

Abstract
Src‐family kinase (SFK) signaling impacts multiple tumor‐related properties, particularly in the context of the brain tumor glioblastoma. Consequently, the pan‐SFK inhibitor dasatinib has emerged as a therapeutic strategy, despite physiologic limitations to its effectiveness in the brain. We investigated the importance of individual SFKs (Src, Fyn, Yes, and Lyn) to glioma tumor biology by knocking down individual SFK expression both in culture (LN229, SF767, GBM8) and orthotopic xenograft (GBM8) contexts. We evaluated the effects of these knockdowns on tumor cell proliferation, migration, and motility‐related signaling in culture, as well as overall survival in the orthotopic xenograft model. The four SFKs differed significantly in their importance to these properties. In culture, Src, Fyn, and Yes knockdown generally reduced growth and migration and altered motility‐related phosphorylation patterns while Lyn knockdown did so to a lesser extent. However the details of these effects varied significantly depending on the cell line: in no case were conclusions about the role of a particular SFK applicable to all of the measures or all of the cell types examined. In the orthotopic xenograft model, mice implanted with non‐target or Src or Fyn knockdown cells showed no differences in survival. In contrast, mice implanted with Yes knockdown cells had longer survival, associated with reduced tumor cell proliferation. Those implanted with Lyn knockdown cells had shorter survival, associated with higher overall tumor burden. Together, our results suggest that Yes signaling directly affects tumor cell biology in a pro‐tumorigenic manner, while Lyn signaling affects interactions between tumor cells and the microenvironment in an anti‐tumor manner. In the context of therapeutic targeting of SFKs, these results suggest that pan‐SFK inhibitors may not produce the intended therapeutic benefit when Lyn is present.
Keywords: Glioblastoma, Src-family kinase, Proliferation, Migration, Dasatinib, Lyn kinase
Highlights
The function of Src, Fyn, Yes, and Lyn was examined in several glioma cell lines.
They differentially affected proliferation and migration in culture and in vivo.
Src and Fyn depletion did not affect survival in an orthotopic xenograft model.
Yes depletion prolonged survival in the orthotopic xenograft context.
Lyn depletion reduced survival in the orthotopic xenograft context.
Abbreviations
- Epidermal Growth Factor Receptor
EGFR
- Extracellular matrix
ECM
- Glioblastoma
GBM
- Human Retinal Microvascular Endothelial Cells
HRMECs
- Non-target
NT
- Platelet-Derived Growth Factor Receptor
PDGFR
- short-hairpin RNA
shRNA
- Src-family kinases
SFKs
- Standard Error of the Mean
SEM
- Vascular-Endothelial Growth Factor
VEGF
1. Introduction
Glioblastoma (GBM) is a WHO‐classified grade IV brain tumor, which is the most common and lethal of primary brain tumors. GBM is characterized by morphological diversity, high rate of mutation, and biologic aggressiveness (Louis et al., 2007, 2007). In particular, GBMs exhibit high rates of tumor cell proliferation, resistance to apoptosis, extensive abnormal neoangiogenesis, and significant tumor cell migration resulting in diffusely infiltrative tumors that preclude complete surgical resection. Recent molecular characterizations of GBM tumors have revealed multiple signaling pathways as potential therapeutic targets because their abnormal activity underlies glioma biology (Wick et al., 2011). These signaling pathways include VEGF, PDGFR, MET, and EGFR family signaling, PI3K and related pathways, and Src‐family kinase (SFK) signaling, among others. SFK signaling plays a central role in many key GBM features.
Src‐family kinases (SFKs) are a series of nine membrane‐associated, non‐receptor tyrosine kinases (c‐Src, Fyn, Yes, Lyn, Lck, Blk, Fgr, Hck, and Yrk), of which c‐Src (henceforth referred to as Src) is the original, prototypical, and best‐studied family member (Thomas and Brugge, 1997; Yeatman, 2004). SFKs have been implicated in most cancer types (Summy and Gallick, 2003), where they play a critical role in a number of tumor‐related properties, including proliferation, regulation of cell–cell and cell‐extracellular matrix (ECM) adhesion, motility, and neoangiogenesis (Guarino, 2010, 2003, 2006). In the central nervous system, Src, Fyn, and Yes are expressed abundantly and ubiquitously, while Lck expression is especially low and limited to certain neurons (Omri et al., 1996; Thomas and Brugge, 1997). Lyn expression is observed in oligodendrocyte precursors and brain endothelial cells during development and the cerebellar granular layer, the thalamus, cerebral cortex, septum, striatum, nucleus accumbens, and olfactory bulb in the adult (Achen et al., 1995, 2004, 2003, 1992). In gliomas, amplification of the epidermal growth factor (EGF) or the platelet‐derived growth factor (PDGF) receptors, or upregulation of integrin receptors such as αvβ3 and αvβ5 result in increased SFK activity that mediates tumorigenesis (Ding et al., 2003; Feng et al., 2011; Lu et al., 2009; Park et al., 2006). Additionally, SFK activation is thought to be critically involved in GBM tumor cell motility and invasion (Angers‐Loustau et al., 2004; Ding et al., 2003; Du et al., 2009; Huveldt et al., 2013; Kleber et al., 2008; Lu et al., 2009; Park et al., 2006; Yeatman, 2004), and neo‐angiogenesis (Lund et al., 2006; Mukhopadhyay et al., 1995; Theurillat et al., 1999; Yeatman, 2004).
Because SFK signaling impacts multiple pathways important in GBM, the small molecule inhibitor dasatinib has emerged as a novel therapeutic option (Ahluwalia et al., 2010; de Groot and Milano, 2009), particularly in the context of recurrent GBM (Huveldt et al., 2013). However its usefulness as a drug has several limitations, including a short half‐life and sub‐optimal central nervous system penetration. In addition, because dasatinib targets all SFKs, as well as BCR‐ABL, c‐KIT, PDGFR, and ephrin A2, it is difficult to know which contribute to its anti‐tumor effects. Although there is some functional redundancy between co‐expressed SFKs, there is also plenty of evidence for non‐overlapping functions. For example, in cultured endothelial cells, Src, Fyn, and Yes have distinct roles in the response to vascular‐endothelial growth factor (VEGF): all three are required for VEGF‐mediated proliferation of human retinal microvascular endothelial cells (HRMECs) but only Fyn is required for tube formation. In addition, Fyn knockdown increased HRMEC migration, while Yes knockdown decreased it and knockdown of Src had no effect at all (Werdich and Penn, 2005). In the tumor context, Lyn is more important than Src to prostate cancer cell proliferation (Park et al., 2008). In colorectal carcinoma, patients whose liver metastases exhibited Yes (but not Src) activity had reduced survival compared to patients exhibiting Src (but not Yes) activity (Han et al., 1996). In the glioma context, Src, Fyn, Yes, and Lyn have all been shown to be involved in GBM‐related signaling (Ding et al., 2003; Han et al., 2014; Lu et al., 2009; Stettner et al., 2005), but the importance of individual family members and their effects vary. For example, Ding et al. showed that PDGF stimulation of U87MG cells resulted in Lyn or Fyn activation, depending on the substrate present (vitronectin vs. laminin) and the integrin receptor (αvβ3 vs. αvβ5) that was engaged (Ding et al., 2003). Stettner et al. analyzed GBM tumors at autopsy and showed that Lyn activity levels were significantly elevated compared to Src and Fyn activity, accounting for >90% of the SFK activity in the GBM samples, vs. 30% of the SFK activity in various non‐GBM brain samples (Stettner et al., 2005). On the other hand, Lu et al. has demonstrated the importance of Fyn and Src to EGFR and EGFRvIII signaling in both cell culture and orthotopic xenograft contexts (Lu et al., 2009). Finally, Han et al. have shown that Src and Yes, but not Fyn, Lyn, or Lck, are important to glioma stem cell migration on laminin in culture (Han et al., 2014).
To begin to understand the molecular details of dasatinib effects in GBM, we used a targeted shRNA approach to knock down expression of Src, Fyn, Yes, and Lyn, in the context of both conventional and xenograft glioma cell line models. We focused on SFK involvement in proliferative and motility‐related tumor properties, examining the effect of these knockdowns on growth, migration, and motility‐related signaling in vitro. We also examined knockdown effects on tumor natural history in vivo, observing characteristics such as survival time, overall tumor burden, density, proliferation, and migration. We found that the effect of single SFK knockdown varied with the cell line and the outcome measured, further supporting the idea that SFKs are not functionally redundant. In addition, we found Lyn and Yes to have opposite effects on survival in our in vivo model. Together, the data raise questions about the usefulness of targeting all SFK activity with a single inhibitor and suggest that the clinical benefit of dasatinib treatment may depend on the relative expression of Lyn and Yes kinases in each tumor.
2. Methods
Animal studies were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and were conducted according to Mayo Clinic IACUC guidelines for animal husbandry.
2.1. Orthotopic xenograft model
The xenograft cell lines used in this study were derived from resected human tumors which are propagated by serial transplantation in the flank of nude mice as has been described previously (Giannini et al., 2005; Sarkaria et al., 2006; Sarkaria et al., 2007; Yang et al., 2008). Tumor cell lines in this model are propagated in vivo in order to preserve molecular and histopathologic features of the primary patient tumor specimens.
2.2. Cell lines and conventional and short‐term xenograft culture
GBM8 cells were harvested from flank xenografts for short‐term culture as described (Carlson et al., 2011). LN229, U87, U251, TP483, SF767 cells and GBM8 cells were cultured on tissue‐culture treated plastic dishes at 37 °C, 5% CO2, in DMEM media containing 10% Fetal Bovine Serum (not heat‐inactivated), an additional 2 mM l‐glutamine, and 1% non‐essential amino acids. Additionally, penicillin and streptomycin was added to the GBM8 culture media.
2.3. Constructs
The MISSION non‐target shRNA control vector pLKO‐non‐target (SHC002) and pLenti‐human Src, Fyn, Yes1, and Lyn shRNA vectors, all expressing a puromycin resistance gene, were purchased from the Mission RNAi Consortium shRNA collection (Sigma–Aldrich) and were obtained from the Mayo Clinic Comprehensive Cancer Center RNA Interference Technology Resource. SFK shRNA product identification numbers are: pLKO‐shSrc, TRCN0000038150; pLKO‐shFyn, TRCN0000196446; pLKO‐shYes1, TRCN0000010004; pLKO‐shLyn, TRCN0000010101. The lentiviral‐based pSinLuc vector expressing Luciferase was obtained from Dr. Yasuhiro Ikeda (Mayo Clinic) and has been described previously (Iankov et al., 2010).
