

Abstract
Immunogenicity of aggregated or otherwise degraded protein delivered from depots or other biopharmaceutical products is an increasing concern, and the ability to deliver stable, active protein is of central importance. We review characterization approaches for solid protein dosage forms with respect to metrics that are intended to be predictive of protein stability against aggregation and other degradation processes. Each of these approaches is ultimately motivated by hypothetical connections between protein stability and the material property being measured. We critically evaluate correlations between these properties and stability outcomes, and use these evaluations to revise the currently standing hypotheses. Based on this we provide simple physical principles that are necessary (and possibly sufficient) for generating solid delivery vehicles with stable protein loads. Essentially, proteins should be strongly coupled (typically through H-bonds) to the bulk regions of a phase-homogeneous matrix with suppressed β relaxation. We also provide a framework for reliable characterization of solid protein forms with respect to stability.
Graphical abstract
1. Introduction
While there are many potential applications for targeted drug delivery of therapeutic proteins, protein instability [1,2] associated with process, storage, and delivery-related stresses remains the primary roadblock for most of these applications. However, nature has found ways to ameliorate stresses similar to many of those encountered in preparation of drug delivery depots. In nature, some organisms protect proteins under desiccation [3] and freezing stress [4] through elevated local concentrations of sugars or sugar-alcohols. Crowe and Carpenter found they could apply this strategy by lyophilizing proteins or lipids [5] in the presence of disaccharides with nearly 100% recovery of protein function or membrane integrity. This approach is now commonly used in the biopharmaceutical industry, and has facilitated formulation development of many dried therapeutic protein products for human use. The use of disaccharides as stabilizers in drug delivery applications has been relatively limited, but has shown promise [6–10].
Since the work of Crowe and Carpenter, scores of biopharmaceutical products have been administered from a lyophilized state to millions of patients with very few obvious adverse health effects. However, it now appears that it is not uncommon for patients to develop antibodies to the therapeutic proteins over time [11], reducing their efficacy [11,12] or occasionally inducing dangerous immunogenic responses [11]. The appearance of anti-drug antibodies has been associated with the presence of protein aggregates [13], which occur at low levels even for these minimally processed biopharmaceutical products lyophilized in the presence of disaccharides [14].
Recent reviews have pointed out potential immunogenicity of various components of delivery vehicles themselves [15] and the enhanced likelihood of finding immunogenic protein aggregates associated with injectable protein delivery systems [16]. Stresses that could lead to protein aggregation or other protein degradation products can occur at many stages of producing a drug delivery product, including encapsulation-related processing, storage, and delivery or release. As with conventional biopharmaceutics, efforts have been made in drug delivery applications to reduce process-related degradation and improve stability of the protein to be delivered. Among these are the use of systems that are amenable to all-aqueous processing such as chitosan [17] and gels [18]. Proteins and nucleic acids have also been pre-encapsulated in sugars, preventing their direct exposure to solvents [10,19,20], and this has also been effective in preventing burst release [10]. Damage at time of release associated with low pH from degrading polymer has been addressed by Schwendeman and colleagues [21]. These improved approaches will no doubt be helpful in efforts to deliver active, stable protein. However, in addition to employment of a protein-friendly delivery platform, an appropriate level of stabilization generally requires significant formulation effort for each payload protein, involving characterization of intended biopharmaceutical payload as well as degradation products that may accumulate [12,16]. Here we focus on characterizing key properties of dry protein formulations and explore the potential of these properties as reliable metrics for predicting protein stability.
Regulatory and other pressures have driven development of methods for detailed characterization of biopharmaceutical products in order to minimize adverse effects on patients, reduce product loss, ensure reproducibility, and predict stability. Thus, even though the connection between degraded protein and immunogenicity is just emerging, there is an established analytical infrastructure in place for characterizing products with respect to stability. Within this infrastructure, a range of analytical methods is in common use, but the field is still developing as a quantitative science. The utility of most analytical methods used in this context is judged on correlation between metrics and functional outcomes. As we will see below, many of these correlations are not completely reliable, demonstrating at least that additional variables are important. The various characterization methods are inspired by decades-old hypotheses regarding causal factors in protein degradation. These hypotheses are reasonable, and probably each holds some validity, but further careful exploration and critical evaluation will likely lead to analytical approaches that are more reliable and possibly quantitatively predictive of protein stability in dried form.
1.1. Potential Mechanisms of Stabilization
Two hypotheses for how a solid matrix (sugars in particular) can stabilize dry-state proteins were articulated soon after the discovery that sugar-glass can stabilize proteins. One idea is that sugars substitute for water at the protein surface, conferring thermodynamic stability [22]. Another idea is that, upon vitrification, sugars simply impede degradation processes [23]. It has also been suggested that water trapped by sugars at the surface of the proteins mediates either the proposed dynamic [24,25] or thermodynamic [26,27] stabilization. It has been difficult to cleanly discriminate among these hypotheses, for practical and fundamental reasons. Among these reasons is the fact that the hypotheses are not mutually exclusive. It is possible that both could play important roles. Also, it is practically impossible to try to disprove the thermodynamic stabilization hypothesis by performing thermodynamic studies on these vitrified systems since the required system ergodicity may not be obtained on a practical timescale. Adding further difficulty is that these questions have been addressed in several different fields, from biophysics to pharmaceutics, and the notion of what “protein stability” means can differ significantly from one report to another. In spite of these difficulties, sufficient data now exist to allow (a somewhat overdue) refinement of the hypotheses. Here we will focus on work that addresses stability against aggregation and chemical degradation.
2. Thermodynamic Considerations
It has been hypothesized that a good matrix material can provide stability against degradation by substituting the thermodynamic role typically played by water and stabilizing the native protein conformation [22]. It is difficult to directly test this hypothesis because it is not feasible to perform thermodynamic conformational studies for proteins in anhydrous sugars, due to the very slow dynamics of these glassy systems. However, we can glean useful information from studies performed for proteins in anhydrous solvents. It has been known for decades that small proteins can perform enzymatic functions in anhydrous media when introduced as a suspension of lyophilized powder [28]. However, when dissolved in anhydrous hydrophilic solvents, protein conformation [29] and often substrate specificity [29,30] change dramatically, indicating that these solvents really cannot be said to replace water. There has been one example of a protein retaining native conformation in anhydrous solvent, and that is lysozyme in glycerol [31]. However, lysozyme is denatured in other hydrophilic solvents [29], and glycerol is known to induce conformational changes in other small proteins such as alcohol dehydrogenase [32]. Thus, it appears that in almost all cases, anhydrous hydrophilic solvents do not replace water by stabilizing the native conformation of even small proteins, although glycerol may do better than most solvents. We can tentatively extend the results of these solvent studies to sugars since enthalpic interactions between sugars and proteins are comparable to those between glycerol and protein, both being weaker than enthalpic interactions between water and proteins.[33,34]. While it is unclear whether stabilization of the native state per se is required for achieving good functional stabilization, or is even generally possible, it is clear that the thermodynamic interactions between the solid matrix and the protein play a critical role in functional stabilization. Crowe and Carpenter [22] first showed that that dehydration-induced shifts in the positions of amide I and amide II infrared absorption bands for lysozyme could be partially or fully reversed if the protein was dried in the presence of sugars. They suggested that sugars formed hydrogen bonds with the native surface groups of the protein, helping to stabilizing the protein in a near-native state. Later, Allison et al. [35] demonstrated that the dehydration-induced spectral shifts could not be completely accounted for by water trapped at the protein surface, indicating that the sugars directly formed hydrogen bonds with the protein surface. It was later noted that proteins dried in presence of disaccharides showed significantly smaller dehydration-induced shifts in the IR spectra than those dried in polymeric sugars [35], demonstrating that polymers do not hydrogen-bond as well with proteins as do disaccharides, presumably due to topological constraints.
Stability studies have mirrored the spectroscopy trends, showing that polymers do not stabilize proteins as well as disaccharides [36,37], and that increasing amounts of disaccharides in lyophilized formulations lead to increasing stability [38–41]. The important role of hydrogen bonding between matrix and protein surface is particularly evidenced by concomitant amelioration of dehydration-induced IR shifts and improvement in stabilization with increasing sugar-to-protein ratio, with most of the improvement occurring up to the point that enough sugar is in the formulation to titrate the H-bonding sites on the protein surface [38,40,41].
2.1. Secondary Structure
Maintenance of native-like secondary protein structure has been the primary surrogate for evaluating the quality of hydrogen bonding between the matrix and protein for lyophilized biopharmaceuticals. As we will see below, the correlation between native-like secondary structure and storage stability is tenuous. It seems to hold for poorly stabilized systems, but not for formulations that are fairly stable. It has been suggested that measures of tertiary structure may provide a more robust correlation [42–46], but it has not yet been possible to access tertiary structure information for dried proteins.
In principle, any spectroscopy sensitive to vibrations in the fingerprint region (600 to 1800 cm−1) can be used to obtain secondary structure information. Raman spectroscopy is beginning to be used in aqueous media processing studies [47–49], but is used only infrequently to characterize protein structure in dry solid formulations [26,50–52]. The dry formulation studies are more challenging since laser power and dwell times required for Raman scattering can locally heat samples [52]. By contrast, FTIR is routinely used to characterize protein structure in the dry state. It is sensitive to secondary structure through spectral features arising from α-helix and β-sheet. Protein-containing powders are analyzed using FTIR by dispersing the powder in pellets of KBr. Once spectra are obtained, either 2nd derivative analysis or deconvolution of the raw spectra is used to enable interpretation of sometimes-subtle spectral differences [42–45] between solution-state spectra and those obtained from solid forms. An early example of such analysis is provided in Figure 1a [42,43] Here, the top trace in each of the four panels is the solution 2nd derivative spectrum, and the bottom trace is the corresponding 2nd derivative spectrum of the protein freeze-dried without excipient. The value of the spectral correlation coefficient, r, denotes the degree of correspondence between the solid and solution spectra. Note that these data show a range in magnitude of spectral changes on freeze-drying, from subtle to quite pronounced.
Fig. 1.
