

Background
Genetic association studies of transplantation outcomes have been hampered by small samples and highly complex multifactorial phenotypes, hindering investigations of the genetic architecture of a range of comorbidities which significantly impact graft and recipient life expectancy. We describe here the rationale and design of the International Genetics & Translational Research in Transplantation Network. The network comprises 22 studies to date, including 16494 transplant recipients and 11669 donors, of whom more than 5000 are of non-European ancestry, all of whom have existing genomewide genotype data sets.
Methods
We describe the rich genetic and phenotypic information available in this consortium comprising heart, kidney, liver, and lung transplant cohorts.
Results
We demonstrate significant power in International Genetics & Translational Research in Transplantation Network to detect main effect association signals across regions such as the MHC region as well as genomewide for transplant outcomes that span all solid organs, such as graft survival, acute rejection, new onset of diabetes after transplantation, and for delayed graft function in kidney only.
Conclusions
This consortium is designed and statistically powered to deliver pioneering insights into the genetic architecture of transplant-related outcomes across a range of different solid-organ transplant studies. The study design allows a spectrum of analyses to be performed including recipient-only analyses, donor-recipient HLA mismatches with focus on loss-of-function variants and nonsynonymous single nucleotide polymorphisms.
iGeneTRAiN is a consortium that has genome-wide genotype datasets. These genomic data allows robust statistically analysis of genetic associations that impact graft and patients variables such as, such as: graft survival, acute rejection, new onset of diabetes after transplantation, and delayed graft kidney function. Supplemental digital content is available in the text.
Recent advances in genomics, including genomewide association studies (GWAS), second-generation sequencing (SGS), and their application within appropriately powered and carefully phenotyped studies, have yielded meaningful insights into the understanding of the molecular basis of a multitude of common and rare diseases.1-3 The GWAS and SGS studies have identified genes inactivated by homozygous loss-of-function (LoF) mutations,4 including both single-nucleotide variants and large homozygous deletion copy number variants (hdCNVs) which can span exons or even entire genes.5-7 Such LoFs have shown clinical importance in the pathophysiology of graft-versus-host disease in hematopoietic cell transplantation (Tx) studies.7 Large-scale SGS studies also show gene-coding regions that are unique population-specific sequences,8 and such population-specific differences may underpin allogenicity in ethnically diverse donor-recipient (D-R) transplant pairs independent of HLA matching.
Association of transplant outcomes and polymorphisms in HLA and natural killer (NK) cell immunoglobulin-like receptor (KIR) regions are well established9-11 HLA Class I molecules are recognized to act as ligands for KIR on NK cells. The KIR plays essential roles in regulating the ability of NK cells to sense and respond to HLA Class I surface expression, which have been shown to be important in allorecognition.12 Many unique KIR haplotypes have been identified, and these are unevenly distributed across human populations in a manner similar to HLA.13 In 10 years of follow-up, 18% of deceased donor kidney graft failures were attributable to HLA factors, whereas more than double that proportion (38% of failures) were due to immunological reactions against non-HLA factors.14 Incorporating our knowledge of known clinical predictors of graft survival and complications, genetic studies could improve our understanding of immunological reactions to both HLA and non-HLA factors.
Next to the identification of immunological factors, genomic studies focusing on a priori gene regions of pharmacological relevance have led to the discovery of polymorphisms underpinning variance in trough blood concentrations of immunosuppressant therapies (ISTs)15-17 Personalizing drug treatment for patients to achieve optimal dosing using predictive panels of pharmacogenomic markers has begun to be realized within general medicine,18 and such pharmacogenomic approaches will undoubtedly promote better outcomes in the post-transplant drug treatment settings by allowing more precise targeting for immunologic tolerance and reduction of IST toxicities and complications.
Although acute rejection (AR), graft survival, and the other posttransplant outcomes have known genetic underpinnings, they are also heavily influenced by nongenetic factors, including ischemia reperfusion injury, recipient waiting list time, noncompliance, and donor organ quality which limit the effect any given genetic variant will have on the identified phenotype. However, such covariates can be integrated into analyses and risk models. The transplant setting could greatly benefit from GWAS and SGS studies for a variety of reasons including discovery of additional HLA and non-HLA D-R genomic incompatibilities that may underpin rejection and insights gained into primary disease recurrence and important comorbidities, such as new onset of diabetes after transplant (NODAT). This is especially important as the majority of recipients require exposure to potent ISTs for the remainder of their lives and as such the identification of gene-drug interactions and development of strategies for avoiding harmful interactions in susceptible individuals is significant. Because transplant phenotypes, such as AR states, are highly complex traits with multifactorial components at the donor and recipient physiological and genomic level, as well as environmental and treatment settings, scientific advances are challenging. Such phenotyping issues further compound GWAS power constraints for discovery (reviewed by Stegall et al19). To date, there have been 2 transplant-related GWAS published in well-phenotyped cohorts,20,21 which showed compelling findings despite modest patient numbers.
The International Genomics & Translational Research in Transplantation Network (iGeneTRAiN) was established to bring together genomic data and well-curated heart, kidney, liver, and lung transplant phenotype data sets. Genome-wide genotype data are available from more than 28000 individuals to date, of whom more than 5000 are of non-European ancestry and afford ample statistical power for meta-analyses of a number of key transplant phenotypes. The initial aims of iGeneTRAiN include the discovery and validation of genomic underpinnings of rejection and Tx complications, with the ultimate goal to translate this information into clinical applications, such as patient-specific IST selection and dosing, better genomic compatibility matching of D-R pairs and improved rejection monitoring. In this paper we present: the study characteristics of the iGeneTRAiN cohorts with existing genotype data; the strategies for harmonization of proposed GWAS using imputation; prioritization and harmonization of the initial phenotypes of interest and analytical strategies for association studies and functional annotation of findings from these studies. iGeneTRAiN provides a required standard framework for the curation of phenotypes, imputation of genotypes and analytical strategies across studies that will yield increased statistical power through aggregation of large-scale data sets compared to traditional smaller single independent studies.