2.4. Virus production and infections
Lentivirus stocks were produced using Virapower lentivirus packaging mix and the 293FT cell line following manufacturer's protocol (ThermoFisher Invitrogen). For in vitro/cell culture‐based assays (RNA extraction, whole cell lysates, cell growth assays, or migration assays), LN229, SF767, and GBM8 cells at 50% confluence were incubated for 24 h in a 1:20–1:50 dilution of pLKO‐based virus:media with 4ug/ml polybrene. For orthotopic xenograft implantation, GBM8 cells were incubated with 1:25 pSinLuc and 1:20 pLKO‐based virus (virus:media) with 4ug/ml polybrene. After a 24 h recovery in normal culture media without virus, cells were selected for 2 days with 5ug/ml puromycin before being used in experiments.
2.5. RNA extraction and qPCR
Total RNA was isolated from puromycin‐selected non‐target or shSFK‐expressing cells using the miRCURY RNA Isolation Kit ‐ Cell and Plant (Exiqon Inc.) following the manufacturer's protocol. RNA was converted to cDNA using the High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems Inc.). qPCR reactions were performed in triplicate with 10 ng of cDNA and TaqMan® FAST Universal PCR master mix (Applied Biosystems). Human Src (Hs00178494_m1), human Fyn (Hs00941600_m1), human Yes1 (Hs00736972_m1), human Lyn (Hs00176719_m1) and human GAPDH (Hs99999905_m1) primer/probe sets were purchased from Applied Biosystems. Amplification data were collected with an Applied Biosystems Prism 7900 sequence detector and analyzed with Sequence Detection System software (Applied Biosystems). Data were normalized to GAPDH, and mRNA abundance was calculated using the ΔΔC T method (Livak and Schmittgen, 2001). The mRNA abundance data was then expressed as a percentage change relative to NT cells.
2.6. Cell growth assay
Cell number in culture was followed over time using the MTT/formazan precipitate‐based assay as described previously (Lewis‐Tuffin et al., 2010).
2.7. Transwell and xCELLigence migration assays
Transwell (Boyden chamber) migration assays of LN229, U87, U251, TP483, and SF767 cells were performed as described previously (Lewis‐Tuffin et al., 2010). Briefly, cells were serum‐starved overnight, then 1 × 105 cells/well in serum‐free media were plated on collagen‐coated, 8um pore size transwell inserts and allowed to migrate at 37 °C, 5% CO2 for 6 h toward the following chemoattractants: 0.5% FBS (LN229 and SF767), 2% FBS (U251 and TP483) or 5% FBS (U87). Dasatinib (10 μM) or 1:1000 of DMSO vehicle was included in both the upper and lower chambers. The number of cells migrating to the lower chamber was quantified; data is expressed as % control (vehicle) migration. Each cell and treatment combination was done in triplicate per experiment, and each experiment was repeated four times.
The xCelligence migration assay (Roche Applied Science) of LN229 and SF767 cells expressing non‐target shRNA (NT) or shRNAs targeting Src, Fyn, Yes, or Lyn was done according to manufacturer instructions. Briefly, on day zero LN229 or SF767 cells were infected with virus expressing NT, shSrc, shFyn, shYes, or shLyn shRNA. The next morning (day 1), the cells were fed with regular culture media; later that day this was replaced with serum‐free media for overnight serum‐starvation. On day 2, 4 × 104 cells/well were plated in the top chambers of an equilibrated 16‐well CIM plate, with chemoattractant media (media with 0.5% FBS) in the bottom chambers. The cells were allowed to settle and adhere for 0.5 h before being placed in the RTCA DP xCelligence machine at 37 °C, 5% CO2, where cells were allowed to migrate for 8 h. All data is expressed as % control (NT) migration. Each cell type was done in triplicate per experiment, and each experiment was repeated four times.
2.8. Immunofluorescence and microscopy
SF767 and LN229 cells were plated on glass coverslips for a minimum of 24 h. Cells were fixed with 3% paraformaldehyde at room temperature for 30 min, washed twice in PBS+10 mM glycine, permeabilized with PBS/0.2% Triton X‐100 for 2.5 min at room temperature, then washed again with PBS/glycine before blocking with 3% milk/PBS for 10 min. Coverslips were incubated with Alexa 594‐conjugated phalloidin (Invitrogen) to visualize actin and DAPI to visualize the nuclei. After washing, coverslips were mounted on glass slides with Aqua Poly/Mount (Polysciences, Inc. Warrington, PA). Cells were imaged on a Zeiss LSM 510 META laser scanning confocal microscope using a Plan‐Apochromat 63x/1.4 Oil DIC objective. Photos were compiled in Adobe Photoshop.
2.9. Preparation of cell lysates
The panel of flank xenograft tissue lysates used in this study is identical to those used in a previous study (Lewis‐Tuffin et al., 2010). Whole cell lysates of LN229, SF767, or short‐term explant GBM8 cells, either expressing NT or SFK shRNAs, or treated for 17–24 h with DMSO vehicle or 10 μM dasatinib, were prepared using RIPA buffer as described previously (Lewis‐Tuffin et al., 2010). At least two independent sets of lysates were made for each cell‐type, SFK shRNA type, and treatment type.
2.10. Western blotting
Equal μg amounts of protein lysates were separated by SDS‐PAGE and transferred to nitrocellulose filters using standard methods. Blots were blocked in 3% BSA +5% nonfat dry milk in Tris‐buffered saline (TBS), pH 7.4 + 0.1% Tween 20 (TBST) before being incubated in primary antibodies in 3% BSA/TBST. Blots were rinsed three times and washed four times for 5 min each in TBST. Blots were then incubated with HRP‐conjugated secondary antibodies in 5% milk/TBS, and rinsed and washed in TBST as before. Proteins were detected using Amersham ECL Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, United Kingdom). Blots in this manuscript were performed at least twice on independent lysates. Results presented in Figures 3B, 3C, and 5C are representative and were carried out using cell lysates prepared in large enough quantity to support the entire panel of signaling blots as well as SFK knockdown confirmation blots. Primary antibodies were as follows: from Cell Signaling Technology: rabbit anti‐Src (clone 32G6), rabbit anti‐Lyn (clone C13F9), rabbit anti‐Fyn, rabbit anti‐GAPDH (clone 14C10), rabbit anti‐phosphoY416 Src (#2101), rabbit anti‐phosphoY410 p130 Cas, rabbit anti‐phosphoY925 FAK, Caspase 3, PARP; from BD Transduction Labs: mouse anti‐p130 Cas, mouse anti‐FAK; from ThermoFisher Invitrogen: mouse anti‐p120 catenin (clone 15D2), rabbit anti‐Vav2; from Millipore: rabbit anti‐Yes; from Epitomics rabbit anti‐phosphoY228 p120 catenin; from Abcam: rabbit anti‐phosphoY172 Vav2. HRP‐conjugated secondary antibodies (anti‐mouse and anti‐rabbit) were obtained from Jackson ImmunoResearch Laboratories.
Figure 3.
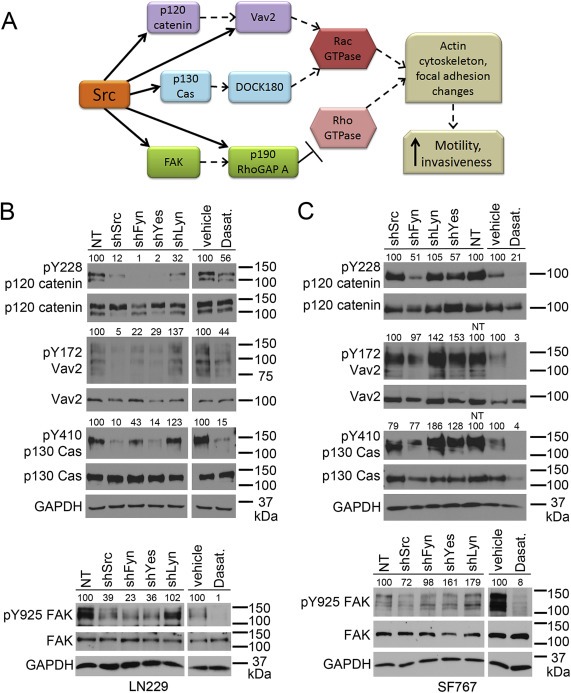
Src‐family kinases differentially target motility‐related proteins. (A) SFKs phosphorylate multiple proteins upstream of the Rac and Rho GTPases, leading to actin cytoskeleton and focal adhesion changes that increase cell motility and invasiveness. In particular, SFKs directly phosphorylate (solid arrows) Y228 on p120 catenin, Y410 on p130 Cas, Y172 on Vav2 (which is also regulated (dashed arrow) by p120 catenin), Y925 on FAK, and Y1105 on p190 RhoGAP A (which is also regulated by FAK). (B,C) Western blot evaluation of motility‐related protein phosphorylation in LN229 (B) and SF767 (C) cells. Lysates were made from cells expressing NT or individual SFK shRNAs, or treated with DMSO vehicle or 10 μM dasatinib for 17–24 h (17–18.5 for LN229, 24 for SF767). PhosphoY228 and total p120 catenin, phosphoY172 and total Vav2, phosphoY410 and total p130 Cas, and phosphoY925 and total FAK were evaluated; GAPDH is the loading control. Numbers above each of the phospho‐protein blots indicate expression relative to the NT lysate (% of NT for NT and shSFK lysates) or the vehicle lysate (% of vehicle for vehicle and dasatinib lysates) and are normalized to the corresponding total protein level as well as to GAPDH expression.
Figure 5.