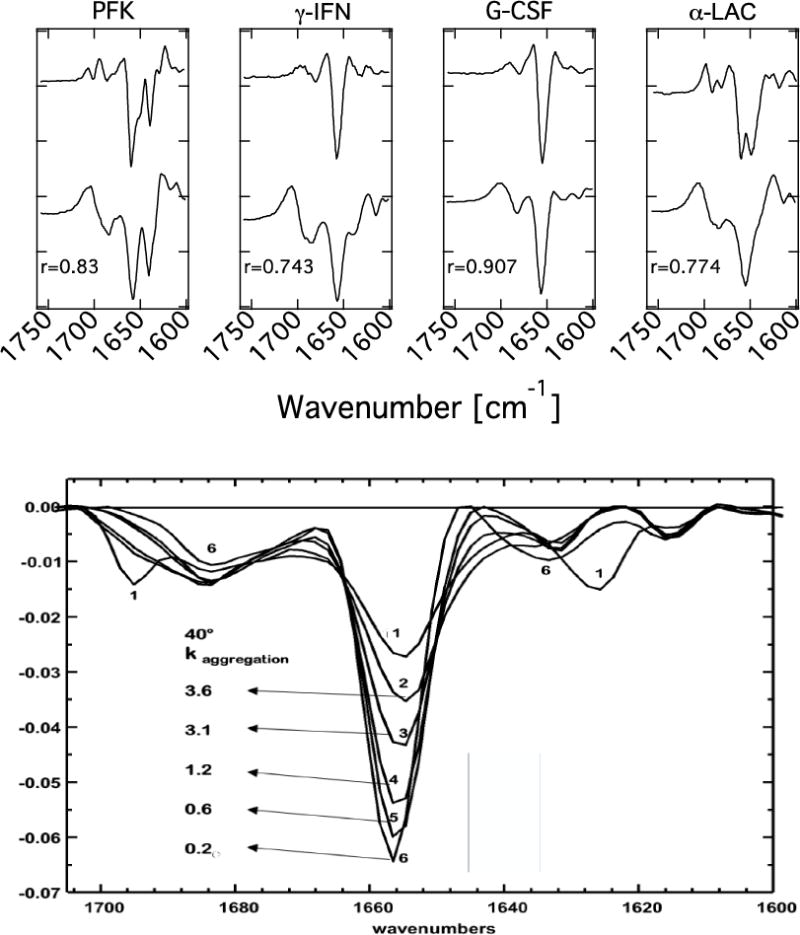
2nd Derivative FTIR data in the Amide I region: top) Solution spectra at top, spectra from freeze-dried proteins without stabilizers at bottom. PFK = phosphofructokinase; γ-IFN = gamma interferon; G-CSF = granulocyte-colony-stimulating factor. “r” is a spectral correlation coefficient [42,43]. bottom) Freeze-dried hGH spectra and rate constants for aggregation at 40 °C. The 1658 cm−1 band indicates α helix content. Curve 1: 50% heat-denatured then freeze-dried. Curve 2: no excipient; 3: HES (1:1 protein: excipient); 4: stachyose (1:1); 5: trehalose (1:1); 6: trehalose (1:6) [44].
Reproduced with permission
A number of studies have demonstrated a correlation between FTIR spectra and storage stability [42–46]. An example is given in Figure 1b for formulations of human growth hormone, hGH, freeze-dried in the presence of a variety of stabilizers [44]. The 1658 cm−1 band, associated with α-helix, is taken as a marker to indicate degree of native structure, and the aggregation rate constants at a storage temperature of 40°C associated with each spectrum are given. Increased amplitude of the α-helix band (thus higher r values) correlates qualitatively with a lower aggregation rate. A similar correlation exists for chemical degradation in these systems [44]. While correlations like this are often observed, it has also been observed for many proteins that secondary structure metrics improve with added sugar (or other stabilizer) only until sufficient stabilizer has been added to interact with all H-bonding cites on the surface of the protein. Further increases in stabilizer: protein ratio lead to increases in stability with no discernable change in markers of secondary structure [38,39].
In part to address this lack of correlation between changes in stability and secondary structure, it was recently suggested that the width of spectral bands may be a more reliable metric than their peak height [46]; however, this seems not to be the case. Figure 2 shows data sets for aggregation of two IGg proteins [39,53] and hGH [44,46], plotted against spectral widths of the 1058 cm−1 or 1040 cm−1 band for α helix in hGH or β sheet in IGg, respectively. When the data are plotted with a linear ordinate axis, as in Figure 2a, they appear to support a nearly linear relationship between aggregation rate and spectral marker width. However, such a presentation underemphasizes the data in the very range where lack of correlation has been observed for peak heights. Plotting the data on a logarithmic ordinate, as in Figure 2b, shows clearly that spectral width is not indicative of aggregation rate if the secondary structure is fairly native-like. Thus, the peak width metric provides the same result as seen using peak heights; changes in spectroscopic measures of secondary structure correlate with changes in stability, but once a nearly native secondary structure has been recovered, there can be significant improvements to stability with no changes in these spectroscopic markers.
Fig. 2.
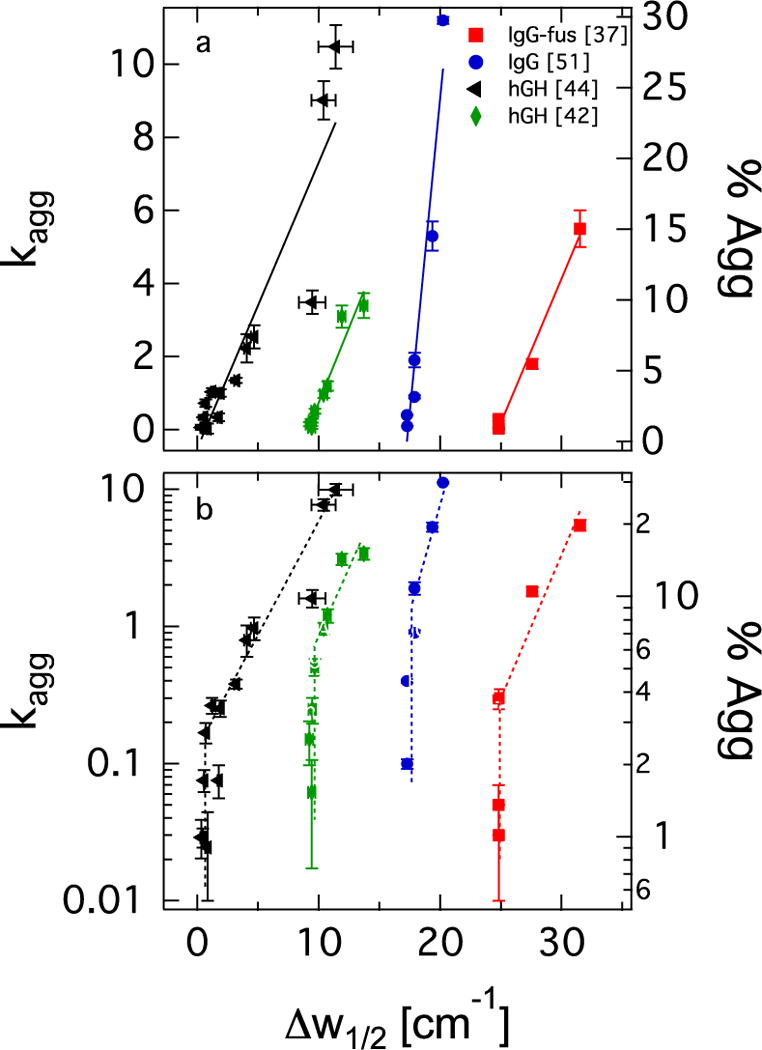
Correlation between spectral width and degradation rates: a) Linear ordinate and best linear fits to data. hGH data plotted against Δw1/2 of 1658 cm−1 α helix marker band [44,46], and IgG data plotted against Δw1/2 of 1640 cm−1 β sheet marker band [39,53]. b) Same data as in a), but plotted against logarithmic ordinate. Dashed lines are guides to the eye.
Annealing studies provide results consistent with our analysis of the data in Figure 2, but without the complication of changes in matrix or protein:matrix ratio between formulations. Chieng et al. [54] found stability improvements in hGH after performing sub-Tg annealing on lyophilized trehalose, sucrose, and hydroxyl-ethyl starch (HES) products that contained the protein. While stability improvements were obtained for all formulations on annealing, improvements in spectral markers were observed only for trehalose and HES formulations; hGH spectra in sucrose resembled that of native hGH before annealing. Similarly aging a sucrose formulation of an IgG1 protein showed no change in spectral markers but did show an improvement in stability [55].
Given the ubiquity in observations of stability change without apparent change in conformation, we can thus conclude that thermodynamic stabilization of the native secondary structure is either not the only causal factor for functional stabilization, or is not a causal factor at all. In absence of more highly detailed conformation information, we are left to consider dynamics information to provide the missing causal factors for protein stabilization in the dry state.
3. Dynamic Considerations
3.1. Materials Dynamics
Matrix materials used to sequester therapeutic proteins, disaccharides such as trehalose or sucrose, polymeric sugars such as HES or dextran, and synthetic polymers such as poly-L-glycolic acid (PLGA), are typically employed in an amorphous state [56]. Amorphous solids have two characteristics that are likely important for stabilizing biological molecules. One is that, compared to the crystalline state, matrix molecule positions are less constrained, so they can interact more strongly with proteins (e.g., through H-bonding). The other is that they are dynamically slow, and have potential to slow degradation processes within them [57,58].
Amorphous solids (i.e., glasses) uniformly exhibit three characteristic dynamic processes covering a wide range of timescales [59]. The fastest motion (βfast relaxation) occurs on a temperature-independent timescale of picoseconds, and corresponds to molecules jostling against one another, either moving independently or collectively [60], where the collective motion comprises the fundamental steps of the diffusion process [61]. The intermediate timescale motion, referred to as Johari-Goldstein β (βJG) relaxation [62] occurs on microsecond to millisecond timescales in the glassy state, and apparently corresponds to transitions between collective and non-collective βfast motion [61]. The slowest motion (α relaxation) is strongly temperature dependent and occurs on (seconds to months) timescales or longer in the glassy state. This terminal relaxation process is sometimes referred to as global or structural relaxation. While it is typically characterized by a single average relaxation time (τα), it is more correctly characterized as having a distribution of timescales that can be as broad as several decades [63]. The average α relaxation time is closely related to viscosity (η) through the relation τα ∝ η/T.
3.1.1. Alpha Relaxation
For the purpose of predicting stability in solid dosage forms, formulators have historically looked to the slowest dynamic process in the material (α relaxation) to correlate with the very slow chemical and physical degradation rates of proteins sequestered in the material [57,64,65]. This is qualitatively supported by observation of changes in characteristics of degradation rates near Tg, where there is a rapid change in α dynamics and rate of degradation processes with temperature [45,53,65–70].
The value of the α relaxation time (τα) can be obtained in a number of ways. The most common in the pharmaceutical field is to assume that log(τα) scales approximately with (Tg – T), as suggested by the Williams-Landel-Ferry (WLF) relationship [71]. One early study [64] demonstrated that for a single matrix, a monoclonal antibody-vinca alkaloid conjugate formulation at temperatures slightly above Tg, degradation rates were proportional to (Tg – T) for all three degradation pathways studied (aggregation, hydrolysis of the antibody-vinca linker, and vinca degradation) as expected from the WLF relation. However, while it is true that log(τα) may scale almost linearly with (Tg – T) over small temperature intervals, it is important to recognize that the scaling will be very different for different materials. In particular, the scaling will depend on the fragility [72] of the glass, its Kauzmann [73] temperature, as well as its thermal history [74]. Neglecting these factors, it was long held that materials with higher Tg would have slower dynamics, more strongly suppressing protein dynamics and degradation processes [53,65,67–70,75]. However, it was recently shown for a collection of more than 100 formulations that using (Tg – T) to predict relative degradation rates could lead to 1000-fold errors [76].