METHODS
iGeneTRAiN Studies
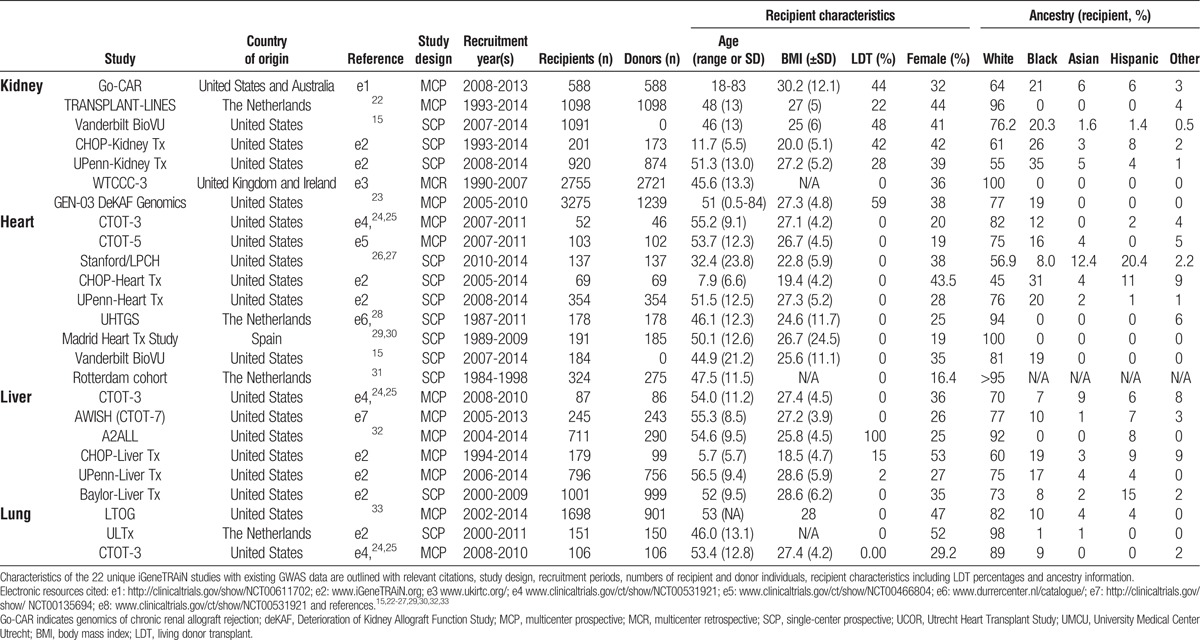
The respective iGeneTRAiN study designs and characteristics for subjects recruited with existing GWAS data are outlined in Table 1. In total, there are over 16494 recipients with 11669 donors across the 4 solid organs. The existing GWAS include participants with a wide geographic representation across the United States, The Netherlands, United Kingdom, Ireland, Spain, and Australia. The majority of transplant recipients in the studies are adults with the exception of 2 cohorts, which comprise approximately 1060 pediatric subjects. The study designs are primarily single or multisite prospective cohorts, and although a small number are retrospective in recruitment approach, most are continuing to accrue longitudinal transplant phenotypes and outcome events. Approximately 18900 samples were subjected to GWAS using a transplant-specific genomewide genotyping array (described below) and approximately 8600 additional samples genotyped using conventional GWAS arrays. The iGeneTRAiN studies range in size from less than 100 D-R pairs to several thousands of recipients. All kidney studies and 2 liver studies have varied proportions of living donor versus deceased donors. Of the 16494 recipients, approximately 81.3%, 12.1%, 1.7%, and 4% are of are of European, African, Asian, and Hispanic ancestry, respectively, with the remainder classified as “other,” and approximately 62% of D-R pairs have conventional 2- or 4-digit HLA typing available. Table S1 (SDC, http://links.lww.com/TP/B191) outlines information regarding specific clinical HLA typing performed, pretransplant anti-HLA immunization status, including peak panel-reactive antibody (PRA), and posttransplant recording of de novo anti-HLA antibodies across each of the 22 studies.
TABLE 1.
Descriptive characteristics of iGeneTRAiN studies contributing genomewide association study data
Phenotypes and Disease Endpoints
Collation and Harmonization of Phenotypes
Four primary phenotypes have been selected for ease of harmonization and of greatest clinical impact: (1) graft survival, (2) AR, (3) NODAT, and (4) delayed graft function (DGF). Length of allograft survival after Tx is arguably the most important clinical outcome, and although many complex factors underpin it, graft survival is a hard outcome which can be unequivocally adjudicated. Acute rejection is an important cause of morbidity and mortality, irrespective of organ, in transplant recipients and has important subtypes: antibody-mediated, cellular, and mixed rejection. Patients who develop AR are at higher risk for chronic graft dysfunction development, which can progress to graft loss.34 Short term (1 year) allograft and patient survival rates are excellent in organs, such as kidney, at 90% to 95%, but long-term outcomes are poor with approximately 50% of kidney allografts failing after 6 to 11 years.35 Kidney transplant patients with chronic graft dysfunction can typically return to dialysis; however, recipients of other organs may die from chronic allograft dysfunction. New onset of diabetes after transplant is a serious complication impacting recipient morbidity with a cumulative incidence of 15% to 30% at 1 year after kidney Tx.36 Cohort studies of solid organ transplant recipients indicate that patients who develop NODAT are at increased risk of fatal and nonfatal CVD events as well as other comorbidities, including infection, graft rejection, and reduced survival.37 Certain ISTs, such as tacrolimus, are considered to be toxic to islet cells, causing NODAT. Delayed graft function is a commonly observed posttransplant adverse event that impacts deceased donor graft survival.38 More complex phenotypes related to pharmacogenomics, disease recurrence, and skin cancer are actively being investigated for the second wave of meta-analyses.
The primary phenotypes described above were collected, arbitrated, and harmonized as follows:
(a) Graft survival is defined as the number of days of functioning organ (inclusive of retransplant metrics and return to dialysis for kidney recipients). We have also measured patient survival which we defined as the number of living days after Tx. Concordance was assessed in each site using internal data coordinating centers (DCC) and/or respective hospital records, and national registry databases.
(b) Acute rejection is defined as biopsy proven rejection (defined by Banff or other international criteria) that was clinically treated with standard IST/steroid regimes. Borderline rejection episodes, mixed, and cellular rejection were systematically recorded in most sites.
(c) NODAT is defined as a new diagnosis of diabetes that occurred after Tx and required continued antidiabetic pharmacotherapy. Where possible, NODAT was strictly defined as a new requirement for oral hypoglycemic agents or insulin for the management of hyperglycemia at a 6-month, 12-month or 2-year period after Tx.
(d) DGF, limited in the first iteration of the iGeneTRAiN program to the kidney cohorts, is defined as the requirement for dialysis within the first 7 days after Tx. The DGF analysis includes the genomics of chronic renal allograft rejection, transplant-LINES Genetics, Vanderbilt, UPenn and The Long-Term Deterioration of Kidney Allograft Function Study Genomics/GEN-03 studies, with incidence rates of 20%, 33%, 9%, 16%, and 9%, respectively.
Analytical Approaches
Association Studies
Our primary analyses will test for genotype differences between recipients and D-R pairs, who encounter the clinical outcomes described above. For graft loss/patient survival, NODAT, and AR, survival analyses will be performed using Cox regression with genotype as the primary exposure, whereas for AR (anytime), NODAT, and DGF, analyses will be performed using logistic regression with genotype as the primary exposure. All models will be adjusted for phenotype-specific covariates (see below). For D-R paired analyses, we will analyze: (1) quantified total number of HLA allele mismatches across the genome; (2) the number and distributions of polymorphisms (and amino acid) discrepancies between D-R pairs; (3) nonsynonymous single-nucleotide polymorphisms; and (4) LoFs, grouped by functional implication. Covariates that will be considered in the regression models include recipient age, sex, body mass index, year of transplantation, diabetes status, primary diagnosis, previous transplant, peak PRA, preemptive Tx, HLA-A, -B, -DR mismatches; donor age, sex, graft status (deceased/living), cause of death (for deceased donors), principal components of ancestry computed from available genotype data (to identify and correct for population substructure), transplant center, cytomegalovirus status, and ischemic time. Where collected for clinical use, PRA measurements will be analyzed for between cohort and between center variation, with the prospective collection made using the Luminex technology where possible. For all D-R pairs, we will compute an HLA-based mismatch score to reflect the genetic compatibility between donor and recipient, which we will test in all models for each D-R pair for association with clinical outcomes.
For the AR analysis, we will examine cause-specific rejection as a categorical variable (antibody-mediated, cellular, and mixed rejection) by organ type and as a collapsed (any rejection) variable, stratified by organ, as well as combined across all organs. We will also perform sensitivity analyses in which borderline rejection, positive protocol, or surveillance biopsies positive for AR and “for-cause” biopsies assessed as nonrejecting are analyzed separately as well as collectively. A separate sensitivity analysis will also be conducted with organ-specific AR assessed alone and subtypes of rejection to determine if any GWAS signals are evident in the combined “all AR types” data sets and/or subsets analyses.