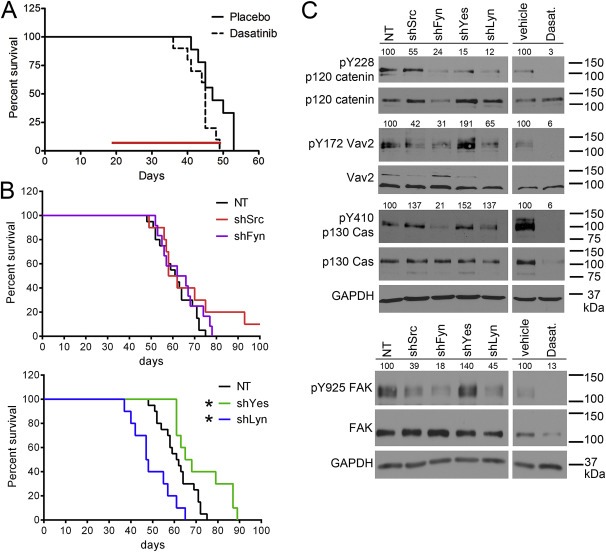
Src‐family kinases differentially influence the survival of mice bearing orthotopic GBM8 tumors as well as motility‐related signaling pathways. (A) Dasatinib treatment had little effect on the survival of mice bearing orthotopic GBM8 tumors. Dasatinib treatment (50 mg/kg, given orally once a day, indicated by the red bar) was begun 19 days after intracranial implantation of GBM8 cells and continued until the mice were moribund. The difference in survival of the two treatment groups (median survival of 47 days for placebo vs. 45 days for dasatinib treatment) was statistically significant (p < 0.05) by Mantel–Cox Log‐rank test but not statistically‐significant by Gehan‐Breslow‐Wilcoxon test. Placebo‐treated n = 9; Dasatinib‐treated n = 10. (B) Individual knockdown of the four SFKs had a differential effect on the survival of mice bearing GBM8 tumors. GBM8 cells were transduced with lentivirus expressing non‐target (NT) shRNA or Src‐, Fyn‐, Yes‐, or Lyn‐specific shRNAs, as well as a luciferase‐expressing lentivirus prior to implantation. There was no difference in the survival of mice implanted with GBM8‐shSrc or –shFyn vs. NT (median survival of 60, 64, and 61.5 days, respectively). Mice implanted with GBM8‐shLyn had shorter survival times vs. NT (median survival of 47.5 days), while mice implanted with GBM8‐shYes had longer survival times vs. NT (median survival of 66.5 days). *indicates statistically significant difference in survival vs. NT‐implanted mice (p < 0.05 by both Mantel–Cox Log‐rank test and Gehan‐Breslow‐Wilcoxon test). NT n = 20; shSrc n = 9; shFyn n = 12; shYes n = 10; shLyn n = 10. (C) Western blot evaluation of motility‐related signaling pathways in GBM8 cells. Lysates were made from cells expressing NT or individual SFK shRNAs, or treated with DMSO vehicle or 10 μM dasatinib for 17 h. PhosphoY228 and total p120 catenin, phosphoY172 and total Vav2, phosphoY410 and total p130 Cas, and phosphoY925 and total FAK were evaluated; GAPDH is the loading control. Numbers above each of the phospho‐protein blots indicate expression relative to the NT lysate (% of NT for NT and shSFK lysates) or the vehicle lysate (% of vehicle for vehicle and dasatinib lysates) and are normalized to the corresponding total protein level as well as to GAPDH expression.
Western blot quantification to determine the relative density of western blot bands was done using ImageJ (National Institutes of Health, U.S.A.). For Figures 2A, 2B and 4C, the goal was to show the relative expression of each SFK in the presence of each shRNA. Therefore SFK band densities were first normalized to GAPDH, and then expressed as a percentage of the NT protein level for that SFK. In Figures 3B, 3C, and 5C, the goal was to show how phosphorylation levels changed in the presence of each shRNA. Therefore, phospho‐protein band densities were first normalized to the corresponding total protein band density. These numbers were then normalized to GAPDH, and finally expressed as a percentage of the NT phospho‐protein level (for NT and shSFK lysates) or as a percentage of the vehicle‐treated phospho‐protein level (for vehicle and Dasatinib lysates).
Figure 2.
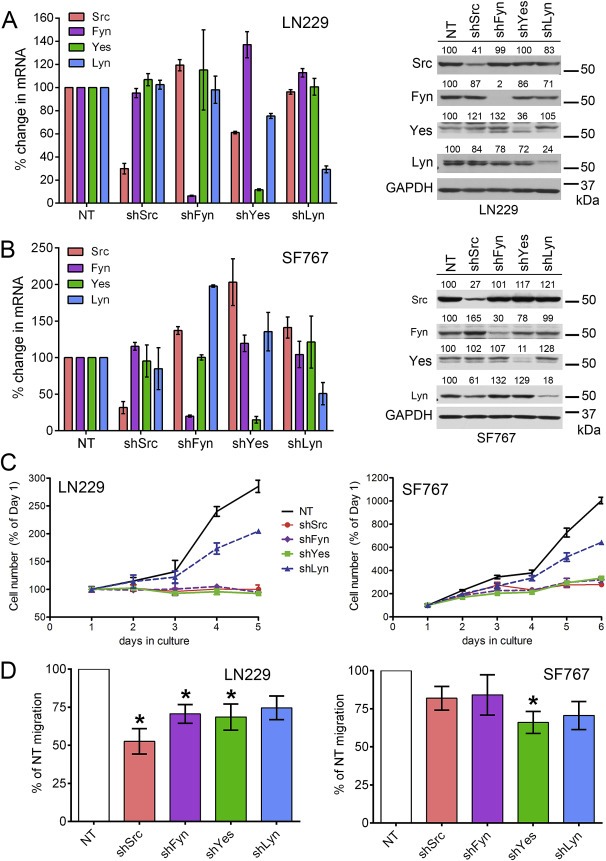
Differential effect of individual Src‐family kinase knockdown on glioma cell line growth and migration. (A) Non‐target (NT) shRNA or shRNA targeted to each of four SFKs (Src, Fyn, Yes, and Lyn) were expressed in LN229 cells. (left panel) QPCR was used to evaluate mRNA levels of all four SFKs in the context of individual SFK knockdown. The selected SFK shRNAs are specific for the mRNA of their targeted kinase without affecting the mRNA levels of the other three kinases. (right panel) Protein expression of all four SFKs in the context of individual SFK knockdown was evaluated by western blot. Numbers above each of the four SFK blots indicate expression relative to the NT lysate (% of NT) and are normalized to GAPDH expression. The blots confirm that each SFK shRNA specifically reduced the protein expression for that kinase with minimal effects on protein levels of the other three kinases. (B) As in (A), but in the context of SF767 cells, confirming that the specificity of the selected shRNAs is cell‐line independent. (C) The effect of individual SFK knockdown on LN229 (left) and SF767 (right) growth in culture varies. LN229‐shSrc, ‐shFyn, and ‐shYes cell number is significantly different from –NT at 3, 4, and 5 days in culture (p < 0.001); LN229‐shLyn is significantly different from –NT at 4 and 5 days in culture (p < 0.001). SF767‐shSrc, ‐shFyn, and ‐shYes cell number is significantly different from –NT at 3, 4, 5, and 6 days in culture (p < 0.001); SF767‐shLyn is significantly different from –NT at 5 and 6 days in culture (p < 0.001). Representative graphs of n = 3 experiments are shown; error bars are SD. (D) The effect of individual SFK knockdown on LN229 (left) and SF767 (right) transwell migration toward media containing 0.5% FBS varies. Migration quantification is normalized to the number of –NT cells that migrated in a particular experiment (set to 100% for each experiment). Error bars are SEM; the data is a combination of n = 4 independent experiments. *significantly different vs. NT (p < 0.05).
Figure 4.
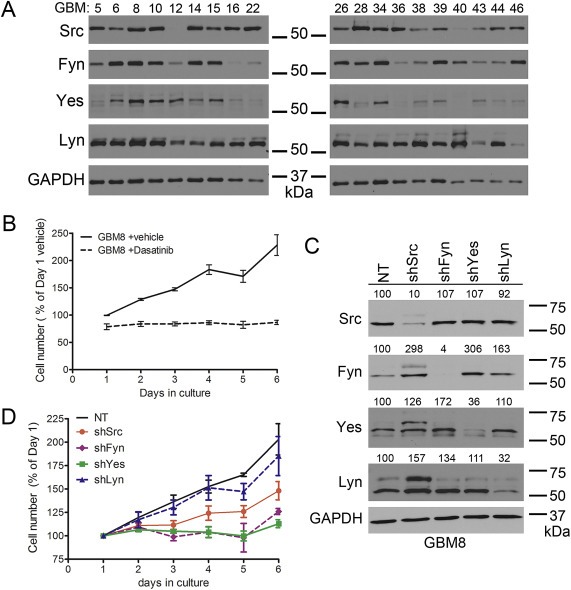
Evaluation of Src‐family kinase expression in GBM xenograft cell lines and effect of individual Src‐family kinase knockdowns on GBM8 cell line growth. (A) Nineteen glioma cell lines propagated as xenografts in mouse flank were examined by western blot for expression of Src, Fyn, Yes, and Lyn. GAPDH serves as a loading control. (B) Dasatinib treatment (10 μM) halts GBM8 growth in culture. GBM8 cell number with dasatinib is significantly different from vehicle‐treated at days 2, 3, 4, 5, and 6 in culture (p < 0.01). Error bars are SEM, the data is a combination of n = 4 independent experiments. (C) The same non‐target (NT) shRNA and Src‐, Fyn‐, Yes‐, and Lyn‐specific shRNAs used with LN229 and SF767 cells were expressed in GBM8 cells. Protein expression of all four SFKs in the context of individual SFK knockdown was evaluated by western blot; GAPDH is the loading control. Numbers above each of the four SFK blots indicate expression relative to the NT lysate (% of NT) and are normalized to GAPDH expression. (D) The effect of individual SFK knockdown on GBM8 growth in culture varies. GBM8‐shSrc cell number is significantly different from –NT at 6 days in culture (p < 0.001). GBM8‐shFyn cell number is significantly different from –NT at 3, 4, 5, and 6 days in culture (p < 0.05). GBM8‐shYes cell number is significantly different from –NT at 4, 5, and 6 days in culture (p < 0.001). GBM8‐shLyn cell number is not different from –NT cell number. Error bars are SEM; the data is a combination of n = 4 independent experiments.
2.11. Orthotopic animal model
Short‐term explant cultures of GBM8 cells derived from flank tumor xenografts, expressing luciferase and either NT or SFK shRNAs, were implanted orthotopically into the brains of 4–5 week‐old female athymic nude mice (HSD: Athymic Nude‐Foxn1 nu) as described previously (Carlson et al., 2011). Tumor cells (3 × 105 per mouse) were implanted 2 mm lateral and 1 mm anterior to bregma, at 3 mm depth. Tumor growth was monitored over time with once/week bioluminescent imaging. Mice were injected intraperitoneally with luciferin at 150 mg/kg in a 0.1 mL volume, then anesthetized with isoflurane and imaged using the IVIS Spectrum (Caliper Life Sciences) at 10–15 min post‐luciferin injection, varying exposure times and sensitivity settings to avoid saturation. For survival experiments, mice were euthanized when they reached a moribund condition. For the fixed tumor growth time experiment, mice were sacrificed four weeks after tumor implantation. In both cases mice were deeply anesthetized with 90 mg/kg pentobarbital delivered intraperitoneally and then euthanized by transcardial perfusion of phosphate‐buffered saline followed by 4% paraformaldehyde. Brains from the mice were resected, cut into four coronal sections of equivalent thickness, and post‐fixed in 4% paraformaldehyde overnight at 4 °C.
