

Abstract
Proteolytic enzymes may serve as promising targets for novel therapeutic treatment strategies seeking to impede cancer progression and metastasis. One such enzyme is cathepsin L (CTSL), a lysosomal cysteine protease. CTSL upregulation, a common occurrence in a variety of human cancers, has been widely correlated with metastatic aggressiveness and poor patient prognosis. In addition, CTSL has been implicated to contribute to cancer associated osteolysis; a debilitating morbidity affecting both life expectancy and the quality of life. In this review we highlight the mechanisms by which CTSL contributes to tumor progression and dissemination and discuss the therapeutic utility of CTSL intervention strategies aimed at impeding metastatic progression and bone resorption.
Keywords: Cathepsin L, metastasis, bone resorption, cancer, protease targeting
1.0. Introduction: Proteases as targets in cancer treatment
Proteolytic enzymes are critical in several stages of malignant tumor progression (Lankelma, et al., 2010; Palermo & Joyce, 2008). Extracellular proteases can promote tumor growth and angiogenesis through proteolytic activation of latent growth factors and pro-angiogenic factors (Bogenrieder & Herlyn, 2002; DeClerck, et al., 2004; S. D. Mason & Joyce, 2011). In the metastatic cascade, proteolytic enzymes are actively involved in several key steps including, tumor cell detachment, degradation of extracellular and interstitial matrices and basement membranes, intravasation and extravasation across the capillary/lymphatic system (Gupta & Massague, 2006; Joyce & Pollard, 2009; Tan, Peng, Lu, & Tang, 2013). Consequently the identification of malignancy promoting proteases and the development of intervention strategies designed to interfere with their activities has gained increasing attention (Lah, Duran Alonso, & Van Noorden, 2006; Palermo & Joyce, 2008). Recognition of the importance of matrix metalloproteases (MMPs) in tumor progression and metastasis subsequently led to the development of a broad range of MMP inhibitors (B. Turk, 2006). However, numerous MMP inhibitor clinical trials had to be discontinued due to adverse side effects and/or lack of clinical benefit (Fingleton, 2007; Overall & Lopez-Otin, 2002; Zucker, Cao, & Chen, 2000). Failure of these early generation MMP inhibitors in patients can be ascribed to a variety of factors but the most critical reason was the lack of specificity of these agents (Kruger, Kates, & Edwards, 2010; Palermo & Joyce, 2008). Most MMP inhibitors tested in the clinic exhibited a broad spectrum activity against many members of the large MMP family which also includes anti-tumoral proteases and proteases that are critical to normal tissue functioning. In light of the clinical failure of these MMP inhibitors, protease targeting as a means of inhibiting tumor progression largely fell out of favor. More recently the concept of proteolytic enzyme targeting is being revived because research exploring other proteolytic targets has shown that if selective inhibitors of pericellular proteolysis can be developed, such agents would have the potential to significantly impair tumor progression and metastasis (Chavarria, et al., 2012; Fisher & Mobashery, 2006; Tomoo, 2010; Zucker & Cao, 2009). For example, several cysteine cathepsins of the papain superfamily of cysteine proteases have been widely implicated as facilitators of neoplastic progression (Gocheva & Joyce, 2007; Joyce, et al., 2004; Mohamed & Sloane, 2006; Palermo & Joyce, 2008). The focus of this review is on one of these, Cathepsin L (CTSL), a family member widely associated with malignant progression and metastatic disease.
2.0 Cathepsin L upregulation in human cancers
CTSL is a ubiquitously expressed lysosomal endopeptidase that is primarily involved in terminal degradation of intracellular and endocytosed proteins (Barrett & Kirschke, 1981; Dennemarker, et al., 2010). However, Gottesman and colleagues observed a strong association between CTSL synthesis and malignancy of Kirsten virus transformed NIH 3T3 cells (Gottesman & Sobel, 1980). Subsequent investigations performed in several different transformation models (different viral oncogenes and chemical carcinogens) reported consistent CTSL over-expression irrespective of the mode of transformation (Doherty, et al., 1985; Gottesman & Sobel, 1980; Lah, et al., 1996; Rabin, Doherty, & Gottesman, 1986). In addition, tumor secreted cytokines that have been widely implicated in malignant progression including VEGF, FGF, PDGF, EGF, NGF, INFγ and IL-6 have also been shown to significantly enhance CTSL promoter activity and synthesis (Asanuma, Shirato, Ishidoh, Kominami, & Tomino, 2002; Frick, Doherty, Gottesman, & Scher, 1985; Gallardo, de Andres, & Illa, 2001; Gerber, Wille, Welte, Ansorge, & Buhling, 2001; Keerthivasan, Keerthivasan, Mittal, & Chauhan, 2007; Lah, Hawley, Rock, & Goldberg, 1995; Nilsen-Hamilton, Hamilton, Allen, & Massoglia, 1981; Scher, Dick, Whipple, & Locatell, 1983). Consistent with these in-vitro observations, CTSL upregulation has been reported in a wide range of human malignancies including ovarian, breast, prostate, lung, gastric, pancreatic and colon cancers (Chauhan, Goldstein, & Gottesman, 1991) (Table 1). Importantly, evidence indicates that CTSL expression may be linked to cancer grade and stage. For example, in endometrial cancer patients, CTSL levels increased 50-fold and positively correlated with tumor grade, growth regulatory genes such as Ki-67, cyclin B1 and p21, and HER2 receptor status (Skrzypczak, et al., 2012). Furthermore, Chauhan and colleagues observed significantly elevated CTSL levels in pancreatic adenocarcinomas compared to benign endocrine tumors and islet cells obtained from adjacent normal tissue thereby suggesting a strong association between CTSL expression levels and pancreatic cancer aggressiveness (Chauhan, et al., 1991). In colorectal cancer, numerous clinical studies have reported upregulated CTSL levels and activities (Adenis, et al., 1995; Herszenyi, et al., 1999; Sheahan, Shuja, & Murnane, 1989). The concomitant upregulation of its downstream target urokinase plasminogen activator-1 in colorectal cancer tissues and the strong correlation with metastatic incidence and overall survival further substantiates the argument that CTSL upregulation is a key factor driving neoplastic progression (Herszenyi, et al., 1999).
Table 1. CTSL upregulation in human cancers.
| Cancer type | Observations | Citations |
|---|---|---|
| Colorectal cancer | Higher expression and activity in tumors, correlation with ras, association with uPA and PAI levels, CTSL levels could be of diagnostic significance | Adenis, et al., 1995; Anestakis Doxakis, 2013; Herszenyi, et al., 1999; Sheahan, et al., 1989; Troy, et al., 2004 |
| Breast cancer | CTSL upregulation and increased activity in high grade tumors | Lah, et al., 1997 |
| Chronic myeloid leukemia | Correlation with disease progression, metastatic incidence and overall survival, upregulation due to promoter hypomethylation | Samaiya, et al., 2011 |
| Pediatric acute myeloid leukemia | CTSL upregulation, inverse correlation with endogenous inhibitor cystatin C, positive association with key AML angiogenesis factor VEGF | Jain, Bakhshi, Shukla, & Chauhan, 2010 |
| Melanoma | Increased CTSL activity in melanocytic tumors compared to pigmented nevi and normal dermis | Frohlich, et al., 2001; Kageshita, et al., 1995 |
| Gastrointestinal stromal tumors and gastric cancers | Overexpression and positive correlation with c-kit expression | Dohchin, et al., 2000; Farinati, et al., 1996; Miyamoto, et al., 2011; Watanabe, et al., 1987; Watanabe, et al., 1989 |
| Endometrial cancer | CTSL upregulation and correlation with growth regulatory genes and HER2 receptor status | Skrzypczak, et al., 2012 |
| Pancreatic cancer | CTSL upregulation in tumor as well as tumor associated macrophages | Niedergethmann, et al., 2004; Singh, et al., 2013; Sulpizio, et al., 2012 |
| Glioma | Correlation with glioma progression with expression profile of low grade glioma < anaplastic astrocytoma < glioblastoma | Sivaparvathi, et al., 1996 |
| Lung cancer | Higher CTSL activity in lung cancer compared to non-malignant tissue, association with tumor grade, upregulated serum levels | Chen, et al., 2011 |
| Nasopharyngeal carcinoma | Overexpression in primary tumor and cervical lymph node metastases | Xu, Yuan, Liu, Zhang, & Chen, 2009 |
| Oral squamous cell carcinoma | Elevated CTSL expression correlated with lymph node metastasis and poor survival, CTSL overexpression increases the likelihood of progression of oral dysplastic lesions into carcinomas | Macabeo-Ong, et al., 2003; Nakashima, Yasumatsu, Masuda, Clayman, & Komune, 2012 |
Chronic myeloid leukemia (CML) patients from both chronic and blast crisis/accelerated disease phases exhibit significantly higher CTSL mRNA and enzymatic activity compared to healthy individuals (Samaiya, Bakhshi, Shukla, Kumar, & Chauhan, 2011). CTSL promoter sequence analysis in CML patients revealed that CTSL expression levels are determined by the methylation status of a CpG island flanking the major transcription initiation site (Jean, Guillaume, & Frade, 2002; Jean, Rousselet, & Frade, 2006). Bisulfite sequencing of the CTSL promoter revealed that promoter flanking CpG dinucleotides were significantly hypomethylated in CML patients compared to healthy individuals thus resulting in CTSL overexpression. Independent investigations performed in melanoma and glioblastoma multiforme further demonstrated that in addition to promoter hypomethylation, CTSL upregulation could also be achieved by other distinct mechanisms such as gene amplification and increased promoter activity (Jean, et al., 2006; Mao & Hamoudi, 2000).
Activity assessments have shown significantly higher CTSL proteolytic activity in neoplastic tissues compared to benign tumors or matched tumor adjacent normal tissues (Frohlich, et al., 2001; Kageshita, et al., 1995). In addition to its expression status, CTSL function is tightly regulated by the concentration of its endogenous inhibitors (V. Turk, et al., 1986). In normal tissues, the extracellular matrix is protected from undesirable proteolytic activities by endogenous cathepsin inhibitors such as cystatins and stefins. Studies in various tumor settings have shown a steep downregulation of these CTSL inhibitors with tumor progression (Chambers, Colella, Denhardt, & Wilson, 1992; Friedrich, et al., 1999; Lah, et al., 1997; Samaiya, et al., 2011; Zajc, Sever, Bervar, & Lah, 2002). Lah and colleagues noted elevated CTSL activity in breast cancer patients due to an imbalance between CTSL to cystatin and stefin ratio; with CTSL activity levels strongly correlating with tumor grade (Lah, et al., 1997). Similarly, significant downregulation of cystatin C levels has been observed in chronic myeloid leukemia patients leading to heightened CTSL proteolytic function (Samaiya, et al., 2011). Consistent with these clinical observations, independent experimental investigations have reported striking imbalance between CTSL and endogenous inhibitor level in highly invasive breast and prostate cancer cells compared to poorly invasive and weakly tumorigenic cells (Zajc, Sever, Bervar, & Lah, 2002; Sudhan & Siemann, 2013). Importantly, Chambers and colleagues demonstrated that this inequity in CTSL and endogenous inhibitor levels in transformed cells is mediated through oncogenic Ras activity (Chambers, Colella, Denhardt, & Wilson, 1992). Thus CTSL upregulation is not paralleled by a commensurate increase in the activity of its endogenous inhibitors and as a result it evades regulation and could engage in unregulated activation of proteolytic cascades.