HLA Imputation and Interaction Analyses
The HLA imputation is carried out using SNP2HLA which has been described in detail elsewhere.39 Two-way statistical gene-gene interactions are defined as a departure from additively on a log odds scale, which can be statistically tested for genetic markers a and b using a logistic regression model of the following form: 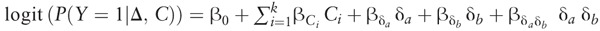 where Y represents a binary phenotype, for example, indicator of transplant rejection; δi is a measure of differential genetic variation between donor and recipient for a specific genetic marker, for example, the difference in the number of risk alleles for an allele (or amino acid derived from SNP2HLA [see below]), and Ci is a clinical covariate measured on the dyad. Given this model, the null hypothesis of no interaction between δa and δb can be specified as
where Y represents a binary phenotype, for example, indicator of transplant rejection; δi is a measure of differential genetic variation between donor and recipient for a specific genetic marker, for example, the difference in the number of risk alleles for an allele (or amino acid derived from SNP2HLA [see below]), and Ci is a clinical covariate measured on the dyad. Given this model, the null hypothesis of no interaction between δa and δb can be specified as 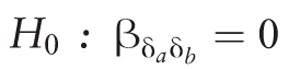 . To improve the statistical power to detect such interactions, the proposed method uses a screening-testing approach inspired by recent gene x environment detection methods.40-42
. To improve the statistical power to detect such interactions, the proposed method uses a screening-testing approach inspired by recent gene x environment detection methods.40-42
Functional Variant Analyses
To identify variants that may be potentially pathogenic, including LoF variants, we will use the Loss of Function Transcript Effect Estimator (http://github.com/konradjk/loftee) which can be used with existing variant effect predictor software packages.43 We will compare the allele frequencies with over 63000 exomes from the Exome Aggregation Consortium to assess rarity of these events, and potential consequences on coding sequence expression. Additionally, we will intersect these variants with functional markers from the ENCODE project44 to assess whether the variants may have an effect at a regulatory level. Finally, we will leverage RNA-Seq data from the Genotype-Tissue Expression project45 to observe whether the variants have an effect on transcription in various tissues.
Organization and Governance
Informed and written consent was obtained independently for each iGeneTRAiN study participant, with appropriate oversight and approvals from respective local institutional review boards/Research Ethics Committees to use either summary-level or anonymized individual-level data.
The iGeneTRAiN network selected 4 solid-organs for collation and harmonization of the appropriate covariates, intermediate phenotypes, and outcomes to an acceptable standardization quality amongst the working groups. Each iGeneTRAiN Phenotyping Committee has been charged with: (a) fully harmonizing the genotype and phenotype data sets across all studies; (b) validating the integrity of these data sets; and (c) annual updates for event accruals and phenotypic measurements (where available). A hematopoietic cell Tx working group is planned for the near future.
Four overarching working groups, whose interests span all solid organ phenotypes, were also formed: (a) HLA working group, (b) genomic and other omics groups (including genomic analysis of SGS/GWAS/other genetic data), (c) pharmacogenomics, and (d) a steering committee comprised of representatives from the working groups (see www.igenetrain.org for committees). The iGeneTRAiN senior investigators, comprising the steering group, have proposed and will carry key projects forward and assess opportunities to include additional collaborators, especially for non-European populations, and to leverage existing phenotype and DNA sets (from organ procurement organizations and/or hospital HLA laboratories). Although each site/study has their own DCC(s), for the purposes of the broader iGeneTRAiN meta-analyses, summary level (or individual level where possible) analyses of GWAS/SGS data for phenotypes of interest are deposited in 3 independent sites, which also coordinate imputation of GWAS data across sites in collaboration with the Center for Systems Genomics, Pennsylvania State University, PA. The overarching iGeneTRAiN DCC is responsible for protocol development and generating statistical designs, including data collection and cleaning and data analysis strategy (in coordination with the iGeneTRAiN Statistical Group). The DCC provides data update collection and data management forms; coordinates statistical analysis across the groups outlined in Table 1; collaborates in manuscript preparation; and provides overall coordination and quality assurance, including coordination of the activities of the data monitoring, the Statistical Group and Phenotyping Committees, and other iGeneTRAiN solid-organ and working groups. In addition, DCC provides continued support of our website (http://www.igenetrain.org) as well as other online resources for the iGeneTRAiN program; continued reporting and management of the awarded funds as they relate to sequencing and other services related to core labs; and the formal release of data to NIH and other sites where appropriate. The iGeneTRAiN Statistical Group is led from University Medical Center Utrecht, The Netherlands and is comprised of at least 1 analyst/statistician and/or principal investigator from each contributing iGeneTRAiN transplant study. This group coordinates the imputation pipeline along with association analyses pipelines, along with Pennsylvania State University, as outlined above.
RESULTS
Tx SNP Array
To maximize power to identify novel loci associated with transplant-related outcomes, we have designed a cost-effective genotyping array to facilitate genomic research studies among the Tx community. The “Tx Array” contains approximately 782,000 genetic variants, and the design was tailored to maximize precise and/or denser coverage in exonic, pharmacogenomic, hdCNV, LoF, MHC, and KIR, CVD/metabolic-related loci while still maintaining strong genomewide content that is compatible with conventional GWAS arrays. The genome-wide coverage was designed to allow accurate imputation of ungenotyped markers using sequencing-based reference panels, such as the 1000 genomes project. We have genotyped 85 HapMap samples (including 8 trios) and show that indeed the genotyping quality is high with a concordance of 99.6%, and ungenotyped SNPs can be imputed with an average accuracy of 96.2%. A more detailed description of the Tx Array can be found in a dedicated design article.46
Genotyping and Imputation
Of the 22 independent genotype data sets, 17 studies were primarily genotyped using the Tx Array (n = 18338 samples were genotyped in a single site). The remaining iGeneTRAiN studies were genotyped across 5 genotyping sites using conventional GWAS arrays (see Table 2).
TABLE 2.
Existing iGeneTRAiN genomics data sets with HLA and new onset of diabetes after transplant and acute rejection outcomes
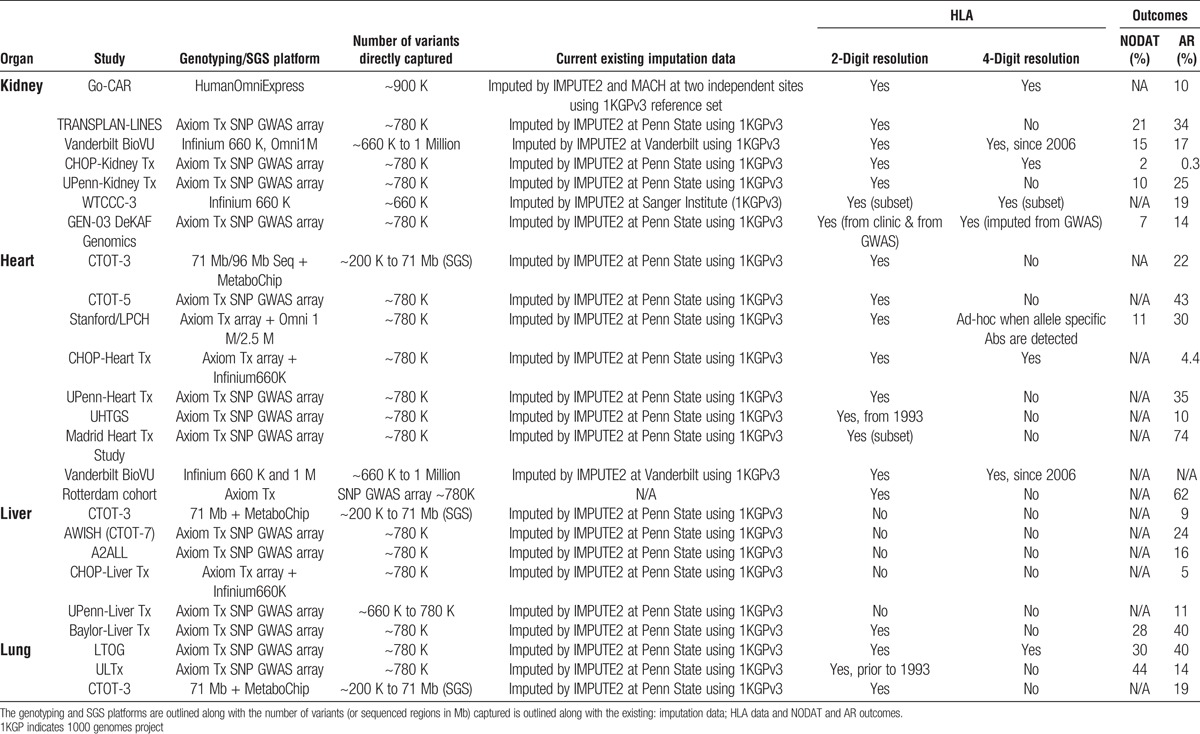
Genotype imputation is the process of inferring unobserved genotypes in a study sample based on the linkage disequilibrium and haplotypes observed in more densely genotyped reference samples of similar genetic background.47 Thoroughly validated and extensively used software packages, including IMPUTE2 and MaCH-Admix, have been used for imputation (see Table 2) of the genomewide content using the following population-based reference data sets: The 1000 genomes project (1KGP), Genome of The Netherlands Consortium,48 and UK10K49 using whole genome sequencing data from 2000 individuals (from 26 different populations), 750 (250 European trios), and 4000 (Europeans), respectively. Comparison of imputation accuracy and metrics from 2 independent pipelines (both using ShapeIT/IMPUTE2 with 1000 genomes project as the reference population) show excellent concordance.46
Because of the highly complex linkage disequilibrium and polymorphic nature of HLA class I/II genes, we will use SNP2HLA, a tool specifically for imputation of HLA Class I/II.39 In addition to standard (typically biallelic) SNP imputation, SNP2HLA also infers multi-allelic markers and amino acids, and most importantly the classical HLA alleles. Testing differences at the aggregate amino acid or HLA allele level is more powerful than testing single SNPs, because multiple SNP markers may have the exact same coding changes or affect the same changes functionally in immune recognition.