2.12. Immunohistochemistry and quantitative analysis
Paraformaldehyde‐fixed brain tissues were embedded in paraffin in a “bread loaf” manner such that four coronal faces of each brain could be stained simultaneously (see Supplemental Figure 2). Paraffin‐embedded tissue was cut into 5 or 10 μm sections, mounted on charged slides, and processed by the Mayo Clinic Cancer Biology Histology Facility for H&E staining or immunohistochemistry using standard procedures. Pathologic examination of H&E slides was done by a neuropathologist (F.J.R.). Properties that were graded included microvasculature (little to none, or increased, when small vessels were easily identifiable) and cell density (low, moderate, or high, depending on the extent of cell crowding). Invasiveness was scored on a three tiered scale as previously described (Lewis‐Tuffin et al., 2010). Briefly, “highly invasive” tumors overtly extended to the contralateral hemispheres through comissures, “moderately invasive” tumors extended to the contralateral hemisphere focally but remained largely localized, and “slightly invasive” remained localized. Immunohistochemical staining for human STEM121 (a human cytoplasmic marker, Stem Cells Inc. #AB‐121‐U‐050, Cambridge, U.K.) and human Lamin A + C (a nuclear envelope marker, Novus Biologicals #NBP1‐95336, Littleton, CO) were done on 10 μm tissue sections following manufacturer instructions; staining for human Ki‐67 (clone MIB‐1, Dako #M7240, Denmark) was done on 5 μm tissue sections. All three were counterstained with hematoxylin. High‐resolution digital images of the staining were obtained by scanning the slides on an Aperio AT2 slide scanner (Leica Biosystems, Vista, CA). Regions of interest were annotated using Aperio ImageScope software. STEM121 and Lamin A + C positivity in these regions was determined using a positive pixel count approach that quantified the % of brown color due to DAB in the context of all color (both DAB (brown) and hematoxylin (blue)). Ki‐67 positivity was determined by counting all the Ki‐67 positive (brown) nuclei and all the negative (blue) nuclei in a 0.25 mm × 0.4 mm region within the core of the tumor.
2.13. Statistical analysis
Migration assays (Figures 1 and 2) and immunohistochemical staining quantifications (Figure 6) were analyzed with one‐way ANOVA followed by Dunnett's multiple comparison test. Cell growth assays (Figures 1, 2, and 4) were analyzed with two‐way ANOVA followed by Bonferroni post hoc testing. Survival analysis (Figure 5) was done using the Mantel–Cox log rank method as well as the Gehan‐Breslow‐Wilcoxon method. Data were considered significant at p < 0.05. All statistical analysis was done using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA).
Figure 1.
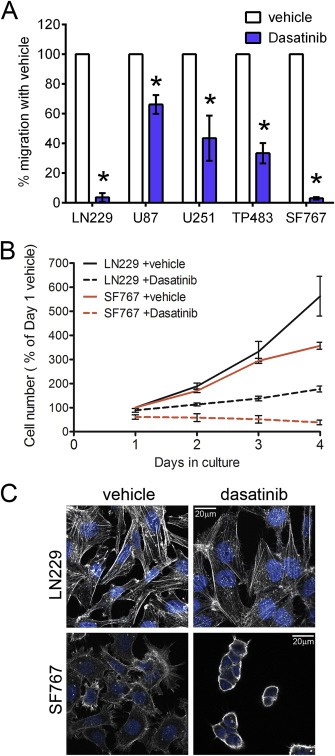
Dasatinib treatment changes conventional glioma cell line migration, growth, and morphology in culture. (A) Dasatinib treatment (10 μM) reduces migration of conventional glioma cell lines toward media containing the following concentrations of FBS: LN229 and SF767: 0.5% FBS; U251 and TP483: 2% FBS; U87: 5% FBS. Error bars are SEM, n = 4. *significantly different vs. vehicle (p < 0.01) (B) Dasatinib treatment (10 μM) reduces growth of LN229 and SF767 glioma cell lines in culture. LN229 cell number with dasatinib is significantly different from vehicle‐treated at days 3 and 4 in culture (p < 0.001). SF767 cell number with dasatinib is significantly different from vehicle‐treated at 2, 3, and 4 days in culture (p < 0.001). Error bars are SEM; the data is a combination of n = 5 independent experiments. (C) Dasatinib treatment (10 μM) changes the morphology of LN229 and SF767 cells by rearranging the actin cytoskeleton, reducing (LN229) or eliminating (SF767) actin stress fibers. LN229 and SF767 cells were plated on coverslips, treated with DMSO vehicle or 10 μM Dasatinib for 24 h, then fixed and stained with phalloidin to visualize the actin cytoskeleton and DAPI to visualize the nucleus. The 20 μm scale bar applies to all images.
Figure 6.
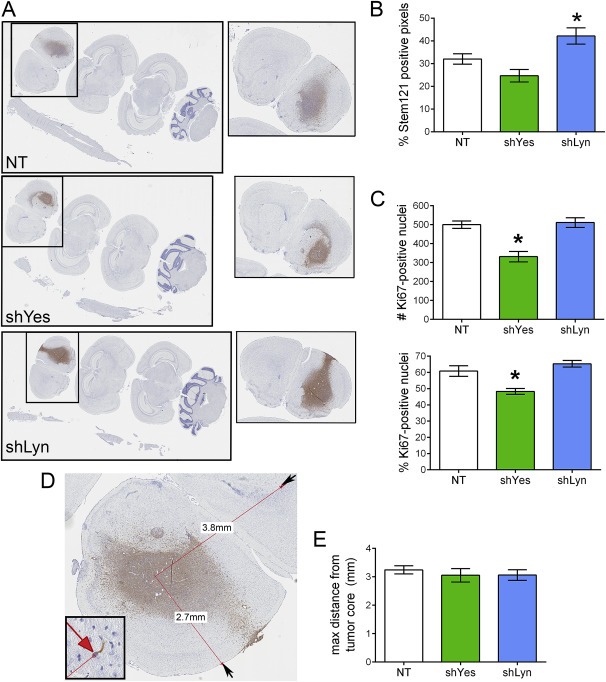
Changes in tumor cell proliferation in mice implanted with GBM8‐shYes but not –shLyn. (A) Brains and spinal cords from mice implanted intracranially with NT, shYes, or shLyn cells were examined histologically for STEM121 (shown), Ki‐67, or Lamin A + C. Single brain slices that showed the most tumor (boxed inset) were used for all follow up analyses. (B) Decreasing Lyn expression resulted in changes to overall tumor burden. The extent of STEM121 staining in a single slice through the main tumor mass was quantified by determining the % positive (brown) pixels of the total pixel number (brown + blue) in high resolution images. Mice implanted with shLyn cells showed a statistically significant increase in the % positive pixels (overall tumor burden) vs. NT (p < 0.05); there was no statistically significant difference between NT and shYes. Data is mean ± SEM; n = 11 brain slices for NT, 8 for shYes, 10 for shLyn. (C) Decreasing Yes expression resulted in decreased proliferation. The total number of Ki‐67 positive nuclei (top), as well as the % Ki‐67 positive (bottom), was determined in a fixed‐size region within the core of each tumor. Mice implanted with shYes cells showed a statistically significant decrease in both measures vs. NT (p < 0.001); there were no differences between NT and shLyn. Data is mean ± SEM; n = 10 brain slices for NT, 8 for shYes, 10 for shLyn. (D) Analysis to determine maximum distance migrated in a single plane. Digital images were created of brains from mice implanted with NT, shYes, or shLyn cells and stained with either the STEM121 human cytoplasm marker (shown) or the human Lamin A/C nuclear marker (Supplemental Figure 2C). On these images, the distance between the center of the main tumor mass and the cell that migrated furthest from this point (red arrow, inset) was determined; data from the two stains was combined for statistical analysis. (E) GBM8 cells migrate away from the main tumor mass to similar extents, regardless of SFK status. There was no statistically significant difference in maximum distance migrated between the NT cells and either knockdown. Data is mean ± SEM; n = 16 brain slices for NT, 11 for shYes, 12 for shLyn.
3. Results
3.1. Dasatinib reduces glioma growth and migration in culture
Src‐family kinases regulate many aspects of cell migration. Therefore, we used a transwell migration assay to screen a panel of five conventionally cultured glioma cell lines for sensitivity to dasatinib (Figure 1A). The migration of all five cell types was significantly inhibited by dasatinib treatment, with the migration of LN229 and SF767 cells being almost completely abolished. We chose these two cell types for further investigation. Both cell types exhibited slowed culture growth (total number of cells over time, reflecting the balance of both cell proliferation and cell death events) in response to dasatinib treatment (Figure 1B). To evaluate the contribution of cell death processes to the dasatinib‐induced growth phenotype, we evaluated the cultures for caspase 3 cleavage and PARP cleavage (Supplemental Figure 1). The results suggest that apoptotic death is not a major contributor to the slowed culture growth observed with dasatinib treatment. In addition, consistent with the known role of SFKs in regulating the actin cytoskeleton, both cell types exhibited morphological changes in response to 24 h of dasatinib treatment (Figure 1C). Dasatinib‐treated LN229 cells became more spread out and had fewer actin stress fibers compared to vehicle‐treated cells. The dasatinib‐treated SF767 cells became completely rounded and adherent to each other, with few of their characteristic filopodial protrusions. Because both cell lines responded to dasatinib treatment with changes in all three of these parameters, we used both for follow up experiments to evaluate the role of individual SFKs in producing the observed changes.
3.2. Individual SFKs differentially affect glioma growth and migration in culture
To examine the role of individual SFKs, we tested five shRNAs against each SFK target (not shown), and identified single shRNA sequences that, when virally transduced into the cells, targeted a single SFK for knockdown while leaving expression of the others unchanged (Figure 2A, B). Using these particular shRNA sequences, we evaluated the effect of single SFK knockdown on overall cell growth (Figure 2C) and migration (Figure 2D). Individual knockdown of Src, Fyn, and Yes protein significantly slowed (SF767) or halted (LN229) culture growth. Knockdown of Lyn also slowed LN229 and SF767 growth, but to a lesser (although still statistically significant) extent (Figure 2C). Interestingly, the effect of single SFK knockdowns on cell migration differed from their effects on growth. Using a transwell migration assay, knockdown of Src, Fyn and Yes significantly reduced migration of LN229 cells, but to different extents, while Lyn knockdown had no effect (Figure 2D). In contrast, only Yes knockdown significantly reduced SF767 migration. Together, these results indicate that the four SFKs have variable roles in cell growth and migration, both within a single cell type and between different cell types. Yes knockdown had the most consistent inhibitory effect across assay and cell type, while Lyn knockdown was noteworthy for its most consistent lack of effect.
