6 | 1 | 2 | 2 | 3 | 0 | 1 | 1 |
| Expecteda) | 2.53 | 5.73 | 0.96 | 2.78 | ||||
| Probabilityb) | p = 0.02 | p = 0.02 | p = 0.05 | p = 0.11 | ||||
Theoretically expected R mutations on the basis of chance alone.
Probability of R mutations arising by chance alone.
2.2 Anti-dsDNA mAb 412.67 IgG1κ expression in F3B6 cells in vitro
The wild-type (mutated) VHDJH and VκJκ gene segments isolated from the anti-dsDNA IgA1κ mAb 412.67-producing cell hybrid [5] were inserted into the pcDNAIG and pSXRDIG vectors, respectively, and expressed as part of an IgG1κ molecule by co-transfection of F3B6 cells (Fig. 3). Five stable clones secreting human IgG1κ were generated, two of which were expanded for antibody production. This “wild-type” recombinant IgG1κ molecule, designated VH67-D(R105, R107)JH67/Vκ67(R97)Jκ67, bound dsDNA and ssDNA as effectively as the original IgA1κ mAb 412.67 produced by the 412.67 somatic cell hybrid (Fig. 4A, B).
Fig. 3.
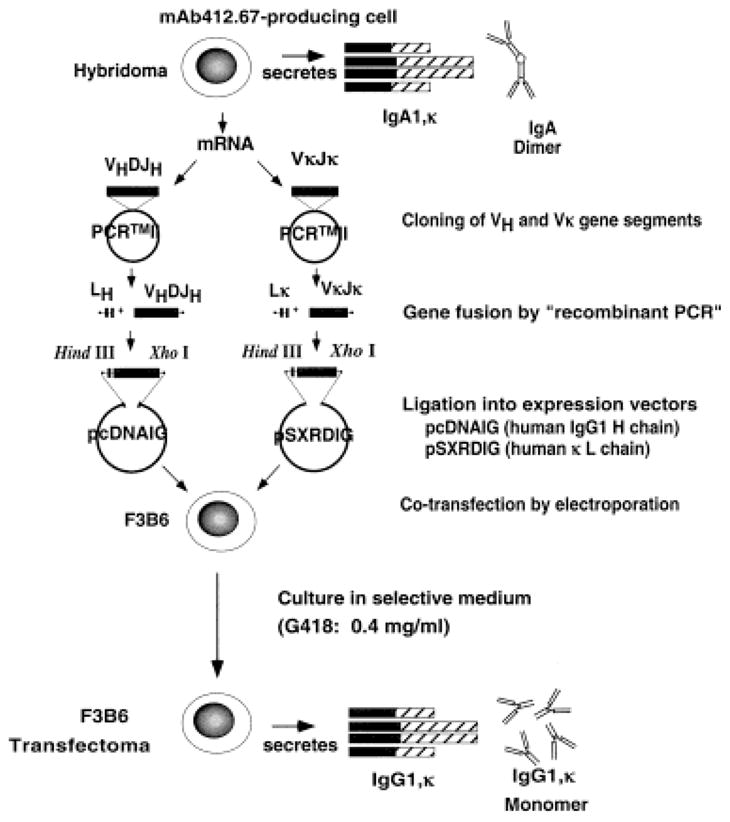
Generation of the germ-line revertant and mutagenized mAb 412.67 VHDJH and VκJκ DNA, their insertion into the pcDNAIG and the pSXRDIG vectors and expression in F3B6 cells.
Fig. 4.

Binding of the original IgA1κ mAb 412.67 (A), recombinant “wild-type” VH67-D(R105, R107)JH67/Vκ67-(R97) Jκ67 (B), “germ-line” VH67gl.5a-D(R105, R107) JH67/VκKv325(R97)Jκ67 (C), “wild-type CDR3 Gly-substituted” VH67-D(G105, G107)JH67/Vκ67-(W97)Jκ67 (D), “Trp-CDR3” revertant VH67-D (R105, R107) JH67/Vκ67(W97)Jκ67 (E), “wild-type CDR3 Gly-substituted” VH67-D(G105, G107) JH67/Vκ67(R97)Jκ67 (F), “single CDR3 Gly-substituted” VH67gl.5a-D(G105, R107)JH67/Vκ67(R97)Jκ67 (G), “single CDR3 Gly-substituted” VH67gl.5a-D(R105, G107)JH67/Vκ67(R97)Jκ67 (H) IgG1κ to ssDNA (■) and dsDNA (□). Bars schematize the composition of original wild-type IgA1κ, the wild-type IgG1κ, and the various recombinant and mutagenized molecules (black rectangles depict the wild-type sequences derived from the original mAb 412.67; white rectangles depict the gene sequence derived from autologous germ-line gene segments or the H-CDR3 Gly-substituted sequences). Antigen-binding activity is expressed as absorbance at 492 nm.
2.3 The “germ-line” revertant binds DNA as efficiently as the wild-type mAb 412.67
To investigate the contribution of the mAb 412.67 VH and Vκ segment somatic point-mutations to DNA binding, we constructed the “germ-line revertant” VH67gl.5a-D(R105, R107)JH67 gene segment by juxtaposing the autologous germ-line 412.67gl.5a VH gene with the mAb 412.67-derived CDR3-FR4 sequence, and the Kv325(R97)Jκ67 gene segment by juxtaposing the previously isolated germ-line HumKv325 Vκ gene [15] with the mAb 412.67-derived κ-CDR3-FR4 sequence. The deduced amino acid sequences of these constructs are depicted in Fig. 2. The recombinant “germ-line” VHDJH and VκJκ gene segments were inserted into pcDNAIG and pSXRDIG, respectively, and expressed in F3B6 cells to yield the “germ-line” recombinant IgG1κ molecule designated VH67gl.5a-D(R105, R107)JH67/JκKv325(R97)Jκ67. This germ-line revertant IgG1κ molecule closely mimicked in both dsDNA and ssDNA binding the “wild-type” VH67-D(R105, R107)JH67/Vκ67(R97)Jκ67 IgG1κ (Fig. 4B and C; Table 2), indicating that the VH and Vκ mutated amino acids contributed nothing to DNA binding by mAb 412.67.