2.1 CTSL secretion by tumor cells
Under normal physiological conditions, CTSL is sequestered within the lysosomes. However, in tumor cells, alterations in expression levels and trafficking pathways result in secretion of CTSL into the extracellular milieu (Denhardt, Greenberg, Egan, Hamilton, & Wright, 1987; Doherty, et al., 1985; Gal, Willingham, & Gottesman, 1985; Gottesman & Sobel, 1980; Rabin, et al., 1986). Comparative analysis of the secretome of transformed cells with that of non-transformed cells revealed that the secretion of a glycoprotein was strikingly enhanced (∼200-fold) in the conditioned media of transformed cells (Gottesman, 1978). This upregulation occurred consistently irrespective of the means of transformation (oncogenic v-ras, v-src or v-mos or chemical tumor promoters) and this mannose-6-phosphate containing glycoprotein was termed as the major excreted protein (MEP) of transformed cells (Doherty, et al., 1985; Gottesman & Sobel, 1980; Rabin, et al., 1986). MEP was subsequently identified as cathepsin L by several independent investigations based on cDNA characterization (Denhardt, et al., 1986; Troen, Gal, & Gottesman, 1987), amino acid sequence homology (Gal & Gottesman, 1988; Troen, et al., 1987), immunolocalization and electron microscopy (Gal, et al., 1985), activity profile (Gal & Gottesman, 1986; R. W. Mason, Gal, & Gottesman, 1987), substrate specificity and cleavage pattern (Gal & Gottesman, 1986).
In agreement with these in-vitro observations, elevated serum CTSL levels have been reported in patients with lung, pancreatic and ovarian cancer when compared to healthy donors (Chen, et al., 2011; Leto, et al., 1997; Nishida, et al., 1995; Siewinski, et al., 2004; Tumminello, et al., 1996; W. Zhang, et al., 2011). Unlike the clinically used CA-125 ovarian cancer biomarker which may also be upregulated in patients with benign tumors yielding false positive results; the selective upregulation of CTSL in aggressive ovarian cancers compared to benign tumors implies significant utility for CTSL as a diagnostic marker (Nishida, et al., 1995). Moreover, its close correlation with tumor grade and tumor invasion suggests that pre-operative serum CTSL levels could serve as indicators of the extent of tumor invasion at the time of surgical resection (W. Zhang, et al., 2011). Similarly, urinary CTSL levels have been shown to be predictive of metastatic spread and tumor recurrence in patients with bladder urothelial cell carcinoma (Svatek, et al., 2008).
Several studies have been undertaken to determine the mechanisms underlying the altered trafficking of this lysosomal enzyme into the extracellular space. During the process of protein translocation, mannose-6-phosphate receptors present within transport vesicles are responsible for accurate and efficient sorting of lysosomal enzymes to the lysosomes. However, (Dong, Prence, & Sahagian, 1989) demonstrated that compared to other lysosomal enzymes, CTSL intrinsically exhibits a low affinity for the mannose-6-phosphate receptor. Thus transformation dependent upregulation in CTSL synthesis combined with poor affinity for the lysosome targeting receptor ultimately results in default secretion of CTSL through the constitutive secretory pathway. Furthermore, growth stimulation limits mannose-6-phosphate receptor concentrations within the golgi through receptor re-distribution which causes the few receptors that are available to be occupied by high affinity lysosomal enzymes whereas, the low affinity CTSL, failing to bind to these receptors is shunted into the secretory pathway (Prence, Dong, & Sahagian, 1990; Stearns, Dong, Pan, Brenner, & Sahagian, 1990). Subsequently, Barbarin and colleagues demonstrated that Rab4a protein is the key regulator of CTSL secretion in melanoma cells. Rab proteins are important regulators of vesicle transport between organelles of the endocytic and secretory system (Barbarin & Frade, 2011). Transfection of highly tumorigenic and metastatic melanoma cells with dominant negative Rab4a mutant, significantly hampered CTSL secretion from these cells and also dramatically impaired their tumorigenic and metastatic potential (Barbarin & Frade, 2011). Collectively these data suggest that upregulated CTSL secretion by tumor cells is achieved through several different mechanisms.
2.2 Nuclear CTSL
In addition to the secreted form, the presence of a truncated nuclear isoform of CTSL has been reported by numerous independent investigations. Nuclear CTSL is translated from a downstream AUG translation initiation site and is consequently devoid of the endoplasmic reticulum targeting signal peptide (Goulet, et al., 2004; Goulet, et al., 2007). However, it does contain a putative nuclear localization signal which might be responsible for its translocation to the nucleus (Hiwasa & Sakiyama, 1996). Nuclear CTSL proteolytically processes and activates the CCAAT-displacement protein/cut homeobox (CDP/Cux) transcription factor which has been strongly associated with poor disease prognosis (Goulet, et al., 2007; Goulet, Truscott, & Nepveu, 2006; Moon, et al., 2002). CTSL processed CDP/Cux exhibits enhanced DNA binding properties which in turn confers a replicative and metastatic advantage to tumor cells (Figure 1). CDP/Cux promotes tumor cell proliferation by accelerating entry into the S phase of cell cycle, and also augments tumor invasion by stimulating hallmark epithelial to mesenchymal transition features such as snail and slug upregulation and E-cadherin repression (Kedinger, et al., 2009).
Figure 1. Distinct roles of secreted and nuclear CTSL.
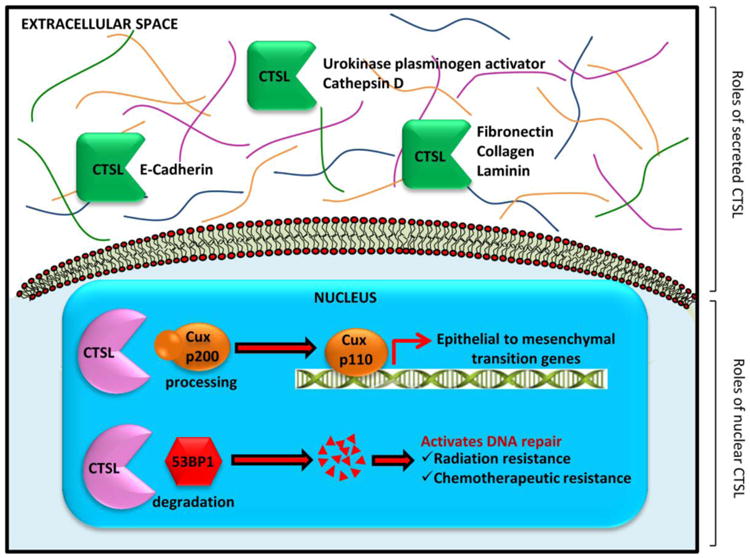
Secreted CTSL aids metastatic dissemination of tumor cells by degrading cell adhesion molecules and extracellular matrix proteins as well as activating other latent proteases. Nuclear CTSL activates the transcription of epithelial to mesenchymal transition genes and also confers therapeutic resistance.
Nuclear CTSL has been documented to promote tumor progression through other CDP/Cux independent mechanisms as well. Nuclear CTSL is frequently upregulated in triple negative breast cancer patients and patients with a either a germline or somatic mutation in BRCA1 tumor suppressor gene (Grotsky, et al., 2013). In fact, increased nuclear CTSL levels serve as a predictive biomarker for treatment response in this subset of breast cancer patients. In the absence of BRCA1, 53BP1 suppresses homologous repair of DNA double strand breaks (DSBs) and instead activates the error prone non homologous end joining (NHEJ) substitute pathway which results in accumulation of unrepaired DNA DSBs, chromosomal translocations and consequently cell death and tumor suppression (Bouwman, et al., 2010; Snouwaert, et al., 1999; Ward, Minn, van Deursen, & Chen, 2003). BRCA1 deficient cells overcome this genomic instability and growth arrest by over-expressing nuclear CTSL which in turn proteolytically degrades 53BP1 (Gonzalez-Suarez, et al., 2011; Grotsky, et al., 2013). Thus nuclear CTSL not only confers a survival advantage on BRCA1 deficient tumor cells, but, by improving their DNA repair capacities it also renders them resistant to radiation and genotoxic chemotherapeutics such as cisplatin, PARP inhibitors and mitomycin C (Grotsky, et al., 2013).
3.1 Role of CTSL in tumor metastasis
In transformed cells extracellular CTSL levels increase up to 200-fold and comprise up to 40 % of total secreted proteins (Gottesman, 1978). Transformation dependent CTSL secretion augments the invasive/metastatic potential of cancer cells through direct degradative proteolysis of several components of the extracellular matrix (ECM) and basement membrane (Baricos, Zhou, Mason, & Barrett, 1988; Chambers, et al., 1992; Colella, Jackson, & Goodwyn, 2004) (Figure 1). In the presence of cell surface glycosaminoglycans, secreted CTSL degrades ECM components such as laminin, Type I and IV collagen, fibronectin and elastin (Gal, et al., 1985; Ishidoh & Kominami, 1995; R. W. Mason, Johnson, Barrett, & Chapman, 1986; Novinec, et al., 2007). Furthermore, it plays a critical role in amplification of the proteolytic cascade by activating latent pro-forms of other key metastasis associated proteases including urokinase plasminogen activator, pro-heparanase, other cathepsins, as well as certain MMPs (Everts, et al., 2006; Goretzki, et al., 1992; Laurent-Matha, Derocq, Prebois, Katunuma, & Liaudet-Coopman, 2006). Cell adhesion protein E-cadherin is significantly reduced during tumor invasion and thus loss of E-Cadherin is widely accepted as a marker of tumor invasiveness. CTSL proteolytically degrades the extracellular domain of E-cadherin and abolishes its adhesive property (Gocheva, et al., 2006). Consistent with these observations, tumor progression studies in a CTSL-/- RIP1-Tag2 pancreatic carcinogenesis model revealed that CTSL deficiency significantly hampers the progression of benign encapsulated tumors into invasive carcinomas thus indicating that CTSL has a non-redundant consequential role in the process of tumor invasion (Gocheva, et al., 2006) (Figure 2). Likewise, numerous studies have reported that an increase in CTSL secretion is a key feature driving the switch of poorly tumorigenic and non-metastatic human melanoma cells to a highly tumorigenic and aggressively metastatic phenotype (Frade, et al., 1998; Jean, et al., 1996; Rozhin, Wade, Honn, & Sloane, 1989). In agreement, treatment with an anti-CTSL antibody resulted in significantly retarded tumor growth and decreased lung metastases incidence (Frade, Rousselet, & Jean, 2008; Jean, et al., 1996). CTSL has also been shown to promote melanoma cell survival and tumor growth by inhibiting complement mediated tumor cell death (Jean, et al., 1996; Jean, et al., 1995). The well-orchestrated modulation of CTSL expression as a metastatic tumor cell transitions through different phases of the metastatic cascade further highlights its significance in the metastatic dissemination process. Metastasizing tumor cells upregulate CTSL expression as they undergo epithelial to mesenchymal transition but once the cells become established at a distant site, CTSL expression is down-regulated as the cells revert back to their epithelial state to promote colonization (Chen, et al., 2013; Kedinger, et al., 2009).