3.3. Individual SFKs differentially affect motility‐related phosphorylation
To understand the differing effects of SFK knockdown on cell growth and migration, we initially examined phosphorylation at the Y416 SFK activation loop site (Supplemental Figure 2). Because this site is conserved in the four SFKs under study, western blots for this marker yield information about total SFK activity in the context of single SFK knockdowns. In general, knockdown of any one SFK produced a decrease in phosphorylation at Y416, but to varying degrees of effect. We next examined a variety of motility‐related signaling proteins known to be downstream targets of SFKs, which can lead to changes in Rho and Rac GTPase activity, alterations in the actin cytoskeleton and focal adhesions, and ultimately cell motility and invasiveness (Frame, 2004; Guarino, 2010; Parsons and Parsons, 2004) (Figure 3A). Specifically, Y228 on p120 catenin, Y172 on Vav2, Y410 on p130 Cas, Y925 on FAK, and Y1105 on p190 RhoGAP A are all known SFK targets (Garrett et al., 2007; Mariner et al., 2001; Mitra and Schlaepfer, 2006; Roof et al., 1998). Dasatinib treatment, which should reduce the activity of all four SFKs, reduced phosphorylation on p120 catenin, Vav2, p130 Cas, and FAK in both cell lines (Figure 3B, C). In contrast, knockdown of individual SFKs differentially altered the phosphorylation pattern of these proteins. Specifically, in LN229 cells knockdown of all four SFKs decreased Y228 phosphorylation on p120 catenin, but only shSrc, shFyn, and shYes were associated with decreased Y172 Vav2, Y410 p130 Cas, and Y925 FAK phosphorylation (Figure 3B). Individual SFK knockdown in SF767 cells produced yet another pattern of phosphorylation changes, with shFyn causing a decrease in the phosphorylation of Y228 p120 catenin, Y172 Vav2, and Y410 p130 Cas, while shSrc was associated with reduced Y925 FAK phosphorylation, and shLyn caused increased Y925 FAK phosphorylation (Figure 3C). In all cases, examination of pY1105 p190 RhoGAP phosphorylation was inconclusive (data not shown). Taken together, these results shed some light on the growth and migration data for the LN299 cells. ShLyn had little effect on LN229 growth and migration and also did not greatly alter the phosphorylation patterns of the motility‐related signaling proteins, while knockdown of Src, Fyn, and Yes altered cell growth, migration, and motility‐related phosphorylation patterns. The SF767 results are less clear, as knockdown of Yes altered the growth and migration of these cells but did not alter the motility‐related phosphorylation patterns, while shFyn did not alter SF767 cell migration but did change the motility‐related phosphorylation patterns. The mixed results suggest that the importance of an individual SFK to glioma growth and migration varies with cell line, and adds further support to the idea that co‐expressed SFKs are not necessarily functionally redundant in a given cell type.
3.4. Src, Fyn, and Yes, but not Lyn affect growth of the GBM8 xenograft line in culture
We next investigated the role of the four SFKs in the context of the flank and orthotopic xenograft model of gliomas. Most of our xenograft cell lines expressed all four SFKs (Figure 4A). Consequently, we focused on the GBM8 xenograft cell line, as it is the most invasive of the cell lines in this model ((Lewis‐Tuffin et al., 2010) and data not shown). When grown under conventional culture conditions, the GBM8 cells responded to dasatinib treatment with an abrupt halt in culture growth (total cell number over time, reflecting both proliferation and death events) (Figure 4B). As with the LN229 and SF767 cells, apoptotic cell death was not a major contributor to this altered culture growth (Supplemental Figure 1). After confirming that our previously identified shRNAs for specific SFKs were equally specific in the GBM8s (Figure 4C), we assessed the impact of individual SFK knockdown on GBM8 growth in conventional culture (Figure 4D). Both shFyn and shYes abrogated GBM8 growth, with statistically significant effects starting at 3 and 4 days, respectively. ShSrc slowed but did not halt GBM8 growth, with significant growth effects only evident at 6 days in culture. ShLyn had no effect on GBM8 growth in conventional culture.
3.5. Knockdown of Yes and Lyn have opposite effects on survival
To further assess the role of the four SFKs on GBM8 growth and to determine effects on tumor cell invasion, we turned to the orthotopic xenograft model of gliomas. Initially we assessed survival time for mice implanted orthotopically with GBM8 and then treated with placebo or dasatinib starting at nineteen days after implantation (Figure 5A). These results indicated that dasatinib treatment had a small but significant negative effect on survival. However, because our results from growth in culture suggested that the individual SFKs could have different effects when compared to dasatinib treatment, we virally transduced GBM8 cells with NT or each of the SFK shRNAs, selected for two days, and then implanted the cells orthotopically into nude mice (Figure 5B). Interestingly, the GBM8‐shLyn and –shYes cells had statistically significant and opposite effects on overall survival: mice implanted with GBM8‐shLyn had a median survival time of 47.5 days and mice with GBM8‐shYes had a median survival time of 66.5 days, vs. 61.5 days for mice implanted with GBM8‐NT. There was no difference in survival times relative to NT for mice implanted with GBM8‐shSrc or –shFyn.
A histological assessment of the brains from these mice revealed that, regardless of their original SFK status, the resulting tumors were highly aggressive. Tumor spread was routinely seen into the contralateral hemisphere and throughout the brain in an anterior‐posterior direction, including into the cerebellum in many cases (Supplemental Figure 3A). Interestingly, during periodic bioluminescent imaging, several mice also showed evidence of tumor spread into the lumbar spinal cord (Supplemental Figure 3B). This spread was confirmed with immunohistochemistry using the STEM121 antibody to visualize the human tumor cells in the context of the mouse nervous system tissue (Supplemental Figure 3A,C). Although this observation confirms the aggressive nature of the GBM8 cell line, there were no differences between SFK status and the number of mice with spinal cord spread (data not shown). Pathological examination of H&E and STEM121‐stained brains sections from these mice was used to evaluate relative tumor density, invasiveness, and the presence of microvasculature (Table 1). There were no differences in the presence of microvasculature based on SFK status of the tumors. There were also no differences in tumor density rating or invasiveness rating between NT and –shSrc or –shFyn tumors. There were observable differences in tumor density rating and invasiveness rating between the NT, ‐shYes, and –shLyn tumors. Specifically, 6 of 12 (50%) –NT tumors were classified as high density, while 100% of the –shYes and –shLyn tumors (6 each) were classified as high density. In addition, 11 of 12 –NT tumors and 5 of 6 shYes tumors were classified as highly invasive, while only 1 of 6 –shLyn tumors was highly invasive.
Table 1.
Pathologic evaluation of brain sections suggests differences in tumor density for GBM8‐shYes and –shLyn vs. –NT tumors, as well as differences in invasiveness for GBM8‐shLyn vs. –NT tumors. Numbers are the number of brains with each classification; N = 12 NT, 7 shSrc, 7 shFyn, 6 shYes, 6 shLyn.
| GBM8‐NT | GBM8‐shSrc | GBM8‐shFyn | GBM8‐shYes | GBM8‐shLyn | |
|---|---|---|---|---|---|
| Microvasculature rating: | |||||
| Little | 5 (42%) | 4 (57%) | 3 (43%) | 4 (67%) | 3 (50%) |
| Increased | 7 (58%) | 3 (43%) | 4 (57%) | 2 (33%) | 3 (50%) |
| Tumor density: | |||||
| Moderate | 6 (50%) | 5 (71%) | 4 (57%) | 0 (0%) | 0 (0%) |
| High | 6 (50%) | 2 (29%) | 3 (43%) | 6 (100%) | 6 (100%) |
| Invasiveness: | |||||
| Moderate | 1 (8%) | 2 (29%) | 1 (14%) | 1 (17%) | 5 (83%) |
| High | 11 (92%) | 5 (71%) | 6 (86%) | 5 (83%) | 1 (17%) |
While the survival experiments were ongoing, we assessed the phosphorylation state of the Y416 SFK activation loop site, as well as the motility‐related signaling proteins in the GBM8 lines. Knockdown of individual SFKs altered the phosphorylation pattern of these proteins, but in different ways (Supplemental Figures 2 and 5C). For example, shFyn decreased pan‐Y416 phosphorylation while shSrc increased it and shYes and shLyn had little effect. ShLyn and shFyn decreased phosphorylation of Y228 on p120 catenin, Y172 on Vav2, Y410 on p130 Cas, and pY925 on FAK. ShSrc had no effect on Y228 p120 catenin, Y172 Vav2, or Y410 p130 Cas phosphorylation, but did decrease Y925 FAK phosphorylation. ShYes had no effect on Y228 p120 catenin, Y410 p130 Cas, or Y925 FAK phosphorylation, but increased Y172 Vav2 phosphorylation. As with the data from the SF767 and LN229 lines, these results failed to explain the in‐culture growth or survival experiment observations. For example, shFyn and shLyn both decreased phosphorylation of all the motility‐related signaling proteins. Yet shFyn abolished growth in culture and had no effect on survival, while shLyn had the opposite effect. Once again, the mixed results support the idea that co‐expressed SFKs are not functionally redundant.
3.6. Mechanisms of Yes and Lyn knockdown effects on survival
Finally, to more closely examine the histological differences and opposite effects on survival of the GBM8‐shLyn vs. ‐shYes tumors, we implanted these cells orthotopically into nude mice and then sacrificed the mice four weeks later. Brains were assessed histologically for overall tumor growth (tumor burden), proliferation, and migration distance in a single plane. Tumor burden was evaluated for each mouse by determining the % of STEM121 positive pixels in the brain section with the most tumor, including both the main tumor mass and any cells that had migrated away from the main tumor within that single plane of section (Figure 6A). Brains from mice implanted with GBM8‐shLyn had a statistically higher % of STEM121‐positive pixels compared to the GBM8‐NT‐implanted brains, indicating that overall tumor burden was higher for these mice (Figure 6B). GBM8‐shYes‐implanted brains had a lower tumor burden vs. the NT mice, but the difference did not reach statistical significance. The tumor burden measure includes a number of features, such as the balance of proliferating vs. dying tumor cells and the number of cells that have migrated away from the main tumor mass. To more specifically examine the effect of shLyn vs. shYes on proliferation, tissue sections were stained for human Ki‐67 (a marker of proliferating cells). An equally‐sized region of interest was identified on images of the tumor cores, and both the total number of Ki‐67‐positive nuclei, and the % Ki67‐positive nuclei, were determined (Figure 6C). By both measures there were significantly fewer proliferating GBM8‐shYes tumor cells compared to GBM8‐NT; there were no differences in proliferation between GBM8‐shLyn vs. –NT. Finally, to analyze migration in the four week tumor implantation experiment, single plane brain sections stained for Stem121 or Lamin A/C were examined. The distance from the center of the main tumor mass to the nucleus of the tumor cell farthest away from this point was determined (in most cases this cell was in the contralateral hemisphere) (Figure 6D). There were no differences based on SFK status in this measure of migration (Figure 6E). Taken together, the data suggest that a reduction in proliferation may contribute to the increased survival of the GBM8‐shYes mice. In addition, a higher overall tumor burden may contribute to the decreased survival of the GBM8‐shLyn mice, but the higher tumor burden in these mice is not due to changes in proliferation or the ability of single cells to migrate.
