

Abstract
Exposure to extreme stress can trigger the development of major depressive disorder (MDD) as well as post-traumatic stress disorder (PTSD). The molecular mechanisms underlying the structural and functional alterations within corticolimbic brain regions, including the prefrontal cortex (PFC) and amygdala of individuals subjected to traumatic stress, remain unknown. In this study, we show that serum and glucocorticoid regulated kinase 1 (SGK1) expression is down-regulated in the postmortem PFC of PTSD subjects. Furthermore, we demonstrate that inhibition of SGK1 in the rat medial PFC results in helplessness- and anhedonic-like behaviors in rodent models. These behavioral changes are accompanied by abnormal dendritic spine morphology and synaptic dysfunction. Together, the results are consistent with the possibility that altered SGK1 signaling contributes to the behavioral and morphological phenotypes associated with traumatic stress pathophysiology.
Activity of the protein kinase SGK1 is reduced in the prefrontal cortex of individuals with post-traumatic stress disorder (PTSD), and SGK1 inhibition can cause PTSD-related behavioral changes in an animal model.
Author Summary
Improper functioning of the brain regions known as prefrontal cortex and amygdala is associated with the development of post-traumatic stress disorder. However, little is known about the molecular mechanisms that underlie this condition. We found that the expression of a protein kinase involved in cellular responses to stress, known as serum and glucocorticoid regulated kinase 1 (SGK1), was decreased in the prefrontal cortex of subjects who had died with post-traumatic stress disorder. Furthermore, we found that experimentally decreasing SGK1 function in the prefrontal cortex of rats resulted in behaviors characteristic of traumatic stress, including the unwillingness to avoid discomfort and the inability to experience pleasure. Finally, reduced SGK1 function in neurons affected their basic electrophysiological properties and caused a decrease in the number of dendritic spines—the specialized protrusions of dendrites that receive synaptic inputs.
Introduction
Major depressive disorder (MDD) and post-traumatic stress disorder (PTSD) are highly comorbid. Approximately 50% of newly diagnosed patients with PTSD meet criteria for MDD [1]. Exposure to traumatic stress is an important factor in the etiology of both, MDD and PTSD. Regardless of the type of trauma, symptoms include increased fear and anxiety, intrusive thoughts and dreams of the trauma, conditioned fear and hyperarousal including increased startle response, avoidance of reminders of the trauma, and emotional numbing and anhedonia [2,3]. Neuroimaging studies in PTSD patients show an impaired function of medial prefrontal cortex (mPFC) [4–6], resulting in decreased inhibitory control of amygdala function that contributes to increased anxiety and fear [7–9]. This general pattern of brain circuit dysfunction also emerges in healthy humans exposed to uncontrollable stress [10]. Decreased responsiveness of the mPFC leads to deficits in the ability to extinguish fear responses, which contributes to the persistence of traumatic memories seen in patients with PTSD [7,8,11].
Inescapable stress, originally recognized as an animal model for depression, produces a syndrome that shares many behavioral, physiologic, and neurochemical features observed in MDD and PTSD [12–15]. In addition, morphological studies conducted in rodent mPFC show that chronic stress causes a reduction of dendrite length and spine density of pyramidal neurons, and these alterations are associated with impairments in mPFC-mediated cognitive tasks [16].
Despite this progress on circuit level analysis of PTSD pathophysiology, there is very little known about the underlying molecular and cellular alterations that cause PTSD. To address this issue, we have established the first PTSD postmortem brain bank with a small cohort of currently available cases (n = 6; [five females and one male] and controls n = 6; (six females) and have conducted a microarray study of these subjects and matched controls. This has resulted in identification of a number of dysregulated genes in the PFC of PTSD subjects, one of which is serum and stress activated kinase 1 (SGK1). Further studies in rodent models demonstrate that disruption of SGK1 signaling results in behavioral and morphological endophenotypes associated with traumatic stress, including helplessness- and anhedonic-like behaviors and decreased spine density in mPFC neurons.
Results
To identify transcriptional alterations that could contribute to traumatic stress in the PFC, we conducted a whole genome array of postmortem PFC samples collected from a small cohort of individuals diagnosed with PTSD and age-matched individuals without psychiatric diagnoses (S1 Table). The microarray study identified 231 down-regulated and 42 up-regulated genes in the PFC of PTSD subjects (S2 Table). One of the most highly dysregulated genes is SGK1, which is reported to be induced by corticosteroid hormones and has been implicated in cellular responses to stress [17,18]. SGK1 belongs to the serine/threonine family of protein kinases consisting of three isoforms, SGK1, SGK2 and SGK3 [18,19]. Our array analysis using the false discovery rate (FDR) method for multiple comparisons [20] shows that SGK1 expression is down-regulated by over 80% in PFC from PTSD subjects relative to the control group (Fig 1A). Levels of SGK2 and SGK3 expression were also decreased relative to controls, although the microarray results were not significant (Fig 1A). Quantitative PCR analysis was used to verify the microarray findings and demonstrates that expression levels of SGK1, as well as SGK2 and SGK3, are significantly decreased in the PFC of PTSD subjects (Fig 1B and S3 Table). Several other stress- and glucocorticoid-regulated proteins were also examined, although only the glucocorticoid receptor DNA binding factor 1 (GRLF1) was significantly altered in the PFC of PTSD subjects (Fig 1C). Previous studies have reported that FKBP5 levels are decreased [21] or increased [22] in blood or lymphocytes of PTSD patients. In our microarray study, FKBP5 expression was down-regulated in PTSD samples, but this result did not pass FDR for significance. Out of three SGK isoforms, we decided to investigate SGK1, as it is the only gene among the three SGK family members that is transcriptionally regulated by glucocorticoids [23–25]. In addition, SGK1 is known to regulate the function of transcription factors, ion channels, and ion carriers and is reported to play a role in cellular and behavioral models of learning and memory [23].
Fig 1. SGK1 expression is significantly decreased in the prefrontal cortex of PTSD patients.
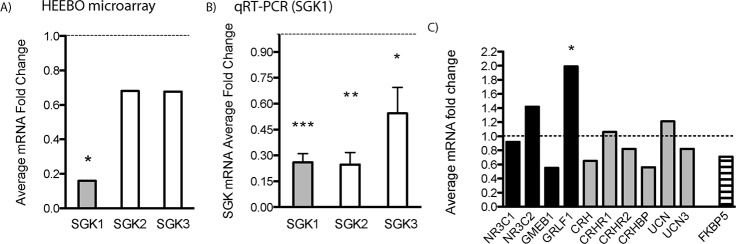
(A) Microarray gene expression analysis of postmortem dorsolateral PFC (DLPFC) samples collected from patients with PTSD shows decreased levels of SGK1 mRNA. Microarray analysis demonstrates that levels of SGK2 and SGK3 isoforms are reduced, but these effects were not significant. (B) Real-time qPCR was conducted to verify the microarray findings. Data are expressed as a mean fold change ± standard error of the mean (SEM) (n = 6), compared to healthy controls (n = 6). [t(10) = 5.966 for SGK1; t(8) = 4.275 for SGK2; t(8) = 2.057 for SGK3, Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001]. (C) Expression levels of stress- and glucocorticoid-regulated genes were also examined. Changes are shown as a mean fold change. Asterisk indicates a significant p-value (*p < 0.05, FDR adjusted). For underlying data, see S1 Data. NR3C1, glucocorticoid receptor; NR3C2, mineralocorticoid receptor; GMEB1, glucocorticoid modulatory element binding protein 1; GRLF1, glucocorticoid receptor DNA binding factor 1; CRH, corticotropin-releasing hormone; CRHR1, corticotropin-releasing hormone receptor 1; CRHR2, corticotropin-releasing hormone receptor 2; CRHBP, corticotropin-releasing hormone binding protein; UCN, urocortin; UCN3, stresscopin (urocortin 3); FKBP5, FK506 binding protein 5.