Table 2.
DNA binding by the wild-type, germ-line revertant and variously substituted mAb 412.67 molecules
| Source | Mutations | CDR3 Arg | Avrel (g/μl) | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Class | VH | Vκ | H | H | κ | ssDNA | dsDNA | |
|
| ||||||||
| 105 | 107 | 97 | ||||||
| VH67-D(R105,R107)JH67 Vκ67(R97)Jκ67 |
IgG1 κ | Yes | Yes | Yes | Yes | Yes | 9.1 × 10−9 | 6.7 × 10−7 |
| VH67gl.5a-(R105,R107)JH67 Vκ Kv326-(R97)Jκ67 |
IgG1 κ | No | No | Yes | Yes | Yes | 9.9 × 10−9 | 6.4 × 10−7 |
| VH67-D(G105,G107)JH67 Vκ67(W97)Jκ67 |
IgG1 κ | Yes | Yes | No | No | No | No bindinga) | No binding |
| VH67-D(R105,R107)JH67 Vκ67(W97)Jκ67 |
IgG1 κ | Yes | Yes | Yes | Yes | No | 9.3 × 10−9 | 4.8 × 10−7 |
| VH67-D(G105,G107)JH67 Vκ67(R97)Jκ67 |
IgG1 κ | Yes | Yes | No | No | Yes | No binding | No binding |
| VH67gl.5a-D(G105,R107)JH67 Vκ67(R97)Jκ67 |
IgG1 κ | No | Yes | No | Yes | Yes | 5.2 × 10−9 | No binding |
| VH67gl.5a-D(R105,G107)JH67 Vκ67(R97)Jκ67 |
IgG1 κ | No | Yes | Yes | No | Yes | 4.4 × 10−9 | No binding |
Higher than 10−4 g/μl.
2.4 The mAb 412.67 CDR3 Arg residues are critical for DNA binding
To determine the role of the H-CDR3 Arg105 and Arg107 and that of the κ-CDR3 Arg97 in mAb 412.67 DNA binding, two recombinant gene segments were constructed: the “Gly105- and Gly107-substituted” VH67-D(G105, G107)JH67, which was identical to the “wild-type” VH67-D(R105, R107)JH67 segment except for the substitution of Arg105 and Arg107 with putative “neutral” Gly [10, 15–17], and the “Trp97 revertant” Vκ67(W97)Jκ67, which was identical to the “wild-type” Vκ67(R97)Jκ67 segment except for the replacement of somatically generated Arg97 by Trp. These recombinant VHDJH and VκJκ gene segments were inserted into the pcDNAIG and pSXRDIG vectors, respectively, and co-expressed in F3B6 cells to yield the “CDR3 Gly-substituted” VH67-D(G105, G107)JH67/Vκ67-(W97)Jκ67 molecule. As shown in Fig. 4D, this IgG1κ molecule failed to bind ssDNA and dsDNA, indicating that the H-CDR3 and/or κ-CDR3 Arg residues are necessary for DNA binding.
2.5 H-CDR3 Arg105 and Arg107 but not κ-CDR3 Arg97 are necessary for DNA binding
To determine the relative contribution of the H-CDR3 and κ-CDR3 Arg to DNA binding, the recombinant “Trp97 revertant” Vκ67(W97)Jκ67 gene segment was inserted into the pSXRDIG vector and expressed in F3B6 cells in conjunction with the “wild-type” H chain gene segment to yield the VH67-D(R105, R107)JH67/Vκ67(W97)Jκ67 IgG1κ molecule. The presence of the H-CDR3 Arg105 and Arg107 was sufficient to confer effective binding activity for both dsDNA and ssDNA to this recombinant IgG1κ molecule. The ssDNA relative avidity (Avrel) value was virtually identical to that of the wild-type mAb 412.67. The Avrel for dsDNA was somewhat higher, but within the experimental variation range (Fig. 4E, Table 2). Having shown that κ-CDR3 Arg97 is not necessary for DNA binding, we explored the relative contribution of H-CDR3 Arg105 and Arg107 to DNA binding by inserting the recombinant “CDR3 Gly-substituted” VH67-D(G105, G107)JH67 gene segment into pcDNAIG, and expressing it in F3B6 cells in conjunction with the “wild-type” κ chain gene segment to yield the VH67-D(G105, G107)JH67/Vκ67-(R97)Jκ67 IgG1κ. The absence of H-CDR3 Arg105 and Arg107 completely abrogated the binding to both ssDNA and dsDNA by this recombinant IgG1κ despite the presence of the “wild-type” κ segment containing the CDR3 Arg97 (Fig. 4F, Table 2).
These experiments showed that the H-CDR3 Arg105 and Arg107 are both necessary and sufficient for effective dsDNA and ssDNA binding by mAb 412.67, while the κ-CDR3 Arg97 is dispensable in this function.