4. Discussion
In this study we examined the roles that Src, Fyn, Yes, and Lyn play in glioma growth, migration, and motility‐related signaling, both in culture and in vivo. Culture studies examined the conventional glioma cell lines LN229 and SF767, and the patient‐derived xenograft cell line GBM8, while in vivo studies focused on GBM8. All three lines express the four SFKs and respond with abrogated cell growth to culture treatment with the SFK inhibitor dasatinib.
In the culture experiments, the four SFKs differed in their importance to cell growth, migration, and motility‐related signaling in two ways. First, while dasatinib treatment abolished cell migration in both LN229 and SF767 cells, depletion of individual SFKs produced marginal effects that reached significance for Yes knockdown in both cell lines, and for Src and Fyn in LN229 cells. The data argue that in the context of cultured cells, SFKs may work together to induce cell migration, and depletion of individual SFKs is not sufficient to block cell migration. Second, while dasatinib treatment suppressed cell growth of all three cell lines, depletion of individual SFKs produced varied effects: depletion of Yes or Fyn phenocopied the effect of dasatinib, while Lyn knockdown was either unable to affect cell growth (GBM8) or had a very modest effect (LN229, SF767) and depletion of Src abolished the growth of the conventional glioma lines, but produced a moderate growth defect in GBM8 cells. The data argue that, unlike cell migration, individual SFKs (Yes, Fyn and Src, but not Lyn) are essential for glioma cell growth in culture. It is important to note that significant differences were observed in the role of the four SFKs both within individual cell lines, and across cell lines. In addition to cell growth and migration, this extended to the effects of individual SFKs on motility‐related signaling effectors. For example, knockdown of Src decreased phosphoY410 p130 Cas in LN229 cells, had little effect on phosphoY410 p130 Cas in SF767 cells, and slightly increased phosphoY410 p130 Cas in GBM8 cells. Furthermore, in no case were conclusions about the role of a particular SFK applicable to all of the measures examined. For example, knockdown of Yes significantly reduced cell growth in all three cell lines as well as migration in the two lines tested (LN229 and SF767). However, when the impact on motility‐related signaling was examined, the across‐cell‐line consistency of Yes knockdown effects disappeared. It is possible that defects in the motility‐related signaling proteins tested here do not reflect the mechanism by which Yes affects glioma cell migration. However, in the context of therapeutic targeting of SFKs, these results create difficulty for predicting therapeutic benefit.
Glioma tumor biology in vivo reflects a combination of growth and migration/invasion processes, as well as signaling processes that stimulate neoangiogenesis and immune system interaction. Based on our in culture examination of the individual SFKs relative to growth and migration, we expected that knockdown of Src, Fyn, or Yes would disrupt the tumor biology of orthotopically‐implanted GBM8 cells; we expected Lyn knockdown to have no effect in this model. In line with our expectations, mice implanted with GBM8‐shYes cells survived longer compared to mice implanted with GBM8‐NT cells. These results suggest that Yes signaling is pro‐tumorigenic. Furthermore, the observed shYes‐associated anti‐tumor effects in both the culture and in vivo environments suggests that Yes signaling impacts tumor cell biology directly, versus impacting interaction between the tumor cells and surrounding microenvironment. There is precedence for pro‐tumor effects of Yes signaling: Kleber et al. showed that Yes mediates promigratory signaling induced by CD95 (Fas)‐CD95L (FasL) interaction (Kleber et al., 2008). Thus, from a therapeutic viewpoint, inhibiting Yes signaling could be an effective GBM treatment strategy.
Surprisingly, given the effects of Src and Fyn knockdown in culture and the previously reported importance of Src and Fyn in other glioma cell lines (Lu et al., 2009), mice implanted with GBM8‐shSrc or –shFyn cells showed no difference in survival compared to GBM8‐NT‐implanted mice. These results make it difficult to classify Src or Fyn signaling as pro‐ or anti‐tumorigenic. The discrepancy between the culture and in vivo results suggests that the role of Src and Fyn signaling may vary depending on the surrounding environment, as well as on the tumor cells themselves. Consistent with this, both Src and Fyn mediate oncogenic EGFR signaling in GBM39 cells (Lu et al., 2009). One obvious difference between the GBM8 cells used in our study and the GBM39 cells is EGFR status: GBM39 express EGFRvIII while GBM8 have only wildtype EGFR (Sarkaria et al., 2006; Sarkaria et al., 2007).
Even more surprising than the Src and Fyn results, mice implanted with GBM8‐shLyn cells did worse than mice implanted with GBM8‐NT or any of the other knockdowns, exhibiting a significant reduction in survival time. This last result is particularly striking, as Lyn knockdown had little to no effect on cell growth in culture for any of the three cell lines, had no significant effect on migration of LN229 or SF767 cells (the lines tested in culture), and also had little effect on motility‐related signaling in these two cell lines, although it did reduce motility‐related phosphorylation in the GBM8 cells. The decreased motility‐related phosphorylation seen in cultured GBM8 cells may underlie the observation that only 1 of 6 shLyn tumors in the moribund mice was classified as highly invasive, possibly linking reduced Lyn activity with reduced glioma cell migration in vivo. Since the highly migratory/invasive nature of gliomas is one of the reasons the human disease has such poor prognosis, at first glance it seems odd that decreasing glioma cell migration would be associated with reduced survival times. However, the shLyn tumors were more likely than NT tumors to be classified as high density in the survival experiment. And in the short‐term in vivo experiment, brains from mice implanted with GBM8‐shLyn had a higher tumor burden than did the NT mice. It is possible that the reduced motility of shLyn tumor cells is the cause of higher tumor density, which could in turn result in faster and greater local accumulation of tumor‐associated damage to the surrounding normal brain tissue. Ultimately this could be more quickly detrimental to the animal than having widespread tumor cells at lower density that do not create as much local damage to the normal brain. The human disease is not cured by surgical resection due to its highly infiltrative nature. Reducing Lyn activity may reduce this infiltrative tendency, but it seems to increase other tumor properties that are somehow worse. It will be important to determine the nature of these properties and Lyn's role in them. The in vivo results suggest that signaling through Lyn can be anti‐tumorigenic. Furthermore, the relative lack of Lyn knockdown effect in culture versus the striking effect on survival in vivo suggest that Lyn's signaling importance lies in interactions between the tumor cells and the surrounding microenvironment, which are not testable in single‐cell‐type, 2‐D culture models. Additional studies are needed to uncover the details of Lyn activity in tumor‐microenvironment interactions.
To follow up these results, we investigated mechanisms that could underlie the survival outcomes for the –shYes and –shLyn mice by repeating the orthotopic implantations, but allowing the tumors to progress for a limited time. Based on our culture results, we predicted that the –shYes tumors would show reduced proliferation and migration at this earlier stage. Using the Ki‐67 proliferation marker we observed a statistically significant decrease in proliferation in –shYes vs. –NT tumors. To assess migration we determined the maximum distance between the main tumor mass and any tumor cell within a single brain slice. This measure likely underestimates the actual migration distance because it is not possible to know the route the cell took. Using this measure, there was no effect on migration of Yes knockdown vs. NT in vivo. Finally, because of the differences in density between ‐shYes and ‐NT tumors in the survival experiment, we evaluated overall tumor burden in the shorter‐term experiment. This measure reflects a combination of processes, including proliferation, cell density in the main tumor mass, as well as how many cells have migrated away from the main tumor. While there was a trend for reduced tumor burden, there was no significant difference between the NT and shYes tumors in this combination measure. Together, the results suggest that changes in proliferation may be the main factor underlying the increased survival of the mice implanted with the GBM8–shYes tumor cells.
In contrast to the results from the short term ‐shYes tumor implantation, there was no difference in proliferation between the short term –shLyn and –NT tumors. Despite this, there was a statistically significant tumor burden increase in mice implanted with –shLyn versus –NT tumor cells. Additionally, we observed no difference in the maximum migration distance between –shLyn and –NT tumor cells in the short‐term setting, although in the survival experiments –shLyn tumors were less invasive. A previous report suggested that Lyn kinase activity accounts for the majority of SFK signaling in GBM (Stettner et al., 2005), but it did not examine the biological effect of Lyn activation. In culture, a stimulatory effect of Lyn on GBM cell migration has been reported in the context of PDGFRβ‐induced motility on vitronectin (Ding et al., 2003). In the context of B cell function, evidence indicates that Lyn is both a positive and a negative regulator, depending on the stimulus and developmental state of the cells (Xu et al., 2005). Thus the consequences of Lyn activation are highly context dependent. Regardless of any possible defects in GBM8 cell migration, the overwhelming effect of Lyn depletion on the GBM8 xenografts is increased cell growth and reduced overall survival.
Importantly, our results suggest that inhibiting Lyn activity may not be desirable, even if inhibiting Yes activity is. Indeed, the opposite effects of Lyn and Yes knockdown in the survival experiment may explain the lack of effect of dasatinb in this same model. Currently there are a few small molecule options for selectively targeting Yes or Lyn. A recent high‐throughput screen identified several potent Yes inhibitors, but did not characterize their specificity with respect to Lyn (Patel et al., 2013). Options for targeting Lyn include the peptide inhibitor KRX‐123 (which does not inhibit Src, Fyn, or Lck, but Yes was not tested, (Goldenberg‐Furmanov et al., 2004)) and the inhibitor NS‐187 (which does not inhibit Src or Yes, but does inhibit Fyn, Abl, and Arg, (Kimura et al., 2005)). Perhaps more useful in the context of our results, MLR‐1023 is an intriguing allosteric activator of Lyn that does not alter Src or Fyn activity (Yes and Lck were not tested), has already shown clinical usefulness as an antiulcer therapeutic, and is currently in pre‐clinical studies for the treatment of Type II diabetes (Saporito et al., 2012).
Taken together, our culture and in vivo results paint a complex picture and add to a growing body of evidence indicating that SFKs are not functionally redundant. Rather, they interact with different upstream and downstream effectors, with different outcomes for cell behavior, that impact both tumor cell biology and tumor cell interaction with the surrounding microenvironment in surprisingly different ways. Furthermore, as therapeutic targets for the treatment of GBM, our results with Yes and Lyn in particular suggest that pan‐SFK inhibitors may be double‐edged swords that silence both anti‐as well as pro‐tumor signaling. As dasatinib is being tested as a treatment for GBM based on evidence that it suppresses GBM cell migration and invasion (Ahluwalia et al., 2010; de Groot and Milano, 2009; Du et al., 2009; Lu et al., 2009), our results suggest that the relative expression and activation levels of Yes vs. Lyn in particular may hold prognostic value for the efficacy of dasatinib therapy.