Figure 2. CTSL deficiency impairs invasiveness of pancreatic islet tumors in RIP-1 Tag-2 mice.
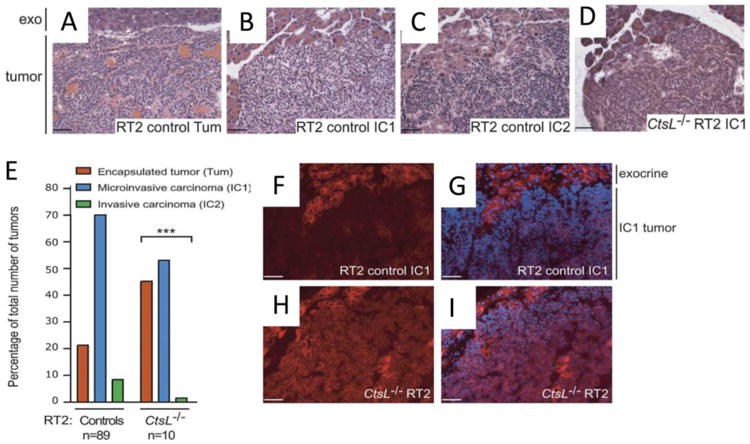
Tumors in RT2 mice can be classified into three different grades A. Encapsulated tumor (Tum), B. microinvasive tumors (IC1) and C. invasive carcinomas (IC2). D. Representative image for the predominant grade in CTSL knockout mice is shown. E. Distribution of tumor types in control and CTSL knockout tumors. Immunofluorescence images indicating increased E-Cadherin levels in CTSL knockout (H and I) compared to control (F and G) tumors (Red, E-cadherin; blue, DAPI). Modified with permission from Gocheva et al (2006). Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes & Development, 20:543-556, under a Creative Commons License (Attribution-NonCommercial 4.0 International License).
In contrast to the well oxygenated and near neutral pH conditions observed within most normal tissues, the tumor microenvironment is frequently marked by hypoxia and acidosis (Jensen, et al., 2010; Lunt, Chaudary, & Hill, 2009; Siemann DW, 2015). It is now a well-recognized fact that these very aberrant microenvironmental conditions are responsible for aggressive tumor progression and metastatic occurrence which ultimately results in poor patient survival (Schwickert, Walenta, Sundfor, Rofstad, & Mueller-Klieser, 1995; Siemann DW, 2015; Walenta, et al., 2000). Hypoxic and acidic conditions are known to result in enhanced tumor cell secretion of CTSL by eliciting two distinct mechanisms, namely, upregulated expression and lysosomal exocytosis (Cuvier, Jang, & Hill, 1997; Sudhan & Siemann, 2013). Lysosomes are normally peri-nuclear in localization. However, in response to hypoxic and acidic exposures, the lysosomes redistributed and undergo anterograde trafficking to the plasma membrane and released their contents (including CTSL) into the extracellular milieu (Sudhan & Siemann, 2013). Since cathepsins exhibit optimal activity under acidic conditions (Ishidoh & Kominami, 1995), CTSL released in an acidic milieu would engage in heightened proteolysis of extracellular matrix components and promote invasiveness. Indeed, hypoxia and acidosis triggered CTSL upregulation closely correlated with metastatic properties of tumor cells thereby suggesting that CTSL may be a key player in microenvironment triggered metastatic aggressiveness (Figure 3) (Sudhan & Siemann, 2013).
Figure 3. Tumor microenvironmental conditions such as hypoxia and acidosis enhance CTSL secretion and tumor cell invasiveness.

A. and C, CTSL secretion by prostate cancer PC-3ML cells upon exposure to hypoxic or acidic conditions. B. and D, PC-3ML invasiveness in response to such conditions. Modified with permission from Sudhan et al (2013). Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clinical & Experimental Metastasis, 30:891-902.
Supporting the observations made in experimental metastasis models, several clinical studies in cancer types including breast, pancreatic, colorectal and lung have shown that tumor CTSL level is a strong prognostic indicator of patient outcome (Table 2). Grotsky and colleagues demonstrated that CTSL along with 53BP1 and vitamin D receptor constitutes a triple biomarker signature for therapeutic resistance to radiation, DNA damaging chemotherapeutics and PARP inhibitors among triple negative breast cancer patients and patients with BRCA1 mutation (Grotsky, et al., 2013). In breast cancer patients, CTSL level inversely correlates with steroid hormone receptor status and is a strong predictor of tumor relapse and poor overall survival (Foekens, et al., 1998; Lah, Cercek, et al., 2000; Lah, Kalman, et al., 2000; Thomssen, et al., 1998; Thomssen, et al., 1995). In fact, Thomssen and colleagues have reported that the prognostic impact of CTSL was comparable to that of axillary lymph node status and tumor histological grading; and could be utilized for identification of node negative patients that are at a high risk of metastatic relapse (Thomssen, et al., 1998; Thomssen, et al., 1995).
Table 2. CTSL in cancer prognosis.
| Cancer type | Observations | Citations |
|---|---|---|
| Breast cancer | Inverse correlation with steroid hormone receptor status and disease free survival and overall survival, prognostic value comparable to axillary lymph node status and tumor grading, biomarker for identification of node negative patients at a high risk of metastatic relapse, CTSL could predict response to surgical resection and adjuvant systemic therapy in patients with operable tumors. | Foekens, et al., 1998; Harbeck, et al., 2001; Jagodic, Vrhovec, Borstnar, & Cufer, 2005; Lah, Cercek, et al., 2000; Lah, Kalman, et al., 2000; Thomssen, et al., 1995 |
| Gastric cancer | Upregulation in tumors locally invading the muscularis propria and tumors with venous invasion, upregulation in chronic atrophic gastritis with intestinal metaplasia and gastric tumors, may participate in gastritis to cancer progression | Dohchin, et al., 2000; Farinati, et al., 1996 |
| Urothelial carcinoma of the bladder | Positive correlation with tumor stage and grade, local and metastatic recurrence and poor survival | Yan, et al., 2010 |
| Pancreatic cancer | Association with tumor stage, lymph node invasion, disease recurrence and overall survival, predictor of disease relapse in patients undergoing curative resection | Niedergethmann, et al., 2004; Singh, et al., 2013 |
| Glioma | correlation with glioma progression in the order of low grade glioma < anaplastic astrocytoma < glioblastoma | Sivaparvathi, et al., 1996 |
| Colorectal cancer | Higher expression and activity in tumors compared to matched normal mucosa, correlation with tumor progression, metastatic incidence and overall survival, high CTSL expression in patients with curative disease is a predictor of disease relapse and poor survival | Anestakis Doxakis, 2013; Herszenyi, et al., 1999; Troy, et al., 2004 |
| Pediatric acute myeloid leukemia | CTSL upregulation exhibited inverse correlation with event free survival and overall survival | Jain, et al., 2010 |
| Oral squamous cell carcinoma | Elevated CTSL expression correlated with lymph node metastasis and poor survival, expression of headpin - endogenous inhibitor to CTSL inversely correlated with tumor grade | Nakashima, et al., 2012 |
| Nasopharyngeal carcinoma | Upregulation correlates with lymph node metastases and distant metastases, tumor stage and overall survival | Xu, et al., 2009 |
3.2 Role of Cathepsin L in bone resorption
The involvement of CTSL in neoplastic bone diseases has been highlighted by numerous experimental and clinical reports of CTSL upregulation in primary and metastatic bone tumors. Several studies have reported elevated CTSL mRNA levels across various human osteosarcoma cell lines and tumor samples (Damiens, et al., 2000; Husmann, et al., 2008; Krueger, Kellner, Buehling, & Roessner, 2001; Park, et al., 1996). In comparison to normal tissue levels, CTSL levels were upregulated in ∼50% of primary bone tumors, and in almost 100% of the bone metastases investigated (Park, et al., 1996). In agreement with these observations Husmann and colleagues have reported that highly metastatic SAOS-2 sublines LM5 and LM7 expressed significantly higher levels of CTSL mRNA compared to parental SAOS-2 osteosarcoma cells thereby highlighting the correlation between CTSL expression levels and metastatic aggressiveness of bone cancers (Husmann, et al., 2008). A serendipitous observation made in CTSL antibody producing hybridoma cells provided one of the first leads to the importance of CTSL in multiple myeloma. Highly tumorigenic and metastatic P3X63Ag8.653 murine myeloma cells secrete high levels of pro-cathepsin L (Weber, Gunther, Laube, Wiederanders, & Kirschke, 1994). Intriguingly, these authors observed that when these CTSL secreting myeloma cells were fused with spleen cells immunized with CTSL to generate CTSL antibody producing hybridoma cells, their tumorigenic and metastatic capacity was remarkably diminished. Compared to the numerous peritoneal tumors and metastatic deposits observed in mice inoculated with either myeloma cells or control hybridoma cells, ∼80% of the mice inoculated with CTSL antibody producing hybridoma cells failed to show signs of primary or metastatic tumor burden. Consistent with these findings, CTSL depletion has been reported to significantly retard the growth of SP and L myeloma tumors (Kirschke, Eerola, Hopsu-Havu, Bromme, & Vuorio, 2000).