2.6 Both Arg105 and Arg107 are required for dsDNA binding; either Arg105 or Arg107 is sufficient for ssDNA binding
To determine the relative contribution of H-CDR3 Arg105 and Arg107 to DNA binding, the recombinant single “CDR3 Gly substitute” VH67gl.5a-D(G105, R107)JH67 and VH67gl.5a-D(R105, G107)JH67 gene segments were independently inserted into pcDNAIG and expressed in F3B6 cells, each as paired with the “wild-type” κ chain gene segment to yield the VH67gl.5a-D(G105, R107) JH67/Vκ67(R97)Jκ67 and VH67gl.5a-D(R105, G107)JH67/Vκ67(R97)Jκ67 IgG1κ molecules. The single Gly-substituted H-CDR3 Arg105 or Arg107 molecule bound ssDNA with an efficiency comparable to that of the original mAb 412.67 (Fig. 4G and H, Table 2); neither single Gly-substituted molecule, however, bound dsDNA, indicating that both H-CDR3 Arg105 or Arg107 are required for dsDNA bindings, but either is sufficient for ssDNA binding.
2.7 Molecular modeling of mAb 412.67 Fv
The fragment variable (Fv) domain molecular model of mAb 412.67 is shown in ribbon (Fig. 5; all H-CDR and κ-CDR Arg and Lys displayed) and α-carbon (Fig. 6) trace. The H-CDR3 Arg105, Arg107 and Lys104 are located around the apex of the H-CDR3 loop, while Lys66 is located at the top of H-CDR2; the κ chain Arg24, Arg55 and Arg97 are not on the top of the respective κ-CDR1, κ-CDR2, and κ-CDR3 loops. Accordingly, Arg105, Arg107, Lys104 and Lys66 in the H chain can contribute to DNA binding while Arg24, Arg55 and Arg97 in the κ chain might not. The modeled combining site of mAb 412.67 features a groove, which can accommodate a DNA double helix (Fig. 6A). The positively charged Arg and Lys in the mAb 412.67 Fv combining site are in a position to effectively interact with DNA via hydrogen bonds and salt bridges (although charge-charge interactions of salt bridges will be weak in an aqueous environment), and their replacement with Gly obliterates dsDNA binding (Fig. 6B and C). The side chains of H-FR3 Arg98 and κ-CDR3 Arg97 appear to be too far from the antigen-binding area to contribute to the binding of dsDNA and ssDNA (Fig. 6B).
Fig. 5.
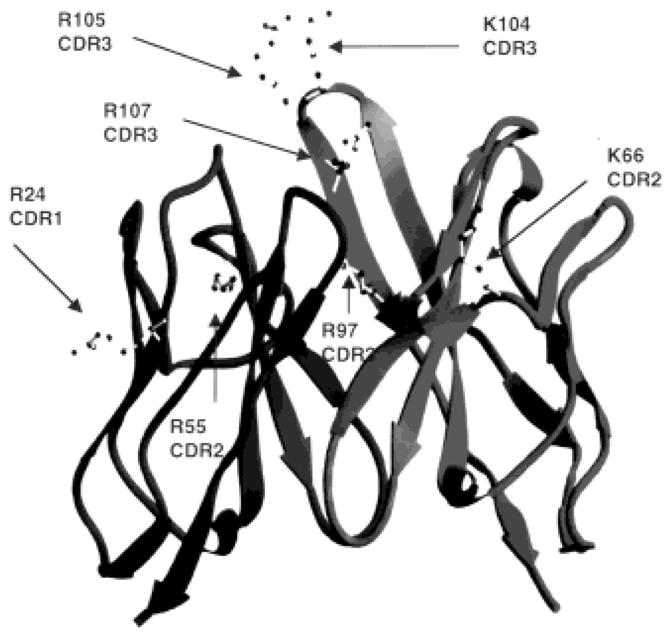
Ribbon drawing of mAb 412.67 Fv showing the κ chain on the left and the H chain on the right. All Arg and Lys in the H-CDR and κ-CDR are indicated.
Fig. 6.
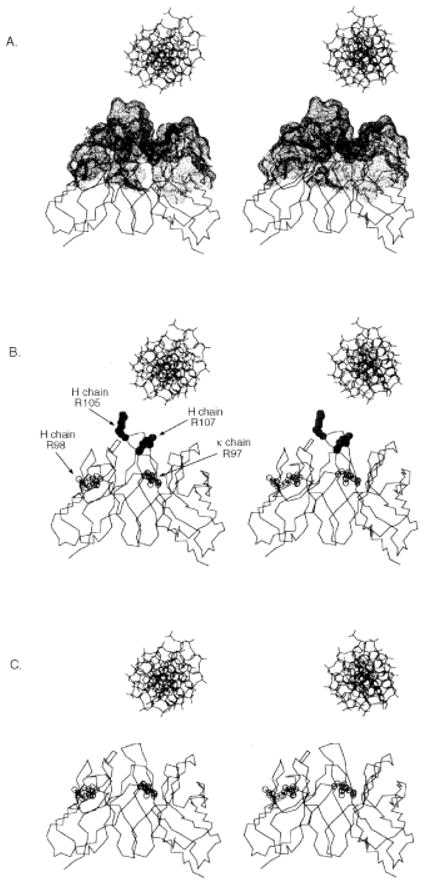
Stereo images of α-carbon trace drawings of the modeled mAb 412.67 Fv with B form DNA positioned above the binding site, drawn to scale. The molecular surface covering the CDR residues is depicted as dots (A). The H-CDR3 Arg 105 (R105) and 107 (R107) are depicted as full circles; the κ-CDR3 Arg97 (R97) and the H-FR3 Arg98 (R98) are depicted as open circles (B). The H-CDR3 Arg105 and Arg107 are substituted with Gly residues (C).