To investigate the effects of SGK1 on rodent behavior, we used a rat inescapable stress paradigm that has been employed in rodent studies of depression but that also models some features of PTSD [23,24]. In this paradigm, exposure to acute foot shock stress results in loss of escape behavior, referred to as helplessness. After a series of inescapable foot shocks on day 1, rats were tested in active avoidance on day 4. Approximately half of the animals failed to escape the foot shocks, consistent with previous reports in outbred animals [25]. These animals are referred to as the low-escape group (>12 or more failures). The remaining animals learned to escape and are referred to as the high-escape group (<12 failures) (Fig 2A). There was a significant decrease in SGK1 protein levels in PFC (dissections included prelimbic, infralimbic, and cingulate PFC) of low-escape rats, compared to naïve, similar to the decrease in PTSD subjects (Fig 2B). We also observed decreased SGK1 mRNA, but not protein levels in the hippocampus (S1 Fig). As SGK1 expression is induced by glucocorticoids, we measured protein levels of glucocorticoid receptor (GR) in the PFC and found no significant changes between naïve, high-, and low-escape rats (S2 Fig). We also found that levels of postsynaptic density protein-95 (PSD-95) were significantly lower in the low-escape group compared to naïve or high-escape rats (Fig 2C). Levels of GluR1, an ionotropic glutamate receptor, were also reduced, but not significantly, in the low-escape group (Fig 2C). Plasma corticosterone levels were slightly elevated in high- and low-escape animals compared to control but did not reach significance (S3 Fig).
Fig 2. SGK1 expression is altered in learned helplessness.
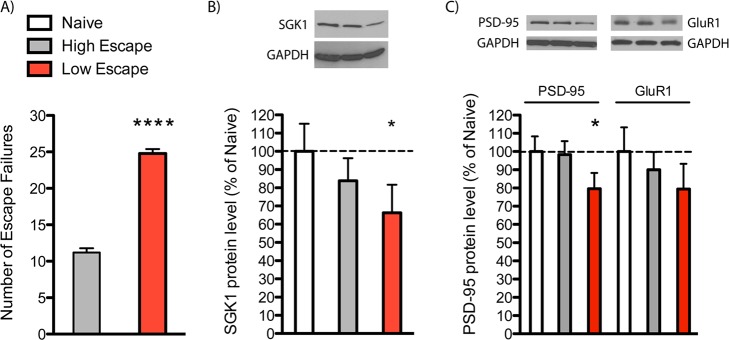
(A) Rats were exposed to inescapable shock (day 1), tested in active avoidance (AA) (day 4), and then sacrificed (day 8) as indicated. Animals were separated into low- and high-escape groups according to their performance in AA test. Data are the mean ± SEM [t(12) = 14.39, Student’s t test, ****p < 0.0001]. (B) SGK1 and (C) PSD-95 protein levels in dissected PFC are decreased in the low-escape group. Data are mean ± SEM percent change over control group (naive, n = 5; high escape, n = 5; low escape, n = 9), One-way ANOVA with post hoc Bonferroni test: F(2,16) = 3.373 for SGK1 and F(2,16) = 3.704 for PSD-95, * p < 0.05. For underlying data, see S1 Data.
To determine whether the behavioral differences between the low- and high-escape groups are due to decreased SGK1 activity, we developed an approach to inhibit SGK1 function by viral expression of a dominant negative form of SGK1 (SGK1 S422A, referred to as dnSGK1). Enhanced green fluorescent protein (EGFP) and/or dnSGK1 were expressed under the regulation of the synapsin I promoter using recombinant adeno-associated virus (rAAV) (Fig 3A and S5 Fig). rAAV-dnSGK1 or rAAV-EGFP control viruses were infused into the mPFC of rats, and after 3 wk to allow for optimal viral expression, they were assessed in behavioral models according to the timeline shown in Fig 3B. Expression and location of the infusion were confirmed by analysis of EGFP (Fig 3C). Infusions of rAAV-dnSGK decreased Ser133 phosphorylation of cAMP response element-binding protein (CREB), a direct SGK1 phosphorylation target, demonstrating the functional consequences of dominant negative SGK expression (Fig 3D and 3E). Studies in primary cultures demonstrate that dnSGK1 does not influence total CREB levels (S6 Fig), consistent with previous reports [26], indicating that decreased phospho-CREB (pCREB) is due to decreased phosphorylation of CREB, not total CREB levels. In the absence of prior foot shock stress, rats infused with rAAV-dnSGK1 displayed significantly fewer escapes compared to rAAV-EGFP controls (Fig 3F), similar to the low-escape group exposed to foot shock stress. In contrast, infusion of rAAV-overexpressing SGK1 (referred to as wtSGK1) resulted in a higher number of escapes in this paradigm (Fig 3J). rAAV-dnSGK1 rats also displayed significantly decreased preference for a sweetened sucrose solution, an indication of anhedonia (Fig 3G). There was no difference in total fluid consumption (Fig 3H) or locomotor activity (Fig 3I) between groups. These studies demonstrate that inhibition of SGK1 is sufficient to cause helplessness- and anhedonic-like behaviors. Infusions of rAAV-dnSGK1 compared to rAAV-EGFP had no significant effects on behavior in anxiety-based models, including the elevated plus maze, novelty-suppressed feeding, and open field (note that rAAV-EGFP control and rAAV-dnSGK1 rats displayed short center time in the open field, which could occlude any further decrease that would be suggestive of increased anxiety) or forced swim test (S4 Fig).
Fig 3. Viral expression of dnSGK1 increases escape failures.

(A) rAAV construct used to express dnSGK1/EGFP or EGFP alone (control). ITR, inverted terminal repeats; IRES, internal ribosomal entry site. (B) Experimental timeline for behavioral analysis after bilateral mPFC infusions of rAAV-EGFP, rAAV-dnSGK1, or rAAV-wtSGK1. SPT, sucrose preference test; LA, locomotor activity. (C) Shown is a representative low-power magnification of green fluorescent protein (GFP) expression in the medial PFC after infusion of rAAV-dnSGK1. Expression was observed in the prelimbic (PrL) and infralimbic (IL) regions of the medial PFC. Scale bar = 500 μm. (D) Colocalization of dnSGK1 and phospho-CREB (pCREB) in the prefrontal cortex: pCREB Ser133 immunostaining in rat PFC from animals infused with rAAV-control and rAAV-dnSGK1. (E) Quantification of pCREB-positive cells showed an 18% reduction in the dnSGK1-overexpressing group (rAAV-control, n = 16 neurons; rAAV-dnSGK1, n = 16 neurons; two rats per group). Data are mean ± SEM percent change over control group. (rAAV-control 100 ± 5.58; rAAV-dnSGK1 82.55 ± 4.09.) (t(30) = 2.094, *p < 0.05, Student’s t test.) Effects of rAAV-dnSGK1 infusion into medial PFC on behavior were tested in (F) AA (escape failures), (G) sucrose preference, (H) total fluid consumption, and (I) locomotor activity. Data are shown as mean ± SEM (controls n = 11; dnSGK1 n = 9). (t(18) = 2.61 for AA and t(18) = 2.795 for SPT, Student’s t test, *p < 0.05]. (J) AA (escape failures) for rAAV-wtSGK1 injected rats (controls n = 10; wtSGK1 n = 10). (t(18) = 1.933, one-tailed Student’s t test, *p < 0.05.) For underlying data, see S1 Data.