While (Tg – T) is experimentally the easiest metric to access, there are a number of other ways to determine τα for a material. Positron annihilation spectroscopy (PALS) [77] provides a measure of nanometer-sized void volume in materials. Within free volume paradigms [78], such voids are thought to influence α relaxation. However, PALS-derived parameters seem not to provide robust correlations with degradation processes [36,54], but this is perhaps not too surprising given the complicated relationship between PALS outputs and relaxation processes [79]. Dielectric relaxation is a more direct and widely used approach to characterize α relaxation dynamics of glassy, small-molecule pharmaceutical systems [80]; however this technique is very challenging in lyophilized solids due to effects of moisture and their porous nature. In one of the very few demonstrations of its use for protein formulations, Duddu et al. [81] showed that aggregation rates of a MAb correlated with τα, but differently for different formulations.
Structural relaxation times (τα) may also be measured by enthalpy relaxation techniques involving either a kinetic analysis of enthalpy recovery data accumulated at a given temperature and selected times, or by analysis of the rate of heat production by a sample undergoing enthalpy relaxation in a thermal activity monitor (TAM) (i.e, an isothermal micro-calorimeter), at a temperature of interest [82]. The Kohlrausch-Williams-Watts (KWW) model is frequently used for analysis of the kinetic enthalpy recovery data:
| (1) |
where t is time, τ is the relaxation time, βKWW is the so-called stretching parameter (βKWW < 1), and 1- φ(t) is the extent of enthalpy relaxation at time t, given by
| (2) |
where ∆Hr(t) is the enthalpy relaxation (or enthalpy recovery) after time, t [82]. Note that for isothermal calorimetry experiments, it is power (the time derivative of the enthalpy change) that is obtained experimentally. The KWW form predicts infinite heat flow at zero annealing time, so a more complex three-parameter “Modified Stretched Exponential (MSE)” [82] function is preferred for analysis of isothermal micro-calorimetry data. The MSE equation provides better fits at short aging times. The value of τ obtained from either the KWW or MSE analysis is variously reported as the 1/e point, the mode, or the first moment average over the distribution of α relaxation times within the glassy system [74,82].
A problem of using either the KWW or MSE equation with enthalpy relaxation experiments is that τ is considered independent of time. In reality, τ increases during the experiment as the glass ages, so τ values determined from data at the start of the experiment [83] are smaller than those obtained from later data. Simulation studies [83] indicate also that βKWW values found by fitting are too small. However, it is found that the parameter is essentially invariant to aging, and thus is normally reported and compared with rate constants for stability. Note that the rate constants are almost always evaluated based on square root of time, which is consistent with comparisons with when βKWW ≈ 0.5 [83]. When plotted this way, a linear relationship is found between log(k), where k is the degradation rate, and log(τα), but with a slope of less than unity, i.e., k ∝ τα−γ, with γ < 1, and γ varying between formulations [44].
Even though correlations of stability with are better than with (Tg – T), the use of this metric can also be problematic for comparing formulations [39,44,46,53]. An example is provided in Figure 3. Here, the logarithm of the rate constant for aggregation, log(kagg), is plotted against Tg and at 40 °C in a series of IgG1-sucrose formulations [53]. At low to moderate sucrose content, the relation between kagg and has the expected sign, but the sign flips at higher sucrose content. Note also that the relationship between (Tg – T) and stability has the opposite sign to that expected; using this metric would lead to the erroneous prediction that stability should decrease with increasing sucrose. The inconsistency in the relationship between or other measures of α relaxation and stability when compared across multiple formulations can be traced back to the fact that γ in the relationship k ∝ τα−γ can be different for each formulation [44,81].
Fig. 3.
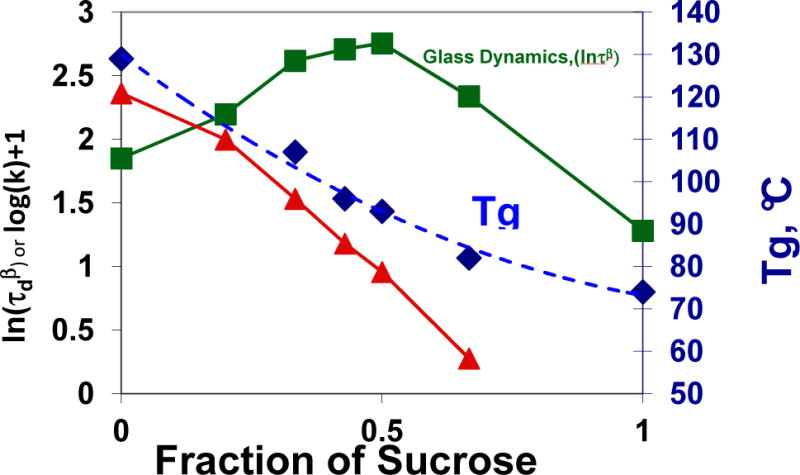
Correlations between Tg, structural relaxation time , and stability: Aggregation in sucrose formulations of an IgG1 antibody. Data from reference [84].
The fact that the relationship between k and τα can change between formulations is underscored by results of antiplasticization studies [76,84,85] where small molecule glass-forming liquids are added to formulations before lyophilization, decreasing Tg and τα, but increasing stability. For example, Chang et al. [84] reported that adding small amounts of an antiplasticizer (sorbitol or water) to sucrose-antibody formulations resulted in a monotonic decrease in Tg and τα, but optimal stabilization was obtained at intermediate levels of added sorbitol or water. In these experiments, changed with antiplastizer [86] in the same non-monotonic way as did stability, suggesting that these faster dynamic processes may be coupled directly to degradation rates.
3.1.2. Beta Relaxation Processes
Evidence for a relationship between β relaxation dynamics and protein degradation rates in lyophilized products was first observed through correlation across a series of formulations between protein stability and mean squared displacement (<u2>) measurements from neutron scattering, where <u2> ∝ amplitude of the βfast process [85]. It was later found that log(k) ∝ <u2>−1 for aggregation or chemical degradation rates over a range of formulations and proteins [39], and that the proportionality constant seems not to vary significantly with changes in protein or formulation [76]. This is shown in Figure 4a where the log of relative aggregation, deamidation or oxidation rates for a large number of proteins and formulations is plotted against <u2>−1. It was also found that k ∝ τβJG−γ, with γ=1 for > 100 enzyme-stabilizing formulations [76], and this data is displayed in Figure 4b. These relationships between measures of β relaxation and degradation rates have been subsequently confirmed for more than a dozen formulations and proteins [39,44,76,87–90], with no evidence for significant formulation-dependent changes in γ. Thus, it seems that the relationship between k and , or the amplitude of βfast is material invariant.
Fig. 4.
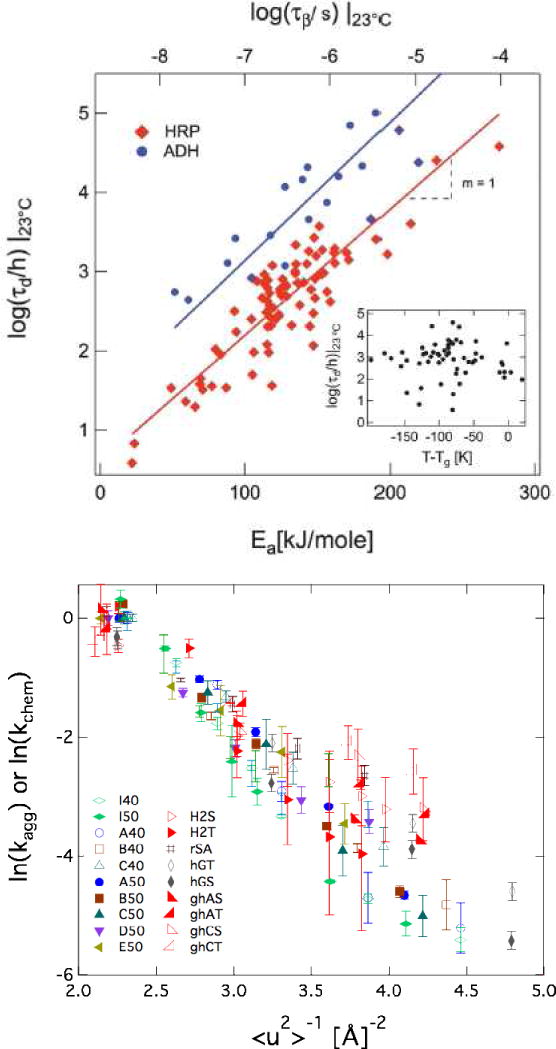
Correlation between stability and β relaxation processes: a) Enzyme degradation rates at 23 °C in >100 plasticized and antiplasticized sugar-glasses. The solid lines are best fits to the data and yield a slope near 1. Inset: T–Tg for most of the glasses shown in the main figure. b) Aggregation and chemical degradation rates of proteins freeze-dried in sucrose or trehalose-based glasses plotted as a function of <u2>−1. Each symbol represents a distinct temperature – protein – disaccharide combination. See reference [76] for details. Reproduced with permission.
While most of the work on β relaxation has been performed with neutron back scattering, this is not an approach that can be used for routine formulation analysis. Solid state NMR can also provide characterization of dried protein formulations on relevant timescales for β relaxation through spin-lattice relaxation times (T1) on a picosecond-nanosecond timescale, and spin-lattice relaxation times in the rotating frame (T1ρ) on a microsecond-millisecond timescale. Several studies have found good correlations between these relaxation times and pharmaceutical stability in solid formulations [57,90,91].
Recently it has been shown that a surrogate for <u2> from neutron scattering could be found in signatures of time-dependent fluorescence from a probe molecule located within the formulation of interest [92]. However, technical challenges prevented this method from being used for powder samples. An experimentally simpler approach, based on a steady-state fluorescence red edge effect, which is amenable to powder samples has recently been developed. Qian et al. showed that the <u2> surrogate extracted using this method matched with <u2> data from neutron scattering for lyophilized powders of trehalose and sucrose at temperatures of interest for formulation development [93].
3.1.3. Degradation Rates and Transport
It is recently becoming clear that the faster relaxation processes observed in glasses, the strength of the βfast process, and the timescale of the βJG process are directly coupled to diffusive motions [61,94]. This provides insight as to why these dynamic parameters should provide a robust metric for predicting protein stability in solid formulations, since essentially all of the degradation processes of concern can be modeled as reactions, and such reactions are often transport limited at viscosity ranges typical for solid protein formulations [95]. In some cases, such as at slightly elevated water content, degradation processes involving water have been found to not be diffusion limited [96–98]. In such cases, one would not expect βfast relaxation metrics to track protein stability. On the other hand, degradation processes involving water often appear to be transport limited at lower water content, and water transport is found explicitly to be coupled to secondary relaxation in sugar glass. [76,94]
Furthermore, the fact that degradation reactions are transport limited provides clarification as to the physical origin of the variable relationship between α relaxation and degradation rates. Through the Stokes-Einstein relation (DT=kB T/6π η rH) and the prediction of Kramers’ theory in the high-friction limit (k∝η−1), one can write a series of generalized relationships between reaction rate, diffusion coefficient, viscosity, and τα:
| (3) |
where γ =1 at low and intermediate viscosity values where one expects reaction rates to be proportional to η or τα. As has been hinted at above, for viscosities that obtain in glasses, γ values are observed over the range 0.7<γ<1 [99], and the relationship between k or DT and τα can be dramatically material dependent [100].
