

Abstract
C4′-oxidized (C4-AP) and C5′-oxidized abasic sites (DOB) that are produced following abstraction of a hydrogen atom from the DNA backbone reversibly form cross-links selectively with dA opposite a 3′-adjacent nucleotide, despite the comparable proximity of an opposing dA. A previous report on UvrABC incision of DNA substrates containing stabilized analogues of the ICLs derived from C4-AP and DOB also indicated that the latter is repaired more readily by nucleotide excision repair [Ghosh, S., and Greenberg, M. M. (2014) Biochemistry 53, 5958–5965]. The source for selective cross-link formation was probed by comparing the reactivity of ICL analogues of C4-AP and DOB that mimic the preferred and disfavored cross-links with that of reagents that indirectly detect distortion by reacting with the nucleobases. The disfavored C4-AP and DOB analogues were each more reactive than the corresponding preferred cross-link substrates, suggesting that the latter are more stable, which is consistent with selective ICL formation. In addition, the preferred DOB analogue is more reactive than the respective C4-AP ICL, which is consistent with its more efficient incision by UvrABC. The conclusions drawn from the chemical probing experiments are corroborated by UV melting studies. The preferred ICLs exhibit melting temperatures higher than those of the corresponding disfavored isomers. These studies suggest that oxidized abasic sites form reversible interstrand cross-links with dA opposite the 3′-adjacent thymidine because these products are more stable and the thermodynamic preference is reflected in the transition states for their formation.
DNA–DNA interstrand cross-links comprise an important family of DNA damage whose members are absolute blocks of replication and transcription.1,2 ICLs are typically produced by exogenous bis-electrophiles that react in the minor or major grooves with staggered nucleobases on opposite strands.3–6 Recently, ICLs that result from reaction of various abasic lesions (Figure 1) with dA, dC, and/or dG on the complementary strand have been characterized.7–10 Such cross-links emanate from AP, as well as the oxidized abasic lesions, C4-AP and DOB, that are in equilibrium with ring-opened, electrophilic aldehyde forms (Figure 1 and Scheme 1). C4-AP yields two types of ICLs, one in which both strands are intact (Scheme 1) and one in which the oxidized abasic site has undergone strand scission.11–14 The former C4-AP ICL and that produced from DOB are formed reversibly and selectively with the dA opposite the 3′-adjacent thymidine (Scheme 2).15 Chemically stable analogues [X–Y and W–V (Scheme 1)] that facilitate the characterization of these reversibly formed ICLs have been prepared.16 ICLs (and analogues) arising from oxidized abasic sites (C4-AP and DOB) are substrates for bacterial nucleotide excision repair, which transforms some of these molecules into double-strand breaks.17,18 The chemically stabilized C4-AP ICL analogue is a poorer substrate than that resulting from DOB, but the structural basis for this difference is unknown.18 Experiments designed to probe the source for the differences in NER efficiency and selective formation of ICLs between C4-AP and DOB and the dA opposite the 3′-adjacent thymidine are described herein.
Figure 1.

Abasic site structures.
Scheme 1.

ICLs and Stabilized ICLs from C4-AP and DOB
Scheme 2.

ICL Positioning from C4-AP and DOB
The right-handed helical nature of duplex DNA provides a rationalization for reaction with the N6-amino group of a dA opposite the 3′-adjacent thymidine compared to the same nucleotide opposite a 5′-thymidine because of the proximity of the C1-aldehyde to the major groove. However, molecular models do not explain why DOB and C4-AP do not readily form ICLs with an opposing nucleotide. Models suggest that the distances between the C1-aldehyde of DOB (see the Supporting Information) or C4-AP (Figure 2) and the N6-amino group of an opposing dA are comparable (within 0.4 Å) to those of the same functionality on a dA opposite the 3′-adjacent thymidine. We considered the possibility that an ICL between C4-AP or DOB and an opposing dA was less stable than one resulting from reaction of an oxidized abasic site with a dA opposite the 3′-adjacent thymidine and that this would be reflected in the kinetics and equilibria of the reversibly formed ICLs. We hypothesized that the structures of the chemically stabilized ICL analogues would correlate with the actual products produced from DOB and C4-AP (Scheme 1), and that the unobserved, putatively less stable molecules consisting of cross-links with opposing dA’s would experience greater base pairing disruption. Relative perturbations of duplex structure may also correlate with ICL susceptibility to NER. Although the mechanism for NER recognition of DNA damage is still under investigation, the proteins are believed to recognize macroscopic duplex distortion.6,19–23 Greater distortion of DOB ICL analogues than of those derived from C4-AP would correlate with comparisons of NER.18 Base pairing disruptions in the ICLs should translate into greater reactivity with chemical agents (e.g., dimethyl sulfate, diethyl pyrocarbonate, potassium permanganate, and hydroxyl radical) that probe nucleobase accessibility, as well as lower UV melting temperatures.24–35
Figure 2.

Molecular model of C4-AP in a 5′-d(TXT)/(AAA) sequence (X = C4-AP).