To further analyze the role of SGK1 in relation to PTSD, we used fear conditioning and extinction as another model of PTSD-related behavior that can be tested in animals [27,28]. First, rAAV-dnSGK1 or rAAV-EGFP control virus were infused into the mPFC. After 4 wk, rats were subjected to an auditory fear conditioning and extinction paradigm (Fig 4A). Rats were fear conditioned (day 3) and then underwent extinction training (day 4) and recall testing (day 5) in a contextually distinct environment from that used for conditioning. During the extinction training session, there was no significant difference in levels of conditioned stimulus (CS)-elicited freezing between the dnSGK1 and EGFP controls (Fig 4B), indicating a similar rate and magnitude of extinction between both groups. Twenty-four hours later, the rats were returned to the extinction context in order to assess extinction recall (day 5, Fig 4C). The dnSGK1-expressing rats did not show significantly higher levels of overall freezing compared to EGFP controls, indicating that inhibition of SGK1 did not affect the acquisition of extinction or impair the consolidation of extinction learning. Twenty-four hours after the extinction recall session, the animals were reexposed to the context in which they were fear conditioned to examine context-dependent fear (day 6, Fig 4D). Rats infused with rAAV-dnSGK1 displayed higher levels of freezing compared to EGFP controls during the first 4 min of contextual memory recall test (Fig 4D), although there was no effect during the second block of 4 min. Taken together, the results demonstrate that inhibition of SGK1 enhances the memory of contextual cues associated with fear conditioning.
Fig 4. Influence of viral expression of dnSGK1 on contextual memory recall.
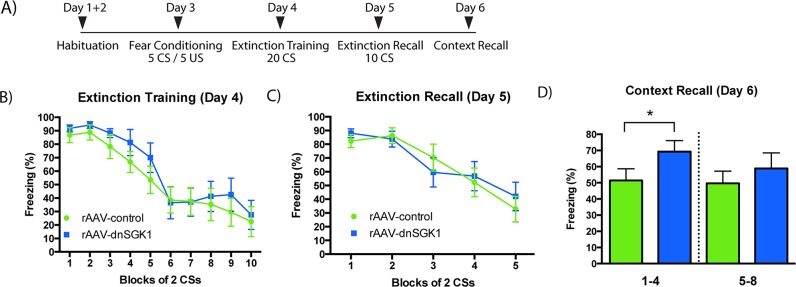
(A) Rats were infused with rAAV-dnSGK1 or rAAV-EGFP into the mPFC. Four weeks post infusion rats underwent auditory fear conditioning and then extinction training and testing according to the timeline schedule shown. (B) During extinction training, CS-elicited freezing was comparable between the dnSGK1 and EGFP groups (effect for group F(1,18) = 0.7033, p = 0.4127; for trial F(9,162) = 23.15, p < 0.0001; interaction F(9,162) = 0.3374, p = 0.9613, two-way repeated measures ANOVA). (C) During extinction recall, there was no significant difference in freezing between dnSGK1 and controls (effect for group F(1,18) = 0.01975, p = 0.8898; for trial F(4,72) = 20.27, p < 0.0001; interaction F(4,72) = 0.7638, p = 0.5523, two-way repeated measures ANOVA). (D) When reexposed to the original context, rats infused with dnSGK1 showed significantly higher freezing during the first 4 min of testing (t(18) = 1.787, one-tailed Student’s t test, *p < 0.05), but not during the second 4-min block. For underlying data, see S1 Data.
To examine the cellular mechanisms underlying these behavioral effects of SGK1 inhibition, levels of synaptic density and function were analyzed in rat mPFC and in primary neuronal cultures. In our initial studies of SGK1 expression in the learned helplessness model, we observed decreased levels of the synaptic protein PSD-95 and a trend for decreased levels of the glutamate receptor GluR1 in the low-escape group (Fig 2C), suggesting that the number of functional spine synapses may be altered in low-escape rats. To directly test this possibility, we examined the influence of SGK1 inhibition on dendritic spines of layer II/III pyramidal neurons in mPFC. Infusion of rAAV-dnSGK1 into the mPFC significantly decreased the density of spines on the dendrites of layer II/III neurons compared to animals receiving control virus (Fig 5A–5C). This indicates that SGK1 is necessary for maintaining the structural integrity of dendritic spines in the PFC. Studies in primary neuronal cultures (14–16 days in vitro [DIV]) further demonstrate that rAAV-dnSGK1 significantly decreases the number of spines on apical dendrites of neurons (Fig 6A and 6B) as well as the function of spine synapses, demonstrated by a reduction of the amplitude (43%) and frequency (36%) of miniature evoked-postsynaptic currents (mEPSCs) (Fig 6C–6E). These in vivo and in vitro findings suggest that the reduction in spine number and function associated with decreased SGK1 expression could contribute to the structural changes of PFC in PTSD patients [29,30] and to the loss of PFC inhibitory control of connected target regions [27,28].
Fig 5. Impact of dnSGK1 overexpression on spine density in the medial PFC.
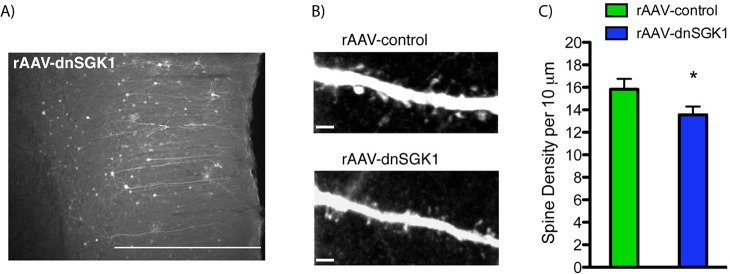
rAAV-dnSGK1was infused into the medial PFC of rats, and 3 wk later, tissue was collected for analysis of spine density. (A) Low-power image of EGFP labeled layer II/III pyramidal neurons. Scale bar = 500 μm. (B) High-power image of representative dendrite segments taken from rAAV-control or rAAV-dnSGK1 showing altered spine number. Scale bar = 4 μm. (C) Spine density analysis of layer II/III pyramidal dendrite segments sampled from rAAV-dnSGK1 injected mPFC (mostly prelimbic cortex) (animals, n = 5; branches, n = 10, total dendritic length = 347.81 μm) or rAAV-EGFP (animals, n = 7; branches, n = 14, total dendritic length = 404.46 μm). Data are shown as mean ± SEM compared to the rAAV-EGFP control group (t(22) = 1.777, Student’s t test, *p < 0.05). For underlying data, see S1 Data.
Fig 6. Impact of dnSGK1 overexpression on dendritic spine density and electrophysiological properties in cultured neurons.

Primary neuronal cultures were incubated with rAAV-control or rAAV-dnSGK1, and spine density and function were analyzed (14–16 DIV). (A) Representative image of EGFP labeling in primary neurons exposed to control virus. Scale bar = 25 μm. (B) Quantitation of spines density showing a decrease in neurons cultured with dnSGK1 (neurons, n = 6; branches, n = 7; total dendritic length = 174.36 μm) as compared to EGFP control (neurons, n = 5; branches, n = 7; total dendritic length = 197.95 μm). Data are shown as mean ± SEM, compared to the control group (t(12) = 3.24, Student’s t test, **p < 0.01]. (C) Analysis of spontaneous mEPSCs (representative recording traces) in hippocampal primary neurons (16 DIV) overexpressing dnSGK1 (n = 7) showed a decrease in amplitude (D) and frequency (E) of mEPSCs compared to EGFP controls (n = 6). Data are shown as mean ± SEM, compared to the control group (t(11) = 4.335, **p < 0.01 for the amplitude; t(11) = 2.738, *p < 0.05 for the frequency, Student’s t test). For underlying data, see S1 Data.