In addition to these primary bone neoplasias, several human cancers such as lung, prostate and breast adenocarcinomas exhibit a marked proclivity to metastasize to the bone (Mundy, 2002; Roodman, 2004). Bone metastases are rarely indolent – they usually inflict devastating skeletal morbidities such as intractable bone pain, pathological fractures, paralysis due to nerve compression and hypercalcemia (Mundy, 2002; Roodman, 2004). These adverse skeletal complications are predominantly triggered by unrestrained osteoblastic and osteoclastic activities of bone metastases. Under normal physiological conditions, bone remodeling is tightly regulated through reciprocal interactions between bone resorbing osteoclasts and bone forming osteoblasts (Raggatt & Partridge, 2010). However, both primary and metastatic bone lesions perturb this equilibrium through sustained activation of osteoclasts or osteoblasts which ultimately leads to excessive osteolysis or bone formation. While it is well-recognized that osteolytic cancers perpetrate bone resorption through osteoclasts; even predominantly osteoblastic cancers such as prostate cancer, have been shown to depend on an initial osteoclastic trigger for subsequent osteoblastic events to occur (Inoue, et al., 2005; Keller & Brown, 2004; Roato, et al., 2008; J. Zhang, et al., 2003). Since inhibition of unrestrained osteoclastic activity can alleviate skeletal morbidities associated with both osteoclastic and osteoblastic metastases, significant efforts have focused on the identification of osteoclast targeting strategies and led to the development of agents including bisphosphonates, denosumab, and Cathepsin K inhibitors (Clezardin, 2011; Neville-Webbe & Coleman, 2010; Onishi, Hayashi, Theriault, Hortobagyi, & Ueno, 2010; Rachner, Hadji, & Hofbauer, 2012).
Cathepsin K (CTSK) is the predominant protease in osteoclasts and is the prime executor of resorption during both normal and pathological bone turnover. Nonetheless, studies in knockout mouse models have shown other proteases; in particular CTSL to closely participate in conjunction with CTSK in the process of pathological bone resorption (Everts, et al., 2006; Potts, et al., 2004). Although osteoclastic CTSL levels are significantly lower than CTSK levels during normal bone remodeling, in the presence of bone resorption promoting inflammatory cytokines such as parathyroid hormone, IL-1α, IL-6 and TNF-α, osteoclastic CTSL activity increases several fold and it plays a significant, non-redundant role in the process of pathological bone resorption (Cox, et al., 2006; Furuyama & Fujisawa, 2000a, 2000b; Kakegawa, et al., 1995; Tagami, et al., 1994). Using immunohistochemical procedures and electron microscopy, Goto and colleagues have demonstrated that in contrast to the low intracellular levels of CTSL observed within osteoclasts, the protease shows a strong extracellular presence and heavily co-localizes with collagen fibrils within resorption pits suggesting its involvement in degradation of the bone matrix (Goto, et al., 1994; Goto, et al., 1993). CTSL also has been widely implicated in the proteolytic degradation of bone and cartilage matrix components (Kakegawa, et al., 1995; Kirschke, Kembhavi, Bohley, & Barrett, 1982; Leto, Sepporta, Crescimanno, Flandina, & Tumminello, 2010; Maciewicz, Wotton, Etherington, & Duance, 1990; Nakase, et al., 2000). In fact, serum CTSL levels were found to be significantly elevated in patients with low bone density conditions such as osteoporosis and osteopenia compared to individuals with normal bone mineral density (Lang, Willinger, & Holzer, 2004) Likewise, treatment with anti-resorptive agents such as bisphosphonates restored serum CTSL levels near normal levels. The close association of CTSL with various bone disorders involving destructive bone loss such as rheumatoid arthritis (Devauchelle, et al., 2004; Lemaire, et al., 1997; Schedel, et al., 2004), osteoporosis (Katunuma, Tsuge, Nukatsuka, & Fukushima, 2002; Lang, et al., 2004; Potts, et al., 2004), osteoarthritis (Cawston & Young, 2010; Solau-Gervais, et al., 2007) and periodontal disease (Cox, et al., 2006; Yamaguchi, Naruishi, Arai, Nishimura, & Takashiba, 2008) further suggests the likelihood of CTSL's participation in cancer associated bone resorption. Indeed, (Katunuma, Tsuge, Nukatsuka, Asao, & Fukushima, 2002; Katunuma, Tsuge, Nukatsuka, & Fukushima, 2002) showed that CTSL inhibition significantly mitigates osteolytic events and hypercalcemia in mice with bone metastases and importantly, this protective effect was shown to be superior to that of bisphosphonate.
In addition to cancer induced osteolysis, bone loss is also inflicted by several “standard of care” anti-cancer agents; a morbidity that severely affects the quality of life and life expectancy of cancer survivors (Holroyd, Cooper, & Dennison, 2008; Khan & Khan, 2008; VanderWalde & Hurria, 2011). Patients receiving chemotherapeutic agents such as methotrexate, cyclophosphamide, dexamethasone, prednisone and doxorubicin as post-operative adjuvant therapy, often suffer from hypogonadism triggered osteoporosis (Pfeilschifter & Diel, 2000; Reid, 1997; VanderWalde & Hurria, 2011). Similarly, surgical or medical hormonal ablation in patients with hormone responsive cancers such as breast, ovarian or prostate cancer can result in a drastic worsening of bone health (Fogelman, et al., 2003; Hadji, et al., 2011; Mincey, et al., 2006). Furthermore, steroidal hormones such as estrogen have been shown to negatively regulate osteoclastic synthesis of cathepsins L and K both directly by binding to osteoclastic estrogen receptor and indirectly by suppressing the expression of resorptive cytokines (Furuyama & Fujisawa, 2000a). Thus ovariectomized mice exhibit a significant upregulation of CTSK and particularly CTSL. In fact, CTSL deficient mice showed a marked resistance to osteoporosis upon ovariectomy (Potts, et al., 2004). Thus CTSL intervention strategies would not only serve to impede the metastatic dissemination of tumor cells but would also alleviate both treatment-induced and cancer-associated osteolysis.
4.0 Cathepsin L targeting
The role of CTSL in promoting tumor progression and metastatic aggressiveness, its contribution to cancer associated bone resorption and its strong association with disease relapse and patient mortality has raised significant interest in the development of CTSL intervention strategies. Since unrestrained CTSL activity stems from an imbalance between CTSL and endogenous inhibitor levels, several investigations have attempted to abolish CTSL proteolytic function through ectopic delivery/overexpression of endogenous inhibitors such as cystatins and stefins (Gianotti, Sommer, Carmona, & Henrique-Silva, 2008; Saleh, et al., 2003; Shridhar, et al., 2004). Notably, Gianotti and colleagues have. reported that the anti-invasive property of cystatin over-expression was mainly mediated through inactivation of extracellular but not intracellular CTSL; further validating the hypothesis that metastatic phenotype of tumor cells is primarily driven by secreted CTSL (Gianotti, et al., 2008). Likewise, CTSL downregulation through RNA interference in different tumor models including glioma, osteosarcoma, myeloma and melanoma resulted in consistent inhibition of tumorigenicity and invasiveness of neoplastic cells (Kirschke, et al., 2000; Krueger, et al., 2001; Levicar, et al., 2003; Yang & Cox, 2007). In addition to its anti-invasive effect, Levicar et al., reported that CTSL suppression in glioblastoma also enhanced the sensitivity of tumor cells to apoptotic agents such as staurosporine. Studies performed in the RIP1-Tag2 pancreatic carcinogenesis model comparing the effect of various cathepsin deficiencies on tumor progression showed that compared to other cathepsins, CTSL knockout led to the largest reduction in tumor burden and a significant impairment of the progression of benign encapsulated tumors to invasive carcinoma (Gocheva, et al., 2006).
Although anti-sense and genetic knockout strategies support the anti-metastatic significance of CTSL abrogation, the development of specific inhibitors of CTSL has been limited by the high degree of structural homology between different members of the cathepsin family (Sadaghiani, et al., 2007; V. Turk, Turk, & Turk, 2001). Indeed most currently studied cathepsin inhibitors have broad activity spectrums (Bell-McGuinn, Garfall, Bogyo, Hanahan, & Joyce, 2007; Elie, et al., 2010; Joyce, et al., 2004; Navab, Mort, & Brodt, 1997; Palermo & Joyce, 2008). While the anti-tumor effects exhibited by pan specific cysteine cathepsin inhibitors appear promising, the dismal outcomes of previous broad spectrum targeting approaches are disconcerting (Palermo & Joyce, 2008; B. Turk, 2006). Since the proteolytic function of all cathepsins has as yet not been deciphered, and given the critical normal tissue functions of some cathepsins, indiscriminate inhibition of all members of the cathepsin family could potentially entail the same negative consequences as reported for the application of broad spectrum MMP inhibitors (V. Turk, et al., 2001). A likely factor underlying the failure of MMP inhibitors in clinical trial was their lack of specificity. These broad spectrum inhibitors indiscriminately inhibited several members of the MMP family which also includes anti-cancer proteases and proteases that are crucial to normal tissue function (Kruger, et al., 2010; Lopez-Otin & Matrisian, 2007; Overall & Lopez-Otin, 2002). For example, owing to their undesired inhibitory activity against the closely related ADAM proteases, several patients experienced severe musculoskeletal toxicity, which necessitated a reduction in dosage to suboptimal concentrations that were ineffective at inhibiting MMPs (Coussens, Fingleton, & Matrisian, 2002; Fingleton, 2007; B. Turk, 2006; Zucker, et al., 2000).
Despite the challenges imposed by the high degree of homology between the S2 substrate recognition domains of cysteine cathepsins, a few CTSL specific inhibitors have been developed and tested in preclinical models (Katunuma, 2011; Katunuma, Tsuge, Nukatsuka, Asao, et al., 2002; Kishore Kumar, et al., 2010; Song, et al., 2013; Torkar, Lenarcic, Lah, Dive, & Devel, 2013). Kumar and colleagues generated and screened a library of functionalized benzophenone, thiophene, pyridine, and fluorene thiosemicarbazone derivatives for potent inhibitors of CTSL (Kishore Kumar, et al., 2010; Kumar, et al., 2010). The lead molecule, KGP94, significantly retarded the growth of both recently implanted and established tumors (Chavarria, et al., 2012). In addition, KGP94 treatment significantly impaired various metastasis associated tumor cell functions such as migration, invasion and lung colonization (Figure 4) (Chavarria, et al., 2012; Sudhan & Siemann, 2013). Similarly, (Katunuma, Tsuge, Nukatsuka, Asao, et al., 2002; Katunuma, Tsuge, Nukatsuka, & Fukushima, 2002) showed that epoxysuccinate based CTSL inhibitors of the CLIK series, namely CLIK-148 and CLIK-195 not only attenuated cancer associated bone resorption and hypercalcemia but also led to a significant reduction in metastatic burden. Bone resorption not only provides room for the expansion of metastatic lesions but also leads to the release of several active growth factors from the bone matrix that supports aggressive neoplastic cell growth. Thus a ‘vicious cycle’ is created between the tumor and the bone: tumor secreted cytokines stimulate bone resorption by upregulating osteoclastic CTSL activity; and the resorbed bone in turn releases growth factors stimulating tumor-cell proliferation and further cytokine release (Leto, et al., 2010; Mundy, 2002). Conceivably, CTSL inactivation disengages this vicious cycle and thus not only alleviates bone resorption, but also decreases the tumor burden in the bone.