We performed a validation of the SNP2HLA imputation pipeline using clinical HLA typing data from European individuals within the Utrecht Heart Transplant Study, using the HLA panel from the TxSNP array platform.46 HLA-A, -B, -C, -DRB1, and -DQB1 were serologically typed between 1987 and 2011 in Utrecht Heart Transplant Study donors and recipients at the University Medical Center Utrecht. We converted the typed broad and split antigens to 2-digit using the Nomenclature for factors of the HLA system, 2010.50 Then, we tested the concordance for each of the alleles between the 2-digit serologically typed and converted alleles and the 2-digit alleles imputed by SNP2HLA.39 In total, 329 samples (donors and recipients combined) passed genotyping QC and were HLA imputed. The HLA alleles were compared in all subjects of which nonmissing data for that allele was available, so for which 2 alleles were serologically measured. In total, between 142 (for HLA C) and 314 (for HLA DRB1) samples were included in each of the comparisons. The imputation accuracies were 96.7%, 95.8%, 89.8%, 95.5%, and 96.1% for HLA-A, -B, -C, DRB1, and DQB1, respectively, as shown in Table S2 (SDC, http://links.lww.com/TP/B191). With the exception of HLA-C, which is modestly lower than the SNP2HLA imputation accuracies within European subjects observed in the SNP2HLA design article, the other HLA Class I/II alleles are imputed to similar accuracies.39
Sample Size and Power
Figure 1 shows our power to detect main effect association signals across a 5-Mb region, such as the MHC. The statistical power calculations based on different MAFs for the first wave of transplant phenotypes of interest: graft survival, AR, NODAT, and DGF (in kidney only) under an additive model in recipients, only assuming approximately 20000 tests for the MHC region. For graft failure, we have very high power to detect a 25% minor allele frequency with a conservatively estimated effect size of 1.2-fold increased risk per allele. Figure 2 shows our power to detect main effect association signals across the entire human genome assuming a similar main effect association signals but using ∼500000 SNP tests. We also have excellent power to detect modest effect sizes across the four initial phenotypes of interest. This indicates the overall iGeneTRAiN data set is well-powered for common genetic variation with modest effect sizes as can be expected in the MHC and across the genome.
FIGURE 1.
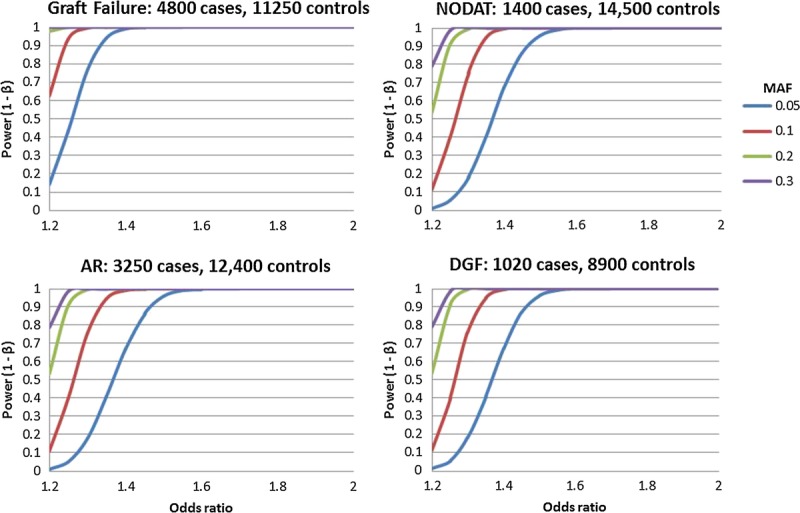
Statistical power calculations to detect main effects for the four main iGeneTRAiN phenotypes across the Major Histocompatibility Complex. Graft Survival in 4800 cases and 11,250 controls (top-left); AR in 3250 cases and 12,400 controls (bottom-left); NODAT in 1400 cases and 14,500 controls (top-right); DGF in 1,020 cases and 8,900 controls (kidney only) (bottom-right). The X-axes shows the OR effect size, and the Y-axes illustrate the statistical power to detect the main effects under different MAFs: 5%, 10%, 20%, and 30% shown in blue, red, green, and purple, respectively. The models are additive model in recipients and assume approximately 20,000 tests (Bonferroni correction 0.05). OR indicates odds ratio; MAFs, minor allele frequencies.
FIGURE 2.