Discussion
Whole genome microarray analysis demonstrates altered expression of a number of genes in postmortem PFC of PTSD subjects. Based on magnitude of change and potential functional impact in response to stress and trauma, we chose SGK1 as a candidate that could contribute to the pathophysiology of PTSD. A recent report describes the role of SGK1 in cortisol-induced reduction of progenitor cell proliferation in the hippocampus and enhancing GR function [31], both of which would be relevant to depression. However, in the PFC, SGK1 expression is not reported to be altered in MDD [32] subjects, indicating possible specificity for PTSD. In addition, the reduction of SGK1 expression in animals that display decreased escape behavior in the learned helplessness model is consistent with the possible role of SGK1 in traumatic stress. However, because of the small cohort of PTSD postmortem subjects (five females and one male) that were analyzed, this finding must be replicated with additional PTSD subjects and better representation of both sexes when samples become available to confirm a role of SGK1 in PTSD. In particular, comparison between PTSD subjects versus those exposed to trauma but with no PTSD diagnosis would be of particular interest, as this comparison would be the most informative in identifying key genes involved in the behavioral deficits observed in PTSD patients.
The results also demonstrate that inhibition of SGK1 in the mPFC causes behavioral deficits characteristic of traumatic stress. We used learned helplessness, a paradigm often employed for studies of depression, because the behavioral deficits are induced by traumatic stress and because helplessness is often associated with PTSD [33].
Given the high comorbidity between MDD and PTSD and the fact that exposure to trauma is a risk factor for MDD as well as PTSD, it has been suggested that pathways mediating learned helplessness are activated in multiple disorders induced by uncontrollable stress, including PTSD and MDD [34].
Supporting the utility of this model, we found that a subgroup of rats that showed decreased avoidance (i.e., helpless-like behavior) also expressed lower levels of SGK1 mRNA and protein, similar to the deficits observed in PTSD postmortem subjects. We directly tested the role of SGK1 in helpless-like behavior using a viral vector and found that overexpression of a dominant negative form of SGK1 in the mPFC reproduced the deficit in active avoidance observed in animals exposed to inescapable stress. In contrast, overexpression of wtSGK1 in mPFC increased the number of escapes in the AA paradigm, compared to control group. We also found that expression of dnSGK1 in the mPFC caused anhedonic-like behavior (i.e., decreased preference for a sweet solution). These studies demonstrate that inhibition of SGK1 is sufficient to cause helplessness- and anhedonic-like behaviors reported in PTSD [33].
Another hallmark of PTSD is the inability to extinguish or inhibit maladaptive fear responses to stimuli previously associated with a traumatic event, even when there is no possibility for recurrence of the trauma. We found that overexpression of dnSGK1 in the PFC enhanced freezing in a context in which the rats had been fear conditioned. These findings demonstrate the functional impact of SGK1 for proper processing of context-associated fear memory. Recent studies showed the importance of mPFC not only for contextual fear conditioning but also for expression of contextual fear [35,36]. These processes require mPFC activity and N-methyl-D-aspartate (NMDA) receptor activation [37,38]. Our findings are consistent with a previous report demonstrating that impaired fear inhibition is a specific marker of PTSD [39].
We also examined the possibility that inhibition of SGK1 causes cellular deficits that could underlie the behavioral changes that were observed. SGK1 is reported to influence the morphology of oligodendrocytes in mouse corpus callosum in response to stress [40]. We found that viral expression of dnSGK1 in the mPFC significantly decreased the density of spines on layer II/III pyramidal neurons compared to controls. Studies in primary neuronal cultures (hippocampus) showed that inhibition of SGK1 produced a similar decrease in the density as well as function of spine synapses (i.e., decreased mEPSCs), although these findings must be confirmed in primary cortical cultures. Together these findings indicate that normal levels of SGK1 are necessary to maintain the structural and functional integrity of spine synapses in the PFC. Moreover, the reduction in spine number and function associated with decreased SGK1 expression could contribute to the structural changes of PFC in PTSD patients [29,30] and to the loss of PFC inhibitory control of amygdala that is required for the extinction of fear memory in animals and humans [27,28]. Further studies will be required to determine the functional impact of decreased SGK1 in vivo and to determine the underlying mechanisms by which SGK1 influences synaptic density and function, such as regulation of the insertion of synaptic glutamate receptors. For example, SGK1 is reported to inhibit stress-induced potentiation of glutamatergic transmission through regulation of NMDA and AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking, as well as to produce more long-term effects via regulation of transcription factors [17,41,42].
In summary, the results of this study demonstrate that SGK1 expression is decreased in the postmortem PFC of a small cohort of PTSD subjects, a finding that must be confirmed in additional PTSD subjects. In addition, studies are currently underway to examine levels of SGK1 in the blood cells of PTSD patients as well as to test for association of SGK1 polymorphisms in PTSD. It is possible that SGK1 may serve as a potential clinical biomarker for PTSD, as recently described for pituitary adenylate cyclase-activating polypeptide (PACAP) [43], which was also shown to contribute to behavioral changes observed in LH [44]. Finally, identification of downstream targets of SGK1 could contribute to a better understanding of the molecular mechanisms underlying the observed behavioral deficits and provide targets for novel PTSD therapeutic medications.
Materials and Methods
Ethics Statement
Postmortem brain samples were obtained from the Stanley Medical Group Brain bank, which was responsible for consent from next of kin. All animal procedures were in accordance with United States National Institutes of Health standards and approved by the Yale University Institutional Animal Care and Use Committee (Protocol number: Yale IACUC #2014–07235).
Human Subjects
The study includes total RNA samples obtained postmortem from six patients diagnosed with PTSD (using DMS-IV criteria) and six age-matched healthy subjects.
Microarray Analysis
Brains from patients diagnosed with PTSD were matched to brains from healthy subjects based on sex, race, and age. Human Exonic Evidence-Based Oligonucleotide (HEEBO) microarrays (Microarrays, Nashville, Tennessee) were used to analyze changes in gene expression within the PFC. Four μg of total RNA (RIN > 5.0, A260/280 > 2.0) from PTSD (n = 6) and matched control human samples (n = 6) were reverse transcribed into cDNA and indirectly labeled with highly sensitive fluorescent technology (Genisphere, Hatfield, Pennsylvania). The whole genome expression analysis was performed using two-channel MI Ready microarrays (Microarray, Huntsville, Alabama). Arrays contained 70-mer oligonucleotide probes from HEEBO. First, cDNA was applied on the array chip overnight to allow hybridization. Next, chips went through a series of washes to minimize nonspecific binding and then were labeled with fluorescent Cy3 and Cy5 dendrimers. After posthybridization washes, the array chips were scanned using a GenePix scanner (Axon Instruments, Union City, California), and image analysis was performed using GenePix Pro 6.0 software (Axon Instruments). Statistical expression analysis of microarray image files was performed using GeneSpring GX 7.3.1 software (Silicon Genetics, Redwood City, California). One sample from the control group had to be excluded from the analysis because of poor hybridization. Raw values were normalized to both control channel values (per spot) and to positive control genes (per chip). The level of regulation for an individual gene was calculated by taking the log ratio of the median experimental channel signal to the median control channel signal. Down-regulated genes were identified by an average expression ratio of <0.77-fold, while up-regulated genes were defined as having an average expression ratio of >1.3-fold. Statistical analysis was performed by an unpaired t-test without using a cross gene error model within the GeneSpring software. Statistical significance was considered to be p ≤ 0.05 with Benjamini and Hochberg FDR as the correction for multiple testing.