Figure 4. Effect of CTSL inhibition on metastasis.

A. Effect of CTSL specific inhibitor KGP94 on migratory capacity of OS-156 osteosarcoma cells. B. Anti-invasive effect of KGP94 across various tumor cell types; osteosarcoma cells (OS-156), androgen dependent prostate cancer cells (LNCaP), androgen resistant bone metastatic prostate cancer cells (C42B), and highly lung metastatic breast cancer cells (LM2-4175). C. Effect of CTSLi KGP94 treatment on lung metastases forming capacity of SCCVII squamous carcinoma cells, mice (N>9) treated with daily 20 mg/kg doses of the CTSL inhibitor KGP94. Line, median; bars 25th and 75th percentiles. D. Representative images of lungs from C.
In addition to participating in bulk protein turnover within the lysosomes, CTSL is involved in numerous other activities critical to normal tissue functioning. CTSL is responsible for activation of several key pituitary neuropeptides including β-endorphin, enkephalin, α-melanocyte stimulating hormone and adrenocorticotropic hormone present within zymogenic granules (Funkelstein, et al., 2008; Hwang, et al., 2007; Yasothornsrikul, et al., 2003). CTSL also plays a cardioprotective role through inhibition of apoptosis promoting Akt signaling pathway in cardiomyocytes (Tang, et al., 2009). The significance of CTSL in normal tissue functioning is reflected by numerous pathological conditions that stem from CTSL deficiency such as dilated cardiomyopathy, metabolic syndromes, brain atrophy, epithelial hyperplasia and hypotrichosis (Felbor, et al., 2002; Huang, et al., 2003; Petermann, et al., 2006; Potts, et al., 2004; Stypmann, et al., 2002). Thus inhibition of nuclear CTSL poses a significant risk of such undesirable side-effects due to inactivation of intracellular CTSL. One key difference between the normal tissue and neoplastic cell functions of CTSL is that while most normal tissue functions are furnished by intracellular CTSL, tumor and metastasis associated functions are mainly mediated through extracellular CTSL. Selective targeting of extracellular CTSL could therefore broaden the therapeutic window of CTSL inhibition strategies by obviating any toxicity resulting from inhibition of intracellular functions. In fact, the cell impermeable JPM-OEt inhibitor was able to achieve a significant inhibition of tumor growth, angiogenesis and invasion without exerting any overt normal tissue toxicity (Sadaghiani, et al., 2007).
Long term administration of selective inhibitors of CTSL poses a potential risk of therapeutic resistance through compensation by other members of the cathepsin family. Thus simultaneous inhibition of CTSL and other critical tumor promoting cathepsins could potentially yield a better outcome. Since CTSB and CTSK have been shown to significantly impact two distinct aspects of tumor progression, tumor metastasis and bone resorption (Jensen, et al., 2010; Roshy, Sloane, & Moin, 2003), respectively, it might be feasible to test the efficacy of CTS L+B or CTS L+K dual targeting strategies Such protease inhibition approaches require careful deliberation supported by studies in conditional knockout models in order to achieve maximum anti-tumor effects while obviating normal tissue toxicity. Studies have shown that CTSL inhibition improves the therapeutic window of various chemotherapeutic agents by lowering the effective concentrations at which they mediate cytotoxic effects (Zheng, Chou, Mirkin, & Rebbaa, 2004; Zheng, et al., 2009). These authors demonstrated that CTSL inhibition not only prevents the emergence of a drug resistant phenotype but also effectively reverses resistance to various cytotoxic and targeted agents including doxorubicin, etoposide, imatinib, trichostatin A and tamoxifen. While administration of highest tolerable dose of doxorubicin had no impact on the growth of doxorubicin resistant tumors, the CTSL inhibitor iCL yielded a 40 % reduction in tumor growth; combination treatment with doxorubin restored the sensitivity to doxorubicin and led to a 90% reduction in tumor growth. Since the majority of anticancer drug therapy failures may be attributed to either inherent or acquired resistance, if drug sensitivity restoration by CTSL inhibition could be successfully translated to the clinic, such a strategy would have significant impact on patient treatment outcome.
Despite the promising results of CTSL inhibition in preclinical models, questions on protease targeting raised by the unanticipated outcomes of MMP inhibitor clinical trials need to be considered. One such issue is the development of reliable tools for the identification of patients who would best benefit from anti-CTSL therapy. While methodologies such as CTSL immunohistochemistry of tumor tissue and measurement of CTSL level/activity in serum samples have been proposed, it is critical to evaluate the reliability of these techniques. Recently, activity based probes (ABPs) have been developed as a non-invasive imaging tool that not only lends itself to patient identification but could also be used for monitoring treatment response (Blum, von Degenfeld, Merchant, Blau, & Bogyo, 2007; Paulick & Bogyo, 2008). ABPs are comprised of three distinct elements namely (i) a reactive site that covalently links to the active site of target enzyme based on its activity status, (ii) a target recognition motif that confers specificity towards its target enzyme and (iii) a reporter tag that enables direct visualization of probe labeled proteins. In contrast to the passive measurement of protein abundance offered by widely used proteomic tools that depend on antibody labeling, ABPs provide a direct readout of the activity status of its target enzyme both in-vivo as well as ex-vivo. These features of ABPs allow longitudinal in-vivo assessment of response to enzyme inhibitors using non-invasive imaging techniques (Paulick & Bogyo, 2008; Verdoes, et al., 2013). However, most ABPs that have been developed for the assessment of cysteine proteinases were designed based on broad spectrum reactive sites and are thus not amenable for detection of specific cathepsins such CTSL (Blum, et al., 2007; Paulick & Bogyo, 2008). Like CTSL inhibitors, the development of CTSL specific ABP has been hindered by the high degree of similarity between the substrate recognition pockets of different members of the cathepsin family. However, Torkar and colleagues recently developed a novel non-cell permeable CTSL specific photo-affinity based probe that could be utilized for the measurement of secreted CTSL activity ex-vivo (Torkar, et al., 2013).
Although issues such as patient identification, therapeutic response assessment and comparison of effectiveness of intracellular versus extracellular CTSL targeting need to be addressed, the abundance of favorable preclinical and clinical findings make CTSL targeting an attractive and promising approach to target tumor cell dissemination and associated skeletal morbidities.
Acknowledgments
The authors' work is supported by the National Cancer Institute (Public Health Service grant R01 CA169300).
Abbreviations
- MMPs
Matrix metalloproteases
- CTSL
Cathepsin L
- VEGF
Vascular endothelial growth factor
- FGF
Fibroblast growth factor
- PDGF
Platelet derived growth factor
- EGF
Epidermal growth factor
- NGF
Neuronal growth factor
- INFγ
Interferon gamma
- IL
Interleukin
- 53BP1
p53 binding protein 1
- TNF-α
Tumor necrosis factor-α
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adenis A, Huet G, Zerimech F, Hecquet B, Balduyck M, Peyrat JP. Cathepsin B, L, and D activities in colorectal carcinomas: relationship with clinico-pathological parameters. Cancer Lett. 1995;96:267–275. doi: 10.1016/0304-3835(95)03930-u. [DOI] [PubMed] [Google Scholar]
- Anestakis Doxakis, A M, Petanidis Savvas, Iakovidou-Kritsi Zafiroula. Assessment of the Roles of Cathepsins B, H and L in the Progression of Colorectal Cancer. Journal of Cancer Therapy. 2013;4:1–7. [Google Scholar]
- Asanuma K, Shirato I, Ishidoh K, Kominami E, Tomino Y. Selective modulation of the secretion of proteinases and their inhibitors by growth factors in cultured differentiated podocytes. Kidney Int. 2002;62:822–831. doi: 10.1046/j.1523-1755.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- Barbarin A, Frade R. Procathepsin L secretion, which triggers tumour progression, is regulated by Rab4a in human melanoma cells. Biochem J. 2011;437:97–107. doi: 10.1042/BJ20110361. [DOI] [PubMed] [Google Scholar]
- Baricos WH, Zhou Y, Mason RW, Barrett AJ. Human kidney cathepsins B and L. Characterization and potential role in degradation of glomerular basement membrane. Biochem J. 1988;252:301–304. doi: 10.1042/bj2520301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AJ, Kirschke H. Cathepsin B, Cathepsin H, and cathepsin L. Methods Enzymol. 1981;80(Pt C):535–561. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- Bell-McGuinn KM, Garfall AL, Bogyo M, Hanahan D, Joyce JA. Inhibition of cysteine cathepsin protease activity enhances chemotherapy regimens by decreasing tumor growth and invasiveness in a mouse model of multistage cancer. Cancer Res. 2007;67:7378–7385. doi: 10.1158/0008-5472.CAN-07-0602. [DOI] [PubMed] [Google Scholar]
- Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat Chem Biol. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- Bogenrieder T, Herlyn M. Cell-surface proteolysis, growth factor activation and intercellular communication in the progression of melanoma. Crit Rev Oncol Hematol. 2002;44:1–15. doi: 10.1016/S1040-8428(01)00196-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawston TE, Young DA. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010;339:221–235. doi: 10.1007/s00441-009-0887-6. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Colella R, Denhardt DT, Wilson SM. Increased expression of cathepsins L and B and decreased activity of their inhibitors in metastatic, ras-transformed NIH 3T3 cells. Mol Carcinog. 1992;5:238–245. doi: 10.1002/mc.2940050311. [DOI] [PubMed] [Google Scholar]