There are perhaps two purposes for engineering dynamics and thermodynamics of a protein preservation matrix. One is to slow transport processes within the matrix. The other is to retard the dynamic processes of the protein itself.
3.2. Protein/Matrix Coupling
From dynamics studies of proteins in a trehalose glass, Hagen et al. [101] first showed that protein internal dynamics could be coupled to dynamic properties of a host matrix. In that work the authors suggested that protein dynamics were coupled with matrix viscosity, i.e., the longest timescale motions of the matrix. The generality of this conclusion was later questioned when it was shown that internal protein dynamics were suppressed more in glycerol at low temperatures than in trehalose [102,103], even at temperatures where glycerol is liquid and trehalose is a glass [102]. Using light scattering, Caliskan et al. [104] subsequently resolved this question by demonstrating that protein dynamics were coupled to high-frequency atomic motions of the solvent rather than the slower motions associated with viscosity. From molecular dynamics simulations, Dirama et al. [105] also found that protein and matrix were most strongly coupled at high frequency. They also found that the timescale and temperature dependence of atomic motions vary from the surface to the core of the protein, with the surface motions being much more sensitive to the solvent dynamics. These results were consistent with experiments of Gottfried et al. [106] who showed that trehalose glass did not completely suppress local dynamic processes deep within the protein, but did prevent conformational changes that involve relatively large-scale motion of surface residues.
The simulations of Dirama et al. [105] also showed that the protein-matrix dynamic coupling could be traced to the hydrogen bonds formed at the protein-matrix interface. Thus, the strength and number of these interactions will determine how well the vitrified matrix can slow the protein’s dynamics. This result provides an important connection between spectroscopic FTIR hydration signatures or secondary structure and protein stability in the glass. Better H-bonding seems to provide for more native-like secondary structure, but we hereby understand that it will also tie host and protein dynamics more tightly, slowing the dynamics of the protein, and more effectively stabilizing it.
FTIR has been the traditional method used to obtain information about the interactions between protein and matrix; however, the strength of these crucial enthalpic interactions may also be obtained through partial volume of mixing [107]. Extraction of interaction energies requires some assumptions, but in qualitatively comparing various mixtures with similar compositions, larger negative volumes of mixing indicate stronger enthalpic interactions. Using high-precision density measurements of lyophilized materials, Chieng et al. [54] found larger negative volume of mixing for hGH and sucrose than for hGH:trehalose systems. Correspondingly, they found that sucrose was a better stabilizer for hGH than trehalose [44,54], even though no difference was found between FTIR spectra of hGH in the sucrose of trehalose formulations. Similar correlation has been found between volumes of mixing and protein stability in formulations containing hydroxyethylstarch, disaccharide, and small amounts of either sorbitol or glycerol [53,108].
3.3. Protein Dynamics
Proteins are known to have a dynamical transition temperature (TD) below which internal motion is severely restricted. In low and moderate hydration conditions, this transition is usually seen in the range (200 to 230) K, and corresponds to onset of function [109,110]. In anhydrous conditions, and in the presence of a host material, TD can be significantly higher [111]. This dynamic transition has been likened to a glass transition temperature (Tg) [112,113], and seems to have phenomenological similarities. For example, differential scanning calorimetry (DSC) data on several formulations of protein dried in a disaccharide matrix showed a broad non-reversible endotherm near 60 °C with features characteristic of an enthalpy recovery peak found near the Tg of the matrix material (> 100 °C). To investigate whether this corresponded to a protein TD, kinetics of hydrogen-deuterium exchange (HDX) in freeze-dried BSA was used to evaluate the temperature dependence of the number of slowly exchanging protons, and it was found that there was a significant change in the temperature dependence of this value near 60 °C, consistent with the location of the DSC endotherm [111], confirming a dynamical transition within the protein. Similar experiments in our laboratory with crystalline insulin powder and freeze dried hGH gave similar results (unpublished data). This TD observed for dried proteins does seem to be an onset temperature for internal protein motion, and its value may reflect the level of stabilization of the protein. However, this measure of dynamics is rather new to the pharmaceutical field and thus far there have been no attempts to correlate functional stability with TD.
HDX has long been employed to study structure and mobility within proteins and recently has been adapted to studies of proteins in the solid state [64,114]. The methodology involves exposing the protein-containing solid at a given temperature to vapor from an aqueous solution of D2O/salt solution with a low water activity [114]. The extent of HDX is measured at various time points by FTIR [64], or by LC-Mass Spectroscopy [114]. Conditions are chosen so that equilibration with regard to water transfer from the solution to the solid protein system is sufficiently fast that a kinetic analysis can be used to separate the relative amounts of fast exchanging protons from slow exchanging protons. We point out that the introduction of small amounts of water (through the low D2O vapor pressure) may alter dynamics of different matrix materials in very different ways [75], so this must be taken into account when comparing results across multiple matrix materials.
Each protein will have some native-state characteristic ratio of fast to slow exchanging residues. An increase in that ratio is taken to indicate loss of native structure stability, and greater access of protein residues to reactive species in the matrix. Figure 5 compares the extent of aggregation after 360 days for various myoglobin formulations during stability studies at 25 °C and 40 °C with the fraction of fast exchanging protons, determined at time zero [114]. The aggregation data is also plotted against α-helix band intensity by FTIR for comparison. While there is some correlation between stability and FTIR structure, the correlation is far better with the HDX data.
Fig. 5.
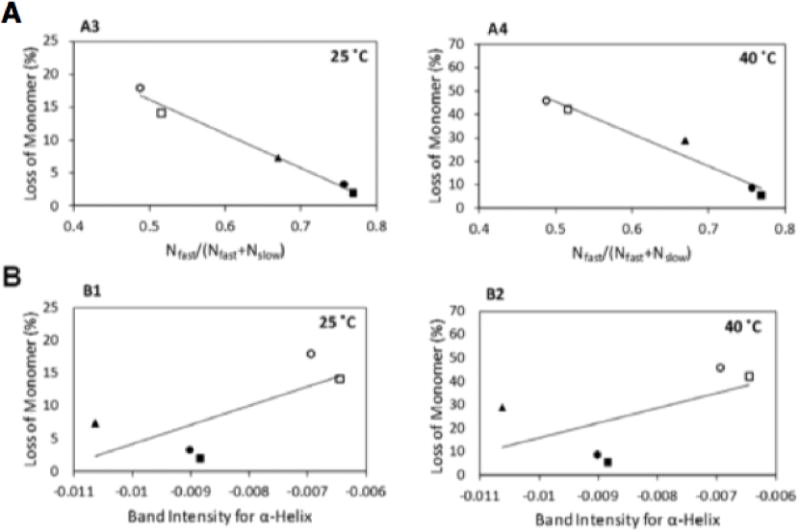
Myoglobin aggregation after storage for 360 days at 25 °C and 40 °C a) Correlation with fraction of rapidly exchanging protons by HDX. b) Correlation with FTIR spectral signature of α helix content [114]. Reproduced with permission
The ratio of fast and slow exchanging residues, averaged over the entire protein, can be determined through the ratio of infrared absorption bands associated with hydrogenated and deuterated vibrational oscillators as was done in Figure 5. However, dynamical information structurally resolved at the residue fragment level can be obtained through a mass spectroscopy based analysis procedure (HDX-MS) [37].
4. Other Factors
4.1. Phase separation
Phase separation can be deleterious to stability when the stabilizer phase separates from the protein, negating the intended benefit of the stabilizer [48,115,116]. Although studies are limited, solid state NMR appears ideal for investigation of phase separation in glassy protein formulations. The classical method for detecting phase separation is observation of multiple Tgs with differential scanning calorimetry (DSC). However, dry solid phases rich in protein normally do not show a Tg, so DSC is of limited utility for protein formulations [117]. Raman line mapping can also be used to detect phase separation in protein formulations, as long as the phase-separation domain size is larger than optical resolution limits of approximately 1 μm. Solid state NMR does not have this domain size limitation, and ssNMR can be extremely useful in demonstrating phase separation even with very small domain sizes well under the Raman line mapping capability [118].
4.2. Specific Surface Area
Dried protein products in common use, such as micrometer- or nanometer-sized [119] drug depots, freeze-dried cakes or spray-dried powders [120] can have very high specific surface areas (SSA). Surface regions tend to be enhanced in surface-active species, such as proteins, and depleted in other species, such as stabilizers. Consequently, significant fractions of the total protein load may be associated with interfaces, as frequently manifested in burst release. These effects could be particularly pronounced for formulations containing very low levels of protein or vaccine and high SSA.
Interfacial regions are also dynamically distinct from the bulk. Dynamics are typically enhanced (sped up) by many orders of magnitude [121] in these regions, which can extend on the order of 10 nm towards the bulk from the surface. Given the very different dynamic environment, and the potential lack of stabilizers, one might expect that protein at the surface would be less stable than that in the bulk, and this is observed [46,89].
Total protein load at the surface can be evaluated using electron spectroscopy for chemical analysis (ESCA) [46,89], and SSA can evaluated by Brunauer–Emmett– Teller (BET) analysis of nitrogen or krypton adsorption isotherm data [89]. The fraction of protein at the surface then follows directly from an estimate of the thickness of the surface region sensed by ESCA, which is typically (50 to 100) nm [46,89]. Using ESCA and BET, Abdul-Fattah et al. [89] evaluated a series of dried protein products containing an IgG(1) with SSA covering a 30-fold range, made by lyophilization, foam drying, and spray drying. They observed strong correlation between aggregation rate and fraction of protein at surface.
In a study on a model protein, BSA, Costantino et al. found a linear relationship between spray-dried powder surface area and monomer loss [20]. Subsequently, Xu et al. [46] published a study of the impact of formulation and processing on stability of freeze-dried hGH, where the stability results were compared with estimated percentage of protein at the surface. Figure 6 summarizes some of the results of that study. They found that aggregation after 16 weeks at 50 °C is correlated with fraction of surface protein. The trend of accumulated aggregation is linearly proportional to the fraction of total protein at the interface, and has a slope of nearly 1, suggesting that protein degradation at the interface is responsible for a significant fraction of the overall degradation. Similar results were found for sucrose and trehalose formulations, but with correlation lines of smaller slope than for HES. Low protein levels were used in this study (2% of the dried product), and efforts were made to generate products with high SSA, so that over 30% of the protein was in the interfacial region for some samples. Given the dominant effects that surface degradation can have, it seems prudent to seriously consider fraction of protein at the surface when interpreting stability trends for freeze dried or spray dried protein products.