Quantitative Real-Time PCR (qPCR)
Five hundred ng of total RNA extracted from human PFC tissue (six PTSD patients and six control subjects) was reverse transcribed into cDNA using oligo-dT primers (Genisphere, Hatfield, Pennsylvania). Real-time PCR (qPCR) was performed utilizing a hot-start SYBR Green method with an ABI 7900 instrument (Applied Biosystems, Foster City, California) with the following conditions: 2 s at 94°C (denaturation), 30 s at 60°C (annealing), and 30 s at 72°C (elongation), for 40 cycles, using a Quantitect SYBR Green PCR kit (Qiagen, Valencia, California). All primers were designed using Primer3 v. 0.4.0 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/; Whitehead Institute for Biomedical Research, Cambridge, Massachusetts), and their specificity was verified using nucleotide blast software (BLAST Interface, NCBI). SGK1 gene fold changes in PTSD subjects versus those in controls were determined using ΔΔCt (Ct = cycle number at threshold) analytical method with normalization against housekeeping genes cyclophilin and GAPDH. The sequences of qPCR primers used are as follows: SGK1 forward 5ʹ- cgccggagtatctcgcacctg-3ʹ and reverse 5ʹ-gcggcaggccatacagcatc-3ʹ; cyclophilin forward 5ʹ-caaatcagaatgggacaggtggag-3ʹ and reverse 5ʹ-gtttgtgttgcggcctgcatttg-3ʹ; GAPDH forward 5ʹ-cgggaaactgtggcgtgatgg-3ʹ and reverse 5ʹ-gccagtgagcttcccgttcagc-3ʹ.
Animals and Behavioral Experiments
Male Sprague-Dawley rats (Charles River Laboratories International) were used for all experiments. All animal procedures were in accordance with US National Institutes of Health standards and approved by the Yale University Institutional Animal Care and Use Committee. For the inescapable foot shock (IFS) and active avoidance (AA) paradigms for rAAV-wtSGK1, rats were habituated to the chamber for 5 min and then subjected to 60 randomized 0.8 mA foot shocks. Three days after IFS or 3 wk after PFC virus infusion, animals were tested in the AA paradigm. The results are expressed as the number of escape failures or the number of times that the animal did not terminate the foot shock by making the appropriate crosses. For the SPT, rats were habituated to 1% sucrose solution (Sigma, St. Louis, Missouri) for 48 h on day 21 post-PFC infusion. The test was performed for 1 h after 4 h deprivation in the presence of two bottles containing 1% sucrose solution and tap water as previously described [45]. The amounts of sucrose solution or water were measured, and the percent sucrose preference was determined.
Novelty suppressed feeding
A novelty suppressed feeding test was conducted as previously described [46]. Briefly, rats were food deprived overnight and, on the test day, placed in an open field (76.5 cm × 76.5 cm × 40 cm, Plexiglas) with food pellets in the center. The animals were given 15 min to approach the food and eat. The test was stopped as soon as the animal took the first bite. The latency to eat was recorded in minutes. Home cage food intake was also measured as a control (results are presented in S4 Fig).
Elevated plus maze
The elevated plus maze consisted of two open arms (50 × 10 cm) and two closed arms (50 × 10 × 40 cm) arranged such that the two open arms were opposite to each other. The maze was elevated to a height of 50 cm. Rats were placed in the center of the maze facing an open arm and were allowed to explore the maze for 10 min. The total time spent in each arm as well as the number of entries into each arm was recorded (results are presented in S4 Fig).
Open field
Open field test was performed in an open field (76.5 cm × 76.5 cm × 40 cm, Plexiglas). Each rat was allowed to freely explore the open field for 10 min. Anxiety-like behavior was assessed as the time spent at the periphery of the open field over a 5-min period (results are presented in S4 Fig).
Forced swim test
A forced swim test (FST) was conducted as previously described [47]. Animals were exposed to preswim on day 1; on the next day, rats were placed in a clear cylinder with water (24 ± 1°C, 45-cm depth) for 15 min and scored for the duration of immobility. Immobility was defined as floating or remaining motionless without leaning against the wall of the cylinder (results are presented in S4 Fig).
Locomotor activity
Ambulatory locomotor activity was recorded for 30 min using automated cage activity meters equipped with photocells (Digiscan animal activity monitor, Omnitech Electronics, Columbus, Ohio) and measured as a number of beam breaks (results are presented in S4 Fig).
Western blot analysis
Fresh rat PFC and hippocampal tissue was homogenized in lysis buffer (25 mM HEPES, 300 mM NaCl pH 7.4, 2% Triton-100) containing proteinase inhibitors (Roche, Indianapolis, Indiana) and phosphatase inhibitors (10 mM NaF, 1 mM NaVO3). The protein concentration was measured using a BCA kit (Pierce, Rockford, Illinois). Then, protein samples were electrophoretically separated on an SDS-PAGE gel (10% Tris-HCl; Bio-Rad, Hercules, California) and transferred overnight to nitrocellulose membranes (0.2 μm pores; Millipore, Bedford, Massachusetts). The membranes were incubated in 5% dry, nonfat milk blocking solution (Tris-buffered saline [TBS], 0.1% Tween 20) for 1 h and then incubated at 4°C overnight with primary anti-SGK1 (1:400, Novagen), anti-PSD-95 (1:1000, Invitrogen), anti-GluR1 (1:1000, Abcam), anti-GR (1:1000, Santa Cruz), and anti-GAPDH (1:10000, Advanced Immunochemical) in 1% dry nonfat milk TBST. After 3 x 15 min washes membranes were incubated for 1 h with secondary, horseradish peroxidase (HRP) conjugated antibodies (1:10000, Vector Laboratories, Burlingame, California). Following 3 x 15 min washes in TBST, blots were developed using a chemilluminescence kit (Pierce, Rockford, Illinois) for 5 min and exposed onto HyBlot CL autoradiography film (Denville Scientific, Metuchen, New Jersey). Developed blots were scanned and analyzed using ImageJ software (Scion Corp, Frederick, Maryland).
For in vitro studies, E19 rat hippocampal or cortical primary cultures lysates were prepared in the same way as described above. After SDS-PAGE, nitrocellulose membranes were incubated with anti-CREB (1:1000, Cell Signaling) antibody at 4°C overnight.
Immunohistochemistry for pCREB
Sections of PFC prepared from perfused (4% paraformaldehyde) brains from rAAV-EGFP and rAAV-dnSGK1 infused rats were blocked in 5% NGS (normal goat serum) for 1 h and incubated overnight at 4°C with anti-pCREB Ser133 (1:800, Upstate) and anti-GFP (Aves Labs) antibodies. After 3 x 15 min washes, the slices were incubated for 2 h with secondary Alexa Fluor 488 and 546-conjugated antibodies (Jacson ImmunoResearch and Invitrogen, respectively), and confocal images were collected. For quantitation of pCREB levels, a region of interest (ROI = GFP positive neurons) was selected using the ImageJ program (ROI manager), first on the GFP image (green channel). The same ROI mask was then applied to the corresponding pCREB image (red channel) for quantitation of pCREB signal intensity. Measured intensity values were corrected for the background nonspecific autofluorescence.