MATERIALS AND METHODS
Materials and General Methods
Oligonucleotides were prepared on an Applied Biosystems Inc. 394 DNA synthesizer. Commercially available DNA synthesis reagents were obtained from Glen Research Inc. DNA substrates used in this study are presented in Figure 3. T4 polynucleotide kinase, Klenow exo–, terminal deoxynucleotide transferase, and T4 DNA ligase were obtained from New England Biolabs. [α-32P]dCTP, [α-32P]-dTTP, [γ-32P]ATP, and [α-32P]cordycepin 5′-triphosphate were purchased from PerkinElmer. Analyses of radiolabeled oligonucleotides were conducted using a Storm 840 Phosphor-imager and ImageQuant TL software. C18-Sep-Pak cartridges were obtained from Waters.
Figure 3.

Cross-linked DNA substrates used in this study.
General Procedure for the Hydroxyl Radical Cleavage Reaction of 1–3 and 8–10
The appropriately 32P-labeled substrate (5000–15000 cpm) was suspended in 8 μL of water and mixed with 10 μL of 2× oxidation buffer [200 mM NaCl, 20 mM sodium phosphate (pH 7.2), 2 mM sodium ascorbate, and 0.5 mM hydrogen peroxide]. An Fe·EDTA solution [2 μL, 1 mM EDTA and 0.5 mM Fe(NH4)2(SO4)2·6H2O] was added, and the reaction was allowed to proceed for 3–5 min. The reaction was quenched by addition of 100 mM thiourea (10 μM) and the sample precipitated with 3 M NaOAc (4 μL), 1 μg/μL calf thymus DNA (5 μL), and cold ethanol (115 μL). After centrifugation, removal of the supernatant, and drying under vacuum, the DNA was suspended in formamide loading buffer [10 μL, 50% formamide, 5 mM EDTA (pH 8.0), 0.1% xylene cyanol, and 0.1% bromophenol blue], heated (90 °C, 2 min), cooled (0 °C, 5 min), and analyzed by polyacrylamide gel electrophoresis (PAGE) (20% denaturing) using a Storm 840 phosphorimager and Imagequant TL software.
General Procedure for DEPC Treatment of 1–3 and 8–12
The appropriately 32P-labeled substrate (1–3) (5000–15000 cpm) was reacted (30 μL) with 1.04 M DEPC in PBS buffer [10 mM sodium phosphate (pH 7.2) and 100 mM NaCl] for 10 min at room temperature. The reaction tubes were briefly vortexed every 2 min during the reaction. Following incubation with DEPC, the samples were precipitated with 5 M NH4OAc (5.2 μL), 1 μg/μL calf thymus DNA (5 μL), water (10 μL), and cold ethanol (155 μL). The precipitated samples were treated with 1 M piperidine (50 μL) for 20 min at 90 °C and dried in vacuo. The samples were resuspended in water (50 μL) and again dried in vacuo. The DNA was resuspended in formamide loading buffer [10 μL, 50% formamide, 5 mM EDTA (pH 8.0), 0.1% xylene cyanol, and 0.1% bromophenol blue], heated (90 °C, 2 min), cooled (0 °C, 5 min), and analyzed by PAGE (20% denaturing) using a Storm 840 phosphorimager and ImageQuant TL software. Reactions with substrates 8–12 were conducted as described above except the concentration of DEPC was 1.75 M and the total volume of the reaction was 35.5 μL.
General Procedure for KMnO4 Treatment of 1–3 and 8–12
The appropriately 32P-labeled substrate (1–3) (5000–15000 cpm) was reacted (20 μL) with 0.3 mM KMnO4 in PBS buffer [10 mM sodium phosphate (pH 7.2) and 100 mM NaCl] for 10 min at room temperature. The reaction tubes were briefly vortexed every 2 min during the reaction. Following incubation with KMnO4, the reactions were quenched by the addition of allyl alcohol (1 μL) and vortexing. The samples were precipitated with 5 M NH4OAc (5.2 μL), 1 μg/μL calf thymus DNA (5 μL), water (20.5 μL), and cold ethanol (155 μL). The precipitated samples were treated with 1 M piperidine (50 μL) for 20 min at 90 °C and dried in vacuo. The samples were resuspended in water (50 μL) and again dried in vacuo. The samples were resuspended in formamide loading buffer [10 μL, 50% formamide, 5 mM EDTA (pH 8.0), 0.1% xylene cyanol, and 0.1% bromophenol blue], heated (90 °C, 2 min), cooled (0 °C, 5 min), and analyzed by PAGE (20% denaturing) using a Storm 840 phosphorimager and Imagequant TL software. Reactions with substrates 8–12 were conducted as described above except the concentration of KMnO4 was 0.6 mM.
General Procedure for DMS Treatment of 1–3
The appropriately 32P-labeled substrate (1–3) (5000–15000 cpm) was reacted (20 μL) with 25 mM DMS (diluted from a 250 nM DMS solution in ethanol) in PBS buffer [10 mM sodium phosphate (pH 7.2) and 100 mM NaCl] for 12 min at room temperature. The reaction tubes were briefly vortexed every 2 min during the reaction. Following incubation, the samples were precipitated with 5 M NH4OAc (5.2 μL), 1 μg/μL calf thymus DNA (5 μL), water (20.5 μL), and cold ethanol (155 μL). The precipitated samples were treated with 1 M piperidine (50 μL) for 20 min at 90 °C and dried in vacuo. The samples were resuspended in water (50 μL) and again dried in vacuo. The samples were resuspended in formamide loading buffer [10 μL, 50% formamide, 5 mM EDTA (pH 8.0), 0.1% xylene cyanol, and 0.1% bromophenol blue], heated (90 °C, 2 min), cooled (0 °C, 5 min), and analyzed by PAGE (20% denaturing) using a Storm 840 phosphorimager and Imagequant TL software.
