

Abstract
The advent of next-generation sequencing for genetic diagnoses of complex developmental disorders, such as intellectual disability (ID), has facilitated the identification of hundreds of predisposing genetic variants. However, there still exists a vast gap in our knowledge of causal genetic factors for ID as evidenced by low diagnostic yield of genetic screening, in which identifiable genetic causes are not found for the majority of ID cases. Most methods of genetic screening focus on protein-coding genes; however, noncoding RNAs may outnumber protein-coding genes and play important roles in brain development. Long noncoding RNAs (lncRNAs) specifically have been shown to be enriched in the brain and have diverse roles in gene regulation at the transcriptional and posttranscriptional levels. LncRNAs are a vastly uncharacterized group of noncoding genes, which could function in brain development and harbor ID-predisposing genetic variants. We analyzed lncRNAs for coexpression with known ID genes and affected biological pathways within a weighted gene coexpression network derived from RNA-sequencing data spanning human brain development. Several ID-associated gene modules were found to be enriched for lncRNAs, known ID genes, and affected biological pathways. Utilizing a list of de novo and pathogenic copy number variants detected in ID probands, we identified lncRNAs overlapping these genetic structural variants. By integrating our results, we have made a prioritized list of potential ID-associated lncRNAs based on the developing brain gene coexpression network and genetic structural variants found in ID probands.
Keywords: lncRNAs, coexpression network analysis, intellectual disability
Introduction
Intellectual disability (ID) is a developmental brain disorder characterized by diminished intellectual functions and adaptive behaviors, with an estimated prevalence in the population between 1% and 3%.1 Currently, in the most cases of ID, an identifiable genetic cause is still unclear.2 However, known ID genes are predominantly involved in synaptic functions, such as cytoskeletal reorganization and synaptic plasticity.3 Disruptions in the synapse function likely cause a cascade of detrimental effects persisting in brain development, indicating the necessity of the precise spatiotemporal gene expression required for normal brain development. Genetic studies on ID have almost exclusively focused on variants in protein-coding genes, such as copy number variants (CNVs) and single nucleotide variants. However, in human cells, the majority of RNA transcripts may not encode proteins, suggesting the need to expand the search for causal factors of ID beyond protein-coding genes.4 In this study, we have examined over 4,000 long noncoding RNAs (lncRNAs) to assess their potential association with ID through the integration of RNA-sequencing (RNA-seq) data and genetic structural variants detected in ID-affected individuals.
LncRNA transcripts are longer than 200 nucleotides with diverse emerging regulatory mechanisms. While some lncRNAs may encode small peptides, lncRNAs are vastly untranslated.5–7 Functionally, lncRNAs have been shown to be involved in transcriptional and posttranscriptional regulation, in addition to the roles in epigenetic mechanisms.8 Specifically in the brain, lncRNAs have been shown to be involved in neural differentiation and synaptic plasticity.8–10 CNVs in the genomic regions of these lncRNAs likely cause disruptive effects through the alteration of gene copy number, thereby leading to aberrant expression and possible downstream effects. The identification of lncRNAs involved in neuronal and developmental processes, which are also affected by ID-predisposing CNVs, may lead to the identification of novel ID genes.
Considering the genetic heterogeneity of ID and largely uncharted molecular roles of lncRNAs, we chose to construct a gene coexpression network using RNA-seq data from the developing brain. This analysis facilitates the functional annotation of uncharacterized lncRNAs by clustering genes based on the correlations of expression levels across brain developmental stages. Weighted gene coexpression network analysis (WGCNA) is a well-established method for biological data mining.11 In previous studies, WGCNA has been used to elucidate convergent molecular pathways, specific brain regions, and developmental periods associated with autism spectrum disorder (ASD), illustrating the functionality of coexpression networks for complex developmental disorders.12–16 Within a coexpression network, a module represents a group of correlated genes, based on expression profiles, which likely share genetic regulation and/or biological function. Thus, by clustering genes into coexpression modules, the biological function of an lncRNA may be inferred from the Gene Ontology enrichment of the known genes in the module and the degree to which the lncRNA correlates within the module. In this study, the gene coexpression network is based on a comprehensive dataset of human brain developmental transcriptomes. We compiled a list of known ID genes from multiple sources and used them to identify the potential ID-associated lnc RNAs in the coexpression network. Moreover, we used a list of CNVs identified in a large cohort of probands with ID to identify lncRNAs residing within the CNVs. This approach has facilitated the prioritization of candidate ID-associated lncRNAs based on the developing brain gene coexpression network seeded with known ID genes and pathogenic genetic variants found in ID probands.