3 Discussion
If DNA was responsible for driving the selection of mAb 412.67, then the VH and Vκ segment somatic point mutations should contribute significantly to this binding activity. This was expected to be the case for H-CDR1 His32, H-CDR2 Asn55, H-CDR2 Asn57, κ-FR3 Lys62, and κ-CDR3 Arg97, as Arg, His, Lys and, to a lesser extent, Asn have been all shown to play a significant role in DNA binding [3, 4]. Contrary to our expectations, reversion to germ-line configuration of all the above point mutations did not significantly change the dsDNA or ssDNA binding activity of mAb 412.67; instead, the whole DNA binding activity was mediated by the H-CDR3 Arg105 and Arg107. There is no paradigm for explaining the primary structure of anti-dsDNA autoantibodies, but the primary role of H-CDR3 Arg in DNA binding has been shown in murine autoantibodies [4, 10]. These autoantibodies bear at least one and in most cases two or three Arg in the H-CDR3 [10, 18–21]. Anti-dsDNA autoantibodies induced by immunization with bacterial DNA in normal BALB/c mice bear only one or no H-CDR3 Arg [22], but these antibodies do not cross-react with mammalian DNA. The presence of H-CDR3 Arg105 and Arg107 and their critical role in mAb 412.67 DNA binding further emphasize the structural difference between the anti-self (mammalian) DNA antibodies arising in an autoimmune context and anti-DNA antibodies induced by microbial DNA.
In the human lupus mAb 412.67 autoantibody, the H-CDR3 Arg105 and the Arg107 were central in dsDNA binding, as reversion of just one of these two Arg to Gly resulted in total loss of dsDNA binding. Molecular modeling of mAb 412.67 Fv showed that the central role of Arg105 and Arg107 in DNA binding reflect the central position of these Arg in the binding site, near the apex of the H-CDR3 chain loop and well suited to fit in the major or minor groove of a dsDNA helix. In addition, the proposed three-dimensional H-CDR3 loop structure supports the results of the experiments involving the Ig V(D)J recombinant constructs and emphasizes the stricter structural requirements for dsDNA binding as compared to ssDNA binding: one of the two H-CDR3 Arg was sufficient for ssDNA binding, but both Arg105 and Arg107 were required for dsDNA binding. The distance of the κ-CDR3 Arg97 from the surface of the binding area of mAb 412.67 Fv provides a structural explanation for the lack of contribution by this Arg to DNA binding, and extends previous observations in the mouse suggesting a limited role for the L chain in DNA binding [23].
As H-CDR3 Arg105 and Arg107 were encoded by N segment additions, it was impossible to establish whether they were constitutive to the Ig expressed by the progenitor of the mAb 412.67-producing cell, or arose as superimposed mutations. If Arg105 and Arg107 were constitutive to the progenitor Ig H-CDR3, then the anti-dsDNA mAb 412.67 autoantibody could have been generated by mere expansion of a pre-existing unmutated high-affinity anti-DNA antibody-producing cell progenitor recruited by bacterial or viral antigens, that later drove the selection of the VHDJH and VκJκ gene segment mutations. Alternatively, H-CDR3 Arg105 and/or Arg107 might have emerged as random mutations during a microbial antigen-driven expansion of the mAb 412.67-producing cell progenitor to be subsequently fixed through selection by DNA. The role of bacteria in the emergence of anti-DNA autoantibodies is consistent with the high affinity for Streptococcus pneumoniae of a murine pathogenic anti-dsDNA IgG1 autoantibody [24] and by the induction of an autoantibody response to dsDNA by peptides that do not bind DNA [25]. The B-1 cell origin of the anti-dsDNA mAb 412.67 [26] would support a role of microbial antigens in the selection of mAb 412.67 somatic R mutations, as these B cells are highly enriched in antibody-producing cell precursors to bacteria and viruses [26, 27].
Another not mutually exclusive possibility in the recruitment and expansion of the mAb 412.67-producing cell precursor is that the selecting antigen was ab initio dsDNA or DNA-associated protein(s), i.e. histones. Naked dsDNA molecules do not elicit adequate antibody responses but become immunogenic when complexed with DNA-binding proteins [28]. Early autoantibodies in autoimmune MRL/MP-lpr/lpr and BXSB mice and lupus patients would recognize discontinuous epitopes on native chromatin, the (H2A-H2B)-DNA subnucleosome, and ssDNA [29]. As the autoimmune response progresses, dsDNA and other chromatin constituents, i.e. (H3-H4)2-DNA, become antigenic, suggesting that anti-dsDNA autoantibodies are a subset of the wider spectrum of anti-chromatin autoantibody population occurring at a mature stage of the response [29–31]. A similar series of events could have driven the selection of the mAb 412.67-producing cell precursor, with dsDNA being responsible for the selection of the mAb 412.67 H-CDR3 Arg105 and/or Arg107, as mutants of originally N segment addition-encoded residues. Structure-function analysis of more human anti-DNA autoantibodies should clarify the nature of the selecting antigens, and provide more clues to the role of somatic mutations in DNA binding.
4 Materials and methods
4.1 Structure of the anti-dsDNA mAb 412.67 VHDJH and the VkJκ gene segments
The mAb 412.67-producing cell line was generated by fusion of circulating B-1 cells from a lupus patient with human-mouse non-secretor F3B6 cells [5, 26]. mAb 412.67 was isolated as an IgA1κ to dsDNA/ssDNA, and was originally referred to as mAb 412.67.F1.3 [5, 26]. The mAb 412.67 VHDJH and VκJκ gene sequences and those of the related germ-line templates isolated from the same patient whose B cells were used to generate this mAb have been reported [5, 26]. The mAb 412.67 V(D)J genes were re-sequenced for the purpose of this study (Figs. 1 and 2). The VHDJH segment consisted of a mutated DP77 gene [32, 33] juxtaposed with DM2 [34] and JH4b [35] genes with intervening N segment additions. These encoded four amino acids, including the CDR3 Arg105 and Arg107 (Fig. 2). The VκJκ segment consisted of a mutated Humkv325 gene [36] juxtaposed with a mutated Jκ1 gene.