- Chauhan SS, Goldstein LJ, Gottesman MM. Expression of cathepsin L in human tumors. Cancer Res. 1991;51:1478–1481. [PubMed] [Google Scholar]
- Chavarria GE, Horsman MR, Arispe WM, Kumar GD, Chen SE, Strecker TE, Parker EN, Chaplin DJ, Pinney KG, Trawick ML. Initial evaluation of the antitumour activity of KGP94, a functionalized benzophenone thiosemicarbazone inhibitor of cathepsin L. Eur J Med Chem. 2012;58:568–572. doi: 10.1016/j.ejmech.2012.10.039. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chen L, Zhao R, Yang XD, Imran K, Xing CG. Microarray analyses reveal liver metastasis-related genes in metastatic colorectal cancer cell model. J Cancer Res Clin Oncol. 2013;139:1169–1178. doi: 10.1007/s00432-013-1424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fei J, Wu L, Jiang Z, Wu Y, Zheng Y, Lu G. Detection of cathepsin B, cathepsin L, cystatin C, urokinase plasminogen activator and urokinase plasminogen activator receptor in the sera of lung cancer patients. Oncol Lett. 2011;2:693–699. doi: 10.3892/ol.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clezardin P. Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res. 2011;13:207. doi: 10.1186/bcr2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella R, Jackson T, Goodwyn E. Matrigel invasion by the prostate cancer cell lines, PC3 and DU145, and cathepsin L+B activity. Biotech Histochem. 2004;79:121–127. doi: 10.1080/10520290400010572. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- Cox SW, Eley BM, Kiili M, Asikainen A, Tervahartiala T, Sorsa T. Collagen degradation by interleukin-1beta-stimulated gingival fibroblasts is accompanied by release and activation of multiple matrix metalloproteinases and cysteine proteinases. Oral Dis. 2006;12:34–40. doi: 10.1111/j.1601-0825.2005.01153.x. [DOI] [PubMed] [Google Scholar]
- Cuvier C, Jang A, Hill RP. Exposure to hypoxia, glucose starvation and acidosis: effect on invasive capacity of murine tumor cells and correlation with cathepsin (L + B) secretion. Clin Exp Metastasis. 1997;15:19–25. doi: 10.1023/a:1018428105463. [DOI] [PubMed] [Google Scholar]
- Damiens C, Grimaud E, Rousselle AV, Charrier C, Fortun Y, Heymann D, Padrines M. Cysteine protease production by human osteosarcoma cells (MG63, SAOS2) and its modulation by soluble factors. Cytokine. 2000;12:539–542. doi: 10.1006/cyto.1999.0593. [DOI] [PubMed] [Google Scholar]
- DeClerck YA, Mercurio AM, Stack MS, Chapman HA, Zutter MM, Muschel RJ, Raz A, Matrisian LM, Sloane BF, Noel A, Hendrix MJ, Coussens L, Padarathsingh M. Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am J Pathol. 2004;164:1131–1139. doi: 10.1016/S0002-9440(10)63200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhardt DT, Greenberg AH, Egan SE, Hamilton RT, Wright JA. Cysteine proteinase cathepsin L expression correlates closely with the metastatic potential of H-ras-transformed murine fibroblasts. Oncogene. 1987;2:55–59. [PubMed] [Google Scholar]
- Denhardt DT, Hamilton RT, Parfett CL, Edwards DR, St Pierre R, Waterhouse P, Nilsen-Hamilton M. Close relationship of the major excreted protein of transformed murine fibroblasts to thiol-dependent cathepsins. Cancer Res. 1986;46:4590–4593. [PubMed] [Google Scholar]
- Dennemarker J, Lohmuller T, Muller S, Aguilar SV, Tobin DJ, Peters C, Reinheckel T. Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol Chem. 2010;391:913–922. doi: 10.1515/BC.2010.097. [DOI] [PubMed] [Google Scholar]
- Devauchelle V, Marion S, Cagnard N, Mistou S, Falgarone G, Breban M, Letourneur F, Pitaval A, Alibert O, Lucchesi C, Anract P, Hamadouche M, Ayral X, Dougados M, Gidrol X, Fournier C, Chiocchia G. DNA microarray allows molecular profiling of rheumatoid arthritis and identification of pathophysiological targets. Genes Immun. 2004;5:597–608. doi: 10.1038/sj.gene.6364132. [DOI] [PubMed] [Google Scholar]
- Dohchin A, Suzuki JI, Seki H, Masutani M, Shiroto H, Kawakami Y. Immunostained cathepsins B and L correlate with depth of invasion and different metastatic pathways in early stage gastric carcinoma. Cancer. 2000;89:482–487. [PubMed] [Google Scholar]
- Doherty PJ, Hua L, Liau G, Gal S, Graham DE, Sobel M, Gottesman MM. Malignant transformation and tumor promoter treatment increase levels of a transcript for a secreted glycoprotein. Mol Cell Biol. 1985;5:466–473. doi: 10.1128/mcb.5.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong JM, Prence EM, Sahagian GG. Mechanism for selective secretion of a lysosomal protease by transformed mouse fibroblasts. J Biol Chem. 1989;264:7377–7383. [PubMed] [Google Scholar]
- Elie BT, Gocheva V, Shree T, Dalrymple SA, Holsinger LJ, Joyce JA. Identification and pre-clinical testing of a reversible cathepsin protease inhibitor reveals anti-tumor efficacy in a pancreatic cancer model. Biochimie. 2010;92:1618–1624. doi: 10.1016/j.biochi.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts V, Korper W, Hoeben KA, Jansen ID, Bromme D, Cleutjens KB, Heeneman S, Peters C, Reinheckel T, Saftig P, Beertsen W. Osteoclastic bone degradation and the role of different cysteine proteinases and matrix metalloproteinases: differences between calvaria and long bone. J Bone Miner Res. 2006;21:1399–1408. doi: 10.1359/jbmr.060614. [DOI] [PubMed] [Google Scholar]
- Farinati F, Herszenyi L, Plebani M, Carraro P, De Paoli M, Cardin R, Roveroni G, Rugge M, Nitti D, Grigioni WF, D'Errico A, Naccarato R. Increased levels of cathepsin B and L, urokinase-type plasminogen activator and its inhibitor type-1 as an early event in gastric carcinogenesis. Carcinogenesis. 1996;17:2581–2587. doi: 10.1093/carcin/17.12.2581. [DOI] [PubMed] [Google Scholar]
- Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A. 2002;99:7883–7888. doi: 10.1073/pnas.112632299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr Pharm Des. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006;25:115–136. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- Foekens JA, Kos J, Peters HA, Krasovec M, Look MP, Cimerman N, Meijer-van Gelder ME, Henzen-Logmans SC, van Putten WL, Klijn JG. Prognostic significance of cathepsins B and L in primary human breast cancer. J Clin Oncol. 1998;16:1013–1021. doi: 10.1200/JCO.1998.16.3.1013. [DOI] [PubMed] [Google Scholar]
- Fogelman I, Blake GM, Blamey R, Palmer M, Sauerbrei W, Schumacher M, Serin D, Stewart A, Wilpshaar W. Bone mineral density in premenopausal women treated for node-positive early breast cancer with 2 years of goserelin or 6 months of cyclophosphamide, methotrexate and 5-fluorouracil (CMF) Osteoporos Int. 2003;14:1001–1006. doi: 10.1007/s00198-003-1508-y. [DOI] [PubMed] [Google Scholar]
- Frade R, Rodrigues-Lima F, Huang S, Xie K, Guillaume N, Bar-Eli M. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998;58:2733–2736. [PubMed] [Google Scholar]
- Frade R, Rousselet N, Jean D. Intratumoral gene delivery of anti-cathepsin L single-chain variable fragment by lentiviral vector inhibits tumor progression induced by human melanoma cells. Cancer Gene Ther. 2008;15:591–604. doi: 10.1038/cgt.2008.51. [DOI] [PubMed] [Google Scholar]
- Frick KK, Doherty PJ, Gottesman MM, Scher CD. Regulation of the transcript for a lysosomal protein: evidence for a gene program modified by platelet-derived growth factor. Mol Cell Biol. 1985;5:2582–2589. doi: 10.1128/mcb.5.10.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich B, Jung K, Lein M, Turk I, Rudolph B, Hampel G, Schnorr D, Loening SA. Cathepsins B, H, L and cysteine protease inhibitors in malignant prostate cell lines, primary cultured prostatic cells and prostatic tissue. Eur J Cancer. 1999;35:138–144. doi: 10.1016/s0959-8049(98)00273-1. [DOI] [PubMed] [Google Scholar]
- Frohlich E, Schlagenhauff B, Mohrle M, Weber E, Klessen C, Rassner G. Activity, expression, and transcription rate of the cathepsins B, D, H, and L in cutaneous malignant melanoma. Cancer. 2001;91:972–982. [PubMed] [Google Scholar]
- Funkelstein L, Toneff T, Mosier C, Hwang SR, Beuschlein F, Lichtenauer UD, Reinheckel T, Peters C, Hook V. Major role of cathepsin L for producing the peptide hormones ACTH, beta-endorphin, and alpha-MSH, illustrated by protease gene knockout and expression. J Biol Chem. 2008;283:35652–35659. doi: 10.1074/jbc.M709010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama N, Fujisawa Y. Regulation of collagenolytic cysteine protease synthesis by estrogen in osteoclasts. Steroids. 2000a;65:371–378. doi: 10.1016/s0039-128x(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Furuyama N, Fujisawa Y. Regulation of collagenolytic protease secretion through c-Src in osteoclasts. Biochem Biophys Res Commun. 2000b;272:116–124. doi: 10.1006/bbrc.2000.2698. [DOI] [PubMed] [Google Scholar]
- Gal S, Gottesman MM. The major excreted protein of transformed fibroblasts is an activable acid-protease. J Biol Chem. 1986;261:1760–1765. [PubMed] [Google Scholar]
- Gal S, Gottesman MM. Isolation and sequence of a cDNA for human pro-(cathepsin L) Biochem J. 1988;253:303–306. doi: 10.1042/bj2530303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal S, Willingham MC, Gottesman MM. Processing and lysosomal localization of a glycoprotein whose secretion is transformation stimulated. J Cell Biol. 1985;100:535–544. doi: 10.1083/jcb.100.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo E, de Andres I, Illa I. Cathepsins are upregulated by IFN-gamma/STAT1 in human muscle culture: a possible active factor in dermatomyositis. J Neuropathol Exp Neurol. 2001;60:847–855. doi: 10.1093/jnen/60.9.847. [DOI] [PubMed] [Google Scholar]