Fig. 6.
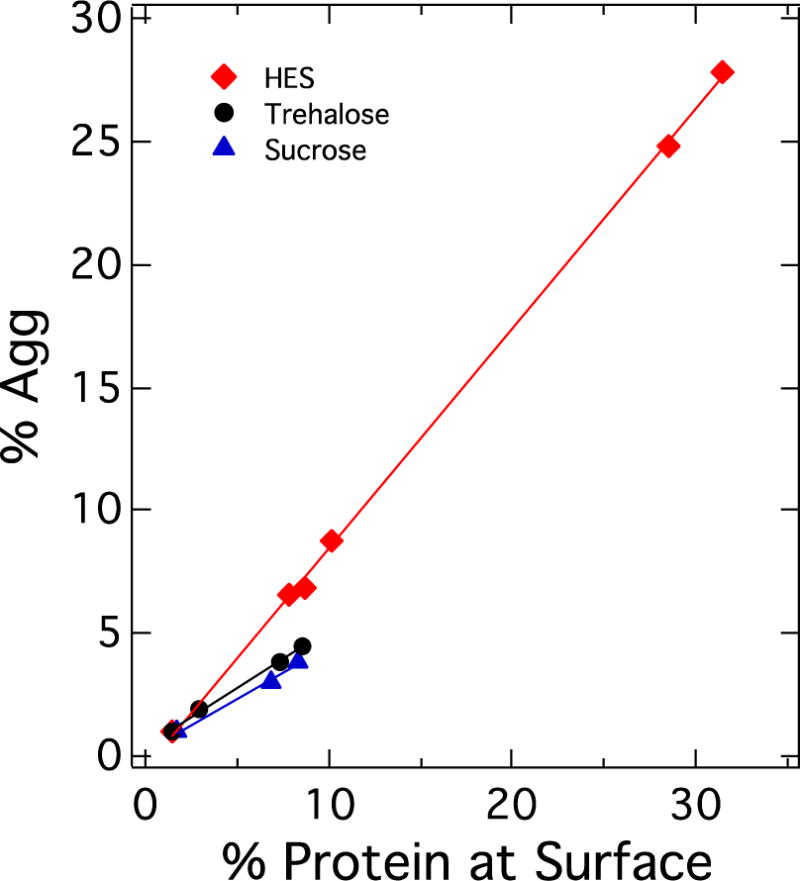
Correlation of hGH aggregation after 16 weeks at 50°C with fraction of protein in the interfacial region. Formulations are in stabilizer indicated, at 2% of total solids. Data points represent different freeze drying methods. Data taken from reference [46].
4.3. pH Effects
Changes in pH can have dramatic effects on conformational stability of proteins in solution, and pH considerations should figure prominently in preparing formulations for lyophilization. It is important to note that the product does not necessarily retain its initial solution pH during the entire lyophilization process, and this can lead to product instability. Differences in temperature-dependence of solubility for various members of a buffer salt family can lead to large swings in pH at low temperatures. Phosphate and succinate buffer systems are known to have these issues, with the latter exhibiting swings of up to 6 pH units during the freezing process [122]. Even if such large changes in pH are avoided during processing, effective pH can change in unexpected ways upon dehydration of the product.
Early studies indicated that molecular ionization states in the dried powders were similar to those in the original aqueous solution [123,124]. This was interpreted as the dried powder having the same effective pH as the solution, and termed “pH memory”, but it was later shown that, in many cases, the degree of ionization in the solid had more to do with the ionic strength of the solution than the pH [125]. Taking ionic strength into account, Li et al [126], showed that H+ activity may in fact change substantially upon drying from aqueous solution. The effective pH of lyophilized materials can be characterized through the Hammett acidity function [127], where the state of an acid-sensitive dye is used to determine the activity of H+ in the solid. Hammett acidity was shown as a good predictor of relative rates of acid-catalyzed chemical reactions in the solid state [127–129], and has been suggested as characterization tool to rank-order formulation stability for acid-sensitive molecules in dry formats [130].
5. Revised and Alternate Hypotheses
Historically the “water replacement” hypothesis has included the idea that the solid matrix (e.g., sugars) can recapitulate the function of water in thermodynamically stabilizing the native state of the protein. While the idea is pleasing and one can make arguments why it may be valid, we have several critiques of the hypothesis as formulated: 1) There is, at present, no convenient way to measure tertiary structure in dried form, making the hypothesis essentially untestable. 2) There is no evidence that anhydrous media can generally stabilize protein structure, with the apparently sole exception of lysozyme in glycerol [31]. 3) Changes in secondary structure and protein stability correlate well only under specific circumstances. There are numerous accounts of changes in protein stability with no change in spectroscopic markers of secondary structure. We can conclude that either stabilization of tertiary (not just secondary) structure is the causal factor in stabilizing proteins, or that there is at least one other causal factor, and native state stabilization may or may not be causal at all.
Given these critiques, we propose a modified hypothesis and several alternate hypotheses. The basis of a modified hypothesis could be that stabilization of secondary structure is necessary but not sufficient for pharmaceutical stabilization. An alternate hypothesis is that observed secondary structure changes are a coincidental side effect of changes in enthalpic coupling between matrix and protein, and it is the coupling that is the key factor vis a vis imposing the slow matrix dynamics on the protein. In this scenario, one would expect significant changes to stability when the fraction of the protein surface H-bonded to the matrix changes. Another alternate hypothesis, stated earlier in this work is that spectroscopic structure markers are simply snapshots of dynamics or degradation in process. In this scenario, one can view results of in situ conformational characterization at a given time as an indicator of how much the protein has unfolded or aggregated up to that point. There seems to be a basis for the idea that dynamic factors figure in results of secondary structure measurements, since time-dependent conformational changes in the solid from have been observed [88].
Modification of the dynamic hypothesis is straightforward. It is clear from fundamental transport theory, decades of experimentation on model systems, and results of many pharmaceutical stability studies that α relaxation does not, and should not, correlate reliably with degradation-related reactions in vitreous media. Rather, measures of transport should scale linearly with reaction rates in these systems. We now know that β relaxation processes are fundamentally connected with these transport processes, and we observe apparently universal relationships between appropriate measures of β relaxation and stablity. We modify the dynamic hypothesis to propose that, in the glassy state, suppressed β relaxation processes in the matrix slow transport of reactive species, and thus slow degradation. Further, suppressed β relaxation processes slow the dynamics of the guest protein further slowing its degradation processes, provided there is good dynamic coupling between the matrix and the protein, which is provided by strong enthalpic interactions – usually H-bonds.
6. Conclusions
It is crucial that proteins be encapsulated, stored, and released from biopharmaceutical formulations or drug delivery vehicles with minimal degradation. Even a few percent of the original protein load being degraded may be unacceptable, and acceptable limits may drop as more becomes known about immunogenicity of degraded protein. Platforms are being developed for drug delivery that shield proteins from harsh environments during processing and delivery. However, even with such measures, careful characterization of the dried protein-containing product is an imperative. Reduction of degraded protein to an acceptable level will undoubtedly involve careful formulation tailoring for each protein, and reliable, predictive metrics for protein stability are a key part of the formulation feedback process.
Based on data and discussion presented above, it appears that appropriate analytic capabilities for characterizing dried protein-containing products with respect to stability against degradation should include techniques for characterizing matrix-protein interaction strength, such as spectroscopic analysis of secondary structure, analysis of matrix β relaxation or transport properties, analysis of matrix phase homogeneity, analysis of protein fraction at the surface of the material, and measurement of protein dynamics in the solid form. Each of these metrics provides key information needed to ensure that the protein is well stabilized. We believe that as advanced approaches are used to formulate and characterize proteins in drug delivery appliations, the instability barrier that impedes most applications [1] can be overcome.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health (Grant: R01 EB006398-01A1)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Official contribution of the National Institute of Standards and Technology; not subject to copyright in the United States
References
- 1.Fu K, Klibanov AM, Langer R. Protein stability in controlled-release systems. Nat Biotech. 2000;18:24–25. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 2.Costantino H, Langer R, Klibanov A. Moisture-Induced Aggregation of Lyophilized Insulin. Pharm Res. 1994;11:21–29. doi: 10.1023/a:1018981208076. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi G, Gamba A, Murelli C, Salamini F, Bartels D. Novel carbohydrate metabolism in the resurrection plant Craterostigma plantagineum. The Plant Journal. 1991;1:355–359. doi: 10.1046/j.1365-313X.1991.t01-11-00999.x. [DOI] [PubMed] [Google Scholar]
- 4.Storey JM, Storey KB. Functional Metabolism: Regulation and Adaptation. John Wiley & Sons, Inc; 2004. pp. 473–503. [Google Scholar]
- 5.Crowe JH, Crowe LM, Carpenter JF, Wistrom CA. Stabilization of dry phospholipid bilayers and proteins by sugars. Biochemical Journal. 1987;242:1. doi: 10.1042/bj2420001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma X, Santiago N, Chen Y-S, Chaudhary K, Milstein SJ, Baughman RA. Stability Study of Drug-loaded Proteinoid Microsphere Formulations during Freeze-drying. Journal of Drug Targeting. 1994;2:9–21. doi: 10.3109/10611869409015889. [DOI] [PubMed] [Google Scholar]
- 7.Cleland J, Jones AS. Stable Formulations of Recombinant Human Growth Hormone and Interferon-γ for Microencapsulation in Biodegradable Mircospheres. Pharm Res. 1996;13:1464–1475. doi: 10.1023/a:1016063109373. [DOI] [PubMed] [Google Scholar]
- 8.Chow AL, Tong HY, Chattopadhyay P, Shekunov B. Particle Engineering for Pulmonary Drug Delivery. Pharm Res. 2007;24:411–437. doi: 10.1007/s11095-006-9174-3. [DOI] [PubMed] [Google Scholar]
- 9.Anchordoquy TJ, Koe GS. Physical stability of nonviral plasmid-based therapeutics. Journal of Pharmaceutical Sciences. 2000;89:289–296. doi: 10.1002/(SICI)1520-6017(200003)89:3<289::AID-JPS1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 10.Giri J, Li W-J, Tuan RS, Cicerone MT. Stabilization of Proteins by Nanoencapsulation in Sugar–Glass for Tissue Engineering and Drug Delivery Applications. Advanced Materials. 2011;23:4861–4867. doi: 10.1002/adma.201102267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clinical therapeutics. 2002;24:1720–1740. doi: 10.1016/s0149-2918(02)80075-3. [DOI] [PubMed] [Google Scholar]