General Procedure for Hydroxylamine Treatment of 1–3
A 2 M stock solution (10 mL) of hydroxylamine (pH 6.0) was prepared by dissolving hydroxylamine hydrochloride (1.34 g) in autoclaved water (5 mL). Diethylamine (approximately 1.2 mL) was added to adjust the pH to 6.0. Autoclaved water was added to this solution to achieve a final volume of 10 mL. The stock solution was diluted with autoclaved water to make a 1.04 M hydroxylamine solution. The 2 M stock solution can be stored in a –20 °C freezer and used for up to 1 week. The appropriately 32P-labeled substrate (1–3) (5000–15000 cpm) was reacted (26 μL) with 0.80 M hydroxylamine (pH 6.0) (diluted from a 1.04 M stock solution) in PBS buffer [10 mM sodium phosphate (pH 7.2) and 100 mM NaCl] for 10 min at room temperature. The reaction tubes were briefly vortexed every 2 min during the reaction. Following incubation, the samples were precipitated with 5 M NH4OAc (5.2 μL), 1 μg/μL calf thymus DNA (5 μL), water (20.5 μL), and cold ethanol (155 μL). The precipitated samples were treated with 1 M piperidine (50 μL) for 20 min at 90 °C and dried in vacuo. The samples were resuspended in water (50 μL) and again dried in vacuo. The samples were resuspended in formamide loading buffer [10 μL, 50% formamide, 5 mM EDTA (pH 8.0), 0.1% xylene cyanol, and 0.1% bromophenol blue], heated (90 °C, 2 min), cooled (0 °C, 5 min), and analyzed by PAGE (20% denaturing) using a Storm 840 phosphorimager and Imagequant TL software.
UV Melting Temperature Experiments
UV melting temperature experiments were conducted in quartz cells with a path length of 1 cm. The samples were prepared by mixing oligonucleotides (1 μM), PIPES (pH 7.0, 10 mM), NaCl (100 mM), and MgCl2 (10 mM). For substrates 4–7, the temperature was held at 13 °C for 2 min and then increased linearly to 85 °C at a rate of 1.0 °C/min while the absorbance was monitored at 260 nm. The cuvette rack was constantly flushed with N2 flow to avoid moisture condensation in the lower temperature range (<17 °C). For substrates 13 and 14, the temperature was varied from 20 to 85 °C. For each experiment, the absorbance (260 nm) of a solution containing the same buffer but without the oligonucleotides was recorded as a function of temperature and subtracted from the absorbance (260 nm) of the oligonucleotide solutions. The data were processed using Origin 6.1 by plotting the absorbance versus temperature and fitting the curve to the equation y = A2 + (A1 – A2)/{1 + exp[(x – x0)/dx]}, where y is the absorbance, A1 is the initial absorbance (left horizontal asymptote), A2 is the final absorbance (right horizontal asymptote), x0 is the center (point of inflection or Tm), and dx is the change in the X value corresponding to the most significant change in the Y value.
RESULTS
Substrate Design and Preparation
Substrates containing ICL analogues of C4-AP and DOB (Figure 3) were synthesized by a combination of solid-phase oligonucleotide synthesis and enzymatic ligation, as previously described.16,18 Substrates containing the cross-link with the abasic sites opposite the 3′-adjacent dT (“observed”) and with the cross-linked dA opposite the abasic site analogues (“unobserved”) were prepared. The “observed” ICLs correspond to those that are (preferentially) formed by C4-AP and DOB (Scheme 2). Specific ligation protocols for previously unreported substrates are described in the Supporting Information. The chemo-enzymatic approach provided a convenient method for controlling which strand and terminus in the cross-linked substrate was labeled with 32P. Labeling each terminus was desirable because analysis of the reactivity of nucleotides flanking the ICLs proceeded with the highest resolution when the nucleotides were between the radiolabel and the cross-link. [Please note that the strand labeled in each substrate is termed “t” for the strand containing the abasic site analogue (top strand in Figure 3) or “b” for the strand containing the dA that is involved in the cross-link (bottom strand in Figure 3).] This approach worked as planned for preparing the desired 32P-labeled cross-linked substrates containing the stabilized DOB ICL [X–Y (Scheme 1 and Figure 3)]. However, slight impurities were detected when 5′-32P-b-8 and 5′-32P-b-9 were prepared. Consequently, substrates 11 and 12 were employed for examining the reactivity of the 5′-bottom portion of ICLs containing the C4-AP analogues. Although the overall lengths of substrates 11 and 12 are greater than the lengths of the others (8 and 9) containing the C4-AP ICL analogue (W–V), the flanking sequences over which reactivity was examined (four or five nucleotides) were identical. In addition, studies conducted on DOB ICLs revealed that the reaction of 2′-deoxycytidines with hydroxylamine was significantly less precise in our hands than that of other chemical probing reagents. Consequently, C4-AP cross-link substrates were designed to be devoid of G·C base pairs near the cross-link.
