

Abstract
The presence of amyloid plaques composed of amyloid beta (Aβ) fibrils is a hallmark of Alzheimer’s disease (AD). The Aβ peptide is present as several length variants with two common alloforms consisting of 40 and 42 amino acids, denoted Aβ1–40 and Aβ1–42, respectively. While there have been numerous reports that structurally characterize fibrils of Aβ1–40, very little is known about the structure of amyloid fibrils of Aβ1–42, which are considered the more toxic alloform involved in AD. We have prepared isotopically 13C/15N labeled AβM01–42 fibrils in vitro from recombinant protein and examined their 13C–13C and 13C–15N magic angle spinning (MAS) NMR spectra. In contrast to several other studies of Aβ fibrils, we observe spectra with excellent resolution and a single set of chemical shifts, suggesting the presence of a single fibril morphology. We report the initial structural characterization of AβM01–42 fibrils utilizing 13C and 15N shift assignments of 38 of the 43 residues, including the backbone and side chains, obtained through a series of cross-polarization based 2D and 3D 13C–13C, 13C–15N MAS NMR experiments for rigid residues along with J-based 2D TOBSY experiments for dynamic residues. We find that the first ∼5 residues are dynamic and most efficiently detected in a J-based TOBSY spectrum. In contrast, residues 16–42 are easily observed in cross-polarization experiments and most likely form the amyloid core. Calculation of ψ and φ dihedral angles from the chemical shift assignments indicate that 4 β-strands are present in the fibril’s secondary structure.
Introduction
Protein misfolding and aggregation and the subsequent formation of amyloid fibrils is established as part of the pathology of over 40 human diseases,1,2 including Creutzfeldt-Jakob disease,3,4 Parkinson’s disease,5−10 dialysis related amyloidosis,11,12 type II diabetes,13,14 Huntington’s disease,15 and Alzheimer’s disease (AD).16−18 Of these, AD is probably the most prevalent and devastating of the neurodegenerative diseases. For example, in the US there are currently about 5.2 million AD patients. In addition to the enormous personal suffering, the cost associated with care for these individuals is $214 billion annually. By 2050 these numbers are projected to increase to 16 million patients and a cost of $1.2 trillion. There is therefore an urgent need for new therapeutic or diagnostic approaches for the treatment of AD and for a fundamental understanding of the underlying chemical and structural biology.
One of the hallmarks of AD is the accumulation of amyloidogenic senile plaques found in Alzheimer’s patients consisting of fibrils composed of β-amyloid protein (Aβ), a peptide with 39–43 residues, that is produced from cleavage of the amyloid precursor protein (APP) by β- and γ-secretases.19,20 Among the most prevalent alloforms are peptides with 40 (Aβ1–40) and 42 (Aβ1–42) amino acid residues with the latter identified as the more toxic species that possesses a significantly higher aggregation propensity and as a result nucleates fibril formation.21−23 In addition to AD, the toxic effects of Aβ are also linked to Down’s syndrome (trisomy 21), a genetic disease leading to intellectual impairment and diminished physical growth and elevated risk of early on-set AD, as the gene for Aβ is located on chromosome 21.24 Aβ1–40 aggregates have also been shown to act as prions possessing transmissibility, with Aβ1–40 prions containing cerebral deposits possessing Aβ1–40 and Aβ1–42, while Aβ1–42 prions form smaller amyloid deposits consisting of mostly Aβ1–42.25−27 Moreover, Aβ fibrils present reactive surfaces for secondary nucleation and generation of toxic species from monomers in a fibril-catalyzed reaction.28−31 Elucidating the structural details of Aβ1–42 fibrils is therefore an important first step toward understanding this autocatalytic process. In addition, structures can guide the rational design of diagnostic and therapeutic tools with which to diagnose and treat AD, and potentially Down’s syndrome as well.
Unfortunately, Aβ fibrils are insoluble and do not diffract to high resolution, rendering conventional tools for biological structure determination such as solution NMR spectroscopy and X-ray diffraction currently incapable of characterizing samples such as these. Fortunately, magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy has proven to be a powerful technique to elucidate the structural details of amyloid fibrils on an atomic level, including backbone conformations, supramolecular organization, and registry of interstrand arrangements of amyloid fibrils.32 Using this approach, we have recently determined the high-resolution structure of amyloid fibrils formed by a small peptide within transthyretin (TTR105–115) utilizing the combination of cryo-electron microscopy (cryo-EM) and MAS NMR spectra.33,34 Similar approaches have been used to determine the structure of Aβ1–40 carrying the Osaka mutation (E22Δ)35,36 and Het-S.37−39
The initial MAS NMR characterization of an amyloid was an Aβ fragment consisting of the residues Aβ34–42 and revealed an antiparallel cross-β structure.40 Subsequently, investigations of Aβ10–3541,42 lead to a structural model where the basic subunit of the fibril consists of two Aβ10–35 monomers that possess parallel in-register organization and is consistent with the EM, STEM, and MAS NMR experiments.43 Following these investigations, a number of studies were devoted to investigating the structural features of Aβ with different fibril morphologies, and several structural models were proposed.40,43−58 One of the commonalities present in these structural models is a hairpin conformation consisting of two β-strands connected by a flexible loop. Tycko and co-workers have utilized mass per unit length (MPL) measurements of different polymorphs of Aβ prepared from peptide synthesized protein with specific amino acids uniformly 13C and 15N labeled finding that different polymorphs were found to be two or 3-fold symmetric.55,59 They have also presented data consistent with a parallel-in-register alignment of the strands in the sheet. In addition, FS-REDOR69 experiments indicate that a salt bridge exists between D23 and K28, which may help to stabilize the loop region within the fibrils.45 More recently, Bertini et al. characterized AβM01–40 fibrils of recombinant protein with all residues 13C and 15N labeled, which were prepared under different conditions from those extensively studied by Tycko and co-workers.49 In that report, the authors observed spectra consistent with a different fibril form, which possessed 2-fold symmetry. Additionally, Reif and co-workers have also characterized AβM01–40 prepared from recombinant material under conditions slightly different from Bertini and Tycko and proposed that an asymmetric dimer is the basic subunit of AβM01–40 fibrils.50
Various site-specific mutations of Aβ1–40 are related to familial or early onset AD, including the Arctic mutant (E22G), the Iowa mutant (D23N), and the Osaka mutant (E22Δ). As a result, significant attention has also been devoted to determining the aggregation mechanism60 and the structure of the various mutants of Aβ1–40. Tycko and co-workers reported results indicating that D23N Aβ1–40 fibrils are polymorphic, with some having parallel interstrand arrangements, while others have antiparallel interstrand arrangements.61,62 Later, they showed that through multiple rounds of seeding the sample yields a single polymorph.63 Recently, a high resolution structure of the E22Δ Osaka mutant was reported by Huber et al.35 and Schuetz et al.36 Following assignments of the amyloid fibrils the authors utilized a variety of isotope labeling schemes and recoupling sequences to generate an excellent high resolution structure with a number of intra- and intermolecular constraints.35,36 The structure proposed for the E22Δ mutant is substantially different from other models proposed for Aβ1–40. In particular, while the Osaka structure has strands that are parallel in register, the strands are intercalated in a “cinnamon roll” arrangement rather than forming a simple hairpin proposed in models for wild-type Aβ1–40