Methods
Brain developmental transcriptome data
The BrainSpan developmental transcriptome dataset contains RNA-seq expression profiles summarized to gene-level reads per kilo-base million mapped reads (RPKM) with GENCODE 10 annotations.17,18 Only samples less than or equal to three postnatal years and from the neocortex were used in this study. This resulted in 210 RNA-seq samples derived from 28 different individuals across 11 regions within the neocortex. LncRNAs were related to the genes of the developmental transcriptome by using the GENCODE v22 long noncoding RNA annotations. Genes were variance-filtered by removing the lowest 25% of genes based on standard deviation. The gene-level RPKM values were then normalized by using the log2 (RPKM + 1) for further analyses.
Gene lists
The full ID gene list was compiled through the combination of three curated gene sets: the ID all gene set by Parikshak et al, the XLID gene panel, and the ID gene database.13,17,18 The ASD gene list was obtained from the SFARI human gene autism database.19 The ID only gene list was created by removing genes from the full ID gene list that are also classified as ASD genes. The ID and ASD gene set contains all overlapping genes from the ID all and ASD gene list. The XLID gene set represents the XLID diagnostic panel.17 All gene lists are provided in Supplementary Table S1.
Weighted gene coexpression network analysis
Signed WGCNA was performed in R, version 3.2.0, utilizing the WGCNA, v1.46, R package.11,27 Readers interested in the mathematical derivations of the technique are recommended to view the excellent WGCNA theory Web site (http://labs.genetics.ucla.edu/horvath/Coexpression-Network/). Traditional coexpression networks are created by filtering a symmetric correlation matrix by a hard threshold that likely results in many false negatives due to the arbitrary cutoff of the threshold. WGCNA does not suffer from this pitfall due to the utilization of a soft threshold that simply emphasizes high correlations. In addition, this soft threshold enables the coexpression network to approximate scale-free topology, an inherent property of biological networks.28 First, a correlation matrix, also known as a symmetric adjacency matrix, was made by calculating the biweight midcorrelation, a robust alternative to the Pearson correlation coefficient, between all gene pairs. This adjacency matrix was then raised to a soft threshold power of 10 to achieve a scale-free topology. The topological overlap measure (TOM) is computed for all genes by taking into account direct pairwise correlations as well as shared correlations between other genes. Gene modules are formed by unsupervised clustering of genes of the hierarchical cluster tree based on the threshold of dissimilarity, 1-TOM. In this study, the minimum module size was set to 50 genes with the module-merging cut height set at 0.20. The WGCNA results are provided in Supplementary Table S2.
Modular gene set enrichment analysis
For each gene set, we determined overrepresentation within each module using the Fisher’s exact test. All resulting P-values from each gene set were adjusted by the false discovery rate method, Benjamini–Hochberg correction.29 We required an adjusted P-value of <0.05 and an odds ratio of >1 to classify modular enrichment of a specific gene list. Heat maps of the −log10 (adjusted P-value) were created using the gplots of R package.30 Results from the ID gene enrichment for all modules are provided in Supplementary Table S3.