- 12.Chirmule N, Jawa V, Meibohm B. Immunogenicity to Therapeutic Proteins: Impact on PK/PD and Efficacy. AAPS J. 2012;14:296–302. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg A. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006;8:E501–E507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cromwell MM, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J. 2006;8:E572–E579. doi: 10.1208/aapsj080366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiskoot W, van Schie RF, Carstens M, Schellekens H. Immunological Risk of Injectable Drug Delivery Systems. Pharm Res. 2009;26:1303–1314. doi: 10.1007/s11095-009-9855-9. [DOI] [PubMed] [Google Scholar]
- 16.Jiskoot W, Randolph TW, Volkin DB, Middaugh CR, Schöneich C, Winter G, Friess W, et al. Protein instability and immunogenicity: Roadblocks to clinical application of injectable protein delivery systems for sustained release. Journal of Pharmaceutical Sciences. 2012;101:946–954. doi: 10.1002/jps.23018. [DOI] [PubMed] [Google Scholar]
- 17.George M, Abraham TE. Polyionic hydrocolloids for the intestinal delivery of protein drugs: Alginate and chitosan — a review. Journal of Controlled Release. 2006;114:1–14. doi: 10.1016/j.jconrel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 18.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. The development of microgels/nanogels for drug delivery applications. Progress in Polymer Science. 2008;33:448–477. [Google Scholar]
- 19.Allison SD, Molina MD, Anchordoquy TJ. Stabilization of lipid/DNA complexes during the freezing step of the lyophilization process: the particle isolation hypothesis. Biochimica et Biophysica Acta (BBA) – Biomembranes. 2000;1468:127–138. doi: 10.1016/s0005-2736(00)00251-0. [DOI] [PubMed] [Google Scholar]
- 20.Costantino HR, Firouzabadian L, Wu C, Carrasquillo KG, Griebenow K, Zale SE, Tracy MA. Protein spray freeze drying. 2. Effect of formulation variables on particle size and stability. Journal of Pharmaceutical Sciences. 2002;91:388–395. doi: 10.1002/jps.10059. [DOI] [PubMed] [Google Scholar]
- 21.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Critical Reviews in Therapeutic Drug Carrier Systems. 2002;19 doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 22.Carpenter JF, Crowe JH. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry. 1989;28:3916–3922. doi: 10.1021/bi00435a044. [DOI] [PubMed] [Google Scholar]
- 23.Franks F, Hatley RHM, Mathias SF. Materials Science and the Production of Shelf-Stable Biologicals. Biopharm-the Technology & Business of Biopharmaceuticals. 1991;4:38–3. [Google Scholar]
- 24.Massari AM, Finkelstein IJ, McClain BL, Goj A, Wen X, Bren KL, Loring RF, et al. The Influence of Aqueous versus Glassy Solvents on Protein Dynamics: Vibrational Echo Experiments and Molecular Dynamics Simulations. J Am Chem Soc. 2005;127:14279–14289. doi: 10.1021/ja053627w. [DOI] [PubMed] [Google Scholar]
- 25.Francia F, Dezi M, Mallardi A, Palazzo G, Cordone L, Venturoli G. Protein−Matrix Coupling/Uncoupling in “Dry” Systems of Photosynthetic Reaction Center Embedded in Trehalose/Sucrose: The Origin of Trehalose Peculiarity. J Am Chem Soc. 2008;130:10240–10246. doi: 10.1021/ja801801p. [DOI] [PubMed] [Google Scholar]
- 26.Belton PS, Gil AM. IR and Raman spectroscopic studies of the interaction of trehalose with hen egg white lysozyme. Biopolymers. 1994;34:957–961. doi: 10.1002/bip.360340713. [DOI] [PubMed] [Google Scholar]
- 27.Cordone L, Cottone G, Giuffrida S. Role of residual water hydrogen bonding in sugar/water/biomolecule systems: a possible explanation for trehalose peculiarity. Journal of Physics: Condensed Matter. 2007;19:205110. [Google Scholar]
- 28.Klibanov AM. Improving enzymes by using them in organic solvents. Nature. 2001;409:241–246. doi: 10.1038/35051719. [DOI] [PubMed] [Google Scholar]
- 29.Knubovets T, Osterhout JJ, Klibanov AM. Structure of lysozyme dissolved in neat organic solvents as assessed by NMR and CD spectroscopies. BIOTECHNOLOGY AND BIOENGINEERING. 1999;63:242–248. [PubMed] [Google Scholar]
- 30.Zaks A, Klibanov AM. Enzymatic catalysis in nonaqueous solvents. Journal of Biological Chemistry. 1988;263:3194–3201. [PubMed] [Google Scholar]
- 31.Rariy RV, Klibanov AM. Correct protein folding in glycerol. Proceedings of the National Academy of Sciences. 1997;94:13520–13523. doi: 10.1073/pnas.94.25.13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myers JS, Jakoby WB. Glycerol as an agent eliciting small conformational changes in alcohol dehydrogenase. Journal of Biological Chemistry. 1975;250:3785–3789. [PubMed] [Google Scholar]
- 33.Arakawa T, Timasheff SN. Stabilization of protein structure by sugars. Biochemistry. 1982;21:6536–6544. doi: 10.1021/bi00268a033. [DOI] [PubMed] [Google Scholar]
- 34.Gekko K, Timasheff SN. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 1981;20:4667–4676. doi: 10.1021/bi00519a023. [DOI] [PubMed] [Google Scholar]
- 35.Allison SD, Chang B, Randolph TW, Carpenter JF. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Archives of Biochemistry and Biophysics. 1999;365:289–298. doi: 10.1006/abbi.1999.1175. [DOI] [PubMed] [Google Scholar]
- 36.Devineni D, Gonschorek C, Cicerone MT, Xu Y, Carpenter JF, Randolph TW. Storage stability of keratinocyte growth factor-2 in lyophilized formulations: Effects of formulation physical properties and protein fraction at the solid–air interface. European Journal of Pharmaceutics and Biopharmaceutics. 2014 doi: 10.1016/j.ejpb.2014.05.012. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Williams T, Topp E. Effects of Excipients on Protein Conformation in Lyophilized Solids by Hydrogen/Deuterium Exchange Mass Spectrometry. Pharm Res. 2008;25:259–267. doi: 10.1007/s11095-007-9365-6. [DOI] [PubMed] [Google Scholar]
- 38.Cleland JL, Lam X, Kendrick B, Yang J, Yang TH, Overcashier D, Brooks D, et al. A specific molar ratio of stabilizer to protein is required for storage stability of a lyophilized monoclonal antibody. Journal of Pharmaceutical Sciences. 2001;90:310–321. doi: 10.1002/1520-6017(200103)90:3<310::aid-jps6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 39.Wang BQ, Tchessalov S, Cicerone MT, Warne NW, Pikal MJ. Impact of Sucrose Level on Storage Stability of Proteins in Freeze-Dried Solids: II. Correlation of Aggregation Rate with Protein Structure and Molecular Mobility. Journal of Pharmaceutical Sciences. 2009;98:3145–3166. doi: 10.1002/jps.21622. [DOI] [PubMed] [Google Scholar]
- 40.Costantino HR, Carrasquillo KG, Cordero RA, Mumenthaler M, Hsu CC, Griebenow K. Effect of excipients on the stability and structure of lyophilized recombinant human growth hormone. Journal of Pharmaceutical Sciences. 1998;87:1412–1420. doi: 10.1021/js980069t. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka K, Takeda T, Miyajima K. Cryoprotective Effect of Saccharides on Denaturation of Catalase by Freeze-Drying. Chemical & Pharmaceutical Bulletin. 1991;39:1091–1094. [Google Scholar]
- 42.Prestrelski SJ, Tedeschi N, Arakawa T, Carpenter JF. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophysical Journal. 1993;65:661–671. doi: 10.1016/S0006-3495(93)81120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prestrelski SJ, Arakawa T, Carpenter JF. Separation of Freezing- and Drying-Induced Denaturation of Lyophilized Proteins Using Stress-Specific Stabilization: II. Structural Studies Using Infrared Spectroscopy. Archives of Biochemistry and Biophysics. 1993;303:465–473. doi: 10.1006/abbi.1993.1310. [DOI] [PubMed] [Google Scholar]
- 44.Pikal MJ, Rigsbee D, Roy ML, Galreath D, Kovach KJ, Wang BQ, Carpenter JF, et al. Solid State Chemistry of Proteins: II. The Correlation of Storage Stability of Freeze-Dried Human Growth Hormone (hGH) with Structure and Dynamics in the Glassy Solid. Journal of Pharmaceutical Sciences. 2008;97:5106–5121. doi: 10.1002/jps.21374. [DOI] [PubMed] [Google Scholar]
- 45.Prestrelski SJ, Pikal KA, Arakawa T. Optimization of lyophilization conditions for recombinant human interleukin-2 by dried-state conformational analysis using Fourier-transform infrared spectroscopy. Pharm Res. 1995;12:1250–1259. doi: 10.1023/a:1016296801447. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Grobelny P, Von Allmen A, Knudson K, Pikal M, Carpenter JF, Randolph TW. Protein Quantity on the Air–Solid Interface Determines Degradation Rates of Human Growth Hormone in Lyophilized Samples. Journal of Pharmaceutical Sciences. 2014;103:1356–1366. doi: 10.1002/jps.23926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Beer TRM, Vercruysse P, Burggraeve A, Quinten T, Ouyang J, Zhang X, Vervaet C, et al. In-line and real-time process monitoring of a freeze drying process using Raman and NIR spectroscopy as complementary process analytical technology (PAT) tools. Journal of Pharmaceutical Sciences. 2009;98:3430–3446. doi: 10.1002/jps.21633. [DOI] [PubMed] [Google Scholar]
- 48.Dong J, Hubel A, Bischof JC, Aksan A. Freezing-Induced Phase Separation and Spatial Microheterogeneity in Protein Solutions. The Journal of Physical Chemistry B. 2009;113:10081–10087. doi: 10.1021/jp809710d. [DOI] [PubMed] [Google Scholar]
- 49.Kauppinen A, Toiviainen M, Aaltonen J, Korhonen O, Järvinen K, Juuti M, Pellinen R, et al. Microscale Freeze-Drying with Raman Spectroscopy as a Tool for Process Development. Anal Chem. 2013;85:2109–2116. doi: 10.1021/ac3027349. [DOI] [PubMed] [Google Scholar]