Construction, Preparation, and Infusion of Recombinant AAV
The rat SGK1 S422A cDNA (dnSGK1), followed by the IRES-EGFP expression cassette, was cloned into an AAV2 backbone containing human Synapsin I promoter. The wtSGK1 cDNA was cloned into AAV2 backbone under the control of Synapsin I promoter. The control rAAV contained Synapsin IRES-EGFP expression cassette. Recombinant AAV2/1 viral stocks were generated as described previously [48]. Briefly, HEK293 cells were transfected using Perfectin reagent (Genlantis, San Diego, California) with AAV cis plasmid together with pDp1 and pDp2 helper plasmids. At post-transfection day 3, cell pellets were collected and lysed in three freeze-thaw cycles in dry ice/ethanol bath. Next, collected lysates were filtered using 0.4 μm gauge filters and applied to HiTrap heparine HP affinity columns (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) for AAV purification. Viral titers were determined in rat hippocampal primary cultures. For PFC virus infusions, rats were anesthetized with xylazine (6 mg/kg, IM Lloyd laboratories, Shenandoar, Iowa) and ketamine (80 mg/kg i.m., Fort Dodge Animal Health, Overland Park, Kansas). The virus was delivered bilaterally at +3.5 mm anterior-posterior, ±0.5 mm lateral, and −4 mm ventral from the bregma [49]. Each PFC hemisphere was infused with 3 μl of titer adjusted viral stocks at a rate of 0.2 μl per minute. Behavioral tests were performed 3 wk after virus infusion.
Auditory Fear Conditioning and Extinction
Four identical conditioning chambers were used in all experiments (30 x 20 x 25 cm; MED-Associates). Fear conditioning and extinction testing occurred in two different contexts. The context was cleaned between animals using a dilute 0.9% acetic acid solution. For extinction training and testing (in a contextually distinct environment), the appearance of the chambers was modified, and in addition, the extinction context was further changed by cleaning the chambers with a 1% almond-scented solution. On days 1 and 2, the rats were habituated to the conditioning chambers for 10-min each. On day 3, the rats were acclimated to the chambers for 3 min. Following this, five auditory CS tones (20 s, 75 dB, 1 kHz) were presented that coterminated with foot shocks (1 s, 1.3 mA). The intertone interval ranged between 60 and 120 s (mean = 78 s). On day 4, the rats underwent extinction training in a different context. After 3 min, the rats received 20 presentations of the CS alone (no US; 60 s intertone interval). Defensive freezing, which was defined as the absence of observable movement except those necessary for respiration, was scored every 3 s during presentation of the tones, and values were averaged across two trials, resulting in ten individual CS blocks for analysis. On day 5, an extinction test was performed in which the rats were returned to extinction context and received ten additional presentation of the CS alone. Again, freezing during the tone was scored every 3 s and averaged across two trials resulting in five individual CS blocks for analysis. Finally, on day 6, the rats were returned to conditioning context and an 8-min extinction test was performed, at which time no tones or foot shocks were delivered. During the context recall test, freezing levels was scored every 8 s during the entire 8-min test. Recordings were scored by an observer blind to the rats’ treatment history.
Immunocytochemistry and Spine Density Analysis
Brains were perfused with 4% paraformaldehyde and sectioned using a Vibrotome (Leica). 60-μm brain sections were blocked in 5% NGS for 1 h and incubated overnight at RT with chicken anti-GFP primary antibody (1:400, Aves Labs). After 3 x 15 min washes, slices were incubated for 2 h with secondary anti-chicken Alexa Fluor 488-conjugated antibody (Jacson ImmunoResearch). Confocal z-stacks of dendrites (distal tuft branches) were collected using Olympus Fluoview confocal microscope with a 100x objective with constant laser intensity. Total branch length and spine density were determined using Volocity software (Improvision) in 3D restored confocal z-stacks (1024 x 1024 resolution) that went through deconvolution to remove noise pixels. The analysis was performed in the blind fashion, and the spines were counted by hand.
In Situ Hybridization
In situ hybridization was conducted according to standard previously published protocol used in our laboratory [50]. (35S)-radiolabeled, SGK1 specific riboprobes were generated by an in vitro transcription reaction using a PCR product-derived template and MAXIscript kit T7 polymerase (Ambion, Austin, Texas). For generation of the probe template, the following primers (forward and reverse) were used in PCR reaction:
rSGK1 L, 5ʹ-cagagcgcaatgttctgttgaag -3ʹ;
rSGK1 L, 5ʹ-CCAAGCCTTCTAATACGACTCACTATAGGGAGAgtcccattgtgctcgatgttctc-3ʹ
The optical density was quantified within three regions of the hippocampus (CA1, CA3, and DG) using ImageJ (NIH). Using the manufacturer’s calibration scale, specific labeling was determined by subtracting nonspecific binding from total binding.
ELISA
Serum was extracted and analyzed using ELISA assay for corticosterone levels according to the manufacturer’s specifications (Assay Designs, Ann Arbor, Michigan).
Hippocampal and Cortical Primary Cultures
E(18) hippocampal and cortical primary cultures were prepared from Sprague Dawley rat tissue as previously described [51,52] and cultured for 2–3 wk in Neurobasal medium supplemented with B27 (Invitrogen).
Electrophysiology
The protocol for measuring the spontaneous postsynaptic currents has been described previously [53]. Briefly, the currents were recorded at −70 mV by whole-cell patch-clamp at room temperature (~25°C). Recording electrodes contained (in mM) 120 potassium gluconate, 8 NaCl, 0.5 EGTA, 10 Hepes, and 2 MgATP (pH 7.3). The external solution contained 150 NaCl, 5 KCl, 2.5 CaCl2, 5.5 Hepes-acid, 4.5 Hepes-Na, 10 glucose, and 0.1 tetrodotoxin (pH 7.3). Hippocampal neurons (DIV 16–18) were perfused continuously over the cells, and the inward currents were recorded at resting potential, using Clampex 8, and the data were analyzed, using pClamp 10.2 (Molecular Devices).
Supporting Information
(XLSX)
(A) Representative autoradiographs and quantitative analysis of hippocampal SGK1 mRNA levels determined by in situ hybridization in high- and low-escape rats. SGK1 mRNA levels in the CA1 and CA3 pyramidal cell layers and the dentate gyrus granule cell layer were determined. (One-way ANOVA with post hoc Bonferroni test [F(2,16) = 6,014 for CA1, F(2,16) = 3,214 for CA3, and F(2,16) = 7,131 for DG, *p < 0.05). (B) Western blot analysis of SGK1 protein levels in rat whole hippocampal lystates prepared from high- and low-escape rats (naïve, n = 5; high escape, n = 5; low escape, n = 9).
(TIF)
(A) Representative western blot and quantitative analysis of SGK1 protein levels in high- and low-escape rats (naïve, n = 5; high escape, n = 5; low escape, n = 9).
(TIF)
Rats were exposed to inescapable shock (day 1), tested in AA (day 4), and then sacrificed and blood samples were collected (day 8) (naïve, n = 5; high escape n = 5; low escape n = 9).