- Gerber A, Wille A, Welte T, Ansorge S, Buhling F. Interleukin-6 and transforming growth factor-beta 1 control expression of cathepsins B and L in human lung epithelial cells. J Interferon Cytokine Res. 2001;21:11–19. doi: 10.1089/107999001459114. [DOI] [PubMed] [Google Scholar]
- Gianotti A, Sommer CA, Carmona AK, Henrique-Silva F. Inhibitory effect of the sugarcane cystatin CaneCPI-4 on cathepsins B and L and human breast cancer cell invasion. Biol Chem. 2008;389:447–453. doi: 10.1515/BC.2008.035. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle. 2007;6:60–64. doi: 10.4161/cc.6.1.3669. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, Joyce JA. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20:543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J. 2011;30:3383–3396. doi: 10.1038/emboj.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goretzki L, Schmitt M, Mann K, Calvete J, Chucholowski N, Kramer M, Gunzler WA, Janicke F, Graeff H. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992;297:112–118. doi: 10.1016/0014-5793(92)80339-i. [DOI] [PubMed] [Google Scholar]
- Goto T, Kiyoshima T, Moroi R, Tsukuba T, Nishimura Y, Himeno M, Yamamoto K, Tanaka T. Localization of cathepsins B, D, and L in the rat osteoclast by immuno-light and -electron microscopy. Histochemistry. 1994;101:33–40. doi: 10.1007/BF00315829. [DOI] [PubMed] [Google Scholar]
- Goto T, Tsukuba T, Kiyoshima T, Nishimura Y, Kato K, Yamamoto K, Tanaka T. Immunohistochemical localization of cathepsins B, D and L in the rat osteoclast. Histochemistry. 1993;99:411–414. doi: 10.1007/BF00717054. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Transformation-dependent secretion of a low molecular weight protein by murine fibroblasts. Proc Natl Acad Sci U S A. 1978;75:2767–2771. doi: 10.1073/pnas.75.6.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Sobel ME. Tumor promoters and Kirsten sarcoma virus increase synthesis of a secreted glycoprotein by regulating levels of translatable mRNA. Cell. 1980;19:449–455. doi: 10.1016/0092-8674(80)90519-x. [DOI] [PubMed] [Google Scholar]
- Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14:207–219. doi: 10.1016/s1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]
- Goulet B, Sansregret L, Leduy L, Bogyo M, Weber E, Chauhan SS, Nepveu A. Increased expression and activity of nuclear cathepsin L in cancer cells suggests a novel mechanism of cell transformation. Mol Cancer Res. 2007;5:899–907. doi: 10.1158/1541-7786.MCR-07-0160. [DOI] [PubMed] [Google Scholar]
- Goulet B, Truscott M, Nepveu A. A novel proteolytically processed CDP/Cux isoform of 90 kDa is generated by cathepsin L. Biol Chem. 2006;387:1285–1293. doi: 10.1515/BC.2006.159. [DOI] [PubMed] [Google Scholar]
- Grotsky DA, Gonzalez-Suarez I, Novell A, Neumann MA, Yaddanapudi SC, Croke M, Martinez-Alonso M, Redwood AB, Ortega-Martinez S, Feng Z, Lerma E, Ramon y Cajal T, Zhang J, Matias-Guiu X, Dusso A, Gonzalo S. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J Cell Biol. 2013;200:187–202. doi: 10.1083/jcb.201204053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hadji P, Aapro MS, Body JJ, Bundred NJ, Brufsky A, Coleman RE, Gnant M, Guise T, Lipton A. Management of aromatase inhibitor-associated bone loss in postmenopausal women with breast cancer: practical guidance for prevention and treatment. Ann Oncol. 2011;22:2546–2555. doi: 10.1093/annonc/mdr017. [DOI] [PubMed] [Google Scholar]
- Harbeck N, Alt U, Berger U, Kruger A, Thomssen C, Janicke F, Hofler H, Kates RE, Schmitt M. Prognostic impact of proteolytic factors (urokinase-type plasminogen activator, plasminogen activator inhibitor 1, and cathepsins B, D, and L) in primary breast cancer reflects effects of adjuvant systemic therapy. Clin Cancer Res. 2001;7:2757–2764. [PubMed] [Google Scholar]
- Herszenyi L, Plebani M, Carraro P, De Paoli M, Roveroni G, Cardin R, Tulassay Z, Naccarato R, Farinati F. The role of cysteine and serine proteases in colorectal carcinoma. Cancer. 1999;86:1135–1142. doi: 10.1002/(sici)1097-0142(19991001)86:7<1135::aid-cncr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Hiwasa T, Sakiyama S. Nuclear localization of procathepsin L/MEP in ras-transformed mouse fibroblasts. Cancer Lett. 1996;99:87–91. doi: 10.1016/0304-3835(95)04041-2. [DOI] [PubMed] [Google Scholar]
- Holroyd C, Cooper C, Dennison E. Epidemiology of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2008;22:671–685. doi: 10.1016/j.beem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Huang X, Vaag A, Carlsson E, Hansson M, Ahren B, Groop L. Impaired cathepsin L gene expression in skeletal muscle is associated with type 2 diabetes. Diabetes. 2003;52:2411–2418. doi: 10.2337/diabetes.52.9.2411. [DOI] [PubMed] [Google Scholar]
- Husmann K, Muff R, Bolander ME, Sarkar G, Born W, Fuchs B. Cathepsins and osteosarcoma: Expression analysis identifies cathepsin K as an indicator of metastasis. Mol Carcinog. 2008;47:66–73. doi: 10.1002/mc.20362. [DOI] [PubMed] [Google Scholar]
- Hwang SR, Garza C, Mosier C, Toneff T, Wunderlich E, Goldsmith P, Hook V. Cathepsin L expression is directed to secretory vesicles for enkephalin neuropeptide biosynthesis and secretion. J Biol Chem. 2007;282:9556–9563. doi: 10.1074/jbc.M605510200. [DOI] [PubMed] [Google Scholar]
- Inoue H, Nishimura K, Oka D, Nakai Y, Shiba M, Tokizane T, Arai Y, Nakayama M, Shimizu K, Takaha N, Nonomura N, Okuyama A. Prostate cancer mediates osteoclastogenesis through two different pathways. Cancer Lett. 2005;223:121–128. doi: 10.1016/j.canlet.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Ishidoh K, Kominami E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem Biophys Res Commun. 1995;217:624–631. doi: 10.1006/bbrc.1995.2820. [DOI] [PubMed] [Google Scholar]
- Jagodic M, Vrhovec I, Borstnar S, Cufer T. Prognostic and predictive value of cathepsins D and L in operable breast cancer patients. Neoplasma. 2005;52:1–9. [PubMed] [Google Scholar]
- Jain M, Bakhshi S, Shukla AA, Chauhan SS. Cathepsins B and L in peripheral blood mononuclear cells of pediatric acute myeloid leukemia: potential poor prognostic markers. Ann Hematol. 89:1223–1232. doi: 10.1007/s00277-010-1012-3. [DOI] [PubMed] [Google Scholar]
- Jain M, Bakhshi S, Shukla AA, Chauhan SS. Cathepsins B and L in peripheral blood mononuclear cells of pediatric acute myeloid leukemia: potential poor prognostic markers. Ann Hematol. 2010;89:1223–1232. doi: 10.1007/s00277-010-1012-3. [DOI] [PubMed] [Google Scholar]
- Jean D, Bar-Eli M, Huang S, Xie K, Rodrigues-Lima F, Hermann J, Frade R. A cysteine proteinase, which cleaves human C3, the third component of complement, is involved in tumorigenicity and metastasis of human melanoma. Cancer Res. 1996;56:254–258. [PubMed] [Google Scholar]
- Jean D, Guillaume N, Frade R. Characterization of human cathepsin L promoter and identification of binding sites for NF-Y, Sp1 and Sp3 that are essential for its activity. Biochem J. 2002;361:173–184. doi: 10.1042/0264-6021:3610173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean D, Hermann J, Rodrigues-Lima F, Barel M, Balbo M, Frade R. Identification on melanoma cells of p39, a cysteine proteinase that cleaves C3, the third component of complement: amino-acid-sequence identities with procathepsin L. Biochem J. 1995;312(Pt 3):961–969. doi: 10.1042/bj3120961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean D, Rousselet N, Frade R. Expression of cathepsin L in human tumor cells is under the control of distinct regulatory mechanisms. Oncogene. 2006;25:1474–1484. doi: 10.1038/sj.onc.1209196. [DOI] [PubMed] [Google Scholar]
- Jensen AB, Wynne C, Ramirez G, He W, Song Y, Berd Y, Wang H, Mehta A, Lombardi A. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: results of a 4-week, double-blind, randomized, controlled trial. Clin Breast Cancer. 10:452–458. doi: 10.3816/CBC.2010.n.059. [DOI] [PubMed] [Google Scholar]
- Jensen AB, Wynne C, Ramirez G, He W, Song Y, Berd Y, Wang H, Mehta A, Lombardi A. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: results of a 4-week, double-blind, randomized, controlled trial. Clin Breast Cancer. 2010;10:452–458. doi: 10.3816/CBC.2010.n.059. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageshita T, Yoshii A, Kimura T, Maruo K, Ono T, Himeno M, Nishimura Y. Biochemical and immunohistochemical analysis of cathepsins B, H, L and D in human melanocytic tumours. Arch Dermatol Res. 1995;287:266–272. doi: 10.1007/BF01105077. [DOI] [PubMed] [Google Scholar]
- Kakegawa H, Tagami K, Ohba Y, Sumitani K, Kawata T, Katunuma N. Secretion and processing mechanisms of procathepsin L in bone resorption. FEBS Lett. 1995;370:78–82. doi: 10.1016/0014-5793(95)00790-g. [DOI] [PubMed] [Google Scholar]
- Katunuma N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:29–39. doi: 10.2183/pjab.87.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katunuma N, Tsuge H, Nukatsuka M, Asao T, Fukushima M. Structure-based design of specific cathepsin inhibitors and their application to protection of bone metastases of cancer cells. Arch Biochem Biophys. 2002;397:305–311. doi: 10.1006/abbi.2001.2709. [DOI] [PubMed] [Google Scholar]
- Katunuma N, Tsuge H, Nukatsuka M, Fukushima M. Structure-based development of cathepsin L inhibitors and therapeutic applications for prevention of cancer metastasis and cancer-induced osteoporosis. Adv Enzyme Regul. 2002;42:159–172. doi: 10.1016/s0065-2571(01)00060-7. [DOI] [PubMed] [Google Scholar]
- Kedinger V, Sansregret L, Harada R, Vadnais C, Cadieux C, Fathers K, Park M, Nepveu A. p110 CUX1 homeodomain protein stimulates cell migration and invasion in part through a regulatory cascade culminating in the repression of E-cadherin and occludin. J Biol Chem. 2009;284:27701–27711. doi: 10.1074/jbc.M109.031849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthivasan S, Keerthivasan G, Mittal S, Chauhan SS. Transcriptional upregulation of human cathepsin L by VEGF in glioblastoma cells. Gene. 2007;399:129–136. doi: 10.1016/j.gene.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. 2004;91:718–729. doi: 10.1002/jcb.10662. [DOI] [PubMed] [Google Scholar]