CNV detection
CNV genomic coordinates were obtained from 213 idiopathic ID subjects using array CGH analysis as described by Qiao et al.22 We extracted the genomic coordinates of all lncRNAs from the human genome assembly NCBI 37 using the R package biomaRt.31 We performed genomic liftover, using the R package rtracklayer for conversion to the assembly NCBI 36 coordinates, to match the coordinates of the CNV dataset.32 All CNV overlaps were quantified using the R package GenomicRanges.33
LncRNA candidate prioritization
The network plot shown in Figure 3 was constructed by selecting the top two lncRNAs with the highest modular membership values within each module that was statistically enriched for the ID only gene list. Modular membership is defined as the correlation of a gene’s expression profile with the eigengene of the module. Next, for each of the chosen lncRNAs, we selected the top three most highly correlated genes based on connectivity. The network plot (Fig. 3) was generated using Cytoscape v3.1.1 package by visualizing all bicor midweight correlations >0.65.34 LncRNA prioritization (Table 1) was performed by ranking all lncRNAs that overlapped a de novo or DECIPHER CNV by the maximal absolute Pearson correlation coefficient to all genes in the full ID gene list. The full ranked lncRNA list is available in Supplementary Table S4, including the genomic location and the type of CNV overlap.
Figure 3.
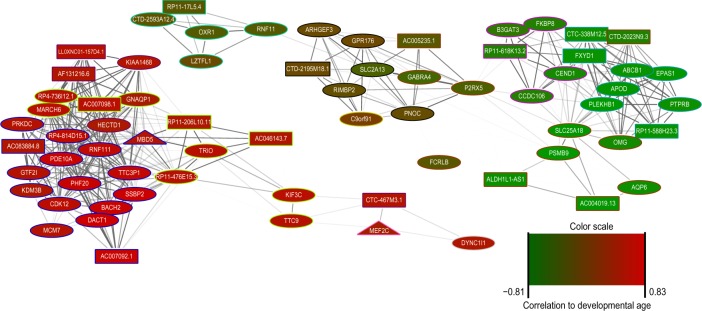
Coexpression network plot of lncRNAs residing in ID-associated CNVs.
Notes: Network nodes represent genes, with the color being representative of the expression correlated with developmental months. Red nodes show expression negatively correlated with developmental months, while green indicates positive correlation. The colored border of the node indicates the module to which this gene belongs. Squares represent lncRNAs, circles indicate protein-coding genes, and triangles are ID genes.
Table 1.
Prioritized list of ID-associated lncRNA candidates. LncRNAs overlapping pathogenic CNVs are ranked based on maximum correlation with known ID genes. The lncRNA module assignment and correlation of expression to developmental age are also provided.
| LncRNA | MODULE | CORRELATION TO DEV AGE | CNV TYPE | CORRELATION TO ID GENE | ID GENE |
|---|---|---|---|---|---|
| AC004019.13 | Brown | 0.80 | DECIPHER | 0.975 | ALDH4A1 |
| RP5-1059M17.1 | Brown | 0.57 | De Novo | 0.960 | PTHLH |
| RP11-486F17.1 | Blue | −0.59 | DECIPHER | 0.957 | SATB2 |
| AC017096.1 | Yellow | −0.53 | DECIPHER | 0.956 | SATB2 |
| RP11-2E17.1 | Brown | 0.85 | DECIPHER | 0.938 | ALDH4A1 |
| AC099508.1 | Blue | −0.79 | De Novo | 0.935 | DCX |
| ATP13A4-AS1 | Brown | 0.75 | DECIPHER | 0.929 | KANK1 |
| AP003039.3 | Brown | 0.78 | De Novo | 0.929 | ASPA |
| RP1-163G9.1 | Turquoise | 0.74 | De Novo | 0.928 | KCNJ10 |
| CTD-2210P24.4 | Brown | 0.79 | DECIPHER | 0.928 | ALDH4A1 |
| RP11-1026M7.2 | Turquoise | 0.57 | De Novo | 0.928 | CACNA1F |
| RP11-588H23.3 | Turquoise | 0.76 | DECIPHER | 0.921 | CA2 |
| AC007098.1 | Greenyellow | −0.65 | De Novo | 0.919 | USP9X |
| CTC-467M3.1 | Purple | −0.62 | De Novo | 0.915 | MEF2C |
Results and Discussion
Brain gene coexpression network analysis and identification of coexpression modules enriched with known ID genes and lncRNAs
For WGCNA, we utilized a comprehensive developmental transcriptome dataset with RNA-seq data from 210 neocortical samples during the early brain development (from eight weeks postconception to three postnatal years). This developmental transcriptome dataset contains more than 50,000 genes, including >9,000 genes currently classified as lncRNAs. To reduce the search space for ID-associated lncRNAs, we chose to remove the genes thought not to be involved in brain development by filtering out genes with the lowest variance across the different stages during the brain development. After removing the bottom quartile of genes based on variance, we retained a total of 39,000 genes, of which 6,000 are classified as lncRNAs. In addition, we utilized high thresholds for module construction for further filtering, resulting in a final gene coexpression network comprising 26,030 genes, including 4,070 lncRNAs, which are distributed among 16 gene modules labeled by colors.