Competing interests
The authors have no competing interests.
Financial statement
This work was supported by National Institutes of Health grants R01 NS069753 and R21 NS070117 to P.Z.A. The funding sources had no role in study design, data collection, analysis or interpretation, manuscript writing, or the decision on when and where to publish this article.
Supporting information
The following are the supplementary data related to this article:
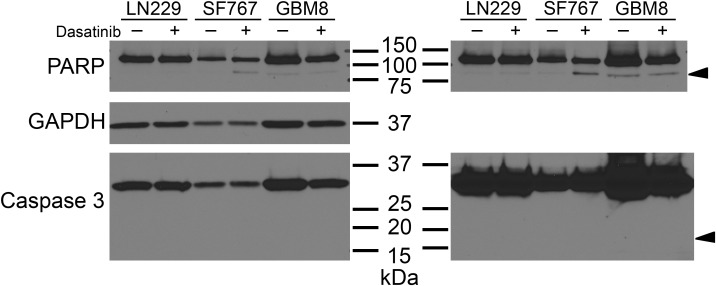
Supplemental Figure 1 Dasatinib treatment does not induce apoptotic cell death in LN229, SF767, or GBM8 tumor cells. Western blot evaluation of lysates from LN229, SF767, or GBM8 cells treated with DMSO vehicle or 10 μM dasatinib for 17 (LN229, GBM8) or 24 (SF767) hours. Cells undergoing apoptotic cell death exhibit cleavage of Caspase 3 and PARP proteins: full length Caspase 3 is 35 kDa while cleaved Caspase 3 is 17 kDa (arrowhead); full length PARP is 116 kDa while cleaved PARP is 89 kDa (arrowhead). For LN229 cells, cleavage is not evident for both proteins in either normal exposure (left panels) or over‐exposed (right panel) blots. SF767 cells show a small amount of dasatinib‐associated cleaved PARP, but do not exhibit any cleaved Caspase 3. GBM8 cells show a small amount of cleaved PARP regardless of treatment, and no cleaved Caspase 3.
{kind=link}
Supplemental Figure 2 Evaluation of SFK activation loop phosphorylation at Y416 with SFK knockdown. Western blot evaluation of phosphoY416 in lysates from cells expressing NT or individual SFK shRNAs, or treated with DMSO vehicle or 10 μM dasatinib. LN229 and SF767 lysates were also used for Figure 3; GBM8 lysates were also used for Figure 5. GAPDH is the loading control. Numbers above each of the phospho‐protein blots indicate expression relative to the NT lysate (% of NT for NT and shSFK lysates) and are normalized to GAPDH expression.
{kind=link}
Supplemental Figure 3 Orthotopically‐implanted GBM8 tumor cells produce aggressive tumors that can spread to the spinal cord. (A) The spread of GBM8‐NT and –shSFK tumor cells implanted intracranially into mice was observed by staining for STEM121 (a human cytoplasm antigen). Tumor cells were seen throughout the brain, including into the cerebellum in many cases, as well as into the spinal cord. (B) GBM8‐NT and –shSFK cells were engineered to co‐express the luciferase enzyme prior to intracranial implantation. Tumors could then be monitored with IVIS imaging for luciferase expression (displayed as color superimposed on the photograph). During the extended time course of the survival experiment, this imaging suggested that tumor cells occasionally spread beyond the brain to the lower thoracic or lumbar region of the mouse spinal cord (boxed). Mice implanted with GBM8‐NT, ‐shFyn, and –shLyn are shown; this pattern was also observed with GBM8‐shYes (not shown). We did not see spinal cord spread in any of the GBM8‐shSrc mice by IVIS imaging. Spread to the spinal cord that was observed in the survival experiment was confirmed with histological staining for the STEM121 human cytoplasm marker. When spinal cord spread was suggested by IVIS (NT, shFyn, shLyn, shYes), tumor cells were macroscopically visible in the resultant STEM121‐stained tissue sections (see images in (A). However, tumor cells were found in all spinal cords that were examined histologically by microscope (for example, boxed area on shSrc image in (A), illustrating the detection limits of the IVIS imaging. (C) Staining for STEM121 (human cytoplasm antigen) and human Lamin A/C (nuclear scaffold protein) were both effective markers of tumor cells, enabling easy visualization of tumor cells that migrated away from the main tumor mass. Scale bar is 50 μm.
{kind=link}
Acknowledgments
We wish to acknowledge the excellent IHC assistance of Brandy Edenfield of the Mayo Clinic Cancer Biology Histology Facility, as well as the Mayo Clinic Comprehensive Cancer Center RNA Interference Technology Resource for providing the shRNA clones used in this work. We wish to thank Al Copland for providing the pSinLuc construct.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.06.001.
Lewis-Tuffin Laura J., Feathers Ryan, Hari Priya, Durand Nisha, Li Zhimin, Rodriguez Fausto J., Bakken Katie, Carlson Brett, Schroeder Mark, Sarkaria Jann N., Anastasiadis Panos Z., (2015), Src family kinases differentially influence glioma growth and motility, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.06.001.
References
- Achen, M.G. , Clauss, M. , Schnurch, H. , Risau, W. , 1995. The non-receptor tyrosine kinase Lyn is localised in the developing murine blood-brain barrier. Differentiation. 59, 15–24. [DOI] [PubMed] [Google Scholar]
- Ahluwalia, M.S. , de Groot, J. , Liu, W.M. , Gladson, C.L. , 2010. Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies. Cancer Lett. 298, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers-Loustau, A. , Hering, R. , Werbowetski, T.E. , Kaplan, D.R. , Del Maestro, R.F. , 2004. SRC regulates actin dynamics and invasion of malignant glial cells in three dimensions. Mol. Cancer Res. 2, 595–605. [PubMed] [Google Scholar]
- Carlson, B.L. , Pokorny, J.L. , Schroeder, M.A. , Sarkaria, J.N. , 2011. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr. Protoc. Pharmacol. (Chapter 14), Unit 14.16, p. 14.16.1–14.16.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato, H. , Ramachandrappa, S. , Olsen, I.M. , ffrench-Constant, C. , 2004. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 167, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, J. , Milano, V. , 2009. Improving the prognosis for patients with glioblastoma: the rationale for targeting Src. J. Neurooncol. 95, 151–163. [DOI] [PubMed] [Google Scholar]
- Ding, Q. , Stewart, J. , Olman, M.A. , Klobe, M.R. , Gladson, C.L. , 2003. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 278, 39882–39891. [DOI] [PubMed] [Google Scholar]
- Du, J. , Bernasconi, P. , Clauser, K.R. , Mani, D.R. , Finn, S.P. , Beroukhim, R. , Burns, M. , Julian, B. , Peng, X.P. , Hieronymus, H. , Maglathlin, R.L. , Lewis, T.A. , Liau, L.M. , Nghiemphu, P. , Mellinghoff, I.K. , Louis, D.N. , Loda, M. , Carr, S.A. , Kung, A.L. , Golub, T.R. , 2009. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 27, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, H. , Hu, B. , Liu, K.W. , Li, Y. , Lu, X. , Cheng, T. , Yiin, J.J. , Lu, S. , Keezer, S. , Fenton, T. , Furnari, F.B. , Hamilton, R.L. , Vuori, K. , Sarkaria, J.N. , Nagane, M. , Nishikawa, R. , Cavenee, W.K. , Cheng, S.Y. , 2011. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumorigenesis in mice and humans. J. Clin. Invest. 121, 4670–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame, M.C. , 2004. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 117, 989–998. [DOI] [PubMed] [Google Scholar]
- Garrett, T.A. , Van Buul, J.D. , Burridge, K. , 2007. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell Res. 313, 3285–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini, C. , Sarkaria, J.N. , Saito, A. , Uhm, J.H. , Galanis, E. , Carlson, B.L. , Schroeder, M.A. , James, C.D. , 2005. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 7, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg-Furmanov, M. , Stein, I. , Pikarsky, E. , Rubin, H. , Kasem, S. , Wygoda, M. , Weinstein, I. , Reuveni, H. , Ben-Sasson, S.A. , 2004. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 64, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Guarino, M. , 2010. Src signaling in cancer invasion. J. Cell Physiol. 223, 14–26. [DOI] [PubMed] [Google Scholar]
- Han, N.M. , Curley, S.A. , Gallick, G.E. , 1996. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res.: Official J. Am. Assoc. Cancer Res. 2, 1397–1404. [PubMed] [Google Scholar]
- Han, X. , Zhang, W. , Yang, X. , Wheeler, C.G. , Langford, C.P. , Wu, L. , Filippova, N. , Friedman, G.K. , Ding, Q. , Fathallah-Shaykh, H.M. , Gillespie, G.Y. , Nabors, L.B. , 2014. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 45, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveldt, D. , Lewis-Tuffin, L.J. , Carlson, B.L. , Schroeder, M.A. , Rodriguez, F. , Giannini, C. , Galanis, E. , Sarkaria, J.N. , Anastasiadis, P.Z. , 2013. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One. 8, e56505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iankov, I.D. , Msaouel, P. , Allen, C. , Federspiel, M.J. , Bulur, P.A. , Dietz, A.B. , Gastineau, D. , Ikeda, Y. , Ingle, J.N. , Russell, S.J. , Galanis, E. , 2010. Demonstration of anti-tumor activity of oncolytic measles virus strains in a malignant pleural effusion breast cancer model. Breast Cancer Res. Treat. 122, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, S. , Naito, H. , Segawa, H. , Kuroda, J. , Yuasa, T. , Sato, K. , Yokota, A. , Kamitsuji, Y. , Kawata, E. , Ashihara, E. , Nakaya, Y. , Naruoka, H. , Wakayama, T. , Nasu, K. , Asaki, T. , Niwa, T. , Hirabayashi, K. , Maekawa, T. , 2005. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood. 106, 3948–3954. [DOI] [PubMed] [Google Scholar]
- Kleber, S. , Sancho-Martinez, I. , Wiestler, B. , Beisel, A. , Gieffers, C. , Hill, O. , Thiemann, M. , Mueller, W. , Sykora, J. , Kuhn, A. , Schreglmann, N. , Letellier, E. , Zuliani, C. , Klussmann, S. , Teodorczyk, M. , Grone, H.J. , Ganten, T.M. , Sultmann, H. , Tuttenberg, J. , von Deimling, A. , Regnier-Vigouroux, A. , Herold-Mende, C. , Martin-Villalba, A. , 2008. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 13, 235–248. [DOI] [PubMed] [Google Scholar]