- 50.Sane SU, Wong R, Hsu CC. Raman spectroscopic characterization of drying-induced structural changes in a therapeutic antibody: Correlating structural changes with long-term stability. Journal of Pharmaceutical Sciences. 2004;93:1005–1018. doi: 10.1002/jps.20014. [DOI] [PubMed] [Google Scholar]
- 51.Yu N-T, Jo BH, Liu CS. Laser Raman spectroscopic study of the effect of solvation on the conformation of ribonuclease A. J Am Chem Soc. 1972;94:7572–7575. doi: 10.1021/ja00776a048. [DOI] [PubMed] [Google Scholar]
- 52.Hedoux A, Paccou L, Achir S, Guinet Y. Mechanism of protein stabilization by trehalose during freeze-drying analyzed by in situ micro-raman spectroscopy. Journal of Pharmaceutical Sciences. 2013;102:2484–2494. doi: 10.1002/jps.23638. [DOI] [PubMed] [Google Scholar]
- 53.Chang LL, Shepherd D, Sun J, Ouellette D, Grant KL, Tang XC, Pikal MJ. Mechanism of protein stabilization by sugars during freeze-drying and storage: native structure preservation, specific interaction, and/or immobilization in a glassy matrix? J Pharm Sci. 2005;94:1427–1444. doi: 10.1002/jps.20364. [DOI] [PubMed] [Google Scholar]
- 54.Chieng N, Cicerone MT, Zhong Q, Liu M, Pikal MJ. Characterization of dynamics in complex lyophilized formulations: II. Analysis of density variations in terms of glass dynamics and comparisons with global mobility, fast dynamics, and Positron Annihilation Lifetime Spectroscopy (PALS) European Journal of Pharmaceutics and Biopharmaceutics. 2013;85:197–206. doi: 10.1016/j.ejpb.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens K Forney. Doctoral Thesis. University of Connecticut; 2013. Extending the Shelf-Life of Lyophilized Protein Formulations: Amino Acids as Stabilizers and Early Detection of Amorphous Phase Separation. [Google Scholar]
- 56.Hancock BC, Zografi G. Characteristics and significance of the amorphous state in pharmaceutical systems. Journal of Pharmaceutical Sciences. 1997;86:1–12. doi: 10.1021/js9601896. [DOI] [PubMed] [Google Scholar]
- 57.Yoshioka S, Aso Y. Correlations between molecular mobility and chemical stability during storage of amorphous pharmaceuticals. J Pharm Sci. 2007;96:960–981. doi: 10.1002/jps.20926. [DOI] [PubMed] [Google Scholar]
- 58.Hill JJ, Shalaev EY, Zografi G. The Importance of Individual Protein Molecule Dynamics in Developing and Assessing Solid State Protein Preparations. Journal of Pharmaceutical Sciences. 2014;103:2605–2614. doi: 10.1002/jps.24021. [DOI] [PubMed] [Google Scholar]
- 59.Debenedetti PG, Stillinger FH. Supercooled liquids and the glass transition. Nature. 2001;410:259–267. doi: 10.1038/35065704. [DOI] [PubMed] [Google Scholar]
- 60.Russina M, Mezei F, Lechner R, Longeville S, Urban B. Experimental evidence for fast heterogeneous collective structural relaxation in a supercooled liquid near the glass transition. Physical Review Letters. 2000;84:3630–3633. doi: 10.1103/PhysRevLett.84.3630. [DOI] [PubMed] [Google Scholar]
- 61.Cicerone MT, Zhong Q, Tyagi M. Picosecond Dynamic Heterogeneity, Hopping, and Johari-Goldstein Relaxation in Glass-Forming Liquids. Physical Review Letters. 2014;113:117801–117801. doi: 10.1103/PhysRevLett.113.117801. [DOI] [PubMed] [Google Scholar]
- 62.Johari GP, Goldstein M. Viscous Liquids and the Glass Transition. III. Secondary Relaxations in Aliphatic Alcohols and Other Nonrigid Molecules. The Journal of Chemical Physics. 1971;55:4245–4252. [Google Scholar]
- 63.Williams G, Watts DC. Non-symmetrical dielectric relaxation behaviour arising from a simple empirical decay function. Trans Faraday Soc. 1970;66:80–85. [Google Scholar]
- 64.Roy ML, Pikal MJ, Rickard EC, Maloney AM. The effects of formulation and moisture on the stability of a freeze-dried monoclonal antibody-vinca conjugate: a test of the WLF glass transition theory. Developments in biological standardization. 1992;74:323–39. discussion 340-. [PubMed] [Google Scholar]
- 65.Streefland L, Auffret A, Franks F. Bond Cleavage Reactions in Solid Aqueous Carbohydrate Solutions. Pharm Res. 1998;15:843–849. doi: 10.1023/a:1011912228954. [DOI] [PubMed] [Google Scholar]
- 66.Duddu S, Dal Monte P. Effect of Glass Transition Temperature on the Stability of Lyophilized Formulations Containing a Chimeric Therapeutic Monoclonal Antibody. Pharm Res. 1997;14:591–595. doi: 10.1023/a:1012144810067. [DOI] [PubMed] [Google Scholar]
- 67.Bell LN, Hageman MJ, Muraoka LM. Thermally induced denaturation of lyophilized bovine somatotropin and lysozyme as impacted by moisture and excipients. Journal of Pharmaceutical Sciences. 1995;84:707–712. doi: 10.1002/jps.2600840608. [DOI] [PubMed] [Google Scholar]
- 68.Sun WQ, Davidson P, Chan HSO. Protein stability in the amorphous carbohydrate matrix: relevance to anhydrobiosis. Biochimica et Biophysica Acta (BBA) – General Subjects. 1998;1425:245–254. doi: 10.1016/s0304-4165(98)00077-4. [DOI] [PubMed] [Google Scholar]
- 69.Buitink J, van den Dries IJ, Hoekstra FA, Alberda M, Hemminga MA. High critical temperature above T<sub>g</sub> may contribute to the stability of biological systems. Biophysical Journal. 2000;79:1119–1128. doi: 10.1016/S0006-3495(00)76365-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinrichs WLJ, Prinsen MG, Frijlink HW. Inulin glasses for the stabilization of therapeutic proteins. International Journal of Pharmaceutics. 2001;215:163–174. doi: 10.1016/s0378-5173(00)00677-3. [DOI] [PubMed] [Google Scholar]
- 71.Williams ML, Landel RF, Ferry JD. The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-forming Liquids. J Am Chem Soc. 1955;77:3701–3707. [Google Scholar]
- 72.Angell CA. Relaxation in liquids, polymers and plastic crystalsstrong/fragile patterns and problems. Journal of Non-Crystalline Solids. 1991;131:13–31. [Google Scholar]
- 73.Kauzmann W. The Nature of the Glassy State and the Behavior of Liquids at Low Temperatures. Chemical Reviews. 1948;43:219–256. [Google Scholar]
- 74.Rey L. Freeze-drying. Marcel Dekker; New York: 2004. [Google Scholar]
- 75.Green JL, Angell CA. Phase relations and vitrification in saccharide-water solutions and the trehalose anomaly. J Phys Chem. 1989;93:2880–2882. [Google Scholar]
- 76.Cicerone MT, Douglas JF. β-Relaxation governs protein stability in sugar-glass matrices. Soft Matter. 2012;8:2983. [Google Scholar]
- 77.Kilburn D, Claude J, Schweizer T, Alam A, Ubbink J. Carbohydrate Polymers in Amorphous States: An Integrated Thermodynamic and Nanostructural Investigation. Biomacromolecules. 2005;6:864–879. doi: 10.1021/bm049355r. [DOI] [PubMed] [Google Scholar]
- 78.Kanaya T, Tsukushi T, Kaji K, Bartos J, Kristiak J. Microscopic basis of free-volume concept as studied by quasielastic neutron scattering and positron annihilation lifetime spectroscopy. Physical Review E. 1999;60:1906–1912. doi: 10.1103/physreve.60.1906. [DOI] [PubMed] [Google Scholar]
- 79.Bartoš J, Iskrová M, Köhler M, Wehn R, Sauša O, Lunkenheimer P, Krištiak J, et al. Positron annihilation response and broadband dielectric spectroscopy: salol. Eur Phys J E Soft Matter. 2011;34:104. doi: 10.1140/epje/i2011-11104-x. [DOI] [PubMed] [Google Scholar]
- 80.Ngai KL. Relaxation and Diffusion in Complex Systems. Springer; Berlin, New York: 2010. [Google Scholar]
- 81.Duddu SP, Zhang GZ, DalMonte PR. The relationship between protein aggregation and molecular mobility below the glass transition temperature of lyophilized formulations containing a monoclonal antibody. Pharmaceutical Research. 1997;14:596–600. doi: 10.1023/a:1012196826905. [DOI] [PubMed] [Google Scholar]
- 82.Liu J, Rigsbee DR, Stotz C, Pikal MJ. Dynamics of pharmaceutical amorphous solids: The study of enthalpy relaxation by isothermal microcalorimetry. Journal of Pharmaceutical Sciences. 2002;91:1853–1862. doi: 10.1002/jps.10181. [DOI] [PubMed] [Google Scholar]
- 83.Kawakami K, Pikal MJ. Calorimetric investigation of the structural relaxation of amorphous materials: evaluating validity of the methodologies. J Pharm Sci. 2005;94:948–965. doi: 10.1002/jps.20298. [DOI] [PubMed] [Google Scholar]
- 84.Chang L, Shepherd D, Sun J, Tang X, Pikal MJ. Effect of sorbitol and residual moisture on the stability of lyophilized antibodies: Implications for the mechanism of protein stabilization in the solid state. Journal of Pharmaceutical Sciences. 2005;94:1445–1455. doi: 10.1002/jps.20363. [DOI] [PubMed] [Google Scholar]
- 85.Cicerone MT, Soles CL. Fast dynamics and stabilization of proteins: Binary glasses of trehalose and glycerol. Biophysical Journal. 2004;86:3836–3845. doi: 10.1529/biophysj.103.035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Psurek T, Soles CL, Page KA, Cicerone MT, Douglas JF. Quantifying Changes in the High-Frequency Dynamics of Mixtures by Dielectric Spectroscopy. Journal of Physical Chemistry B. 2008;112:15980–15990. doi: 10.1021/jp8034314. [DOI] [PubMed] [Google Scholar]
- 87.Yoshioka S, Miyazaki T, Aso Y. beta-relaxation of insulin molecule in lyophilized formulations containing trehalose or dextran as a determinant of chemical reactivity. Pharmaceutical Research. 2006;23:961–966. doi: 10.1007/s11095-006-9907-3. [DOI] [PubMed] [Google Scholar]
- 88.Wang B, Cicerone MT, Aso Y, Pikal MJ. The Impact of Thermal Treatment on the Stability of Freeze-Dried Amorphous Pharmaceuticals: II. Aggregation in an IgG1 Fusion Protein. Journal of Pharmaceutical Sciences. 2010;99:683–700. doi: 10.1002/jps.21960. [DOI] [PubMed] [Google Scholar]
- 89.Abdul-Fattah AM, Truong-Le V, Yee L, Nguyen L, Kalonia DS, Cicerone MT, Pikal MJ. Drying-induced variations in physico-chemical properties of amorphous pharmaceuticals and their impact on stability (I): Stability of a monoclonal antibody. Journal of Pharmaceutical Sciences. 2007;96:1983–2008. doi: 10.1002/jps.20859. [DOI] [PubMed] [Google Scholar]