General Aspects of Chemical Reactivity Studies
Reactivities at all sites were calibrated in two ways. Reactions were conducted to low conversion (single-hit conditions), and ratios of cleavage at a nucleotide of interest proximal to the ICL relative to a distal position were recorded. In addition, reactivity at a given position within an ICL was calibrated by comparison to that of the other ICL and a control substrate lacking a cross-link. Reactions at dA, dG, and T were examined using diethyl pyrocarbonate (DEPC), dimethyl sulfate (DMS), and potassium permanganate (KMnO4), respectively. Reactivity at dC using hydroxylamine was used sparingly because of unsatisfactory precision in many instances. (Please note that for the sake of convenience the deoxy descriptor is dropped in the discussion below.) Similarly, hydroxyl radical cleavage was not very useful for detecting differences in structure, and results using this reagent are reported only for substrates that provide information about the reactivity of the abasic site analogues themselves.
Chemical Reactivity of the DOB ICL Analogue
Reactivity of 5′-32P-b-1–3 was examined using DEPC and DMS (Figure 4). Reaction at A14 of the unobserved ICL (2) with DEPC (Figure 4C) was significantly greater than in the duplex control, indicating a high degree of distortion at this position. In contrast, reaction at A14 in the observed ICL (1), which is the cross-linked purine, was negligible. This suggests that the abasic site analogue bonded to the N6 position hinders DEPC reaction at the N7 position. Per below, this was a general observation for the cross-linked positions in all of the substrates. Reaction at G13 in the unobserved ICL (2) was still greater than that at the corresponding nucleotide in 1, despite the greater distance from the cross-linking position (Figure 4B). However, there was no enhanced reactivity in either cross-linked substrate compared to that of the control duplex (3) at A11, indicating that any structural distortion induced by the ICLs dissipated at that position (Figure 4A).
Figure 4.
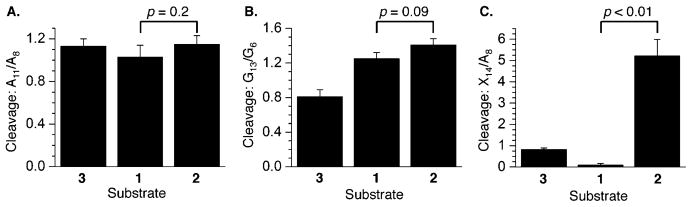
Chemical reactivity of DOB ICL analogues in 5′-32P-b-1–3 with (A) DEPC at A11 relative to A8, (B) DMS at G13 relative to G6, and (C) DEPC at X14 relative to A8 (X = A in 2 and 3, and X = cross-linked A in 1). P values were calculated using the Student’s t test (four replicates).
As mentioned above, reaction of 3′-32P-b-1–3 (Figure 5) with DEPC indicated that the 2′-deoxyadenosine involved in the unobserved ICL (A15 in 2) was inaccessible. However, the 2′-deoxyadenosine adjacent to the cross-linked purine nucleotide (A15) in observed ICL 1 was ~2-fold more reactive than the comparable nucleotide in duplex DNA (3). At the same time, the 2′-deoxyadenosine adjacent to the cross-linked purine nucleotide (A16) in the unobserved cross-linked substrate (2) was more reactive than the comparable position (A15) in the observed ICL (1) or A16 in the duplex (3). The greater reactivity at A16 in unobserved ICL 2 compared to that of A15 of 1, both of which are adjacent to the cross-linked dA, is even more striking when one considers that the latter is opposite the abasic site, which provides no opportunity for Watson–Crick base pairing. The difference in reactivity between ICLs at A17 was negligible, but both were more susceptible to DEPC than the control duplex (3). Enhanced reactivity at these positions could be due to fraying of the base paired complement in 1 and 2. Fraying of the noncovalently bound strand may also contribute to the enhanced reactivity at T44 and T45 with KMnO4 in cross-linked substrates 1 and 2 (Figure 6). This is particularly apparent at the 3′-terminus of the strand (T45) where reactivity in either ICL is >6-fold greater than in control duplex 3. However, the reactivity in the unobserved ICL (2) remains >6 times higher when the nucleotide (T44) is one nucleotide further removed from the ICL. In comparison, reactivity at T44 in 5′-32P-t-1 is reduced to ~3-fold greater than in duplex 3.
Figure 5.
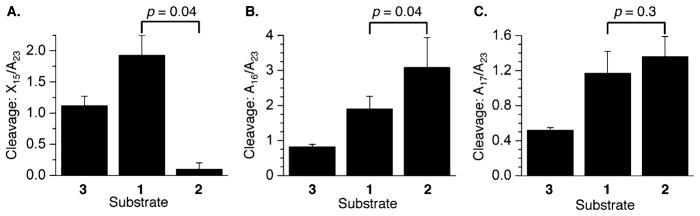
Chemical reactivity of DOB ICL analogues in 3′-32P-b-1–3 with DEPC at (A) X15 relative to A23 (X = A in 1 and 3, and X = cross-linked A in 2), (B) A16 relative to A23, and (C) A17 relative to A23. P values were calculated using the Student’s t test (four replicates).
Figure 6.