In comparison with the extensive studies of Aβ1–40, little is known about the structure of Aβ1–42 fibrils,64−68 which are established to be more toxic,69 and possess different aggregation properties in comparison with Aβ1–40.30 Furthermore, genetic mutations that correlate with a predisposition to AD are known to result in elevated levels of Aβ1–42.70 To date, the characterization of Aβ1–42 has largely been limited to H/D exchange and cryo-EM studies,71 which provide valuable information about the fibrils, but do not provide atomic level structural and distance information. Although a sparse number of MAS NMR studies have been reported on Aβ1–42 fibrils,71 a complete structure has yet to be elucidated. Because of the propensity for Aβ1–42 to aggregate rapidly, it is challenging to produce homogeneous fibril samples that can yield high quality MAS NMR spectra with sufficient spectral resolution permitting structural characterization. Additionally, the differences in behavior of the two alloforms, their distinct morphologies as observed by cryo-EM and low propensity to form joint fibrils72 may be indicative of a different structure at short length scale, which propagates into the higher order morphology with shorter and more tightly twisted AβM01–42 compared to AβM01–40 fibrils,72 and alternate pathways for aggregation.30 We have therefore focused our efforts on studies of AβM01–42 and employed a preparation method that utilized size exclusion chromatography (SEC) to isolate monomeric AβM01–42. This is followed by fibrilization yielding samples that exhibit high quality MAS NMR spectra that allow for a more complete characterization of the structure of the fibrils. Furthermore, this approach to sample preparation has recently been shown to result in reproducible aggregation kinetics.73 Herein, we use MAS NMR along with a new sample preparation to explore the structure of AβM01–42 fibrils with the goals of first assigning the MAS spectra and, subsequently, in a future publication reporting the fibril structure.
Results and Discussion
The procedures used in the protein expression and purification are described in the methods section below with details provided elsewhere.73 Briefly, the peptide was purified from inclusion bodies using ion exchange chromatography in batch format followed by two rounds of size exclusion chromatography to isolate pure AβM01–42 monomers. Fibrils formed spontaneously when this solution was kept at room temperature at pH 8 overnight. An EM of the resultant fibrils, which have been lyophilized to allow for resuspension in a smaller volume before packing into the rotor, is shown in Figure 1. The fibrils have relatively uniform twist distance and thickness, with an average length of ca. 300 nm, with no amorphous aggregates present and only one morphology observed. No significant difference is found relative to fresh fibril samples.29,72 For a typical β-sheet interpeptide spacing of ca. 0.5 nm, this means that less than 0.2% of the peptide monomer units are at the fibril ends, and the NMR spectra acquired will reliably report on the core structure of the fibrils. The length of the fibrils observed in the EM is consistent with the observation that Aβ1–42 fibrils usually are shorter than Aβ1–40 fibrils.27,68
Figure 1.

Negative stain transmission micrograph of AβM01–42 fibrils.
We began structurally characterizing the AβM01–42 fibrils by recording 1D spectra at ω0H/2π = 800 MHz illustrated in Figure 2, which shows (a) 13C cross-polarization, (b) 13C J-based INEPT transfers, and (c) 15N cross-polarization. We found that seeding subsequent samples is successful and we observe the identical chemical shifts from one sample preparation to another, producing identical 1D and 2D spectra. The appearance of significant intensity in both the CP- and J-based experiments indicate that both rigid and dynamic residues are present within the AβM01–42 fibrils. Similar to the 13C spectra, the 15N 1D spectra display sharp peaks, particularly within the amide region, as well as the side chains of lysine and arginine. The breadth of the 15Nδ1/ε2 His side chain lines in our metal-ion depleted samples is likely due to those residues undergoing dynamics on the NMR time scale, most likely proton exchange. In preliminary experiments at 150 K we observe that the line widths decrease consistent with this hypothesis. The 15N spectrum is typically a sensitive indicator of the order within amyloid proteins, and taken in its totality suggests that our samples are microscopically well ordered, and typically associated with high quality MAS NMR spectra.
Figure 2.

1D MAS NMR spectra of AβM01–42 recorded at ω0H/2π = 800 MHz at 277 K and ωr/2π = 20 kHz with 83 kHz 1H decoupling during acquisition. (a) Cross-polarization 1D 13C spectrum recorded with 512 transients. (b) 13C-INEPT spectrum recorded with 1024 transients. (c) Cross-polarization 1D 15N spectrum recorded with 512 transients.
The expected spectral quality was confirmed by the excellent resolution observed in the 2D RFDR74 (Figure 3A) and ZF-TEDOR75 (Figure 3B) spectra where line widths of ∼0.5 ppm are observed for well resolved resonances. Interestingly, and in contrast to previous reports, only a single set of resonances was observed, and is well illustrated by the three isoleucine, two serines, etc. cross peaks present in the RFDR spectrum indicating that only a single fibril morphology is present within our sample.
Figure 3.

(a) 1.6 ms mixing RFDR spectrum recorded at 800 MHz, ωr/2π = 20 kHz, VT gas regulated to 277 K with 83 kHz CW 1H decoupling during evolution, and 83 kHz TPPM 1H decoupling during acquisition. (b) 1.6 ms mixing ZF-TEDOR spectrum recorded at 800 MHz, ωr/2π = 20 kHz, VT gas regulated to 277 K with 83 kHz TPPM during acquisition.
Until recently, most studies of Aβ involved protein prepared via peptide synthesis and consequently 13C/15N labeling of individual amino acids. This approach has yielded a good deal of useful structural data; however, because of the cost of labeled amino acids it has not been feasible to prepare uniformly labeled Aβ and therefore to obtain a sufficiently large number of structural constraints to calculate an atomic resolution structure (>5 constraints per residue). In addition, it has recently been reported that synthetic Aβ is not as neurotoxic as biosynthetic preparations,69 and the aggregation of synthetic peptides is slowed.69 Finally, MAS NMR line widths are broader in synthetic samples. For example, the spectra in prior publications40,44−46 are significantly broader than those published recently by Bertini et al.49 and Reif, and co-workers.54 These differences could be possibly due to racemization that occurs during synthesis and impurities that are difficult to separate, whereas recombinant expression in Escherichia coli has excellent fidelity with respect to both sequence homogeneity and chirality. We also note that that biosynthetic preparations of PI3-SH3,76 β2-microglobulin,11,12,77 and α-synuclein7,78−94 yield well resolved spectra. In addition, recombinant expression of protein opens the possibilities of utilizing sparse isotopic labeling schemes, which can aid in producing high quality MAS NMR spectra (i.e., 1,3-13C2 -glycerol, 2-13C1-glycerol,95−98 and 1,6-13C2- and 2-13C1-glucose),99−102 which have recently been employed for fibrils of Aβ1–40 E22Δ, β2-microglobulin and its ΔN6 variant.33,79,83
In addition, we recorded 3D spectra (NCOCX, NCACX, and CONCA)103−107 with representative strip plots shown in Figure 4 to complete unambiguous assignments of the backbone and side chain resonances (Table S1). The most intense signals observed in the NCOCX, CONCA, and NCACX spectra correspond to residues 25–34 indicating that these residues are particularly rigid relative to the other residues. We also recorded a 2D 13C–13C-TOBSY108 to assign resonances belonging to dynamic portions of the molecule, (Figure 5). The residues at the N-terminus appear in the J-based experiments, which has been previously observed in a variety of fibrils.11,12,49,77
Figure 4.