Next, we asked if the known ID genes converge onto specific coexpression modules. Because coexpression implies shared function and/or regulation, ID-gene-enriched modules would suggest that the genes in these modules are potentially involved in ID-affected biological pathways. We compiled a comprehensive list of known ID genes (Supplementary Table S1) by combining gene lists from three curated sources, a diagnostic X-linked ID (XLID) gene panel, the ID gene database project, and the ID gene list from a recent publication.13,17,18 The identification of ID-related modules also requires taking into account ASDs, which are present in up to 20% of ID cases.2 Utilizing the ASD gene list from the Simons Foundation Autism Research Initiative (SFARI), a human gene database, we found that our compiled ID gene list had a 20% overlap with the ASD gene list.19 This is not surprising because of the shared genetic components between the disorders, such as the synaptic plasticity and transmission pathways.20 To assess the enrichment of nonsyndromic ID, we created an ID-specific gene set, called ID only, by removing the genes present in both the ID and ASD gene lists, and the overlapping genes are referred to as ID and ASD.
By mapping the gene sets to the developmental coexpression network, we have identified two out of eight modules that are enriched for ID only genes without also being enriched for ID and ASD genes or ASD genes (Fig. 1 and Supplementary Table S2). These two ID-specific modules are labeled as magenta and turquoise. The highest scoring term of biological processes from the Gene Ontology enrichment analysis for the turquoise module is immune response (P-value <0.001), whereas the magenta module does not show any significant functional term enrichment (Fig. 2B). Interestingly, the blue, black, and purple modules are enriched for all four ID gene sets, in addition to the ASD gene list. These modules are likely involved in core synaptic and regulatory pathways that are affected in both ID and ASD. Gene Ontology functional analysis finds enrichment for transcriptional regulation, synaptic transmission, and protein localization, respectively (Fig. 2B). Furthermore, the blue, black, brown, and turquoise modules possess a significant amount of lncRNAs (Supplementary Table S3) and thus are interesting modules to examine the relationships between lncRNAs and known ID genes.
Figure 1.
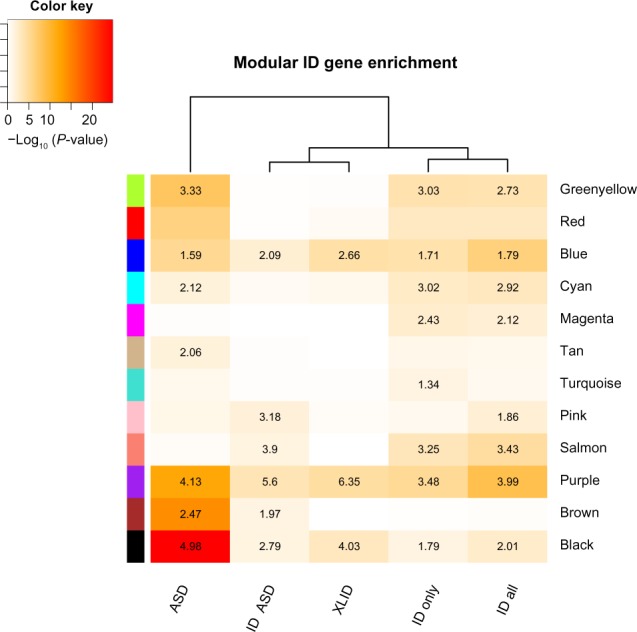
ID gene enrichment in coexpression modules. ID gene overrepresentation is shown in a heat map displaying the −log10 adjusted P-values of the Fisher’s exact test. Odds ratio values are overlaid onto the heat map if they are >1. Only modules with an adjusted P-value of <0.05 in at least one gene set are displayed, resulting in 12 out of the 16 modules shown.