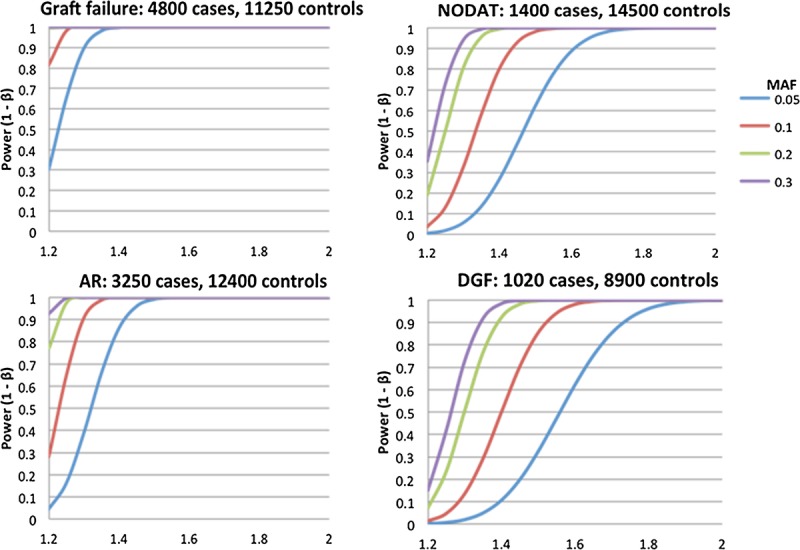
Genome-wide statistical power calculations to detect main effects for the four main iGeneTRAiN phenotypes. Graft Survival in 4800 cases and 11,250 controls (top-left); AR in 3250 cases and 12,400 controls (bottom-left); NODAT in 1400 cases and 14,500 controls (top-right); DGF in 1020 cases and 8900 controls (kidney only) (bottom-right). The X-axes shows the OR effect size, and the Y-axes illustrate the statistical power to detect the main effects under different MAFs: 5%, 10%, 20%, and 30% shown in blue, red, green, and purple, respectively. Significance is assessed at 5% level using Bonferroni correction, assuming 500,000 SNP tests
DISCUSSION
Discovery of genetic factors associated with graft loss and patient survival can generate fundamental insights into the biological and immunological factors underpinning posttransplant diseases and graft survival. Although barriers exist for harmonizing retrospective and prospective transplant study designs beyond single-site independent studies, international guidelines and hard outcomes allow for standardization of phenotypes, such as graft and patient survival, AR, NODAT, and DGF. In addition, where feasible, participating studies will share genotype and phenotype information between centers to fully maximize power to detect novel biological findings with the greater goal of conferring clinical impact. The collapsing of phenotypes across solid-organ types may not be ideal for organ-specific phenotypes such as kidney DGF, for other phenotypes, in particular NODAT and pharmacogenomic-related phenotypes, aggregation will collectively improve statistical power. The current absence of adequately powered data sets makes our consortium a unique force for GWAS efforts in Tx. The data sets in iGeneTRAiN constitute the largest genomic and phenotypic Tx data sets aggregated to date, with genomewide genotyping and phenotypes collected for more than 27500 subjects (with >11300 D-R pairs) recruited from 1993 to 2014.
The results from iGeneTRAiN will create clinical knowledge and applications in a number of specific areas: (1) Knowledge gained from genomewide as well as MHC/KIR variants, which may be highly penetrant, will facilitate novel insight into the biology of genomic incompatibility of D-R pairings. This may inform better patient care through improved risk assessment and monitoring of higher risk HLA D-R pairs, and/or more tailored IST. (2) More appropriate D-R matching before Tx may be possible based on MHC/HLA and KIR genotype combinations, or LoF compatibility in D-R pairs (eg, where 0 gene copies exist, ie, hdCNV, in the recipient and 1 or 2 gene copies exist in the respective donor), especially in the living donor transplant setting where multiple potential donors may exist. Consortia such as The Electronic Medical Records and Genomics (eMERGE) Network have very effective models for the development of genotype/phenotype algorithms from electronic medical records. The eMERGE is currently implementing dissemination of clinical genomic tests into electronic medical records and returning results back to physicians and patients in a clinical care setting,51 and initial efforts in clinically guided genotyping of tacrolimus has now begun in one of the eMERGE/iGeneTRAiN sites.15 (3) Genomic signals associated with clinical outcomes have been shown to be “druggable” through repositioning of existing drugs, or through targeting of defined small molecules known to interact with genes of interest.52,53 Results from iGeneTRAiN GWAS thus offers the potential for facilitating identification of new therapeutics for use after Tx. (4) Genetic loci associated with response to immunosuppressive agents (eg, calcineurin inhibitors, mycophenolate or thymoglobulin) may also enable personalized medicine through means, such as genotype guided dosing of ISTs or identification of genetic variants associated with idiosyncratic (eg, hypersensitivity) drug reactions.
The potential limitations of our study are also common to many GWAS efforts, including complex phenotypes with nongenetic confounders as well as limited power to identify novel loci and variants which may have modest effect sizes. We aim to overcome these issues by setting up large, well-powered studies with manually curated and harmonized phenotypes. Nongenetic factors such as cold ischemic time and use of immunosuppressive drugs heavily influence clinical outcomes, and we will use all available modelling techniques to account for these covariates where available. However, evidence from many phenotypes has taught us that genetic polymorphisms with even modest independent effect sizes can uncover key biological mechanisms in solid organ Tx outcomes.
The iGeneTRAiN aggregation of existing GWAS efforts is essential to amass and harmonize large numbers of highly curated genotype and phenotype data sets,54 which can then be transitioned to deep sequencing studies to gain nucleotide resolution coverage of regions of interest. Such large initial numbers are crucial for accrual of transplant outcome events to inform the sequencing studies for refinement of putative causal signals, which we believe will ultimately advance the field.
The concept of the iGeneTRAiN network structure as a model in Tx research is extremely powerful on a number of fronts. First, the ability to harmonize cohorts within the same solid-organ studies affords the ability to perform replication look-ups of putative genetic associations in independent cohorts, which is an absolute requirement for any large-scale genomic study. There are minimal transplant genomic studies in existence to date for such replication efforts. The ability to discover cross-organ as well as organ-specific associations using such large sample sizes is a unique strength of iGeneTRAiN. We wish to encourage other studies to join this consortium and use genome-wide genotyping arrays with well-phenotyped samples, which can be harmonized with the networks data sets. More appropriately phenotyped studies will increase the statistical power through meta-analysis to discover new loci underpinning phenotypes such as NODAT, immunosuppression-related outcomes, DGF and rejection. A major focus of the consortium is in-depth analyses of HLA and KIR polymorphisms with a range of transplant outcomes. A number of the iGeneTRAiN studies including A-WISH, CTOT-3, Deterioration of Kidney Allograft Function Genomics/Gen03, genomics of chronic renal allograft rejection have also performed functional biomarker studies using micro RNA, messenger RNA, proteomics, and/or metabolomics as follow-up surveillance studies to detect rejection and other complications of Tx.
In conclusion, iGeneTRAiN is a large consortium of solid-organ transplant studies that aims to lead genomic investigations into clinical outcomes after organ Tx. Our consortium seeks to significantly advance our understanding of the genetic architecture of transplant-related outcomes and in doing so, improve outcomes for these high-risk patients. The GWAS analyses will include recipient-only analyses, D-R mismatches with focus on LoF variants and nonsynonymous single-nucleotide polymorphisms, and interaction analyses between D-R pairs. Findings from this consortium are expected to provide unique insights into genomic incompatibility of D-R pairs, and fundamental incremental improvements in knowledge of the biology underpinning rejection and complications of Tx, with the ultimate goal of novel therapeutic targets, informing personalized prescribing of immunosuppressive therapies, and prolonging graft and patient survival.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients and their families for their participation in the genotyping studies. The authors are also very thankful for the contributions of the all of the study coordinators and clinicians from the respective studies that made collection of these DNA samples and phenotyping possible.