4.2 Analysis of the somatic point mutations
The number of expected R mutations in the Ig V gene CDR or FR was calculated taking into account the inherent susceptibility to R substitutions of the DP77 and the DM2 sequences [14]. A binomial probability model was used to verify whether the excess and scarcity of R mutations in the CDR and FR, respectively, were due to chance alone: p = {n!/[k!(n−k)!]} × qk × (1−q)n−k, where q is the probability that an R mutation will localize to CDR or FR (q = CDRrel × CDRRf or FRrel × FRRf), and k is the number of observed R mutations in the CDR or FR [14].
4.3 Introduction of the mAb 412.67 VHDJH DNA into the pcDNAIG human γ1 vector
The features of the pcDNAIG plasmid vector used for the in vitro expression of rearranged Ig VHDJH in conjunction with Cγ and the experimental approach used for the construction of germ-line revertants and recombinant Ig VHDJH gene segments using “recombinant” PCR are detailed in [15–17]. The H-leader (5′ gggaagcttctcaccatgggatgg 3′; underlined sequence denotes restriction site) and H-Ov1 (5′ GCTGCACCTCacactggacacctgcagagaag 3′; capital letters denote mAb 412.67 nucleotides; lowercase letters denote vector nucleotides) primers were used to amplify the vector leader sequence; the H-Ov2 (5′ ttctctgcaggtgtccagtgtGAGGTGCAGCTG 3′) and H-FR4 (5′ gggctcgagactcaccTGAGGAGACGGTGACCA 3′) primers were used to amplify the mAb 412.67 VHDJH gene segments. The primers used at the ends to be joined (H-Ov1 and H-Ov2) were made complementary to one another by including a sequence complementary to the 3′ portion of the other primer. This made the products of PCR 1 and PCR 2 overlap at the ends to be joined, and allowed for the performance of a recombinant PCR (PCR 3) by addition of excess H-leader and H-FR4 primers. The recombinant fragment was sequenced to ensure that no unintended mutations were introduced during PCR amplification [5, 8, 37], digested with Hind III and Xho I, and ligated into pcDNAIG, previously digested with Hind III and Xho I and freed of its original VHDJH gene segment. The recombinant pcDNAIG plasmids were amplified by transformation of competent MC1061/P3 cells (Invitrogen Corp., La Jolla, CA) and selection with ampicillin. Plasmid DNA was isolated using a plasmid kit (Qiagen Inc., Chatsworth, CA).
4.4 Introduction of the mAb 412.67 VκJκ DNA into the pSXRDIG human κ vector
The features of the pSXRDIG vector used for the in vitro expression of rearranged Ig VκJκ gene segment and the approach adopted for the construction of germ-line revertants and recombinant Ig VκJκ gene segments were as we detailed in [15–17]. The κ-leader (5′ gggaagcttatcaagatgaagtca 3′) and κ-Ov1 (5′ CAACACAATTTCgcatctggaacctgcagtcagaga 3′) primers were used to amplify the vector leader DNA; the κ-Ov2 (5′ gcaggttccagatgcGAAATTGTGTTGACGCAGTCT 3′) and κ-FR4 (5′ gggctcgagacttACGTTTGATTT-CCACCTTGG 3′) primers were used to amplify the mAb 412.67 VkJk DNA. The recombinant fragment, leader-Vκ67(R97)Jκ67, was inserted into pSXRDIG after digestion with Hind III and Xho I. Recombinant pSXRDIG plasmids were amplified by transformation of competent DH5α cells (Invitrogen Corp., La Jolla, CA) and selection with ampicillin.
4.5 Construction of the “germ-line revertant” VH67gl.5a-D(R105,R107)JH67 and VkKv325(R97)Jk67 gene segments
The FR1 through FR3 area of the autologous germ-line VH 412.67gl.5a gene was PCR amplified using the H-Ov2 and 67cFR3 (5′ CAGTGAGTTCTTGGCGTTGTC 3′) primers (Fig. 1). The mAb 412.67 VHDJH gene segment FR3 through FR4 area was amplified using the H-FR3 (5′ CCAGAGACAACGCCAAGAACTCACTGTATCTGCAAATGAACAGCC 3′) and H-FR4 primers (Fig. 1). The two amplified fragments were joined by recombinant PCR, and then juxtaposed with the mouse H-leader sequence by another recombinant PCR to yield the leader-VH67gl.5a-D(R105, R107)JH67 gene segment. Analogously, the FR1 through FR3 area of a cDNA copy of the germ-line HumKv325 gene segment was amplified using the κ-Ov2 and κIII-FR3 B (5′ GCTGACAGTAATACACTGCAAAATCTTC 3′) primers (Fig. 1), while the FR3 through FR4 area of mAb 412.67 VκJκ gene segment was amplified using the (reverse complement of κIII-FR3 B) κIII-FR3 A (5′ GAAGATTTTGCAGTGTATTACTGTCAGC 3′) and κ-FR4 primers. The two amplified fragments were purified, joined by recombinant PCR, and then juxtaposed with the mouse κ leader DNA by another recombinant PCR to yield the leader-VκKv325(R97)Jκ67 gene segment.