(TIF)
(A) Behavioral evaluation after bilateral PFC infusions of rAAV-EGFP (n = 8) or rAAV-dnSGK1 (n = 10). Rats were tested in several different anxiety-based animal models, including (B) novelty suppressed feeding (NSF), (C) elevated plus maze (EPM), (D) open field test (OFT), as well as a model of behavioral despair (E), the forced swim test (FST). (F) There was no difference in the locomotor activity between the groups (LA).
(TIF)
(TIF)
Experiment was replicated three times.
(TIF)
F, female; M, male; C, Caucasian; H, Hispanic; PMI, postmortem interval (hours); RIN, RNA integrity number.
(DOCX)
Complete list of up- and down-regulated genes after FDR adjustment for p-values. Healthy controls, n = 5; PTSD patients, n = 6.
(DOCX)
Healthy controls, n = 5; PTSD patients, n = 6. For the microarray, asterisk indicates significant p-value (*p < 0.05, FDR adjusted). Real-time qPCR for FGFR2 t(10) = 3.272, Student’s t test, **p < 0.01.
(DOCX)
Acknowledgments
We would like to thank Steven Southwick, MD, for support and valuable discussions, Samuel S. Newton for help with microarrays, and Rosemary Terwilliger for technical assistance. dnSGK1 and wtSGK1 cDNA was kindly provided by Eminy Lee, PhD, from Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
Traumatic Stress Brain Study Group: David Benedek and Harry C. Holloway, Carol S. Fullerton, Lei Zhang, He Li, and Robert Ursano, Dept of Psychiatry & Center for the Study of Traumatic Stress, USUHS, School of Medicine, Bethesda, MD; Christopher J. Hough, FDA, Silver Spring, MD; Matthew Friedman, National Center for PTSD, US Dept of Veterans Affairs, White River Junction, VT & Dartmouth Medical School, Hanover, NH; John H. Krystal, Clinical Neuroscience Division, VA National Center for PTSD, VA Connecticut Healthcare System, West Haven, CT & Dept of Psychiatry, Yale University School of Medicine, New Haven, CT; Gregory Leskin, National Center for PTSD, VA Palo Alto Health Care System, Palo Alto, CA; James Meyerhoff, Dept of Psychiatry USUHS, School of Medicine, Bethesda, MD and Georgetown University School of Medicine, Washington, DC and Division of Psychiatry & Neurosciences, Walter Reed Army Institute of Research, Silver Spring, MD; Elizabeth A. Osuch, Dept of Psychiatry, University of Western Ontario and Schulich School of Medicine and Dentistry, London, ON, Canada; Maree J. Webster, Stanley Medical Research Institute, Chevy Chase, MD.
Abbreviations
- AA
active avoidance
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid;
- CREB
cAMP response element-binding protein
- CS
conditioned stimulus
- DIV
days in vitro
- DLPFC
dorsolateral PFC
- dnSGK1
dominant negative form of SGK1
- EGFP
enhanced green fluorescent protein
- FDR
false discovery rate
- FST
forced swim test
- GFP
green fluorescent protein
- GR
glucocorticoid receptor
- HEEBO
Human Exonic Evidence-Based Oligonucleotide
- HRP
horseradish peroxidase
- IFS
inescapable foot shock
- IM
intramuscular
- IRES
internal ribosomal entry site
- ITR
inverted terminal repeats
- LA
locomotor activity
- LH
learned helplessness
- mPFC
medial prefrontal cortex
- MDD
major depressive disorder
- mEPSC
miniature excitatory postsynaptic current
- NGS
normal goat serum
- NMDA
N-methyl-D-aspartate
- PACAP
pituitary adenylate cyclase-activating polypeptide
- pCREB
phospho-CREB
- PFC
prefrontal cortex
- PSD-95
postsynaptic density protein-95
- PTSD
post-traumatic stress disorder
- qPCR
quantitative PCR
- rAAV
recombinant adeno-associated virus
- ROI
region of interest
- SEM
standard error of the mean
- SGK1
serum and glucocorticoid regulated kinase 1
- SPT
sucrose preference test
- TBS
Tris-buffered saline
- wk
weeks
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by a VA PTSD Center grant (JHK and RSD), the State of Connecticut, Department of Mental Health and Addiction Services (JHK and RSD), National Institutes of Health (NIH) Ruth L. Kirschstein National Research Service Award (PL), and the National Alliance for Research on Schizophrenia and Depression (MB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Shalev AY, Freedman S, Peri T, Brandes D, Sahar T, Orr SP, et al. Prospective study of posttraumatic stress disorder and depression following trauma. The American journal of psychiatry. 1998;155(5):630–7. [DOI] [PubMed] [Google Scholar]
- 2. Association AP. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, D.C.: American Psychiatric Association; 2000. [Google Scholar]
- 3. Chung H, Breslau N. The latent structure of post-traumatic stress disorder: tests of invariance by gender and trauma type. Psychol Med. 2008;38(4):563–73. 10.1017/S0033291707002589 [DOI] [PubMed] [Google Scholar]
- 4. Bremner JD, Narayan M, Staib LH, Southwick SM, McGlashan T, Charney DS. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry. 1999;156(11):1787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999;45(7):806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168–76. [DOI] [PubMed] [Google Scholar]
- 7. Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73(1):61–71. [DOI] [PubMed] [Google Scholar]
- 8. Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60(4):376–82. [DOI] [PubMed] [Google Scholar]
- 9. Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. [DOI] [PubMed] [Google Scholar]
- 10. Yun RJ, Krystal JH, Mathalon DH. Working memory overload: fronto-limbic interactions and effects on subsequent working memory function. Brain Imaging Behav. 2010;4(1):96–108. 10.1007/s11682-010-9089-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2010;35(1):169–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Kolk B, Greenberg M, Boyd H, Krystal J. Inescapable shock, neurotransmitters, and addiction to trauma: toward a psychobiology of post traumatic stress. Biol Psychiatry. 1985;20(3):314–25. [DOI] [PubMed] [Google Scholar]
- 13. Charney DS, Deutch AY, Krystal JH, Southwick SM, Davis M. Psychobiologic mechanisms of posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):295–305. [DOI] [PubMed] [Google Scholar]
- 14. Neumann ID, Wegener G, Homberg JR, Cohen H, Slattery DA, Zohar J, et al. Animal models of depression and anxiety: What do they tell us about human condition? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(6):1357–75. 10.1016/j.pnpbp.2010.11.028 [DOI] [PubMed] [Google Scholar]
- 15. Zhang L, Li H, Su TP, Barker JL, Maric D, Fullerton CS, et al. p11 is up-regulated in the forebrain of stressed rats by glucocorticoid acting via two specific glucocorticoid response elements in the p11 promoter. Neuroscience. 2008;153(4):1126–34. 10.1016/j.neuroscience.2008.03.022 [DOI] [PubMed] [Google Scholar]
- 16. Licznerski P, Duman RS. Remodeling of axo-spinous synapses in the pathophysiology and treatment of depression. Neuroscience. 2013;251:33–50. 10.1016/j.neuroscience.2012.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lang F, Strutz-Seebohm N, Seebohm G, Lang UE. Significance of SGK1 in the regulation of neuronal function. The Journal of physiology. 2010;588(Pt 18):3349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13(4):2031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Webster MK, Goya L, Firestone GL. Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J Biol Chem. 1993;268(16):11482–5. [PubMed] [Google Scholar]
- 20. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate—a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- 21. Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M, et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol Psychiatry. 2009;66(7):708–11. 10.1016/j.biopsych.2009.02.034 [DOI] [PubMed] [Google Scholar]
- 22. Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA, et al. Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2010;35(8):1684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Kolk B, Greenberg M, Boyd H, Krystal J. Inescapable shock, neurotransmitters, and addiction to trauma: Toward a psychobiology of post traumatic stress. Biological Psychiatry. 1985;20:314–25. [DOI] [PubMed] [Google Scholar]