APPENDIX
The International Genetics & Translational Research in Transplantation Network (iGeneTRAiN):
Brendan J. Keating, DPhil, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA, Department of Pediatrics, Perleman School of Medicine, University of Pennsylvania, Philadelphia, PA, and The Children's Hospital of Philadelphia, Philadelphia, PA; Jessica van Setten, PhD, Division of Heart and Lungs, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands; Pamala A. Jacobson, PharmD, FCCP, College of Pharmacy, University of Minnesota, Minneapolis, MN; Michael V. Holmes, MD, PhD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Shefali S. Verma, MSc, Center for SystemsGenomics, The Pennsylvania State University, University Park, PA; Hareesh R. Chandrupatla, MSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Nikhil Nair, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Hui Gao, PhD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Yun R. Li, PhD, The Children's Hospital of Philadelphia, Philadelphia, PA and Medical Scientist Training Program, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; Bao-Li Chang, PhD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Chanel Wong, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Randy Phillips, MD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Brian S. Cole, PhD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and Department of Biostatistics and Epidemiology, Institute for Biomedical Informatics, Perelman School of Medicine, University of Pennsylvania, PA; Eyas Mukhtar, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Weijia Zhang, PhD, Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY; Hongzhi Cao, PhD, BGI-Shenzhen, Shenzhen, China and Department of Biology, University of Copenhagen,Copenhagen, Denmark; Maede Mohebnasab, MD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Cuiping Hou, MSc, The Children's Hospital of Philadelphia, Philadelphia, PA; Takesha Lee, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Laura Steel, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Oren Shaked, MD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; James Garifallou, BSc, The Children's Hospital of Philadelphia, Philadelphia, PA; Michael B. Miller, PhD, Department of Psychology, University of Minnesota, Minneapolis, MN; Konrad J. Karczewski, PhD, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA and Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, MA; Abdullah Akdere, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Ana Gonzalez, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Kelsey M. Lloyd, BA, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Daniel McGinn, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Zach Michaud, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Abigail Colasacco, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Monkol Lek, PhD, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA and PrograminMedical and PopulationGenetics, Broad Institute of Harvard and MIT, Cambridge, MA; Yao Fu, BSc, Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT; and Mayur Pawashe, BSc, Molecular Biophysics and Biochemistry Department, Yale University, New Haven, CT; Toumy Guettouche, PhD, The Children's Hospital of Philadelphia, Philadelphia, PA; Aubree Himes, BSc, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA and The Children's Hospital of Philadelphia, Philadelphia, PA; Leat Perez, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Weihua Guan, PhD, Division of Biostatistics, University of Minnesota, Minneapolis, MN; Baolin Wu, PhD, Division of Biostatistics, University of Minnesota, Minneapolis, MN; David Schladt, MS, Minneapolis Medical Research Foundation, Hennepin County Medical Center, Minneapolis, MN; Madhav Menon, MD, Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY; Zhongyang Zhang, PhD, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, NY; Vinicius Tragante, PhD, Division of Heart and Lungs, Department of Cardiology, University, Medical Center Utrecht, Utrecht, The Netherlands; Nicolaas de Jonge, MD PhD, Division of Heart and Lungs, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands; Henny G. Otten, PhD, Department of Immunology, University Medical Center, Utrecht, the Netherlands; Roel A. de Weger, PhD, Department of Pathology, University Medical Center, Utrecht, The Netherlands; Ed A. van de Graaf, MD, PhD, Department of Pulmonary Disease, University Medical Center Utrecht, The Netherlands; Carla C. Baan, PhD, Department of Internal Medicine, Erasmus University Medical Center, Rotterdam, The Netherlands; Olivier C. Manintveld, MD, PhD, Department of Cardiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Iwijn De Vlaminck, PhD, Department of Bioengineering and Applied Physics, Stanford University, Stanford, CA and Howard Hughes Medical Institute, Stanford, CA; Brian D. Piening, PhD, Howard Hughes Medical Institute, Stanford, CA and Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, CA; Calvin Strehl, BSc, Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, CA; Mary Shaw, RN, BBA, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Harold Snieder, PhD, Department of Epidemiology, Unit of Genetic Epidemiology and Bioinformatics, University of Groningen, University Medical Center Groningen, The Netherlands; Goran B. Klintmalm, MD, PhD, FACS, Baylor Simmons Transplant Institute, Baylor University Medical Center, Dallas, TX; Jacqueline G. O'Leary, MD, MPH, Division of Hepatology, Annette C. & Harold C. Simmons Transplant Institute, Baylor University Medical Center, Dallas, TX; Sandra Amaral, MD, MHS, Division of Nephrology, The Children's Hospital of Philadelphia, Philadelphia, PA and Department of Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, PA; Samuel Goldfarb, MD, Division of PulmonaryMedicine, The Children's Hospital of Philadelphia, Philadelphia, PA; Elizabeth Rand, MD, Division of Gastroenterology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA; Joseph W. Rossano, MD, MS, FAAP, FAAC, Division of Pediatric Cardiology, Children's Hospital of Philadelphia, Philadelphia, PA; Utkarsh Kohli, MD¸ Division of Pediatric Cardiology, Children's Hospital of Philadelphia, Philadelphia, PA; Peter Heeger, MD, Division of Nephrology, Icahn School of Medicine at Mount Sinai, New York, NY and Department of Medicine, Icahn School of Medicine at Mount Sinai, NY; Eli Stahl, PhD, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, NY; Jason D. Christie, MD, MSCE, Division of Pulmonary, Allergy & Critical Care, University of Pennsylvania, Philadelphia, PA; Maria Hernandez Fuentes, PhD, Division of Transplantation Immunology and Mucosal Biology, Guy’s Hospital & King’s College London, United Kingdom; John E. Levine, MD, MS, Blood and Marrow Transplant Program, University of Michigan, Ann Arbor, MI; Richard Aplenc, MD, PhD, Division of Oncology, The Children's Hospital of Philadelphia, Philadelphia, PA; Eric E. Schadt, PhD, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, NY; Barbara E. Stranger PhD, Section of Genetic Medicine, University of Chicago, Chicago, IL; Jolanda Kluin, MD, PhD, Department of Cardiothoracic Surgery, University Medical Center Utrecht, The Netherlands; Luciano Potena MD, PhD, Heart Failure and Heart Transplant Unit, Cardiovascular Department, University of Bologna, Italy; Andreas Zuckermann, MD, Department of Cardiac Surgery, Medical University of Vienna, Austria; Kiran Khush, MD, MAS, Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, CA; Alhusain J. Alzahrani, PhD, College of Applied Medical Sciences, King Saud University, Riyadh, Saudi Arabia; Fahad A. Al-Muhanna, MD, College of Medicine, University of Dammam, Dammam, Kingdom of Saudi Arabia; Amein K. Al-Ali, PhD, College of Medicine, University of Dammam, Dammam, Kingdom of Saudi Arabia; Rudaynah Al-Ali, MBBS, Department of Internal Medicine, King Fahd Hospital of the University, University of Dammam, Dammam, Kingdom of Saudi Arabia; Abdullah M. Al-Rubaish, MD, College of Medicine, University of Dammam, Dammam, Kingdom of Saudi Arabia; Samir Al-Mueilo, MBBS, College of Medicine, University of Dammam, Dammam, Kingdom of Saudi Arabia; Edna M. Byrne, PhD, The University of