4.6 Construction of the “wild-type CDR3 Gly-substituted” of the mAb 412.67 VHDJH gene segments
The leader throughout CDR3 area of the leader-VH67-D(R105, R107)JH67 DNA segment was PCR amplified using the H-leader and 67H-CDR3 B (5′ CAAAGTTCCCGCTTCCTTT 3′; underlined bold nucleotides denote substitutions giving rise to R mutations) primers. The CDR3 through FR4 of the leader-VH67-D(R105, R107)JH67 gene segment was amplified using the 67H-CDR3 A (5′ AAAGGAAGCGGGAACTTTG 3′) and the H-FR4 primers. The sense primer, 67-CDR3 A, and anti-sense primer, 67H-CDR3 B, reverse complements of one another, were utilized to substitute the first nucleotide (cytosine) of the CGA and CGG triplets, which encode for Arg residues, to guanine, thus creating GGA and GGG triplets, respectively, to encode for Gly residues. The two amplified DNA fragments were joined by recombinant PCR and inserted into the pcDNAIG expression vector.
4.7 Construction of the single “wild-type and germ-line CDR3 Gly-substituted” of the mAb 412.67 VHDJH gene segment
The leader through CDR3 area of the leader-VH67g1.5a-D(R105, R107)JH67 DNA segment was PCR amplified using the H-leader and 67H-G107 B (5′ CAAAGTTCCCGCTTCGTTT 3′) or 67H-G105 B (5′ CAAAGTTCCGGCTTCCTTT 3′) primers. The CDR3 through FR4 area was amplified using the 67H-G107 A (5′ AAACGAAGCGGGAACTTTG 3′) or 67H-G105 A (5′ AAAGGAAGCCGGAACTTTG 3′) and the H-FR4 primers. The sense primer 67H-G107 A (or 67H-G105 A) and anti-sense primer 67H-G107 B (or 67H-G105 B) were reverse complements of one another and were utilized to substitute the first nucleotide (cytosine) of the CGA (or CGG) triplet, which encodes for an Arg, to guanine, thus creating GGA (or GGG) triplet to encode for a Gly. The two amplified fragments were purified, joined by recombinant PCR, and inserted into the pcDNAIG expression vector.
4.8 Construction of the mAb 412.67 VκJκ gene segment “wild-type-Trp97 revertant”
The leader through CDR3 area of the leader-Vκ67(W97)Jκ67 gene segment was PCR amplified using the κ-leader and 67 κ-CDR3 B (5′ GGCCGAACGTCCAAGGTG 3′) primers. The CDR3 through FR4 area was PCR amplified using the 67 κ-CDR3 A (5′ CACCTTGGACGTTCGGCC 3′) and the κ-FR4 primers. The sense, 67 κ-CDR3 A, and anti-sense, 67 κ-CDR3 B, primers were reverse complements of one another and were utilized to substitute the first nucleotide (cytosine) of the CGG triplet, which encoded for an Arg, to thymine, thus creating a TGG triplet coding for a Trp. The two amplified fragments were joined by recombinant PCR and inserted into the pSXRDIG expression vector.
4.9 Transfection, cell culture and purification of antibody molecules
The recombinant and/or mutagenized mAb 412.67 V(D)J genes were expressed by transfection of F3B6 cells as reported [15–17]. Neomycin alone was used as selecting agent because in transfectants expressing only pcDNAIG (H chain) the accumulation of unsecretable H chain molecules leads to cell death. Double γ1 and κ chain producer cells were identified by specific ELISA [5, 7, 15], expanded in culture and frozen. IgGκ were purified by ammonium sulfate precipitation, absorption of solubilized IgG onto a HiTrap protein G-Sepharose column (Pharmacia LKB Biotechnology, Piscataway, NJ), elution with 100 mM glycine-HCl buffer (pH 2.7), followed by neutralization (pH 9.0). mAb IgGκ concentration, binding to and Avrel for ssDNA and dsDNA were measured by ELISA [5, 15]. Avrel represents the concentration of soluble dsDNA or ssDNA that inhibited antibody binding to solid-phase DNA by 50%.
4.10 Molecular modeling of mAb 412.67 Fv
Comparison of the mAb 412.67 deduced amino acid sequences with those of other antibodies for which three-dimensional structures are available (Protein Data Bank, PDB) showed that the closest VHDJH structure was that of human antibody 3D6 (PDB entry 1DFB) and the closest VκJκ structure was that of human TR1.9 (PDB entry 1VGE). The number of residues in mAb 412.67 H-CDR1 and H-CDR2 is the same as in the corresponding CDR in 3D6, while the length of mAb 412.67 H-CDR3 is the same as that in the murine antibodies F9.13.7 (PDB entry 1FBI), 40–50 (PDB entry 1IBG), and L5MK16 (PDB entry 1LMK), with the H-CDR3 of 40–50 being most similar in sequence to that of mAb 412.67. The number of residues in mAb 412.67 κ-CDR2 and κ-CDR3 is the same as in the corresponding CDR in TR1.9, while the length of mAb 412.67 κ-CDR1 is the same as that in the murine antibodies 1F7 (PDB entry 1FIG) and 409.5.3 (PDB entry 1IAI), with the L-CDR1 of 1F7 being more similar in sequence to that of mAb 412.67. A composite of the FR of 3D6, its H-CDR1 and H-CDR2, and the H-CDR3 of 40–50 served as the template for the modeling of mAb 412.67 VHDJH. A composite of the FR of TR1.9, its L-CDR2 and L-CDR3, and the L-CDR1 of 1F7 served as the template for the modeling of mAb 412.67 VκJκ. The VHDJH:VκJκ quaternary structure of TR1.9 was assumed for mAb 412.67. The template structures were mutated according to the sequence of mAb 412.67 using the graphics program LOOK (Molecular Applications Group, Palo Alto, CA) and maximizing the overlap of the amino acid side chains of the model and template structures. Unfavorable steric contacts were minimized by rotating around single bonds and energetically favored torsion angles [38] were imposed. The modeled Fv was subjected to energy minimization using X-PLOR [39], while restrained by artificial structure factors, and placed in a primitive crystal lattice with unit cell dimensions of 100 Å on each side and cell angle of 90 °. Crystallographic refinement by simulated annealing was performed against the artificially computed structure factors with a resolution of 10−4 Å, and a starting temperature of 2000 °K. This artificial refinement achieved energy minimization while restraining the structure to be close to the starting model. Surface calculations were performed using the program MS [40]. Figures were generated using the programs RIBBONS [41] and ORTEP [42].