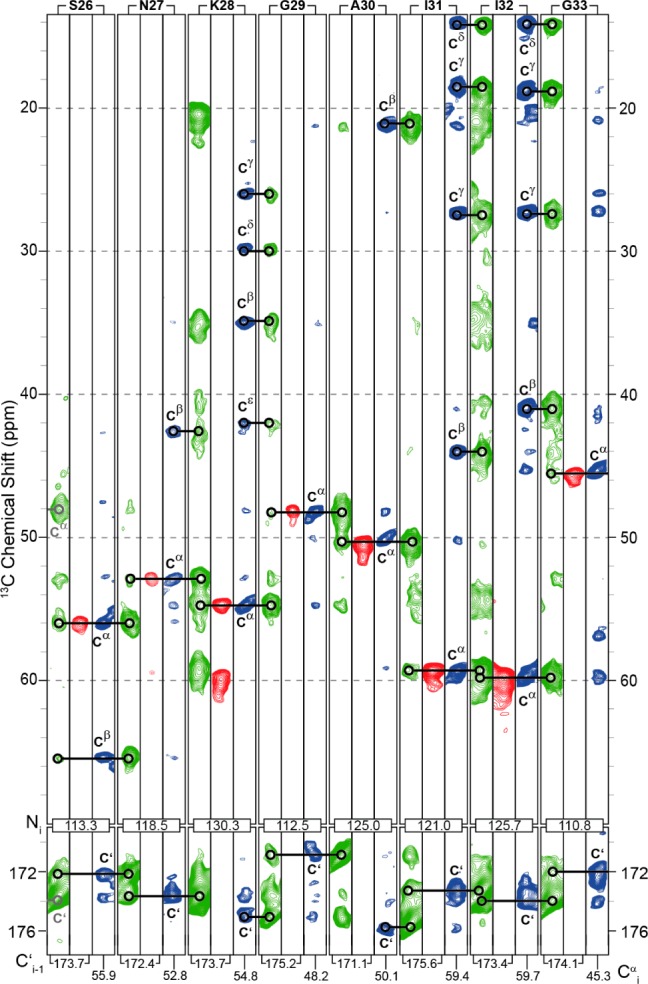
Representative strip plot of NCOCX (green), CONCA (red), and NCACX (blue) spectra (recorded at 750 and 800 MHz, respectively). ωr/2π = 12.5 kHz, T = 277 K, τmix(DARR) = 80 ms. A 83 kHz 1H decoupling field was applied during acquisition.
Figure 5.
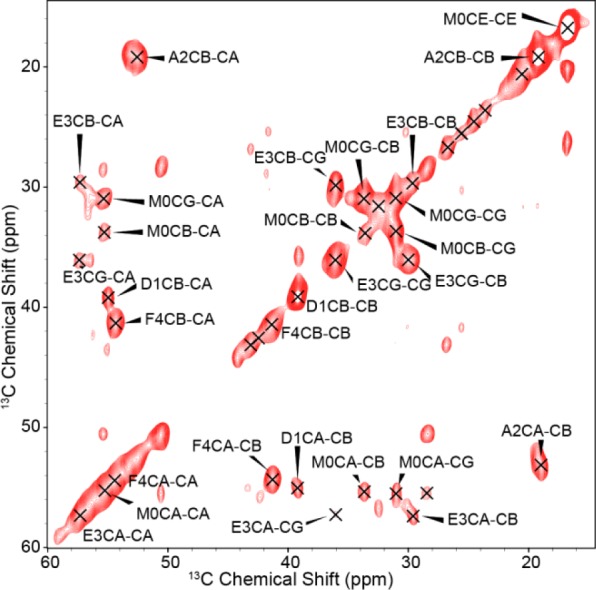
2D 13C–13C-TOBSY recorded at T = 277 K, ω0H/2π = 800 MHz, ωr/2π = 20 kHz, and τmix(TOBSY) = 9.6 ms. A 83 kHz 1H decoupling field was applied during acquisition.
Initially, essentially all of the peaks present in the 2D TOBSY and the 3D NCOCX and NCACX were assigned, the exceptions being R5-D7 and Y10-Q15. Since these residues could be in an intermediate exchange regime, they may be observed with longer mixing times, or alternatively lower temperatures to quench the relevant motions. With this in mind we recorded an 80 ms mixing DARR (Figure 6) and were able to assign R5, Y10, E11, and V12. We note that these resonances are significantly weaker than the resonances observed in the 3D experiments lending support to our speculation that the resonances absent are in an intermediate motional regime.
Figure 6.

2D 13C–13C MAS spectrum of AβM01–42 fibrils using DARR mixing recorded at a field strength corresponding to ω0H/2π = 800 MHz, T = 277 K and ωr/2π = 20 kHz. τmix = 80 ms, with a 83 kHz 1H decoupling field applied during acquisition.
The residues that have been assigned include both the backbone and side chains with the methods used summarized as follows: we were able to identify residues M0-F4 through the J-based 2D TOBSY, and residues S8 and G9 and K16-A42 through the NCOCX, CONCA, and NCACX. Additionally, we have assigned R5, Y10, E11 and V12 from 2D DARR spectra, but were unable to identify five residues, H6, D7, H13, H14 and Q15, that are likely in an intermediate motional regime, shown schematically in Figure 7.
Figure 7.
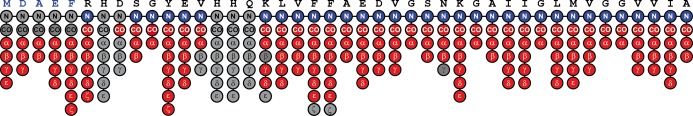
Schematic showing the amino acids and side chains with the side chains filled in. Red circles correspond to assigned 13C’s, blue circles correspond to assigned 15N’s, and gray circles correspond to resonances that are not observed.