Queensland, St. Lucia, Brisbane, Australia; David Miller, PhD, Queensland Centre for Medical Genomics, IMB, University of Queensland, Queensland, Australia and Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia; Stephen I. Alexander, MB, BS, MPH, Centre for Kidney Research, The Children's Hospital at Westmead, University of Sydney, NSW, Australia; Suna Onengut-Gumuscu, PhD; Center for Public Health Genomics, Department of Public Health Sciences, University of Virginia, VA; Stephen S. Rich, PhD, Center for Public Health Genomics, Department of Public Health Sciences, University of Virginia, VA; Manikkam Suthanthiran, MD, Division of Nephrology and Hypertension, Department of Medicine, Weill Cornell Medical College, New York, NY; Helio Tedesco, MD, Division of Nephrology, Hospital do Rim e Hipertensão, São Paulo, Brazil; Chee L. Saw, PhD, HCLD, HLA Laboratory, Hematology Division, McGill University Health Centre, Montreal, Canada; Jiannis Ragoussis, PhD, McGill University and Genome Quebec Innovation Centre, Montreal, Quebec, Canada; Abdallah G. Kfoury, MD, Intermountain Heart Institute, Murray, UT; Benjamin Horne, MPH, PhD, Intermountain Heart Institute, Murray, UT; John Carlquist, PhD, Intermountain Heart Institute, Murray, UT; Mark B. Gerstein, PhD, The University of Queensland, St. Lucia, Brisbane, Australia and Intermountain Heart Institute, Murray, UT; Roman Reindl-Schwaighofer, MD, Division of Nephrology Dialysis, Department of Internal Medicine III, University of Vienna, Austria; Rainer Oberbauer, MD, Division of Nephrology Dialysis, Department of Internal Medicine III, University of Vienna, Austria; Cisca Wijmenga, PhD, University of Groningen, University Medical Center Groningen, Department of Genetics, Groningen, The Netherlands; Scott Palmer, MD, MHS, Department of Medicine, Duke University School of Medicine, Durham, NC; Alexandre C. Pereira, MD, PhD, Laboratory of Genetics & Molecular Cardiology, Heart Institute (InCor), University of São Paulo Medical School, São Paulo, Brazil; Javier Segovia, MD, PhD, Heart Transplant Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro Majadahonda,Madrid, Spain; Luis A. Alonso-Pulpon, MD, PhD, Heart Transplant Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro Majadahonda,Madrid, Spain; Manuel Comez-Bueno, MD, Heart Transplant Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro Majadahonda,Madrid, Spain; Carlos Vilches, MD, Department of Immunology, Hospital Universitario Puerta de Hierro Majadahonda,Madrid, Spain; Natalia Jaramillo, MD, Heart Transplant Unit, Department of Cardiology, Hospital Universitario Puerta de HierroMajadahonda, Madrid, Spain; Martin H. de Borst, MD, PhD, Division of Nephrology, Department of Internal Medicine, University Medical Center, University of Groningen, Groningen, The Netherlands; Maarten Naesens, MD, PhD, Department of Nephrology and Renal Transplantation, University Hospitals Leuven, Leuven, Belgium; Ke Hao, PhD, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, NY; Daniel G. MacArthur, PhD, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA; Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, MA; Suganthi Balasubramanian, PhD, Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT and Molecular Biophysics and Biochemistry Department, Yale University, New Haven, CT; Peter J. Conlon, MD, Department of Transplantation and Renal Medicine, Beaumont Hospital, Dublin, Ireland; Graham M. Lord, MD, PhD, Division of Transplantation Immunology and Mucosal Biology, Guy’s Hospital & King’s College London, United Kingdom; Marylyn D. Ritchie, PhD, Center for Systems Genomics, The Pennsylvania State University, University Park, PA; Michael Snyder, PhD, Department of Genetics, Stanford University School of Medicine, Stanford, CA and Stanford Cardiovascular Insti tute, Stanford University School of Medicine, Stanford, CA; Kim M. Olthoff, MD, FACS, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Jason H. Moore, PhD, Center for Public Health Genomics, Department of Public Health Sciences, University of Virginia, VA; Effie W. Petersdorf, MD, Division of Medical Oncology, Fred Hutchinson Cancer Research Center, University of Washington, School of Medicine Seattle, WA; Malek Kamoun, MD, PhD, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; Jun Wang, PhD, BGI-Shenzhen, Shenzhen, China; Dimitri S. Monos, PhD, The Children's Hospital of Philadelphia, Philadelphia, PA and Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; Paul I.W de Bakker, PhD, Department of Medical Genetics, Center for Molecular Medicine, University Medical Center Utrecht, Utrecht, The Netherlands and Department of Epidemiology, Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht, The Netherlands; Hakon Hakonarson, MD, PhD, The Children's Hospital of Philadelphia, Philadelphia, PA; Barbara Murphy, MD, Division of Nephrology and Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY; Matthew B. Lankree, MD, PhD, Department of Medicine, McMaster University, Hamilton, Ontario, Canada; Pablo Garcia-Pavia, MD, PhD, Heart Transplant Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain; William S. Oetting, PhD, Experimental and Clinical Pharmacology, University of Minnesota, Minneapolis, MN; Kelly A. Birdwell, MD, MSCI, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN; Stephan J. L Bakker, MD, PhD, Division of Nephrology, Department of Internal Medicine, University Medical Center, University of Groningen, Groningen, The Netherlands; Ajay K. Israni, MD, Hennepin County Medical Center, University of Minnesota, Minneapolis, MN; Abraham Shaked, MD PhD, Penn Transplant Institute, Hospital of the University of Pennsylvania, Philadelphia, PA; Folkert W. Asselbergs, MD, PhD, Division of Heart and Lungs, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands, Institute of Cardiovascular Science, University College London, London, United Kingdom, and Durrer Center for Cardiogenetic Research, ICIN-Netherlands Heart Institute, Utrecht, The Netherlands.
Footnotes
This project was funded in part by Fundación Mutua Madrileña, Spain. Part of this work is also supported by the Dutch PLN Foundation (www.stichtingpln.nl). Folkert W. Asselbergs is supported by UCL Hospitals NIHR Biomedical Research Centre and by a Dekker scholarship-Junior Staff Member 2014T001—Netherlands Heart Foundation. Partial funding was also provided by the Deanship of Scientific Research at King Saud University, Riyadh. We also acknowledge funding from the following NIH grants: U01 HG006830; U01-DK062494; UM1AI109565; U01-AI63589 and U19-AI070119.
The authors declare no funding or conflicts of interest.
A.K.I., A.S., and F.W.A. contributed equally to this article.
B.J.K., A.K.I., W.O., P.A.J., A.S., and F.W.A. conceived the study. Y.R.L., J.vS., Y.L., M.V.H., S.S.V., M.L., N.N., H.G., H.C., K.J.K., B.A., H.C., W.G., T.G., D.S.M., T.W., D.G.M.A., M.B.L., H.H., K.B., M.D.R., P.A.J., F.W.A., A.K.I., A.S., and B.J.K. were involved in acquisition of data, analysis, and/or interpretation of data. B.J.K., P.A.J., M.V.H., J.v.S., W.S.O., K.A.B., S.J.L.B., A.K.I., M.B.L., A.S., and F.W.A. were involved in drafting the manuscript and critical revisions. E.M., V.T., L.S., T.L., J.G., B.A., H.C., A.H., J.v.H., A.P., R.U., E.C., A.A.A., F.A.A., A.M.A., B.M., Y.R.L., J.v.S., Y.L., M.V.H., S.S.V., M.L., N.N., H.G., H.C., K.J.K., C.W., M.M., K.M.O., C.W., T.W., M.K., S.B., M.B.L., W.S.O., P.G.P., D.G.M.A., P.A.J., F.W.A., A.K.I., A.S., B.J.K., and all other coauthors were involved in collection of phenotype data and/or critical contributions to the framework of the consortium. All coauthors read and approved the final article, and are accountable for all aspects and integrity of the work.
Supplemental digital content (SDC) is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal's Web site (www.transplantjournal.com).
Contributor Information
Collaborators: The International Genetics & Translational Research in Transplantation Network (iGeneTRAiN)
REFERENCES
- 1. Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009; 106: 9362– 9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011; 12: 745– 755. [DOI] [PubMed] [Google Scholar]
- 3.http://www.genome.gov/gwastudies/index.cfm?pageid=26525384#searchForm.