- 24. Krystal JH. Animal models for post-traumatic stress disorder In: Giller EL Jr., editor. The Biological Assessment and Treatment of PTSD. Washington, D.C.: American Psychiatric Press; 1990. p. 3–26. [Google Scholar]
- 25. Maier SF, Seligman MEP. Learned helplessness: Theory and evidence. Journal of Experimental Psychology: General. 1976;105:3–46. [Google Scholar]
- 26. David S, Kalb RG. Serum/glucocorticoid-inducible kinase can phosphorylate the cyclic AMP response element binding protein, CREB. FEBS letters. 2005;579(6):1534–8. [DOI] [PubMed] [Google Scholar]
- 27. Delgado MR, Nearing KI, Ledoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59(5):829–38. 10.1016/j.neuron.2008.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sah P, Westbrook RF. Behavioural neuroscience: The circuit of fear. Nature. 2008;454(7204):589–90. 10.1038/454589a [DOI] [PubMed] [Google Scholar]
- 29. Yamasue H, Kasai K, Iwanami A, Ohtani T, Yamada H, Abe O, et al. Voxel-based analysis of MRI reveals anterior cingulate gray-matter volume reduction in posttraumatic stress disorder due to terrorism. Proc Natl Acad Sci U S A. 2003;100(15):9039–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodward SH, Kaloupek DG, Streeter CC, Martinez C, Schaer M, Eliez S. Decreased anterior cingulate volume in combat-related PTSD. Biological psychiatry. 2006;59(7):582–7. [DOI] [PubMed] [Google Scholar]
- 31. Anacker C, Cattaneo A, Musaelyan K, Zunszain PA, Horowitz M, Molteni R, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2013;110(21):8708–13. 10.1073/pnas.1300886110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang HJ, Adams DH, Simen A, Simen BB, Rajkowska G, Stockmeier CA, et al. Gene expression profiling in postmortem prefrontal cortex of major depressive disorder. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(48):13329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hammack SE, Cooper MA, Lezak KR. Overlapping neurobiology of learned helplessness and conditioned defeat: implications for PTSD and mood disorders. Neuropharmacology. 2012;62(2):565–75. 10.1016/j.neuropharm.2011.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neuroscience and biobehavioral reviews. 2005;29(4–5):829–41. [DOI] [PubMed] [Google Scholar]
- 35. Resstel LB, Correa FM, Guimaraes FS. The expression of contextual fear conditioning involves activation of an NMDA receptor-nitric oxide pathway in the medial prefrontal cortex. Cereb Cortex. 2008;18(9):2027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knapska E, Maren S. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn Mem. 2009;16(8):486–93. 10.1101/lm.1463909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gilmartin MR, Helmstetter FJ. Trace and contextual fear conditioning require neural activity and NMDA receptor-dependent transmission in the medial prefrontal cortex. Learn Mem. 2010;17(6):289–96. 10.1101/lm.1597410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gilmartin MR, Kwapis JL, Helmstetter FJ. NR2A- and NR2B-containing NMDA receptors in the prelimbic medial prefrontal cortex differentially mediate trace, delay, and contextual fear conditioning. Learn Mem. 2013;20(6):290–4. 10.1101/lm.030510.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, et al. Impaired fear inhibition is a biomarker of PTSD but not depression. Depress Anxiety. 2010;27(3):244–51. 10.1002/da.20663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miyata S, Koyama Y, Takemoto K, Yoshikawa K, Ishikawa T, Taniguchi M, et al. Plasma corticosterone activates SGK1 and induces morphological changes in oligodendrocytes in corpus callosum. PloS one. 2011;6(5):e19859 10.1371/journal.pone.0019859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu W, Yuen EY, Yan Z. The stress hormone corticosterone increases synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors via serum- and glucocorticoid-inducible kinase (SGK) regulation of the GDI-Rab4 complex. J Biol Chem. 2010;285(9):6101–8. 10.1074/jbc.M109.050229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, et al. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Molecular psychiatry. 2011;16(2):156–70. 10.1038/mp.2010.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–7. 10.1038/nature09856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hammack SE, Cheung J, Rhodes KM, Schutz KC, Falls WA, Braas KM, et al. Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology. 2009;34(6):833–43. 10.1016/j.psyneuen.2008.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biological psychiatry. 2007;62(5):496–504. [DOI] [PubMed] [Google Scholar]
- 46. Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64(10):863–70. 10.1016/j.biopsych.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Son H, Banasr M, Choi M, Chae SY, Licznerski P, Lee B, et al. Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proc Natl Acad Sci U S A. 2012;109(28):11378–83. 10.1073/pnas.1201191109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther. 2003;7(6):839–50. [DOI] [PubMed] [Google Scholar]
- 49. Paxinos G, Watson C. The rat brain in stereotaxic coordinates Fourth Edition ed. CA SD, editor: Academic Press; 1998. [Google Scholar]
- 50. Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of MAP kinase causes depressive behavior. Nature medicine. 2010;16(11):1328–32. 10.1038/nm.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35(5):567–76. [DOI] [PubMed] [Google Scholar]
- 52. Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nature medicine. 2012;18(9):1413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105(6):2169–74. 10.1073/pnas.0711647105 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(A) Representative autoradiographs and quantitative analysis of hippocampal SGK1 mRNA levels determined by in situ hybridization in high- and low-escape rats. SGK1 mRNA levels in the CA1 and CA3 pyramidal cell layers and the dentate gyrus granule cell layer were determined. (One-way ANOVA with post hoc Bonferroni test [F(2,16) = 6,014 for CA1, F(2,16) = 3,214 for CA3, and F(2,16) = 7,131 for DG, *p < 0.05). (B) Western blot analysis of SGK1 protein levels in rat whole hippocampal lystates prepared from high- and low-escape rats (naïve, n = 5; high escape, n = 5; low escape, n = 9).
(TIF)
(A) Representative western blot and quantitative analysis of SGK1 protein levels in high- and low-escape rats (naïve, n = 5; high escape, n = 5; low escape, n = 9).
(TIF)
Rats were exposed to inescapable shock (day 1), tested in AA (day 4), and then sacrificed and blood samples were collected (day 8) (naïve, n = 5; high escape n = 5; low escape n = 9).
(TIF)
(A) Behavioral evaluation after bilateral PFC infusions of rAAV-EGFP (n = 8) or rAAV-dnSGK1 (n = 10). Rats were tested in several different anxiety-based animal models, including (B) novelty suppressed feeding (NSF), (C) elevated plus maze (EPM), (D) open field test (OFT), as well as a model of behavioral despair (E), the forced swim test (FST). (F) There was no difference in the locomotor activity between the groups (LA).
(TIF)
(TIF)
Experiment was replicated three times.
(TIF)
F, female; M, male; C, Caucasian; H, Hispanic; PMI, postmortem interval (hours); RIN, RNA integrity number.
(DOCX)
Complete list of up- and down-regulated genes after FDR adjustment for p-values. Healthy controls, n = 5; PTSD patients, n = 6.
(DOCX)
Healthy controls, n = 5; PTSD patients, n = 6. For the microarray, asterisk indicates significant p-value (*p < 0.05, FDR adjusted). Real-time qPCR for FGFR2 t(10) = 3.272, Student’s t test, **p < 0.01.
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.