Figure 2.

Characteristics of ID-gene-enriched coexpression modules. (A) The gene expression of lncRNAs from eight weeks postconception to one postnatal year, grouped by module, was normalized and plotted across developmental time using a scatter smoothing function. The shaded adjacent regions represent 95% confidence intervals. (B) Gene functional enrichment was analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, available at https://david.ncifcrf.gov/), and the top five Gene Ontology terms of biological processes were displayed. The red vertical lines represent the significance threshold of P-value = 0.01.
Besides modular functional annotation, we also examined the developmental expression patterns of the coexpressed lncR-NAs in these modules, which may be used to infer biological function (Fig. 2A). Interestingly, the two modules, turquoise and magenta, which are enriched specifically for nonsyndromic ID genes, show a very similar developmental expression trajectory. LncRNAs of both modules become highly expressed in the late fetal period, a developmental period known for axonal and dendritic outgrowth.21 In contrast, the developmental expression profiles of the blue, black, and purple modules enriched for all ID gene sets show a different developmental trend in which they are downregulated in the late fetal period and rise during the early postnatal stage known for synaptic overproduction before the pruning stage.21 The blue module is enriched for transcriptional regulation, a known biological role of lncRNAs (Supplementary Table S3). In addition, both the blue and black modules are also enriched for synaptic transmission (both P-values <0.00001). Interestingly, the expression patterns of the modules enriched solely for ID genes and the modules enriched for all ID gene sets appear to mirror each other, in terms of upregulation and downregulation (Fig. 2A). The fact that these modules are enriched for ID genes, lncRNAs, and known pathways in ID disorders suggests that these modules are likely essential contributors to normal cognitive development.
LncRNAs overlap with ID-associated CNVs
Next, we asked if lncRNAs could reside within ID-associated CNVs and if the lncRNAs were clustered into ID-gene-enriched modules. To answer this question, we examined the genomic overlaps of lncRNAs with CNVs observed in idiopathic ID from 213 probands.22 Four subtypes of CNVs, de novo, familial, common in ID cases, and common in controls, were experimentally classified.22 In addition, the fifth subtype of known pathogenic CNVs associated with ID was added as a positive control from Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) (http://decipher.sanger.ac.uk/). We found that the lncRNAs within the turquoise, blue, and brown modules had the highest number of genomic overlaps with both the de novo CNVs and the known pathogenic CNVs from DECIPHER. These three modules had over three times the amount of ID-predisposing CNVs than that of the gray module, although the gray module was over two times the size of any other module. The gray module contained genes that failed to merge with any other modules due to low topological overlap.11 Thus, the gray module could effectively serve as a randomized control for modular CNV overlap analysis.
Prioritization of candidate lncRNAs associated with ID
Focused on modules with ID gene enrichment and CNV overlap, we asked whether lncRNAs residing in likely pathogenic CNVs were strongly coexpressed with known ID genes. We examined the coexpression network neighborhood of the top CNV-harboring lncRNAs based on the modular membership (Fig. 3). In particular, the blue module, which is enriched for the functional term of transcriptional regulation, contains two known ID genes, MBD5 and MEF2C, both of which are highly coexpressed with lncRNAs residing in likely pathogenic CNVs. MBD5 is required for methyl-CpG-binding specificity to methylated DNA, and haploinsufficiency of MBD5 is associated with ID.23 Moreover, the expression of the blue module is negatively correlated with developmental time, suggesting its involvement in early neurodevelopmental processes. It is possible that these highly coexpressed lncRNAs may be involved in the transcriptional regulation of ID genes, such as MBD5 and MEF2C. Interestingly, the lncRNA CTC-467M3.1 is highly correlated with MEF2C (Pearson’s correlation coefficient of 0.915). CTC-467M3.1 is located on the antisense strand relative to MEF2C, suggesting the possibility that CTC-467M.3 might be involved in the cis-regulation of MEF2C. Natural anti-sense transcripts have been shown to be involved in altering the gene expression of their protein-coding counterparts, typically by suppression at the epigenetic level, but their roles in transcriptional activation and alternative splicing have also been observed.24 Thus, our approach has found that lncR-NAs can be highly coexpressed with known ID genes and also overlap possibly pathogenic CNVs. These findings have allowed us to prioritize a list of potential ID-associated lncR-NAs for further analysis as candidates for novel ID genes (Table 1). These lncRNAs reside within ID-predisposing CNVs and are ranked by the highest correlation with known ID genes. Notably, the lncRNA CTC-467M3.1 is among the top 15 ranked candidates.