- 4. MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012; 335: 823– 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sudmant PH, Kitzman JO, Antonacci F, et al. Diversity of human copy number variation and multicopy genes. Science. 2010; 330: 641– 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Handsaker RE, Korn JM, Nemesh J, et al. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011; 43: 269– 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCarroll SA, Bradner JE, Turpeinen H, et al. Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nat Genet. 2009; 41: 1341– 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li R, Li Y, Zheng H, et al. Building the sequence map of the human pan-genome. Nat Biotechnol. 2010; 28: 57– 63. [DOI] [PubMed] [Google Scholar]
- 9. Venstrom JM, Pittari G, Gooley TA, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med. 2012; 367: 805– 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vampa ML, Norman PJ, Burnapp L, et al. Natural killer-cell activity after human renal transplantation in relation to killer immunoglobulin-like receptors and human leukocyte antigen mismatch. Transplantation. 2003; 76: 1220– 1228. [DOI] [PubMed] [Google Scholar]
- 11. Zou Y, Stastny P. The role of major histocompatibility complex class I chain-related gene A antibodies in organ transplantation. Curr Opin Organ Transplant. 2009; 14: 414– 418. [DOI] [PubMed] [Google Scholar]
- 12. Moretta A, Bottino C, Vitale M, et al. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. 1996; 14: 619– 648. [DOI] [PubMed] [Google Scholar]
- 13. Norman PJ, Stephens HA, Verity DH, et al. Distribution of natural killer cell immunoglobulin-like receptor sequences in three ethnic groups. Immunogenetics. 2001; 52: 195– 205. [DOI] [PubMed] [Google Scholar]
- 14. Terasaki PI. Deduction of the fraction of immunologic and non-immunologic failure in cadaver donor transplants. Clin Transpl. 2003: 449– 452. [PubMed] [Google Scholar]
- 15. Birdwell KA, Grady B, Choi L, et al. The use of a DNA biobank linked to electronic medical records to characterize pharmacogenomic predictors of tacrolimus dose requirement in kidney transplant recipients. Pharmacogenet Genomics. 2012; 22: 32– 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobson PA, Schladt D, Israni A, et al. Genetic and clinical determinants of early, acute calcineurin inhibitor-related nephrotoxicity: results from a kidney transplant consortium. Transplantation. 2012; 93: 624– 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clatworthy MR, Matthews RJ, Doehler B, et al. Defunctioning polymorphism in the immunoglobulin G inhibitory receptor (FcγRIIB-T/T232) does not impact on kidney transplant or recipient survival. Transplantation. 2014; 98: 285– 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.https://www.zotero.org/groups/emerge_network/items/collectionKey/68QW2EUD Ulop.
- 19. Stegall MD, Park WD, Dierkhising R. Genes and transplant outcomes: the search for “associations”. Transplantation. 2014; 98: 257– 258. [DOI] [PubMed] [Google Scholar]
- 20. O'Brien RP, Phelan PJ, Conroy J, et al. A genome-wide association study of recipient genotype and medium-term kidney allograft function. Clin Transplant. 2013; 27: 379– 387. [DOI] [PubMed] [Google Scholar]
- 21. McCaughan JA, McKnight AJ, Maxwell AP. Genetics of new-onset diabetes after transplantation. J Am Soc Nephrol. 2014; 25: 1037– 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Damman J, Daha MR, Leuvenink HG, et al. Association of complement C3 gene variants with renal transplant outcome of deceased cardiac dead donor kidneys. Am J Transplant. 2012; 12: 660– 668. [DOI] [PubMed] [Google Scholar]
- 23. Gourishankar S, Leduc R, Connett J, et al. Pathological and clinical characterization of the ‘troubled transplant’: data from the DeKAF study. Am J Transplant. 2010; 10: 324– 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cantu E, Lederer DJ, Meyer K, et al. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. Am J Transplant. 2013; 13: 1898– 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olthoff KM, Kulik L, Samstein B, et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl. 2010; 16: 943– 949. [DOI] [PubMed] [Google Scholar]
- 26. Li L, Khush K, Hsieh SC, et al. Identification of common blood gene signatures for the diagnosis of renal and cardiac acute allograft rejection. PLoS One. 2013; 8: e82153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Vlaminck I, Valantine HA, Snyder TM, et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med. 2014; 6: 241ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zijlstra LE, Constantinescu AA, Manintveld O, et al. Improved long-term survival in Dutch heart transplant patients despite increasing donor age: the Rotterdam experience. Transpl Int. 2015; 28: 962– 971. [DOI] [PubMed] [Google Scholar]
- 29. Garcia-Pavia P, Syrris P, Salas C, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart. 2011; 97: 1744– 1752. [DOI] [PubMed] [Google Scholar]
- 30. Garcia-Pavia P, Vazquez ME, Segovia J, et al. Genetic basis of end-stage hypertrophic cardiomyopathy. Eur J Heart Fail. 2011; 13: 1193– 1201. [DOI] [PubMed] [Google Scholar]
- 31. Holweg CT, Balk AH, Uitterlinden AG, et al. Functional heme oxygenase-1 promoter polymorphism in relation to heart failure and cardiac transplantation. J Heart Lung Transplant. 2005; 24: 493– 497. [DOI] [PubMed] [Google Scholar]
- 32. Shaked A, Ghobrial RM, Merion RM, et al. Incidence and severity of acute cellular rejection in recipients undergoing adult living donor or deceased donor liver transplantation. Am J Transplant. 2009; 9: 301– 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diamond JM, Akimova T, Kazi A, et al. Genetic variation in the prostaglandin E2 pathway is associated with primary graft dysfunction. Am J Respir Crit Care Med. 2014; 189: 567– 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meier-Kriesche HU, Ojo AO, Hanson JA, et al. Increased impact of acute rejection on chronic allograft failure in recent era. Transplantation. 2000; 70: 1098– 1100. [DOI] [PubMed] [Google Scholar]
- 35. Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2012 Annual Data Report: kidney. Am J Transplant. 2014; 14 (Suppl 1): 11– 44. [DOI] [PubMed] [Google Scholar]
- 36. Chakkera HA, Pham PT, Pomeroy J, et al. Response to comment on: chakkera et al. can new-onset diabetes after kidney transplant be prevented? Diabetes Care 2013;36:1406–1412. Diabetes Care. 2013; 36: e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rakel A, Karelis AD. New-onset diabetes after transplantation: risk factors and clinical impact. Diabetes Metab. 2011; 37: 1– 14. [DOI] [PubMed] [Google Scholar]
- 38. Kamoun M, Holmes JH, Israni AK, et al. HLA-A amino acid polymorphism and delayed kidney allograft function. Proc Natl Acad Sci U S A. 2008; 105: 18883– 18888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia X, Han B, Onengut-Gumuscu S, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013; 8: e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu L, Jiao S, Dai JY, et al. Powerful cocktail methods for detecting genome-wide gene-environment interaction. Genet Epidemiol. 2012; 36: 183– 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murcray CE, Lewinger JP, Conti DV, et al. Sample size requirements to detect gene-environment interactions in genome-wide association studies. Genet Epidemiol. 2011; 35: 201– 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kooperberg C, Leblanc M. Increasing the power of identifying gene x gene interactions in genome-wide association studies. Genet Epidemiol. 2008; 32: 255– 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010; 26: 2069– 2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489: 57– 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013; 45: 580– 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yun R, Li JvS, Verma SS, et al. Concept and design of a genome-wide association genotyping array tailored for transplantation-specific studies. Genome Med. 2015; 7: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Y, Willer C, Sanna S, et al. Genotype imputation. Annu Rev Genomics Hum Genet. 2009; 10: 387– 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014; 46: 818– 825. [DOI] [PubMed] [Google Scholar]
- 49. Muddyman D, Smee C, Griffin H, et al. Implementing a successful data-management framework: the UK10K managed access model. Genome medicine. 2013; 5: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marsh SG, Albert ED, Bodmer WF, et al. Nomenclature for factors of the HLA system, 2010. Tissue Antigens. 2010; 75: 291– 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gottesman O, Kuivaniemi H, Tromp G, et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013; 15: 761– 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Makley LN, Gestwicki JE. Expanding the number of ‘druggable’ targets: non-enzymes and protein-protein interactions. Chem Biol Drug Des. 2013; 81: 22– 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tragante V, Barnes MR, Ganesh SK, et al. Gene-centric meta-analysis in 87,736 individuals of European ancestry identifies multiple blood-pressure-related loci. Am J Hum Genet. 2014; 94: 349– 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Panagiotou OA, Willer CJ, Hirschhorn JN, et al. The power of meta-analysis in genome-wide association studies. Annu Rev Genomics Hum Genet. 2013; 14: 441– 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.