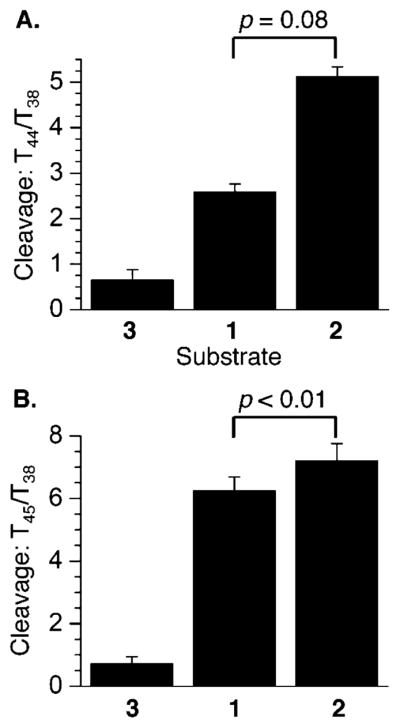
Chemical reactivity of DOB ICL analogues in 5′-32P-t-1–3 with KMnO4 at (A) T44 relative to T38 and (B) T45 relative to T38. P values were calculated using the Student’s t test (four replicates).
In contrast, potassium permanganate reacts much more readily at T47 in the observed ICL (3′-32P-t-1) than it does in the duplex control or the unobserved ICL (3′-32P-t-2). However, T47 is still >6-fold reactive in 2 than it is in duplex 3 (Figure 7B). Evaluation of reactivity at T47 was unusual in that it was calibrated against cleavage at a different nucleotide (G54). This was because cleavage at other thymidines distal from the cross-link within the 3′-arm of the top strand in 1–3 was too low to reliably measure. The abasic site (X46) also reacts more readily with hydroxyl radical in the unobserved cross-link (3′-32P-t-2) than it does in the analogue of the observed molecule (Figure 7A). However, both abasic sites react more readily with hydroxyl radical than does the control duplex. Although reaction at C48 is indistinguishable in 3′-32P-t-1–3 due to the larger error (Figure 7C), the unobserved ICL (2) is more susceptible to DMS treatment at G49 (Figure 7D) than either of the other substrates. Reaction of T50 is indistinguishable in the three substrates (data not shown), indicating that any structural alteration in the ICLs has dissipated by that position.
Figure 7.

Chemical reactivity of DOB ICL analogues in 3′-32P-t-1–3 at (A) X46 relative to G54 (X = T in 3, and X = cross-linked abasic site in 1 and 2) with hydroxyl radical, (B) T47 relative to G54 with KMnO4, (C) C48 relative to C55 with hydroxylamine, and (D) G49 relative to G54 with DMS. P values were calculated using the Student’s t test (four replicates).
Chemical Reactivity of the C4-AP ICL Analogue
As discussed above, reactivity on the 5′-arm in the bottom strand of the cross-linked C4-AP analogue was examined (Figure 8) using longer substrates (11 and 12) but compared against a shorter duplex (10) containing the same sequence in the relevant region. The reactivity of the thymidines (KMnO4) and A’s (DEPC) bonded via the 5′-phosphate of the cross-linked A was quantified. The cross-linked A (A29) in the analogue of the observed C4-AP ICL (11) is protected against DEPC, whereas the respective nucleotide in the analogue of the unobserved cross-link (12) is adjacent to the cross-linked position and is very reactive (Figure 8C). The difference in reactivity between the observed (11) and unobserved (12) ICL analogues rapidly dissipates. The unobserved ICL is only slightly more reactive at T28 (Figure 8B), and the difference is negligible just two nucleotides further removed from the cross-linked position [A27 (Figure 8A)].
Figure 8.
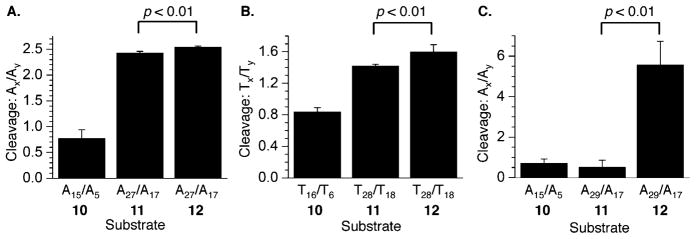
Chemical reactivity of C4-AP ICL analogues in 5′-32P-b-10–12 upon (A) reaction with DEPC, (B) reaction with KMnO4, and (C) reaction with DEPC. P values were calculated using the Student’s t test (four replicates).
The nucleotides bonded to the 3′-phosphate of the cross-linked dA [A18 in 8 (Figure 9A) and T19 in 9 (Figure 9B)] were significantly more reactive than the control duplex (10). In each instance, these are the positions adjacent to the cross-linked dA. However, the reactivity at T19 in the observed C4-AP ICL analogue (8) was anomalously high (Figure 9B), and far greater than that of the 5′-adjacent A18, which is opposite the cross-linked abasic site. Reaction at T19 of the observed ICL is also significantly greater than at T19 in the unobserved C4-AP cross-link product. The reason for the hyperreactivity of T19 in 3′-32P-b-8 is unknown. More enhanced reactivity in the unobserved ICL (9) relative to the analogue of the observed ICL (8) is restored upon moving one nucleotide farther from the ICL [A20 (Figure 9C)], but the reactivities of the cross-linked substrates are indistinguishable at T21 (data are not shown).
Figure 9.