Our results show the high coexpression between lnc-RNAs and known ID genes, suggesting the association of lncRNAs with ID. This is the first time that an assessment of associations between lncRNAs and ID has been performed on a genomic scale. However, there have been a few specific lncRNAs associated with ID, such as the lncRNA Evf2, also known as DlX6-AS1, which is involved in a negative feedback loop of active chromatin remodeling leading to transcriptional repression.25 Interestingly, mutations associated with Coffin–Siris syndrome were found to localize to components of the chromatin remodeling complex, such as DLX1, which upregulates DLX6-AS1.25 In our brain gene coexpression network, DLX6-AS1 belongs to the blue module, which is enriched for transcriptional regulation. Interestingly, DLX6-AS1 possesses the highest correlation (Pearson’s correlation coefficient = 0.87) with DLX2, which has been shown to be functionally redundant to DLX1.26
Conclusion
In this study, we have identified the potential ID-associated lncRNAs based on coexpression with known ID genes. Some coexpression modules enriched for known ID genes are also enriched for lncRNAs. The lncRNAs in these modules show specific developmental expression patterns in the brain. We have observed two distinct expression patterns of lncRNAs in ID-gene-enriched modules, showing inverse relationships most noticeably with regard to the mid-to-late fetal period. The coexpression modules show high level of connectivity between lncRNAs and known ID genes, and affected pathways and developmental periods. Moreover, we have identified lncRNAs residing within de novo and pathogenic CNVs, which are major risk factors in ID. We have shown that lncR-NAs overlapping the CNVs are also highly coexpressed with known ID genes. For instance, we have identified lncRNAs that show strong connections with MDB5 and MEF2C within the brain gene coexpression network. Finally, we have prioritized the lncRNAs overlapping ID-associated CNVs based on their coexpression to known ID genes in the developing brain. This prioritized list was constructed through ranking lncR-NAs by their maximal correlation with known ID genes. The lncRNAs selected in this study can serve as a starting point for a new direction of inquiry for expanding upon the causal genetic factors of ID.
Supplementary Materials
Supplementary Table S1. ID gene lists.
Supplementary Table S2. WGCNA results.
Supplementary Table S3. Modular ID gene enrichment.
Supplementary Table S4. Ranked lncRNAs.
Footnotes
ACADEMIC EDITOR: J.T. Efird, Associate Editor
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1,133 words, excluding any confidential comments to the academic editor.
FUNDING: This work is supported, in part, by Cooperative State Research, Education, and Extension Service/U.S. Department of Agriculture under project number SC-1000675 and a grant from the Self Regional Healthcare Foundation. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions madeby independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived the study: LW. Designed the experiments: BLG, LW. Analyzed the data: BLG. Wrote the first draft of the manuscript: BLG. Contributed to the writing of the manuscript: LW. Both authors reviewed and approved the final manuscript.