The chemical shifts of assigned residues were then used as input to TALOS+109 to estimate the backbone torsion angles (ψ, φ) (Figure 8), which were in turn utilized to predict the secondary structures within AβM01–42 fibrils (Table S2). The dihedral angles predict that four β-strands are present within each monomer in the secondary structure of the fibrils (Figure 8). The location of the four predicted β-strands is similar but slightly different from other Aβ fibrils that have been reported in the literature (see Figure 9 and S1), and consistent with the intensity of the cross-polarization based spectra, in which the signal intensity is an indicator of rigidity. These β-strands are expected to be within the core of the fibrils and therefore rigid. We note that G37 and G38 are potential locations for a loop or turn but these residues could also be part of a longer β-strand between M35 and I41. Elucidating further structural details will determine which of these options is correct. The chemical shifts of the AβM01–42 fibrils we have studied here do not seem to correlate with those reported by Bertini et al., who had characterized AβM01–40 fibrils prepared from recombinant protein expressed by a similar method to the one we have employed. This suggests that there may be significant structural differences between AβM01–42 fibrils and AβM01–40 fibrils, which may be a result of their different hydrophobicities and steric constraints related to accommodating two extra residues within the fibril.71 Furthermore, we note that the location of the β-strands in this work are in slightly different locations compared to many of those reported for Aβ1–40 fibrils. Currently it is unclear what impact this has on the intra- and intermolecular structure of AβM01–42. Efforts to address this important question are ongoing. This suggestion is consistent with the cryo-EM images reported by Fandrich et al. and Schmidt et al. where the electron density can be mapped into a long strand and a N-terminus whose structure was not well resolved.65,110
Figure 8.
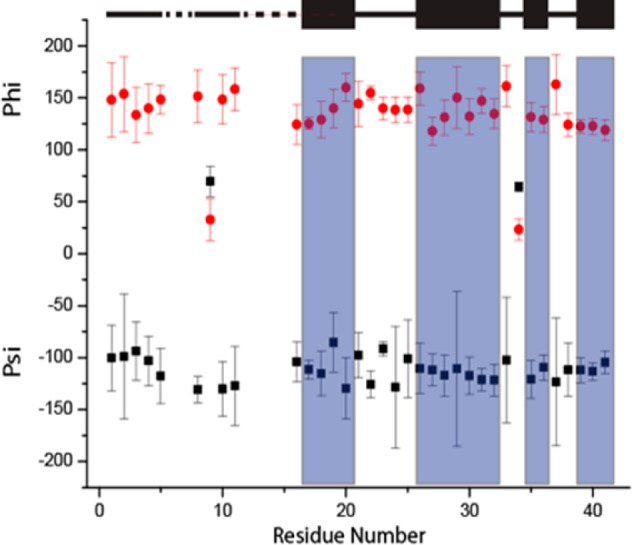
Predicted ψ and φ angles using TALOS. Shaded blue regions indicate predicted β-strands. On the top of the figure the black boxes indicate the location of the beta strands, the solid black line indicate regions that are in loop or random coil conformations and the dashed line indicates regions which have not yet been assigned.
Figure 9.
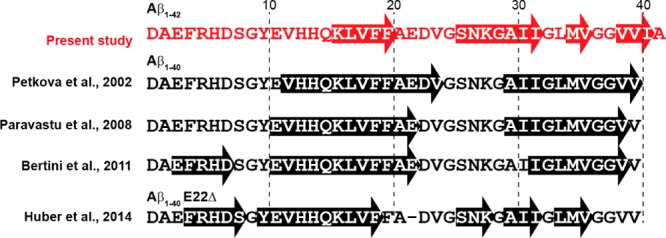
Comparison of the locations of predicted β-strands in various Aβ fibrils. Here, the comparison is limited to NMR chemical shift data (see Figure S1).
We also note that aggregation of Aβ monomers has been found to produce polymorphic fibrils, with the fibrilization conditions (i.e., pH, temperature, salt concentration, monomer concentration, etc.) as some of the variables that affects the polymorphs observed.111 The polymorphism is believed to be responsible for broad lines observed in many Aβ MAS spectra and has limited the structural constraints that can be gleaned from the spectra. Recently, Lu et al. and Pravastu et al. have provided evidence that the differences in morphologies may be linked to disease symptoms by structurally characterizing fibrils prepared by seeding fibrils with material extracted from the brains of patients exhibiting varying presentations of the disease.47,48 This surprising result provides impetus to characterize the structure of fibrils with many different morphologies that can be prepared under varying fibrilization conditions.
Conclusions
We have prepared uniformly 13C/15N labeled AβM01–42 fibrils consisting of a single polymorph and have structurally characterized them with chemical shift assignments obtained from MAS NMR spectra. The backbone and side chains resonances have been assigned for 38 of the 43 residues through a series of 2D and 3D CP and J-based transfer experiments. TALOS+ prediction of ψ and φ angles resulting from the chemical shifts indicate the presence of four β-strands within the fibril structure.
These results also indicate that the residues within the protein have significantly different mobilities. The first five residues are the most dynamic and are most easily detected in J-based TOBSY transfers. In contrast residues 16–42 are relatively rigid, are detected with CP based transfers and likely form the core of the AβM01–42 fibrils. Connecting those two sections is a region of intermediate flexibility, where we observe weak signals in CP-based experiments for R5, S8, G9, Y10, E11, and V12, with their intensity being inversely correlated with temperature. In contrast we do not observe signals from either CP-based and J-based experiments for H6, D7, H13, H14 amd-Q15. However, preliminary experiments at 150 K indicate that the His resonances are modulated by H+ exchange. It seems likely that additional low temperature experiments will permit observation and assignment of these residues.
Figure 9 shows a comparison of the location of β-strands in various Aβ fibrils that have been studied with NMR.35,45,49,55 Many show that the residues from 15 to 40/42 are most likely to possess β-strand character. Although several studies predict that β-strands can occur in the first 15 residues, it has only been proposed to be true for Aβ40 fibrils.