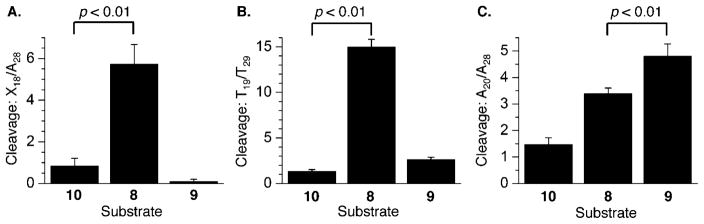
Chemical reactivity of C4-AP ICL analogues in 3′-32P-b-8–10 at (A) X18 relative to A28 (X = A in 8 and 10, and X = cross-linked A in 9) with DEPC, (B) T19 relative to T29 with KMnO4, and (C) A20 relative to A28 with DEPC. P values were calculated using the Student’s t test (four replicates).
The nucleotides in the 5′-arm of the top strands in the C4-AP ICL analogues are consistently more reactive in the unobserved (5′-32P-t-9) cross-link substrate than in 8, which is designed to model the observed cross-link (Figure 10). The differential reactivity was greatest at the 5′-adjacent nucleotide to the abasic site [A54 (Figure 10C)], where the extent of DEPC-induced cleavage was more than 6 times greater in 9 than in the control duplex. With the exception of reaction with hydroxyl radical (Figure 11A), the top strand in the unobserved analogue (9) was also consistently more reactive than 8 on the 3′-side of the cross-link (Figure 11). However, the differences in cleavage in 3′-32P-t-8 and 3′-32P-t-9 are considerably smaller, and reactions of the abasic site analogues (Figure 11A) are indistinguishable from one another, as well as with respect to the control duplex (3′-32P-t-10). Overall, the C4-AP ICL analogues are significantly less reactive than the respective DOB compounds (Figure 7A) with hydroxyl radical.
Figure 10.
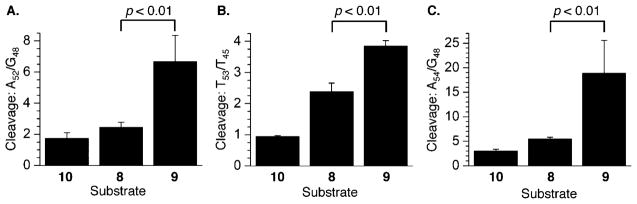
Chemical reactivity of C4-AP ICL analogues in 5′-32P-t-8–10 at (A) A52 relative to G48 with DEPC, (B) T53 relative to T45 with KMnO4, and (C) A54 relative to G48 with DEPC. P values were calculated using the Student’s t test (four replicates).
Figure 11.

Chemical reactivity of C4-AP ICL analogues in 3′-32P-t-8–10 at (A) V55 relative to A67 (X = T in 10, and X = cross-linked abasic site in 8 and 9) with hydroxyl radical, (B) T56 relative to T68 with KMnO4, (C) A57 relative to A67 with DEPC, and (D) T58 relative to T68 with KMnO4. P values were calculated using the Student’s t test (four replicates).
UV Melting Comparison of ICL Analogues
The UV melting temperatures of the observed and unobserved cross-links were measured at 1 μM at pH 7.0 in PIPES (10 mM), MgCl2 (10 mM), and NaCl (100 mM). Two transitions were observed for the DOB ICL analogue substrates [4 and 5 (Figure 12A)]. The lower-temperature transition (TM, 31.0 ± 0.3 °C for 4 and 28.1 ± 0.6 °C for 5) corresponds to melting of the noncovalently bound strand. As expected, the presence of this strand does not affect the melting of the cross-linked strands [4 and 5 (Figure 12A) and 6 and 7 (Supporting Information and Table 1)]. Importantly, the melting of the cross-link required a temperature (~4 °C) for the observed ICL (4) considerably higher than that of the cross-link in which the dA and abasic site are opposite one another (unobserved, 5).
Figure 12.

UV melt of (A) DOB ICL analogues (4 and 5) and (B) C4-AP ICL analogues (13 and 14).
Table 1.
UV Melting Temperatures of ICL Analogues
| ICL analogue | ICL TM (°C)a |
|---|---|
| DOB observed (4) | 69.8 ± 0.8 |
| DOB unobserved (5) | 65.3 ± 0.1 |
| C4-AP observed (13) | 59.1 ± 0.2 |
| C4-AP unobserved (14) | 57.4 ± 0.4 |
Measurements are the average ± the standard deviation of three independent measurements.
A similar trend was observed when the C4-AP ICL analogues (13 and 14) were examined (Figure 12B and Table 1). The substrate containing the cross-linked components opposite one another (14), which represents the orientation that is not observed experimentally, melted at a temperature ~1.7 °C lower than that for the orientation in which the cross-linked dA is opposite the thymidine that is bonded to the 3′-phosphate of the abasic site (13). Although the absolute temperatures for melting of the C4-AP analogue ICLs are lower than those containing the DOB cross-link, the difference in TM between the observed (13) and unobserved (14) is smaller in the former. This suggests that the C4-AP ICL analogue imparts a smaller perturbation on the structure and stability of the DNA than the DOB cross-link analogue.
DISCUSSION
Spontaneous interstrand cross-link formation by abasic sites, which require NER, increases the potential biochemical impact of these DNA lesions.36 Reversible cross-linking to a dA opposite a 3′-adjacent thymidine by C4-AP and DOB oxidized abasic sites occurs with very high selectivity over other sequences (Scheme 2).12–15 Inspection of molecular models (Figure 2) does not provide an obvious rationalization for this selectivity. Consequently, UV melting experiments and chemical reactivity studies were conducted with stabilized analogues of the ICLs to discern a difference in the stabilities of the isomeric cross-linked molecules.