- Lewis-Tuffin, L.J. , Rodriguez, F. , Giannini, C. , Scheithauer, B. , Necela, B.M. , Sarkaria, J.N. , Anastasiadis, P.Z. , 2010. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS One. 5, e13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. , Schmittgen, T.D. , 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Louis, D.N. , Ohgaki, H. , Wiestler, O.D. , Cavenee, W.K. , 2007. WHO Classification of Tumours of the Central Nervous System fourth ed. IARC; Lyon: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, D.N. , Ohgaki, H. , Wiestler, O.D. , Cavenee, W.K. , Burger, P.C. , Jouvet, A. , Scheithauer, B.W. , Kleihues, P. , 2007. The 2007 WHO classification of tumours of the central nervous system.[erratum appears in Acta Neuropathol. 2007 Nov;114(5):547]. Acta Neuropathol. 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K.V. , Zhu, S. , Cvrljevic, A. , Huang, T.T. , Sarkaria, S. , Ahkavan, D. , Dang, J. , Dinca, E.B. , Plaisier, S.B. , Oderberg, I. , Lee, Y. , Chen, Z. , Caldwell, J.S. , Xie, Y. , Loo, J.A. , Seligson, D. , Chakravari, A. , Lee, F.Y. , Weinmann, R. , Cloughesy, T.F. , Nelson, S.F. , Bergers, G. , Graeber, T. , Furnari, F.B. , James, C.D. , Cavenee, W.K. , Johns, T.G. , Mischel, P.S. , 2009. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 69, 6889–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, C.V. , Nguyen, M.T. , Owens, G.C. , Pakchoian, A.J. , Shaterian, A. , Kruse, C.A. , Eliceiri, B.P. , 2006. Reduced glioma infiltration in Src-deficient mice. J. Neurooncol. 78, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariner, D.J. , Anastasiadis, P. , Keilhack, H. , Bohmer, F.D. , Wang, J. , Reynolds, A.B. , 2001. Identification of Src phosphorylation sites in the catenin p120ctn. J. Biol. Chem. 276, 28006–28013. [DOI] [PubMed] [Google Scholar]
- Mitra, S.K. , Schlaepfer, D.D. , 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, D. , Tsiokas, L. , Zhou, X.M. , Foster, D. , Brugge, J.S. , Sukhatme, V.P. , 1995. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 375, 577–581. [DOI] [PubMed] [Google Scholar]
- Omri, B. , Crisanti, P. , Marty, M.C. , Alliot, F. , Fagard, R. , Molina, T. , Pessac, B. , 1996. The Lck tyrosine kinase is expressed in brain neurons. J. Neurochem. 67, 1360–1364. [DOI] [PubMed] [Google Scholar]
- Park, C.M. , Park, M.J. , Kwak, H.J. , Lee, H.C. , Kim, M.S. , Lee, S.H. , Park, I.C. , Rhee, C.H. , Hong, S.I. , 2006. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 66, 8511–8519. [DOI] [PubMed] [Google Scholar]
- Park, S.I. , Zhang, J. , Phillips, K.A. , Araujo, J.C. , Najjar, A.M. , Volgin, A.Y. , Gelovani, J.G. , Kim, S.J. , Wang, Z. , Gallick, G.E. , 2008. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 68, 3323–3333. [DOI] [PubMed] [Google Scholar]
- Parsons, S.J. , Parsons, J.T. , 2004. Src family kinases, key regulators of signal transduction. Oncogene. 23, 7906–7909. [DOI] [PubMed] [Google Scholar]
- Patel, P.R. , Sun, H. , Li, S.Q. , Shen, M. , Khan, J. , Thomas, C.J. , Davis, M.I. , 2013. Identification of potent Yes1 kinase inhibitors using a library screening approach. Bioorg. Med. Chem. Lett. 23, 4398–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof, R.W. , Haskell, M.D. , Dukes, B.D. , Sherman, N. , Kinter, M. , Parsons, S.J. , 1998. Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol. Cell Biol. 18, 7052–7063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporito, M.S. , Ochman, A.R. , Lipinski, C.A. , Handler, J.A. , Reaume, A.G. , 2012. MLR-1023 is a potent and selective allosteric activator of Lyn kinase in vitro that improves glucose tolerance in vivo. J. Pharmacol. Exp. Ther. 342, 15–22. [DOI] [PubMed] [Google Scholar]
- Sarkaria, J.N. , Carlson, B.L. , Schroeder, M.A. , Grogan, P. , Brown, P.D. , Giannini, C. , Ballman, K.V. , Kitange, G.J. , Guha, A. , Pandita, A. , James, C.D. , 2006. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin. Cancer Res. 12, 2264–2271. [DOI] [PubMed] [Google Scholar]
- Sarkaria, J.N. , Yang, L. , Grogan, P.T. , Kitange, G.J. , Carlson, B.L. , Schroeder, M.A. , Galanis, E. , Giannini, C. , Wu, W. , Dinca, E.B. , James, C.D. , 2007. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol. Cancer Ther. 6, 1167–1174. [DOI] [PubMed] [Google Scholar]
- Stettner, M.R. , Wang, W. , Nabors, L.B. , Bharara, S. , Flynn, D.C. , Grammer, J.R. , Gillespie, G.Y. , Gladson, C.L. , 2005. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 65, 5535–5543. [DOI] [PubMed] [Google Scholar]
- Summy, J.M. , Gallick, G.E. , 2003. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 22, 337–358. [DOI] [PubMed] [Google Scholar]
- Summy, J.M. , Gallick, G.E. , 2006. Treatment for advanced tumors: SRC reclaims center stage. Clin. Cancer Res.: Official J. Am. Assoc. Cancer Res. 12, 1398–1401. [DOI] [PubMed] [Google Scholar]
- Theurillat, J.P. , Hainfellner, J. , Maddalena, A. , Weissenberger, J. , Aguzzi, A. , 1999. Early induction of angiogenetic signals in gliomas of GFAP-v-src transgenic mice. Am. J. Pathol. 154, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S.M. , Brugge, J.S. , 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev Biol. 13, 513–609. [DOI] [PubMed] [Google Scholar]
- Umemori, H. , Ogura, H. , Tozawa, N. , Mikoshiba, K. , Nishizumi, H. , Yamamoto, T. , 2003. Impairment of N-methyl-D-aspartate receptor-controlled motor activity in LYN-deficient mice. Neuroscience. 118, 709–713. [DOI] [PubMed] [Google Scholar]
- Umemori, H. , Wanaka, A. , Kato, H. , Takeuchi, M. , Tohyama, M. , Yamamoto, T. , 1992. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res. Mol. Brain Res. 16, 303–310. [DOI] [PubMed] [Google Scholar]
- Werdich, X.Q. , Penn, J.S. , 2005. Src, Fyn and Yes play differential roles in VEGF-mediated endothelial cell events. Angiogenesis. 8, 315–326. [DOI] [PubMed] [Google Scholar]
- Wick, W. , Weller, M. , Weiler, M. , Batchelor, T. , Yung, A.W. , Platten, M. , 2011. Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro Oncol. 13, 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Harder, K.W. , Huntington, N.D. , Hibbs, M.L. , Tarlinton, D.M. , 2005. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 22, 9–18. [DOI] [PubMed] [Google Scholar]
- Yang, L. , Clarke, M.J. , Carlson, B.L. , Mladek, A.C. , Schroeder, M.A. , Decker, P. , Wu, W. , Kitange, G.J. , Grogan, P.T. , Goble, J.M. , Uhm, J. , Galanis, E. , Giannini, C. , Lane, H.A. , James, C.D. , Sarkaria, J.N. , 2008. PTEN loss does not predict for response to RAD001 (Everolimus) in a glioblastoma orthotopic xenograft test panel. Clin. Cancer Res. 14, 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeatman, T.J. , 2004. A renaissance for SRC. Nat. Rev. Cancer. 4, 470–480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplemental Figure 1 Dasatinib treatment does not induce apoptotic cell death in LN229, SF767, or GBM8 tumor cells. Western blot evaluation of lysates from LN229, SF767, or GBM8 cells treated with DMSO vehicle or 10 μM dasatinib for 17 (LN229, GBM8) or 24 (SF767) hours. Cells undergoing apoptotic cell death exhibit cleavage of Caspase 3 and PARP proteins: full length Caspase 3 is 35 kDa while cleaved Caspase 3 is 17 kDa (arrowhead); full length PARP is 116 kDa while cleaved PARP is 89 kDa (arrowhead). For LN229 cells, cleavage is not evident for both proteins in either normal exposure (left panels) or over‐exposed (right panel) blots. SF767 cells show a small amount of dasatinib‐associated cleaved PARP, but do not exhibit any cleaved Caspase 3. GBM8 cells show a small amount of cleaved PARP regardless of treatment, and no cleaved Caspase 3.
Supplemental Figure 2 Evaluation of SFK activation loop phosphorylation at Y416 with SFK knockdown. Western blot evaluation of phosphoY416 in lysates from cells expressing NT or individual SFK shRNAs, or treated with DMSO vehicle or 10 μM dasatinib. LN229 and SF767 lysates were also used for Figure 3; GBM8 lysates were also used for Figure 5. GAPDH is the loading control. Numbers above each of the phospho‐protein blots indicate expression relative to the NT lysate (% of NT for NT and shSFK lysates) and are normalized to GAPDH expression.
Supplemental Figure 3 Orthotopically‐implanted GBM8 tumor cells produce aggressive tumors that can spread to the spinal cord. (A) The spread of GBM8‐NT and –shSFK tumor cells implanted intracranially into mice was observed by staining for STEM121 (a human cytoplasm antigen). Tumor cells were seen throughout the brain, including into the cerebellum in many cases, as well as into the spinal cord. (B) GBM8‐NT and –shSFK cells were engineered to co‐express the luciferase enzyme prior to intracranial implantation. Tumors could then be monitored with IVIS imaging for luciferase expression (displayed as color superimposed on the photograph). During the extended time course of the survival experiment, this imaging suggested that tumor cells occasionally spread beyond the brain to the lower thoracic or lumbar region of the mouse spinal cord (boxed). Mice implanted with GBM8‐NT, ‐shFyn, and –shLyn are shown; this pattern was also observed with GBM8‐shYes (not shown). We did not see spinal cord spread in any of the GBM8‐shSrc mice by IVIS imaging. Spread to the spinal cord that was observed in the survival experiment was confirmed with histological staining for the STEM121 human cytoplasm marker. When spinal cord spread was suggested by IVIS (NT, shFyn, shLyn, shYes), tumor cells were macroscopically visible in the resultant STEM121‐stained tissue sections (see images in (A). However, tumor cells were found in all spinal cords that were examined histologically by microscope (for example, boxed area on shSrc image in (A), illustrating the detection limits of the IVIS imaging. (C) Staining for STEM121 (human cytoplasm antigen) and human Lamin A/C (nuclear scaffold protein) were both effective markers of tumor cells, enabling easy visualization of tumor cells that migrated away from the main tumor mass. Scale bar is 50 μm.