- 90.Yoshioka S, Forney K, Aso Y, Pikal M. Effect of Sugars on the Molecular Motion of Freeze-Dried Protein Formulations Reflected by NMR Relaxation Times. Pharm Res. 2011;28:3237–3247. doi: 10.1007/s11095-011-0512-8. [DOI] [PubMed] [Google Scholar]
- 91.Luthra SA, Hodge IM, Utz M, Pikal MJ. Correlation of annealing with chemical stability in lyophilized pharmaceutical glasses. Journal of Pharmaceutical Sciences. 2008;97:5240–5251. doi: 10.1002/jps.21391. [DOI] [PubMed] [Google Scholar]
- 92.Cicerone MT, Zhong Q, Johnson J, Aamer KA, Tyagi M. Surrogate for Debye– Waller Factors from Dynamic Stokes Shifts. The Journal of Physical Chemistry Letters. 2011;2:1464–1468. doi: 10.1021/jz200490h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qian KK, Grobelny PJ, Tyagi M, Cicerone MT. Using the Fluorescence Red Edge Effect to Assess the Long-Term Stability of Lyophilized Protein Formulations. Mol Pharmaceutics. 2015;12:1141–1149. doi: 10.1021/mp500641f. [DOI] [PubMed] [Google Scholar]
- 94.Molinero V, Goddard WA. Microscopic Mechanism of Water Diffusion in Glucose Glasses. Phys Rev Lett. 2005;95:045701. doi: 10.1103/PhysRevLett.95.045701. [DOI] [PubMed] [Google Scholar]
- 95.Craig ID, Parker R, Rigby NM, Cairns P, Ring SG. Maillard Reaction Kinetics in Model Preservation Systems in the Vicinity of the Glass Transition: Experiment and Theory. J Agric Food Chem. 2001;49:4706–4712. doi: 10.1021/jf0100752. [DOI] [PubMed] [Google Scholar]
- 96.Yoshioka S, Aso Y, Kojima S. Temperature dependence of bimolecular reactions associated with molecular mobility in lyophilized formulations. Pharmaceutical research. 2000;17:925–929. doi: 10.1023/a:1007566919000. [DOI] [PubMed] [Google Scholar]
- 97.Luthra SA, Shalaev EY, Medek A, Hong J, Pikal MJ. Chemical stability of amorphous materials: specific and general media effects in the role of water in the degradation of freeze-dried zoniporide. J Pharm Sci. 2012;101:3110–3123. doi: 10.1002/jps.23128. [DOI] [PubMed] [Google Scholar]
- 98.Ohtake S, Shalaev E. Effect of water on the chemical stability of amorphous pharmaceuticals: I. Small molecules. J Pharm Sci. 2013;102:1139–1154. doi: 10.1002/jps.23440. [DOI] [PubMed] [Google Scholar]
- 99.Guo YS, Bryn SR, Zografi G. Physical characteristics and chemical degradation of amorphous quinapril hydrochloride. Journal of Pharmaceutical Sciences. 2000;89:128–143. doi: 10.1002/(SICI)1520-6017(200001)89:1<128::AID-JPS13>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 100.Cicerone MT, Wagner PA, Ediger MD. Translational diffusion on heterogeneous lattices: A model for dynamics in glass forming materials. Journal of Physical Chemistry B. 1997;101:8727–8734. [Google Scholar]
- 101.Hagen SJ, Hofrichter J, Eaton WA. Protein reaction kinetics in a room-temperature glass. Science. 1995;269:959–962. doi: 10.1126/science.7638618. [DOI] [PubMed] [Google Scholar]
- 102.Sastry GM, Agmon N. Trehalose Prevents Myoglobin Collapse and Preserves Its Internal Mobility†. Biochemistry. 1997;36:7097–7108. doi: 10.1021/bi9626057. [DOI] [PubMed] [Google Scholar]
- 103.Schlichter J, Friedrich J, Herenyi L, Fidy J. Trehalose Effect on Low Temperature Protein Dynamics: Fluctuation and Relaxation Phenomena. Biophysical Journal. 2001;80:2011–2017. doi: 10.1016/S0006-3495(01)76171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caliskan G, Mechtani D, Roh JH, Kisliuk A, Sokolov AP, Azzam S, Cicerone MT, et al. Protein and solvent dynamics: How strongly are they coupled? Journal of Chemical Physics. 2004;121:1978–1983. doi: 10.1063/1.1764491. [DOI] [PubMed] [Google Scholar]
- 105.Dirama TE, Curtis JE, Carri GA, Sokolov AP. Coupling between lysozyme and trehalose dynamics: Microscopic insights from molecular-dynamics simulations. Journal of Chemical Physics. 2006;124 doi: 10.1063/1.2159471. [DOI] [PubMed] [Google Scholar]
- 106.Gottfried DS, Peterson ES, Sheikh AG, Wang J, Yang M, Friedman JM. Evidence for Damped Hemoglobin Dynamics in a Room Temperature Trehalose Glass. J Phys Chem. 1996;100:12034–12042. [Google Scholar]
- 107.Bhatia AB, Singh RN. Volume of mixing of compound forming molten alloys. Physics Letters A. 1980;78:460–462. [Google Scholar]
- 108.Cicerone MT, Soles CL, Chowdhuri Z, Pikal MJ, Chang L. Fast dynamics as a diagnostic for excipients in preservation of dried proteins. American Pharmaceutical Review. 2005;8:197–206. [Google Scholar]
- 109.Doster W, Cusack S, Petry W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 1989;337:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 110.Tsai AM, Neumann DA, Bell LN. Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study. Biophysical journal. 2000;79:2728–2732. doi: 10.1016/S0006-3495(00)76511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mizuno M, Pikal MJ. Is the pre-Tg DSC endotherm observed with solid state proteins associated with the protein internal dynamics? Investigation of bovine serum albumin by solid state hydrogen/deuterium exchange. European Journal of Pharmaceutics and Biopharmaceutics. 2013;85:170–176. doi: 10.1016/j.ejpb.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 112.Iben IET, Braunstein D, Doster W, Frauenfelder H, Hong MK, Johnson JB, Luck S, et al. Glassy behavior of a protein. Physical Review Letters. 1989;62:1916–1919. doi: 10.1103/PhysRevLett.62.1916. [DOI] [PubMed] [Google Scholar]
- 113.Hill JJ, Shalaev EY, Zografi G. Thermodynamic and dynamic factors involved in the stability of native protein structure in amorphous solids in relation to levels of hydration. Journal of Pharmaceutical Sciences. 2005;94:1636–1667. doi: 10.1002/jps.20333. [DOI] [PubMed] [Google Scholar]
- 114.Moorthy BS, Schultz SG, Kim SG, Topp EM. Predicting Protein Aggregation during Storage in Lyophilized Solids Using Solid State Amide Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS) Mol Pharmaceutics. 2014;11:1869–1879. doi: 10.1021/mp500005v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Piedmonte D, Summers C, McAuley A, Karamujic L, Ratnaswamy G. Sorbitol Crystallization Can Lead to Protein Aggregation in Frozen Protein Formulations. Pharm Res. 2007;24:136–146. doi: 10.1007/s11095-006-9131-1. [DOI] [PubMed] [Google Scholar]
- 116.Schwegman JJ, Carpenter JF, Nail SL. Evidence of partial unfolding of proteins at the ice/freeze-concentrate interface by infrared microscopy. Journal of Pharmaceutical Sciences. 2009;98:3239–3246. doi: 10.1002/jps.21843. [DOI] [PubMed] [Google Scholar]
- 117.Padilla AM, Pikal MJ. The study of phase separation in amorphous freeze-dried systems, part 2: Investigation of raman mapping as a tool for studying amorphous phase separation in freeze-dried protein formulations. Journal of Pharmaceutical Sciences. 2011;100:1467–1474. doi: 10.1002/jps.22380. [DOI] [PubMed] [Google Scholar]
- 118.Clauss J, Schmidt-Rohr K, Spiess HW. Determination of domain sizes in heterogeneous polymers by solid-state NMR. Acta Polymerica. 1993;44:1–17. [Google Scholar]
- 119.Jawahar N, Reddy G. Nanoparticles: A Novel Pulmonary Drug Delivery System for Tuberculosis. J Pharm Sci Res. 2012;4:1901–1906. [Google Scholar]
- 120.Ameri M, Maa Y-F. Spray Drying of Biopharmaceuticals: Stability and Process Considerations. Drying Technology. 2006;24:763–768. [Google Scholar]
- 121.Fakhraai Z, Forrest JA. Measuring the Surface Dynamics of Glassy Polymers. Science. 2008;319:600–604. doi: 10.1126/science.1151205. [DOI] [PubMed] [Google Scholar]
- 122.Sundaramurthi P, Shalaev E, Suryanarayanan R. “pH Swing” in Frozen Solutions—Consequence of Sequential Crystallization of Buffer Components. J Phys Chem Lett. 2010;1:265–268. doi: 10.1021/jp100531v. [DOI] [PubMed] [Google Scholar]
- 123.Klibanov AM. Enzymatic catalysis in anhydrous organic solvents. Trends in Biochemical Sciences. 1989;14:141–144. doi: 10.1016/0968-0004(89)90146-1. [DOI] [PubMed] [Google Scholar]
- 124.Costantino HR, Griebenow K, Langer R, Klibanov AM. On the pH memory of lyophilized compounds containing protein functional groups. Biotechnology and bioengineering. 1997;53:345–348. doi: 10.1002/(SICI)1097-0290(19970205)53:3<345::AID-BIT14>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 125.Zacharis E, Halling PJ, Rees DG. Volatile buffers can override the “pH memory” of subtilisin catalysis in organic media. Proceedings of the National Academy of Sciences. 1999;96:1201–1205. doi: 10.1073/pnas.96.4.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li J, Chatterjee K, Medek A, Shalaev E, Zografi G. Acid–base characteristics of bromophenol blue–citrate buffer systems in the amorphous state. Journal of pharmaceutical sciences. 2004;93:697–712. doi: 10.1002/jps.10580. [DOI] [PubMed] [Google Scholar]
- 127.Chatterjee K, Shalaev EY, Suryanarayanan R, Govindarajan R. Correlation between chemical reactivity and the Hammett acidity function in amorphous solids using inversion of sucrose as a model reaction. Journal of pharmaceutical sciences. 2008;97:274–286. doi: 10.1002/jps.21081. [DOI] [PubMed] [Google Scholar]
- 128.Alkhamis KA. Influence of solid-state acidity on the decomposition of sucrose in amorphous systems (I) International journal of pharmaceutics. 2008;362:74–80. doi: 10.1016/j.ijpharm.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 129.Hailu SA, Bogner RH. Solid-state surface acidity and pH-stability profiles of amorphous quinapril hydrochloride and silicate formulations. Journal of pharmaceutical sciences. 2010;99:2786–2799. doi: 10.1002/jps.22051. [DOI] [PubMed] [Google Scholar]
- 130.Shalaev EY, Wang W, Gatlin LA. Rational choice of excipients for use in lyophilized formulations. Drugs and the pharmaceutical sciences. 2008;175:197. [Google Scholar]