In general, the cross-linked 2′-deoxyadenosine was unreactive with DEPC, and certainly less reactive than a comparable nucleotide in duplex DNA. One possible explanation for this is that the cross-linked abasic site model hinders approach to the N7 position of the cross-linked dA and/or reduces its nucleophilicity. Another general observation is that the “top” strands in the cross-linked substrates, which contain the abasic site analogues, tend to be more reactive than the “bottom” strands containing the cross-linked dAs. This suggests significant solvent exposure and structural disruption in the cross-linked strands containing the abasic site analogues.
Although there are some exceptions, a comparison of the reactivity of the “observed” (e.g., 1, 8, and 11) and “unobserved” (e.g., 2, 8, and 12) stabilized cross-link substrates (see also Scheme 2) at individual nucleotides showed that the latter were consistently more reactive. This is consistent with greater distortion of the unobserved cross-links than of the analogues of observed ICLs. These inferential observations were corroborated by UV melting measurements (Table 1) of stabilized analogues of C4-AP (13 and 14) and DOB (4 and 5) ICLs. Substrates containing cross-links between opposing nucleotides melted at temperatures 1.7–4.5 °C lower than those of the analogues of their “observed” counterparts. Overall, UV melting temperatures and chemical reactivity of stabilized analogues of these ICLs (Scheme 1) revealed that the preferred cross-links are more stable and their structures less perturbed than those of comparable unobserved cross-linked substrates, providing a rationale for their preferential formation.
A comparison of the reactivity in the respective “observed” ICL substrates relative to the corresponding duplexes lacking the cross-link indicates that the DOB analogue induces a greater overall enhancement (Figure 13A) than does the C4-AP-like lesion (Figure 13B). The anomalous reactivity at T19 in 3′-32P-b-8 is the one outlier. This is especially true upon comparison of the reactivity of the strands containing the abasic site analogues (“top” strands). In particular, the abasic site analogue in the DOB substrate (Figure 7A) is considerably more reactive than the respective C4-AP-type abasic sites (Figure 11A) with hydroxyl radical. In addition, the thymidine opposite the cross-linked dA [X14 (Figure 13A)] and the first two nucleotides in the flanking strand (T45 and T44) are far more reactive than in the duplex. The greater reactivity at T44 and T45 in the DOB ICL may be attributed to their higher level of breathing because they are in effect at the 3′-terminus of a duplex. Overall, the greater reactivity suggests that cross-links derived from DOB perturb the structure of DNA more than does C4-AP. This is consistent with the repair incision of DNA containing the DOB ICL being more facile than that involving C4-AP by UvrABC, which recognizes macroscopic distortions in the nucleic acid substrate.18–22
Figure 13.

Nucleotide by nucleotide comparison of ICL reactivity relative to analogous duplex: (A) DOB-containing ICL and (B) C4-AP-containing ICL.
Supplementary Material
Acknowledgments
Funding
We are grateful for support of this research by the National Institute of General Medical Sciences (GM-063028).
ABBREVIATIONS
- AP
apurinic/apyrimidinic site
- C4-AP
C4′-oxidized abasic site
- BER
base excision repair
- DOB
5′-(2-phosphoryl-1,4-dioxobutane)
- dsb
double-strand break
- ICL
interstrand cross-link
- NER
nucleotide excision repair
- DMS
dimethyl sulfate
- DEPC
diethyl pyrocarbonate
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.5b00860.
Procedures for preparing ICL substrates, UV melting plots of analogues of 6 and 7 lacking the flanking strand, ESI-MS of cross-linked oligonucleotides, and sample autoradiograms of chemical probing reactions (PDF)
References
- 1.Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozekov ID, Turesky RJ, Alas GR, Harris CM, Harris TM, Rizzo CJ. Formation of deoxyguanosine cross-links from calf thymus DNA treated with acrolein and 4-hydroxy-2-nonenal. Chem Res Toxicol. 2010;23:1701–1713. doi: 10.1021/tx100179g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stone MP, Cho Y-J, Huang H, Kim H-Y, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008;41:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schärer OD. DNA interstrand crosslinks: Natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 6.Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harbor Perspect Biol. 2013;5:a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price NE, Catalano MJ, Liu S, Wang Y, Gates KS. Chemical and structural characterization of interstrand cross-links formed between abasic sites and adenine residues in duplex DNA. Nucleic Acids Res. 2015;43:3434–3441. doi: 10.1093/nar/gkv174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catalano MJ, Liu S, Andersen N, Yang Z, Johnson KM, Price NE, Wang Y, Gates KS. Chemical structure and properties of interstrand cross-links formed by reaction of guanine residues with abasic sites in duplex DNA. J Am Chem Soc. 2015;137:3933–3945. doi: 10.1021/jacs.5b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS. Interstrand DNA-DNA crosslink formation between adenine residues and abasic sites in duplex DNA. J Am Chem Soc. 2014;136:3483–3490. doi: 10.1021/ja410969x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson KM, Price NE, Wang J, Fekry MI, Dutta S, Seiner DR, Wang Y, Gates KS. On the formation and properties of interstrand DNA,DNA cross-links forged by reaction of an abasic site with the opposing guanine residue of 5′-CAp sequences in duplex DNA. J Am Chem Soc. 2013;135:1015–1025. doi: 10.1021/ja308119q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regulus P, Duroux B, Bayle P-A, Favier A, Cadet J, Ravanat J-L. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc Natl Acad Sci U S A. 2007;104:14032–14037. doi: 10.1073/pnas.0706044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sczepanski JT, Jacobs AC, Greenberg MM. Self-promoted DNA interstrand cross-link formation by an abasic site. J Am Chem Soc. 2008;130:9646–9647. doi: 10.1021/ja8030642. [DOI] [PubMed] [Google Scholar]