Acknowledgments
This work was supported by the USPHS NIH grant AR 40908 and by a Research Grant of the S.L.E. Foundation, Inc., New York, New York.
Abbreviations
- Avrel
Relative avidity
- CDR
Complementarity-determining region
- Fv
Fragment variable
- FR
Framework region
- R
Replacement
- S
Silent
- ds
Double stranded
- ss
Single stranded
References
- 1.Zan H, Cerutti A, Dramitinos P, Schaffer A, Li Z, Casali P. Induction of Ig somatic hypermutation and class switching in a human monoclonal IgM+IgD+ B cell line in vitro: Definition of the requirements and modalities of hypermutation. J Immunol. 1999;162:3437–3447. [PMC free article] [PubMed] [Google Scholar]
- 2.Zan H, Li Z-D, Dramitinos P, Schaffer A, Cerutti A, Casali P. BCR engagement and T cell contact induce bcl-6 somatic hypermutation in human B cells: association with initiation of transcription and identity with Ig hypermutation. J Immunol. 2000 doi: 10.4049/jimmunol.165.2.830. in press. [DOI] [PubMed] [Google Scholar]
- 3.Diamond B, Katz JB, Paul E, Aranow C, Lustgarten D, Scharff MD. The role of somatic mutation in the pathogenic anti-DNA response. Annu Rev Immunol. 1992;10:731–757. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- 4.Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. 1994;12:487–520. doi: 10.1146/annurev.iy.12.040194.002415. [DOI] [PubMed] [Google Scholar]
- 5.Kasaian MT, Ikematsu H, Balow JE, Casali P. Structure of the VH and VL segments of monoreactive and polyreactive IgA autoantibodies to DNA in patients with systemic lupus erythematosus. J Immunol. 1994;152:3137–3151. [PMC free article] [PubMed] [Google Scholar]
- 6.Berek C, Milstein C. The dynamic nature of the antibody repertoire. Immunol Rev. 1988;105:5–26. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 7.Ueki Y, Goldfarb IS, Harindranath N, Gore M, Koprowski H, Notkins AL, Casali P. Clonal analysis of a human antibody response. Quantitation of precursors of antibody-producing cells and generation and characterization of monoclonal IgM, IgG, and IgA to rabies virus. J Exp Med. 1990;171:19–34. doi: 10.1084/jem.171.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikematsu H, Harindranath N, Ueki Y, Notkins AL, Casali P. Clonal analysis of a human antibody response. II. Sequences of the VH genes of human monoclonal IgM, IgG and IgA to rabies virus reveal preferential utilization of the VH3 segments and somatic hypermutation. J Immunol. 1993;150:1325–1337. [PMC free article] [PubMed] [Google Scholar]
- 9.Ikematsu W, Kobarg J, Ikematsu H, Ichiyoshi Y, Casali P. Clonal analysis of a human antibody response. III. Nucleotide sequences of monoclonal IgM, IgG, and IgA to rabies virus reveal restricted Vκ gene utilization, junctional VκJκ and VλJλ diversity, and somatic hypermutation. J Immunol. 1998;161:2895–2905. [PubMed] [Google Scholar]
- 10.Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, Weigert M. Residues that mediate DNA binding of autoimmune antibodies. J Immunol. 1993;150:4966–4977. [PubMed] [Google Scholar]
- 11.Seeman NC, Rosember JM, Rich A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc Natl Acad Sci USA. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbas SM, Ditzel HJ, Salonen EM, Yang WP, Silverman GJ, Burton DR. Human autoantibody recognition of DNA. Proc Natl Acad Sci USA. 1995;92:2529–2533. doi: 10.1073/pnas.92.7.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Schettino EW, Casali P. Structure-function analysis of a human anti-DNA autoantibody: role of H chain CDR3 Arg residues in DNA binding. FASEB J. 1998;12:A913. [Google Scholar]
- 14.Chang B, Casali P. The CDR1 sequences of a major proportion of human germline Ig VH genes are inherently susceptible to amino acid replacement. Immunol Today. 1994;15:367–373. doi: 10.1016/0167-5699(94)90175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichiyoshi Y, Casali P. Analysis of the structural correlates for antibody polyreactivity by multiple reassortments of chimeric human immunoglobulin heavy and light chain V segments. J Exp Med. 1994;180:885–895. doi: 10.1084/jem.180.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichiyoshi Y, Casali P. Analysis of the structural correlates for self-antigen binding by natural and disease-related autoantibodies. In vitro expression of recombinant and/or mutagenized human IgG. Ann N Y Acad Sci. 1995;764:328–341. doi: 10.1111/j.1749-6632.1995.tb55844.x. [DOI] [PubMed] [Google Scholar]
- 17.Ichiyoshi Y, Zhou M, Casali P. A human anti-insulin IgG autoantibody apparently arises through clonal selection from an insulin-specific “germ-line” natural antibody template. Analysis by V gene segment reassortment and site-directed mutagenesis. J Immunol. 1995;154:226–238. [PMC free article] [PubMed] [Google Scholar]
- 18.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radic MZ, Mascelli MA, Erikson J, Shan H, Shlomchik M, Weigert M. Structural patterns in anti-DNA antibodies from MRL/lpr mice. Cold Spring Harb Symp Quant Biol. 1989;2:933–946. doi: 10.1101/sqb.1989.054.01.108. [DOI] [PubMed] [Google Scholar]