Experimental Section
Sample Preparation
The AβM01–42 peptide was expressed and purified as previously reported.73 Briefly, AβM01–42 was expressed in 12 L of M9 minimal medium containing 2 g of U–13C glucose and 1 g of 15NH4Cl per liter. Cells were grown at 37 °C, induced at an OD600 = 0.7–1.0 and harvested 5 h later by centrifugation at 6000g for 8 min. The cells were stored at −20 °C. Upon purification, the pellet was thawed and resuspended in 100 mL buffer containing 10 mM Tris, 1 mM EDTA, pH 7.5 (buffer A), sonicated for 1.5 min (half horn, max output, 50% duty cycle, 50 mL at a time) and centrifuged (18000g for 7 min) to pellet the inclusion bodies. The inclusion bodies were sonicated two more times (15 s) in 50 mL buffer A and pelleted as above. The inclusion bodies were then dissolved in buffer containing 10 mM Tris, 1 mM EDTA, pH 8.5 (buffer B) with 8 M urea, diluted with three volumes of buffer B and added to 100 mL DEAE cellulose resin equilibrated in buffer B with 2 M urea added, and purified via ion exchange chromatography in batch format, followed by two size exclusion chromatography steps in 20 mM sodium phosphate, 0.2 mM EDTA, 0.02% NaN3, pH 8.0, using a 26 × 600 mm Superdex 75 column, which ensured that only monomeric AβM01–42 was present when fibrilization began. AβM01–42 fibrils were prepared by incubating 10–50 μM (0.05–0.25 mg/mL) solutions of AβM01–42 in 15 mL Falcon tubes at room temperature overnight, and the formation of fibrils confirmed by withdrawing a small aliquot for fluorescence spectroscopy in the presence of thioflavin-T (ThT). The first fibril sample was lyophylized, and resuspended in a smaller volume of water (milli-Q) before packing into the rotor. Aliquots of this first fibril sample were set aside prior to lyophilization and were added as seeds to subsequent monomer samples, to ensure formation of fibrils of the same morphology in all tubes. Subsequent samples were always kept hydrated and we find that the chemical shifts are identical to samples that had been lyophilized. Thus, in the case of AβM1–42 the lyophilization and rehydrations steps do not significantly alter the fibril structure. 13C–13C and 13C–15N spectra demonstrating this are shown in Figure S2. Typically, ∼30 mg of hydrated AβM01–42 fibrils were packed into a 3.2 mm Bruker rotor (Bruker Biospin, Billerica, MA) using a home-built centrifugal packing tool.
Transmission Electron Microscopy (TEM)
TEM images were acquired using a Philips CM120 BioTWIN electron microscope equipped with a postcolumn energy filter (Gatam GIF100) and a CCD camera. The acceleration voltage was 120 kV. 300 mesh carbon-coated Formvar grids (Electron Microscopy Sciences, Hatfield, PA) were placed up-side down on a buffer droplet for 2 min, then on a droplet of the fibril samples for 3 min, followed by a quick (20 s) rinse on a droplet of water (Milli-Q). The grid was then placed on a droplet of 1.5% uranyl acetate (Merck) for another 3 min to provide the negative stain.
MAS NMR Spectroscopy
Spectra were acquired on either a Cambridge Instruments 750 MHz spectrometer operating under RNMR (courtesy of Dr. David Ruben) or a Bruker 800 MHz AVANCE III spectrometer equipped with a 3.2 mm triple channel HCN Bruker probe (Bruker Biospin, Billerica, MA). Spectra were recorded at ωr/2π = 20 kHz and regulated to ±10 Hz using a Bruker spinning frequency controller for INEPT-TOBSY,112,113 DARR,114 RFDR,74,115 and ZF-TEDOR,75 and 12.5 kHz ± 10 Hz for 3D NCOCX, NCACX, CONCA experiments. All experiments were conducted at 277 K. Spectra recorded at ω0H/2π = 750 MHz (3D NCOCX and CONCA) were processed, displayed and assigned using the NMRPipe software package,116 while spectra recorded at ω0H/2π = 800 MHz (all other) were processed using TopSpin 3.1, and all spectra were analyzed in Sparky.117
1D 15N spectra were recorded with a CP contact time of 2 ms and 1H/15N fields of 62.5 kHz, respectively, and a recycle delay of 3 s. During acquisition, a TPPM1181H decoupling field of 83.3 kHz was applied. The FID containing 2048 points was averaged over 512 transients. Subsequently, a Gaussian window function with 10 Hz line broadening was applied and the FID was zero filled to 8192 points and Fourier transformed. A 1D refocused carbon insensitive nuclei enhanced by polarization transfer (INEPT) spectrum was recorded with an INEPT delay of 1.92 ms (equals a 1JHC coupling of 130 Hz), and a recycle delay of 2.5 s. The refocusing delay was set to 1.28 ms (= 1/61JHC) selecting all multiplicities (CH, CH2, and CH3). During acquisition, a WALTZ-16 proton decoupling field of 2.5 kHz was applied. The FID containing 2048 points was averaged over 1024 transients. Subsequently, a Gaussian window function with 10 Hz line broadening was applied and the FID was zero filled to 8192 points and Fourier transformed. 13C and 15N chemical shifts were referenced using the published shifts of adamantine relative to DSS for 13C referencing and the IUPAC relative frequency ratios between DSS (13C) and liquid ammonia (15N).
2D DARR spectra were recorded with a CP contact time of 1.2 ms and 1H/13C fields of 62.5 kHz, respectively, and a recycle delay of 2.5 s. For DARR mixing, an 80 ms proton field at 20 kHz was applied. During acquisition, a TPPM 1H decoupling field of 83.3 kHz was applied. The FID matrix containing 2048 × 1024 points was averaged over 16 transients. Subsequently, a squared sine window function with a sine bell shift of 3.5 was applied and the FID matrix was zero filled to 4096 × 2048 points and Fourier transformed.
2D RFDR spectra were recorded with a CP contact time of 1.2 ms and 1H/13C fields of 62.5 kHz, respectively, and a recycle delay of 2.5 s. For RFDR mixing, a 1.6 ms 13C RFDR recoupling field was applied using rotor synchronized π pulses at 83 kHz with 100 kHz 1H decoupling. During acquisition, a TPPM 1H decoupling field of 83.3 kHz was applied. The FID matrix containing 2048 × 1024 points was averaged over 16 transients. Subsequently, a squared sine window function with a sine bell shift of 3.5 was applied and the FID matrix was zero filled to 4096 × 2048 points and Fourier transformed.
Z-filtered transferred echo double resonance (ZF-TEDOR) were acquired using 3.2 ms mixing with 50 kHz 13C and 15N π-pulses with 83 kHz 1H TPPM decoupling during acquisition and a 3 s recycle delay. The FID matrix of 1024 × 512 points were averaged for 32 scans.
3D NCOCX used 62.5 kHz and 50 kHz fields respectively for 1H to 15N transfer (2 ms contact time) with 50 kHz and 62.5 kHz fields for 15N to 13C magnetization transfer (4 ms contact) with 100 kHz decoupling during double cross polarization (DCP) transfer and a carrier frequency of 165 ppm, followed by DARR mixing for 80 ms with a 12.5 ms 1H DARR field and 83 kHz 1H decoupling during acquisition all with a recycle delay of 3s. The FID acquired had 1024 points, with 240 and 260 points in the second and third dimension, respectively. Similarly, the CONCA 3D began with cross-polarization from 1H to 13C with fields of 71 kHz and 83 kHz respectively, with 100 kHz decoupling during the DCP transfer and 4 ms contact time during the DCP with 160 and 240 points in the second and third dimension. The 3D NCACX spectrum was recorded with an initial CP contact time of 1.2 ms and 1H/13C fields of 62.5 kHz, respectively, and a recycle delay of 2.5 s. For DARR mixing, an 80 ms proton field at 20 kHz was applied. During acquisition, a TPPM 1H decoupling field of 83.3 kHz was applied. The FID matrix containing 2048 × 1024 points was averaged over 16 transients. Subsequently, a squared sine window function with a sine bell shift of 3.5 was applied and the FID matrix was zero filled to 4096 × 2048 points and Fourier transformed.