REFERENCES
- 1.Leonard H, Wen X. Epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;134(8):117–34. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman L, Ayub M, Vincent JB. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord. 2010;2(4):182–209. doi: 10.1007/s11689-010-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verpelli C, Montani C, Vicidomini C, Heise C, Sala C. Mutations of the synapse genes and intellectual disability syndromes. Eur J Pharmacol. 2013;719(1–3):112–6. doi: 10.1016/j.ejphar.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 4.Yoon J, Abdelmohsen K, Gorospe M. Functional interactions among micro-RNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014;34:9–14. doi: 10.1016/j.semcdb.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X, Wu Z, Fu X, Han W. lncRNAs: insights into their function and mechanics in underlying disorders. Mutat Res. 2014;762:1–21. doi: 10.1016/j.mrrev.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Slavoff SA, Mitchell AJ, Schwaid AG, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9(1):59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lander ES. RNAs do not encode proteins. Cell. 2014;154(1):240–51. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaukowitch K, Kim TK. Emerging epigenetic mechanisms of long non-coding RNAs. Neuroscience. 2014;264:25–38. doi: 10.1016/j.neuroscience.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyengar BR, Choudhary A, Sarangdhar MA, Venkatesh KV, Gadgil CJ, Pillai B. Non-coding RNA interact to regulate neuronal development and function. Front Cell Neurosci. 2014;8(47):1–10. doi: 10.3389/fncel.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull. 2013;97:69–80. doi: 10.1016/j.brainresbull.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ander BP, Barger N, Stamova B, Sharp FR, Schumann CM. Atypical miRNA expression in temporal cortex associated with dysregulation of immune, cell cycle, and other pathways in autism spectrum disorders. Mol Autism. 2015;6:1–13. doi: 10.1186/s13229-015-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–21. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L, Lei J, Sanders SJ, et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol Autism. 2014;5(22):1–18. doi: 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugathan A, Biagioli M, Golzio C, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A. 2014;111(42):E4468–77. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenwood Genetic Center Expanded NGS X-Linked Intellectual Disability Panel. [Accessed June 30, 2015]. Available at: http://www.ggc.org/images/Expanded_NGS_XLID_Panel_Gene_List.pdf.
- 18.Gardiner K. ID Database Gene List. [Accessed June 30, 2015]. Available at: http://gfuncpathdb.ucdenver.edu/iddrc/iddrc/data/IDgenelist_chr.html. 0000.
- 19.Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37(Database issue):832–6. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava AK, Schwartz CE. Neuroscience and Biobehavioral Reviews Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci Biobehav Rev. 2014;46:161–74. doi: 10.1016/j.neubiorev.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen SL. Trajectories of brain development: point of vulnerability or window of opportunity? Neurosci Biobehav Rev. 2003;27:3–18. doi: 10.1016/s0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 22.Qiao Y, Badduke C, Mercier E, Lewis SM, Pavlidis P, Rajcan-Separovic E. miRNA and miRNA target genes in copy number variations occurring in individuals with intellectual disability. BMC Genomics. 2013;14(1):1. doi: 10.1186/1471-2164-14-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams SR, Mullegama SV, Rosenfeld JA, et al. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet. 2009;18(4):436–41. doi: 10.1038/ejhg.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vadaie N, Morris KV. Long antisense non-coding RNAs and the epigenetic regulation of gene expression. Biomol Concepts. 2013;4(4):411–5. doi: 10.1515/bmc-2013-0014. [DOI] [PubMed] [Google Scholar]
- 25.Cajigas I, Leib DE, Cochrane J, et al. Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development. 2015;142:2641–52. doi: 10.1242/dev.126318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petryniak MA, Potter GB, Rowitch DH, Rubenstein JL. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron. 2007;55(3):417–33. doi: 10.1016/j.neuron.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2015. Available at: http://www.r-project.org/ [Google Scholar]
- 28.Albert R. Scale-free networks in cell biology. J Cell Sci. 2005;118(21):4947–57. doi: 10.1242/jcs.02714. [DOI] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57(1):289–300. [Google Scholar]
- 30.Warnes GR, Bolker B, Bonebakker L, et al. gplots: Various R Programming Tools for Plotting Dat. 2015. Available at: http://cran.r-project.org/package=gplots.
- 31.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–91. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence M, Huber W, Pagès H, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence M, Gentleman R, Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25(14):1841–2. doi: 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. ID gene lists.
Supplementary Table S2. WGCNA results.
Supplementary Table S3. Modular ID gene enrichment.
Supplementary Table S4. Ranked lncRNAs.