- 13.Sczepanski JT, Jacobs AC, Majumdar A, Greenberg MM. Scope and mechanism of interstrand cross-link formation by the C4′-oxidized abasic site. J Am Chem Soc. 2009;131:11132–11139. doi: 10.1021/ja903404v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sczepanski JT, Hiemstra CN, Greenberg MM. Probing DNA interstrand cross-link formation by an oxidized abasic site using nonnative nucleotides. Bioorg Med Chem. 2011;19:5788–5793. doi: 10.1016/j.bmc.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan L, Greenberg MM. DNA interstrand cross-link formation by the 1,4-dioxobutane abasic lesion. J Am Chem Soc. 2009;131:15225–15231. doi: 10.1021/ja9061695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S, Greenberg MM. Synthesis of cross-linked DNA containing oxidized abasic site analogues. J Org Chem. 2014;79:5948–5957. doi: 10.1021/jo500944g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sczepanski JT, Jacobs AC, Van Houten B, Greenberg MM. Double strand break formation during nucleotide excision repair of a DNA interstrand cross-link. Biochemistry. 2009;48:7565–7567. doi: 10.1021/bi901006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh S, Greenberg MM. Nucleotide excision repair of chemically stabilized analogues of DNA interstrand cross-links produced from oxidized abasic sites. Biochemistry. 2014;53:5958–5965. doi: 10.1021/bi500914d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspect Biol. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pakotiprapha D, Samuels M, Shen K, Hu JH, Jeruzalmi D. Structure and mechanism of the UvrA,UvrB DNA damage sensor. Nat Struct Mol Biol. 2012;19:291–298. doi: 10.1038/nsmb.2240. [DOI] [PubMed] [Google Scholar]
- 21.Pakotiprapha D, Liu Y, Verdine GL, Jeruzalmi D. A structural model for the damage-sensing complex in bacterial nucleotide exision repair. J Biol Chem. 2009;284:12837–12844. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, Verdine GL. Crystal structure of bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yakubovskaya MG, Belyakova AA, Gasanova VK, Belitsky GA, Dolinnaya NG. Comparative reactivity of mismatched and unpaired bases in relation to their type and surroundings. Chemical cleavage of DNA mismatches in mutation detection. Biochimie. 2010;92:762–771. doi: 10.1016/j.biochi.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Jarem DA, Huckaby LV, Delaney S. AGG interruptions in (CGG)(n) DNA repeat tracts modulate the structure and thermodynamics of non-B conformations in vitro. Biochemistry. 2010;49:6826–6837. doi: 10.1021/bi1007782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun D-A, Hurley LH. Biochemical techniques for the characterization of G-quadruplex structures: EMSA, DMA footprinting, and DNA polymerase stop assay. Methods Mol Biol. 2010;608:65–79. doi: 10.1007/978-1-59745-363-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–513. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 28.Ambrose BJB, Pless RC. DNA sequencing: Chemical methods. Methods Enzymol. 1987;152:522–539. doi: 10.1016/0076-6879(87)52059-6. [DOI] [PubMed] [Google Scholar]
- 29.Fox KR, Grigg GW. Diethylpyrocarbonate and permanganate provide evidence for an unusual DNA conformation induced by binding of the antitumor antibiotics bleomycin and phleomycin. Nucleic Acids Res. 1988;16:2063–2075. doi: 10.1093/nar/16.5.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shcherbakova I, Mitra S. Hydroxyl-radical footprinting to probe equilibrium changes in RNA tertiary structure. Methods Enzymol. 2009;468:31–46. doi: 10.1016/S0076-6879(09)68002-2. [DOI] [PubMed] [Google Scholar]
- 31.Price MA, Tullius TD. Using hydroxyl radical to probe DNA structure. Methods Enzymol. 1992;212:194–219. doi: 10.1016/0076-6879(92)12013-g. [DOI] [PubMed] [Google Scholar]
- 32.Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Thermodynamic consequences of an abasic lesion in duplex DNA are strongly dependent on base sequence. Biochemistry. 1998;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy JG, Williams LD, Rich A. Chemical reactivity of potassium permanganate and diethyl pyrocarbonate with B DNA: Specific reactivity with short A-tracts. Biochemistry. 1990;29:6071–6081. doi: 10.1021/bi00477a027. [DOI] [PubMed] [Google Scholar]
- 34.Bishop EP, Rohs R, Parker SCJ, West SM, Liu P, Mann RS, Honig B, Tullius TD. A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem Biol. 2011;6:1314–1320. doi: 10.1021/cb200155t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tullius TD, Greenbaum JA. Mapping nucleic acid structure by hydroxyl radical cleavage. Curr Opin Chem Biol. 2005;9:127–134. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg MM. Abasic and oxidized abasic site reactivity in DNA: Enzyme inhibition, crosslinking, and nucleosome catalyzed reactions. Acc Chem Res. 2014;47:646–655. doi: 10.1021/ar400229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.