Detailed acquisition and processing parameters can be found in Table S4 and S5, respectively.
Acknowledgments
The authors wish to thank Dr. Kendra K. Fredrick for her assistance and constructive comments. The authors also wish to thank Dr. David Ruben, and Mr. Ajay Thakkar for their assistance with instrumentation, and Gunnel Karlsson for her help with the EM analysis. This work was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institute of Health under Grants EB-003151, EB-001960 and EB-002026, the Swedish Research Council (VR) and a European Research Council (ERC) Advanced Grant.
Supporting Information Available
A table of assignments along with additional experimental information and additional TALOS + secondary structure predictions. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b03997.
Author Contributions
∥ M.T.C. and R.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. Nat. Rev. Mol. Cell Biol. 2014, 15, 384. [DOI] [PubMed] [Google Scholar]
- Helmus J. J.; Surewicz K.; Apostol M. I.; Surewicz W. K.; Jaroniec C. P. J. Am. Chem. Soc. 2011, 133, 13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmus J. J.; Surewicz K.; Surewicz W. K.; Jaroniec C. P. J. Am. Chem. Soc. 2010, 132, 2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Nat. Rev. Neurosci. 2001, 2, 492. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Overk C. R.; Oueslati A.; Masliah E. Nat. Rev. Neurosci. 2013, 14, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G.; Lemkau L. R.; Nieuwkoop A. J.; Kloepper K. D.; Ladror D. T.; Ebisu R.; Woods W. S.; Lipton A. S.; George J. M.; Rienstra C. M. J. Mol. Biol. 2011, 411, 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise H.; Hoyer W.; Becker S.; Andronesi O. C.; Riedel D.; Baldus M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow C. W.; Brundin P. Mov. Disord. 2013, 28, 31. [DOI] [PubMed] [Google Scholar]
- Vilar M.; Chou H. T.; Luhrs T.; Maji S. K.; Riek-Loher D.; Verel R.; Manning G.; Stahlberg H.; Riek R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina G. T.; Platt G. W.; Bayro M. J.; Radford S. E.; Griffin R. G. J. Am. Chem. Soc. 2010, 132, 17077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina G. T.; Platt G. W.; Bayro M. J.; Radford S. E.; Griffin R. G. J. Am. Chem. Soc. 2010, 132, 10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzban L.; Park K.; Verchere C. B. Exp. Gerontol. 2003, 38, 347. [DOI] [PubMed] [Google Scholar]
- Westermark P.; Andersson A.; Westermark G. T. Physiol. Rev. 2011, 91, 795. [DOI] [PubMed] [Google Scholar]
- McGowan D. P.; van Roon-Mom W.; Holloway H.; Bates G. P.; Mangiarini L.; Cooper G. J. S.; Faull R. L. M.; Snell R. G. Neuroscience 2000, 100, 677. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Chem. Rev. 2012, 112, 5147. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Protein Pept. Lett. 2006, 13, 219. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Nat. Rev. Mol. Cell Biol. 2007, 8, 101. [DOI] [PubMed] [Google Scholar]
- De Strooper B.; Saftig P.; Craessaerts K.; Vanderstichele H.; Guhde G.; Annaert W.; Von Figura K.; Van Leuven F. Nature 1998, 391, 387. [DOI] [PubMed] [Google Scholar]
- Vassar R.; Bennett B. D.; Babu-Khan S.; Kahn S.; Mendiaz E. A.; Denis P.; Teplow D. B.; Ross S.; Amarante P.; Loeloff R.; Luo Y.; Fisher S.; Fuller L.; Edenson S.; Lile J.; Jarosinski M. A.; Biere A. L.; Curran E.; Burgess T.; Louis J. C.; Collins F.; Treanor J.; Rogers G.; Citron M. Science 1999, 286, 735. [DOI] [PubMed] [Google Scholar]
- Jarrett J. T.; Berger E. P.; Lansbury P. T. Biochemistry 1993, 32, 4693. [DOI] [PubMed] [Google Scholar]
- Berger E. P.; Jarrett J. T.; Lansbury P. T. Abstr. Pap. Am. Chem. Soc. 1993, 205, 76. [Google Scholar]
- Jarrett J. T.; Berger E. P.; Lansbury P. T. Ann. N. Y. Acad. Sci. 1993, 695, 144. [DOI] [PubMed] [Google Scholar]
- Teller J. K.; Russo C.; Debusk L. M.; Angelini G.; Zaccheo D.; DagnaBricarelli R.; Scartezzini P.; Bertolini S.; Mann D. M. A.; Tabaton M.; Gambetti P. Nat. Med. 1996, 2, 93. [DOI] [PubMed] [Google Scholar]
- Stohr J.; Condello C.; Watts J. C.; Bloch L.; Oehler A.; Nick M.; Bhate M.; DeArmond S. J.; Giles K.; DeGrado W. F.; Prusiner S. B. Prion 2014, 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr J.; Condello C.; Watts J. C.; Bloch L.; Oehler A.; Nick M.; DeArmond S. J.; Giles K.; DeGrado W. F.; Prusiner S. B. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J. C.; Condello C.; Stohr J.; Oehler A.; Lee J.; DeArmond S. J.; Lannfelt L.; Ingelsson M.; Giles K.; Prusiner S. B. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P.; Cukalevski R.; Frohm B.; Knowles T. P. J.; Linse S. J. Am. Chem. Soc. 2014, 136, 219. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G.; Yang X. T.; Hellstrand E.; Frohm B.; Kirkegaard J. B.; Cohen S. I. A.; Dobson C. M.; Linse S.; Knowles T. P. J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A.; Arosio P.; Presto J.; Kurudenkandy F. R.; Biverstål H.; Dolfe L.; Dunning C.; Yang X.; Johansson B. F. M. V. J.; Dobson C. M.; Fisahn A.; Knowles T. P. J.; Linse S. Nat. Struct. Mol. Biol. 2015, 22, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Annu. Rev. Phys. Chem. 2011, 62, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina G. T.; Bayro M. J.; Fitzpatrick A. W.; Ladizhansky V.; Colvin M. T.; Caporini M. A.; Jaroniec C. P.; Bajaj V. S.; Rosay M.; MacPhee C. E.; Vendruscolo M.; Maas W. E.; Dobson C. M.; Griffin R. G. J. Am. Chem. Soc. 2013, 135, 19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P.; Debelouchina G. T.; Bayro M. J.; Clare D. K.; Caporini M. A.; Bajaj V. S.; Jaroniec C. P.; Wang L. C.; Ladizhansky V.; Muller S. A.; MacPhee C. E.; Waudby C. A.; Mott H. R.; De Simone A.; Knowles T. P. J.; Saibil H. R.; Vendruscolo M.; Orlova E. V.; Griffin R. G.; Dobson C. M. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber M.; Ovchinnikova O. Y.; Schutz A. K.; Glockshuber R.; Meier B. H.; Bockmann A. Biomol. NMR Assignments 2015, 9, 7. [DOI] [PubMed] [Google Scholar]