- 20.Tillman DM, Jou NT, Hill RJ, Marion TN. Both IgM and IgG anti-DNA antibodies are the products of clonally selective B cell stimulation in (NZB × NZW)F1 mice. J Exp Med. 1992;176:761–779. doi: 10.1084/jem.176.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Behar SM, Lustgarten DL, Corbet S, Scharff MD. Characterization of somatically mutated S107 VH11-encoded anti-DNA autoantibodies derived from autoimmune (NZB × NZW)F1 mice. J Exp Med. 1991;173:731–741. doi: 10.1084/jem.173.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilkeson GS, Bloom DD, Pisetsky DS, Clarke SH. Molecular characterization of anti-DNA antibodies induced in normal mice by immunization with bacterial DNA. Differences from spontaneous anti-DNA in the content and location of VH CDR3 arginines. J Immunol. 1993;151:1353–1364. [PubMed] [Google Scholar]
- 23.Ibrahim SM, Weigert M, Basu C, Erikson J, Radic MZ. Light chain contribution to specificity in anti-DNA antibodies. J Immunol. 1995;155:3223–3233. [PubMed] [Google Scholar]
- 24.Putterman C, Limpanasithikul W, Edelman M, Diamond B. The double edged sword of the immune response: mutational analysis of a murine anti-pneumococcal, anti-DNA antibody. J Clin Invest. 1996;97:2251–2259. doi: 10.1172/JCI118666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med. 1998;188:29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasaian M, Casali P. B-1 cellular origin and VH segment structure of anti-DNA IgG, IgM, and IgA autoantibodies in patients with systemic lupus erythematosus. Ann NY Acad Sci. 1995;764:410–423. doi: 10.1111/j.1749-6632.1995.tb55856.x. [DOI] [PubMed] [Google Scholar]
- 27.Casali P, Haughton G, Kasaian MT. B-1 (CD 5) cells. In: Coutinho A, Kazatchkine MD, editors. Autoimmunity: Physiology and Disease. Wiley-Liss, Inc; New York: 1994. pp. 57–88. [Google Scholar]
- 28.Desai DD, Krishnan MR, Swindle JT, Marion TN. Antigen-specific induction of antibodies against native mammalian DNA in nonautoimmune mice. J Immunol. 1993;151:1614–1626. [PubMed] [Google Scholar]
- 29.Burlingame RW, Rubin RL, Balderas RS, Theofilopulos AN. Genesis and evolution of antichromatin autoantibodies in murine lupus implicates T-dependent immunization with self-antigen. J Clin Invest. 1993;91:1687–1696. doi: 10.1172/JCI116378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burlingame RW, Boey ML, Starkebaum G, Rubin LR. The central role of chromatin in autoimmune responses to histones and DNA in systemic lupus erythematosus. J Clin Invest. 1994;94:184–192. doi: 10.1172/JCI117305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brard F, Shannon M, Prak EL, Litwin S, Weigert M. Somatic mutation and light chain rearrangement generate auto-immunity in anti-single-stranded DNA transgenic MRL/lpr mice. J Exp Med. 1999;190:691–704. doi: 10.1084/jem.190.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook GP, Tomlinson IM. The human immunoglobulin VH repertoire. Immunol Today. 1995;16:237–242. doi: 10.1016/0167-5699(95)80166-9. [DOI] [PubMed] [Google Scholar]
- 33.Matsuda F, Ishii K, Bourvagnet P, Kuma K, Hayashida H, Miyata T, Honjo T. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med. 1998;188:2151–2162. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravetch JV, Siebenlist U, Korsmeyer S, Waldmann T, Leder P. Structure of the human immunoglobulin mu locus: characterization of embryonic and rearranged J and D genes. Cell. 1981;27:583–591. doi: 10.1016/0092-8674(81)90400-1. [DOI] [PubMed] [Google Scholar]
- 35.Yamada M, Wasserman R, Reichard BA, Shane S, Caton AJ, Rovera G. Preferential utilization of specific immunoglobulin heavy chain diversity and joining segments in adult human peripheral blood B lymphocytes. J Exp Med. 1991;173:395–407. doi: 10.1084/jem.173.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomlinson IM, Cox JP, Gherardi E, Lesk AM, Chothia C. The structural repertoire of the human V kappa domain. EMBO J. 1995;14:4628–4638. doi: 10.1002/j.1460-2075.1995.tb00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantovani L, Wilder RL, Casali P. Human rheumatoid B-1a (CD5+ B) cells make somatically hypermutated high affinity IgM rheumatoid factors. J Immunol. 1993;151:473–488. [PMC free article] [PubMed] [Google Scholar]
- 38.Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 39.Bruenger AT. X-PLOR Version 3.1: A System for X-Ray Crystallography and NMR. Yale University Press; New Haven: 1992. [Google Scholar]
- 40.Connolly ML. Analytical molecular surface calculation. J Appl Crystallogr. 1983;16:548–558. [Google Scholar]
- 41.Carson M. RIBBONS 2.0. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 42.Johnson CK. ORTEP, A Fortran Thermal-Ellipsoids Plot Program for Crystal Structure Illustrations. Oak Ridge National Laboratory; Oak Ridge: 1965. [Google Scholar]