- Schutz A. K.; Vagt T.; Huber M.; Ovchinnikova O. Y.; Cadelbert R.; Wall J.; Guntert P.; Bockmann A.; Glockshuber R.; Meier B. H. Angew. Chem., Int. Ed. 2015, 54, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemer A. B.; Ritter C.; Ernst M.; Riek R.; Meier B. H. Angew. Chem., Int. Ed. 2005, 44, 2441. [DOI] [PubMed] [Google Scholar]
- Van Melckebeke H.; Wasmer C.; Lange A.; AB E.; Loquet A.; Bockmann A.; Meier B. H. J. Am. Chem. Soc. 2010, 132, 13765. [DOI] [PubMed] [Google Scholar]
- Wasmer C.; Lange A.; Van Melckebeke H.; Siemer A. B.; Riek R.; Meier B. H. Science 2008, 319, 1523. [DOI] [PubMed] [Google Scholar]
- Lansbury P. T.; Costa P. R.; Griffiths J. M.; Simon E. J.; Auger M.; Halverson K. J.; Kocisko D. A.; Hendsch Z. S.; Ashburn T. T.; Spencer R. G. S.; Tidor B.; Griffin R. G. Nat. Struct. Biol. 1995, 2, 990. [DOI] [PubMed] [Google Scholar]
- Benzinger T. L. S.; Gregory D. M.; Burkoth T. S.; Miller-Auer H.; Lynn D. G.; Botto R. E.; Meredith S. C. Biochemistry 2000, 39, 3491. [DOI] [PubMed] [Google Scholar]
- Burkoth T. S.; Benzinger T. L. S.; Urban V.; Morgan D. M.; Gregory D. M.; Thiyagarajan P.; Botto R. E.; Meredith S. C.; Lynn D. G. J. Am. Chem. Soc. 2000, 122, 7883. [Google Scholar]
- Antzutkin O. N.; Leapman R. D.; Balbach J. J.; Tycko R. Biochemistry 2002, 41, 15436. [DOI] [PubMed] [Google Scholar]
- Antzutkin O. N.; Balbach J. J.; Leapman R. D.; Rizzo N. W.; Reed J.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Tycko R. Biophys. J. 2002, 82, 320a. [Google Scholar]
- Lu J. X.; Qiang W.; Yau W. M.; Schwieters C. D.; Meredith S. C.; Tycko R. Cell 2013, 154, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu A. K.; Qahwash I.; Leapman R. D.; Meredith S. C.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Gonnelli L.; Luchinat C.; Mao J. F.; Nesi A. J. Am. Chem. Soc. 2011, 133, 16013. [DOI] [PubMed] [Google Scholar]
- Amo J. M. L. d.; Schmidt M.; Fink U.; Dasari M.; Fandrich M.; Reif B. Angew. Chem., Int. Ed. 2012, 51, 6136. [DOI] [PubMed] [Google Scholar]
- Antzutkin O. N.; Balbach J. J.; Tycko R. Biophys. J. 2003, 84, 3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbach J. J.; Petkova A. T.; Oyler N. A.; Antzutkin O. N.; Gordon D. J.; Meredith S. C.; Tycko R. Biophys. J. 2002, 83, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari M.; Espargaro A.; Sabate R.; del Amo J. M. L.; Fink U.; Grelle G.; Bieschke J.; Ventura S.; Reif B. ChemBioChem 2011, 12, 407. [DOI] [PubMed] [Google Scholar]
- del Amo J. M. L.; Schneider D.; Loquet A.; Lange A.; Reif B. J. Biomol. NMR 2013, 56, 359. [DOI] [PubMed] [Google Scholar]
- Paravastu A. K.; Leapman R. D.; Yau W. M.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Yau W. M.; Tycko R. Biophys. J. 2004, 86, 506a.14695295 [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. Biochemistry 2006, 45, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R.; Ishii Y. J. Am. Chem. Soc. 2003, 125, 6606. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Guo Z. H.; Yau W. M.; Mattson M. P.; Tycko R. Science 2005, 307, 262. [DOI] [PubMed] [Google Scholar]
- Yang X.; Meisl G.; Knowles T. P. J.; Linse S., in preparation.
- Tycko R.; Sciarretta K. L.; Orgel J. P. R. O.; Meredith S. C. Biochemistry 2009, 48, 6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W.; Yau W. M.; Luo Y. Q.; Mattson M. P.; Tycko R. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W.; Yau W. M.; Tycko R. J. Am. Chem. Soc. 2011, 133, 4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. Nat. Struct. Mol. Biol. 2010, 17, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrich M.; Schmidt M.; Grigorieff N. Trends Biochem. Sci. 2011, 36, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y.; Ma B. Y.; Tsai C. J.; Nussinov R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Hu X. Y.; Khant H.; Ludtke S. J.; Chiu W.; Schmid M. F.; Frieden C.; Lee J. M. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay W. M.; Huang D. T.; Rosenberry T. L.; Paravastu A. K. J. Mol. Biol. 2013, 425, 2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finder V. H.; Vodopivec I.; Nitsch R. M.; Glockshuber R. J. Mol. Biol. 2010, 396, 9. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T. Acta Histochem. Cytochem. 1999, 32, 13. [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Doeli H.; Schubert D.; Riek R. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukalevski R.; Yang X.; Meisl G.; Weininger U.; Frohm B.; Knowles T. P. J.; Linse S. Chem. Sci. 2015, 10.1039/C4SC02517B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Thulin E.; Minogue A. M.; Gustavsson N.; Pang E.; Teplow D. B.; Linse S. FEBS J. 2009, 276, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett A. E.; Ok J. H.; Griffin R. G.; Vega S. J. Chem. Phys. 1992, 96, 8624. [Google Scholar]
- Jaroniec C. P.; Filip C.; Griffin R. G. J. Am. Chem. Soc. 2002, 124, 10728. [DOI] [PubMed] [Google Scholar]
- Bayro M. J.; Maly T.; Birkett N. R.; MacPhee C. E.; Dobson C. M.; Griffin R. G. Biochemistry 2010, 49, 7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. C.; Sarell C. J.; Eddy M. T.; Debelouchina G. T.; Andreas L. B.; Pashley C. L.; Radford S. E.; Griffin R. G. J. Am. Chem. Soc. 2014, 136, 6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G.; Lemkau L. R.; Zhou D. H. H.; George J. M.; Rienstra C. M. J. Am. Chem. Soc. 2012, 134, 5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath J.; Bousset L.; Habenstein B.; Melki R.; Bockmann A.; Meier B. H. PLoS One 2014, 10.1371/journal.pone.0090659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath J.; Bousset L.; Habenstein B.; Melki R.; Meier B. H.; Bockmann A. Biomol. NMR Assignments 2014, 8, 395. [DOI] [PubMed] [Google Scholar]
- Gath J.; Habenstein B.; Bousset L.; Melki R.; Meier B. H.; Bockmann A. Biomol. NMR Assignments 2012, 6, 51. [DOI] [PubMed] [Google Scholar]
- Kloepper K. D.; Hartman K. L.; Ladror D. T.; Rienstra C. M. J. Phys. Chem. B 2007, 111, 13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloepper K. D.; Woods W. S.; Winter K. A.; George J. M.; Rienstra C. M. Protein Expression Purif. 2006, 48, 112. [DOI] [PubMed] [Google Scholar]
- Kloepper K. D.; Zhou D. H.; Hartman K. L.; Ladror D. T.; George J. M.; Rienstra C. M. Biophys. J. 2007, 561a. [Google Scholar]
- Kloepper K. D.; Zhou D. H.; Li Y.; Winter K. A.; George J. M.; Rienstra C. M. J. Biomol. NMR 2007, 39, 197. [DOI] [PubMed] [Google Scholar]
- Lemkau L. R.; Comellas G.; Kloepper K. D.; Woods W. S.; George J. M.; Rienstra C. M. J. Biol. Chem. 2012, 287, 11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkau L. R.; Comellas G.; Lee S. W.; Rikardsen L. K.; Woods W. S.; George J. M.; Rienstra C. M. PLoS One 2013, 10.1371/journal.pone.0049750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods W. S.; Boettcher J. M.; Zhou D. H.; Kloepper K. D.; Hartman K. L.; Ladror D. T.; Qi Z.; Rienstra C. M.; George J. M. J. Biol. Chem. 2007, 282, 34555. [DOI] [PubMed] [Google Scholar]
- Zhou D. H.; Woods W. S.; Kloepper K. D.; Ladror D.; Hartman K.; George J. M.; Rienstra C. M. Biophys. J. 2007, 149a. [Google Scholar]
- Bousset L.; Pieri L.; Ruiz-Arlandis G.; Gath J.; Jensen P. H.; Habenstein B.; Madiona K.; Olieric V.; Bockmann A.; Meier B. H.; Melki R. Nat. Commun. 2013, 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Cho M. K.; Riedel D.; Kurmar A.; Maier E.; Siebenhaar C.; Becker S.; Fernandez C. O.; Lashuel H. A.; Benz R.; Lange A.; Zweckstetter M. Biophys. J. 2010, 98, 654a. [Google Scholar]
- Lv G. H.; Kumar A.; Giller K.; Orcellet M. L.; Riedel D.; Fernandez C. O.; Becker S.; Lange A. J. Mol. Biol. 2012, 420, 99. [DOI] [PubMed] [Google Scholar]
- Mollenhauer B.; Trautmann E.; Otte B.; Ng J.; Spreer A.; Lange P.; Sixel-Doring F.; Hakimi M.; VonSattel J. P.; Nussbaum R.; Trenkwalder C.; Schlossmacher M. G. J. Neural Transm. 2012, 119, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Cho M. K.; Kumar A.; Maier E.; Siebenhaar C.; Becker S.; Fernandez C. O.; Lashuel H. A.; Benz R.; Lange A.; Zweckstetter M. J. Am. Chem. Soc. 2009, 131, 17482. [DOI] [PubMed] [Google Scholar]
- LeMaster D. M.; Kushlan D. M. J. Am. Chem. Soc. 1996, 118, 9255. [Google Scholar]
- Hong M.; Jakes K. J. Biomol. NMR 1999, 14, 71. [DOI] [PubMed] [Google Scholar]
- Castellani F.; van Rossum B.; Diehl A.; Schubert M.; Rehbein K.; Oschkinat H. Nature 2002, 420, 98. [DOI] [PubMed] [Google Scholar]
- Higman V. A.; Flinders J.; Hiller M.; Jehle S.; Markovic S.; Fiedler S.; van Rossum B. J.; Oschkinat H. J. Biomol. NMR 2009, 44, 245. [DOI] [PubMed] [Google Scholar]
- Hong M. J. Magn. Reson. 1999, 139, 389. [DOI] [PubMed] [Google Scholar]
- Teilum K.; Brath U.; Lundstrom P.; Akke M. J. Am. Chem. Soc. 2006, 128, 2506. [DOI] [PubMed] [Google Scholar]
- Loquet A.; Giller K.; Becker S.; Lange A. J. Am. Chem. Soc. 2010, 132, 15164. [DOI] [PubMed] [Google Scholar]
- Lundstrom P.; Teilum K.; Carstensen T.; Bezsonova I.; Wiesner S.; Hansen D. F.; Religa T. L.; Akke M.; Kay L. E. J. Biomol. NMR 2007, 38, 199. [DOI] [PubMed] [Google Scholar]
- Pauli J.; Baldus M.; van Rossum B.; de Groot H.; Oschkinat H. ChemBioChem 2001, 2, 272. [DOI] [PubMed] [Google Scholar]
- Marulanda D.; Tasayco M. L.; McDermott A.; Cataldi M.; Arriaran V.; Polenova T. J. Am. Chem. Soc. 2004, 126, 16608. [DOI] [PubMed] [Google Scholar]
- Igumenova T. I.; Wand A. J.; McDermott A. E. J. Am. Chem. Soc. 2004, 126, 5323. [DOI] [PubMed] [Google Scholar]
- Shi L. C.; Ahmed M. A. M.; Zhang W. R.; Whited G.; Brown L. S.; Ladizhansky V. J. Mol. Biol. 2009, 386, 1078. [DOI] [PubMed] [Google Scholar]
- Sperling L. J.; Berthold D. A.; Sasser T. L.; Jeisy-Scott V.; Rienstra C. M. J. Mol. Biol. 2010, 399, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronesi O. C.; Becker S.; Seidel K.; Heise H.; Young H. S.; Baldus M. J. Am. Chem. Soc. 2005, 127, 12965. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Vernon R.; Baker D.; Bax A. J. Biomol. NMR 2009, 43, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Sachse C.; Richter W.; Xu C.; Fandrich M.; Grigorieff N. Proc. Natl. Acad. Sci. U. S. A. 2010, 106, 19813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R.; Wickner R. B. Acc. Chem. Res. 2013, 46, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldus M.; Meier B. H. J. Magn. Reson. 1996, 121, 65. [Google Scholar]
- Baldus M.; Iuliucci R. J.; Meier B. H. J. Am. Chem. Soc. 1997, 119, 1121. [Google Scholar]
- Takegoshi K.; Nakamura S.; Terao T. Chem. Phys. Lett. 2001, 344, 631. [Google Scholar]
- Bennett A. E.; Rienstra C. M.; Griffiths J. M.; Zhen W. G.; Lansbury P. T.; Griffin R. G. J. Chem. Phys. 1998, 108, 9463. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. University of California, San Francisco.
- Bennett A. E.; Rienstra C. M.; Auger M.; Lakshmi K. V.; Griffin R. G. J. Chem. Phys. 1995, 103, 6951. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.