- Khan MN, Khan AA. Cancer treatment-related bone loss: a review and synthesis of the literature. Curr Oncol. 2008;15:S30–40. doi: 10.3747/co.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschke H, Eerola R, Hopsu-Havu VK, Bromme D, Vuorio E. Antisense RNA inhibition of cathepsin L expression reduces tumorigenicity of malignant cells. Eur J Cancer. 2000;36:787–795. doi: 10.1016/s0959-8049(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Kirschke H, Kembhavi AA, Bohley P, Barrett AJ. Action of rat liver cathepsin L on collagen and other substrates. Biochem J. 1982;201:367–372. doi: 10.1042/bj2010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore Kumar GD, Chavarria GE, Charlton-Sevcik AK, Arispe WM, Macdonough MT, Strecker TE, Chen SE, Siim BG, Chaplin DJ, Trawick ML, Pinney KG. Design, synthesis, and biological evaluation of potent thiosemicarbazone based cathepsin L inhibitors. Bioorg Med Chem Lett. 2010;20:1415–1419. doi: 10.1016/j.bmcl.2009.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger S, Kellner U, Buehling F, Roessner A. Cathepsin L antisense oligonucleotides in a human osteosarcoma cell line: effects on the invasive phenotype. Cancer Gene Ther. 2001;8:522–528. doi: 10.1038/sj.cgt.7700341. [DOI] [PubMed] [Google Scholar]
- Kruger A, Kates RE, Edwards DR. Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. Biochim Biophys Acta. 2010;1803:95–102. doi: 10.1016/j.bbamcr.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Kumar GD, Chavarria GE, Charlton-Sevcik AK, Yoo GK, Song J, Strecker TE, Siim BG, Chaplin DJ, Trawick ML, Pinney KG. Functionalized benzophenone, thiophene, pyridine, and fluorene thiosemicarbazone derivatives as inhibitors of cathepsin L. Bioorg Med Chem Lett. 2010;20:6610–6615. doi: 10.1016/j.bmcl.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Lah TT, Calaf G, Kalman E, Shinde BG, Somers R, Estrada S, Salero E, Russo J, Daskal I. Cathepsins D, B, and L in transformed human breast epithelial cells. Breast Cancer Res Treat. 1996;39:221–233. doi: 10.1007/BF01806189. [DOI] [PubMed] [Google Scholar]
- Lah TT, Cercek M, Blejec A, Kos J, Gorodetsky E, Somers R, Daskal I. Cathepsin B, a prognostic indicator in lymph node-negative breast carcinoma patients: comparison with cathepsin D, cathepsin L, and other clinical indicators. Clin Cancer Res. 2000;6:578–584. [PubMed] [Google Scholar]
- Lah TT, Duran Alonso MB, Van Noorden CJ. Antiprotease therapy in cancer: hot or not? Expert Opin Biol Ther. 2006;6:257–279. doi: 10.1517/14712598.6.3.257. [DOI] [PubMed] [Google Scholar]
- Lah TT, Hawley M, Rock KL, Goldberg AL. Gamma-interferon causes a selective induction of the lysosomal proteases, cathepsins B and L, in macrophages. FEBS Lett. 1995;363:85–89. doi: 10.1016/0014-5793(95)00287-j. [DOI] [PubMed] [Google Scholar]
- Lah TT, Kalman E, Najjar D, Gorodetsky E, Brennan P, Somers R, Daskal I. Cells producing cathepsins D, B, and L in human breast carcinoma and their association with prognosis. Hum Pathol. 2000;31:149–160. doi: 10.1016/s0046-8177(00)80214-2. [DOI] [PubMed] [Google Scholar]
- Lah TT, Kos J, Blejec A, Frkovic-Georgio S, Golouh R, Vrhovec II, Turk VV. The Expression of Lysosomal Proteinases and Their Inhibitors in Breast Cancer: Possible Relationship to Prognosis of the Disease. Pathol Oncol Res. 1997;3:89–99. doi: 10.1007/BF02907801. [DOI] [PubMed] [Google Scholar]
- Lang TH, Willinger U, Holzer G. Soluble cathepsin-L: a marker of bone resorption and bone density? J Lab Clin Med. 2004;144:163–166. doi: 10.1016/j.lab.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Lankelma JM, Voorend DM, Barwari T, Koetsveld J, Van der Spek AH, De Porto AP, Van Rooijen G, Van Noorden CJ. Cathepsin L, target in cancer treatment? Life Sci. 2010;86:225–233. doi: 10.1016/j.lfs.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Laurent-Matha V, Derocq D, Prebois C, Katunuma N, Liaudet-Coopman E. Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem. 2006;139:363–371. doi: 10.1093/jb/mvj037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire R, Huet G, Zerimech F, Grard G, Fontaine C, Duquesnoy B, Flipo RM. Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells. Br J Rheumatol. 1997;36:735–743. doi: 10.1093/rheumatology/36.7.735. [DOI] [PubMed] [Google Scholar]
- Leto G, Sepporta MV, Crescimanno M, Flandina C, Tumminello FM. Cathepsin L in metastatic bone disease: therapeutic implications. Biol Chem. 2010;391:655–664. doi: 10.1515/BC.2010.069. [DOI] [PubMed] [Google Scholar]
- Leto G, Tumminello FM, Pizzolanti G, Montalto G, Soresi M, Carroccio A, Ippolito S, Gebbia N. Lysosomal aspartic and cysteine proteinases serum levels in patients with pancreatic cancer or pancreatitis. Pancreas. 1997;14:22–27. doi: 10.1097/00006676-199701000-00004. [DOI] [PubMed] [Google Scholar]
- Levicar N, Dewey RA, Daley E, Bates TE, Davies D, Kos J, Pilkington GJ, Lah TT. Selective suppression of cathepsin L by antisense cDNA impairs human brain tumor cell invasion in vitro and promotes apoptosis. Cancer Gene Ther. 2003;10:141–151. doi: 10.1038/sj.cgt.7700546. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- Lunt SJ, Chaudary N, Hill RP. The tumor microenvironment and metastatic disease. Clin Exp Metastasis. 2009;26:19–34. doi: 10.1007/s10585-008-9182-2. [DOI] [PubMed] [Google Scholar]
- Macabeo-Ong M, Shiboski CH, Silverman S, Ginzinger DG, Dekker N, Wong DT, Jordan RC. Quantitative analysis of cathepsin L mRNA and protein expression during oral cancer progression. Oral Oncol. 2003;39:638–647. doi: 10.1016/s1368-8375(03)00034-4. [DOI] [PubMed] [Google Scholar]
- Maciewicz RA, Wotton SF, Etherington DJ, Duance VC. Susceptibility of the cartilage collagens types II, IX and XI to degradation by the cysteine proteinases, cathepsins B and L. FEBS Lett. 1990;269:189–193. doi: 10.1016/0014-5793(90)81151-d. [DOI] [PubMed] [Google Scholar]
- Mao X, Hamoudi RA. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet Cytogenet. 2000;122:87–92. doi: 10.1016/s0165-4608(00)00278-8. [DOI] [PubMed] [Google Scholar]
- Mason RW, Gal S, Gottesman MM. The identification of the major excreted protein (MEP) from a transformed mouse fibroblast cell line as a catalytically active precursor form of cathepsin L. Biochem J. 1987;248:449–454. doi: 10.1042/bj2480449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RW, Johnson DA, Barrett AJ, Chapman HA. Elastinolytic activity of human cathepsin L. Biochem J. 1986;233:925–927. doi: 10.1042/bj2330925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21:228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mincey BA, Duh MS, Thomas SK, Moyneur E, Marynchencko M, Boyce SP, Mallett D, Perez EA. Risk of cancer treatment-associated bone loss and fractures among women with breast cancer receiving aromatase inhibitors. Clin Breast Cancer. 2006;7:127–132. doi: 10.3816/CBC.2006.n.021. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Iwadate M, Yanagisawa Y, Ito E, Imai J, Yamamoto M, Sawada N, Saito M, Suzuki S, Nakamura I, Ohki S, Saze Z, Kogure M, Gotoh M, Omicronbara K, Ohira H, Tasaki K, Abe M, Goshima N, Watanabe S, Waguri S, Takenoshita S. Cathepsin L is highly expressed in gastrointestinal stromal tumors. Int J Oncol. 2011;39:1109–1115. doi: 10.3892/ijo.2011.1127. [DOI] [PubMed] [Google Scholar]
- Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Moon NS, Rong Zeng W, Premdas P, Santaguida M, Berube G, Nepveu A. Expression of N-terminally truncated isoforms of CDP/CUX is increased in human uterine leiomyomas. Int J Cancer. 2002;100:429–432. doi: 10.1002/ijc.10510. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Nakase T, Kaneko M, Tomita T, Myoui A, Ariga K, Sugamoto K, Uchiyama Y, Ochi T, Yoshikawa H. Immunohistochemical detection of cathepsin D, K, and L in the process of endochondral ossification in the human. Histochem Cell Biol. 2000;114:21–27. doi: 10.1007/s004180000162. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Yasumatsu R, Masuda M, Clayman GL, Komune S. Prognostic value of cathepsin L and its inhibitor headpin in oral squamous cell carcinoma. J Laryngol Otol. 2012;126:1134–1137. doi: 10.1017/S0022215112001995. [DOI] [PubMed] [Google Scholar]
- Navab R, Mort JS, Brodt P. Inhibition of carcinoma cell invasion and liver metastases formation by the cysteine proteinase inhibitor E-64. Clin Exp Metastasis. 1997;15:121–129. doi: 10.1023/a:1018496625936. [DOI] [PubMed] [Google Scholar]
- Neville-Webbe HL, Coleman RE. Bisphosphonates and RANK ligand inhibitors for the treatment and prevention of metastatic bone disease. Eur J Cancer. 2010;46:1211–1222. doi: 10.1016/j.ejca.2010.02.041. [DOI] [PubMed] [Google Scholar]
- Niedergethmann M, Wostbrock B, Sturm JW, Willeke F, Post S, Hildenbrand R. Prognostic impact of cysteine proteases cathepsin B and cathepsin L in pancreatic adenocarcinoma. Pancreas. 2004;29:204–211. doi: 10.1097/00006676-200410000-00005. [DOI] [PubMed] [Google Scholar]
- Nilsen-Hamilton M, Hamilton RT, Allen WR, Massoglia SL. Stimulation of the release of two glycoproteins from mouse 3T3 cells by growth factors and by agents that increase intralysosomal pH. Biochem Biophys Res Commun. 1981;101:411–417. doi: 10.1016/0006-291x(81)91275-4. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Kohno K, Kawamata T, Morimitsu K, Kuwano M, Miyakawa I. Increased cathepsin L levels in serum in some patients with ovarian cancer: comparison with CA125 and CA72-4. Gynecol Oncol. 1995;56:357–361. doi: 10.1006/gyno.1995.1063. [DOI] [PubMed] [Google Scholar]
- Novinec M, Grass RN, Stark WJ, Turk V, Baici A, Lenarcic B. Interaction between human cathepsins K, L, and S and elastins: mechanism of elastinolysis and inhibition by macromolecular inhibitors. J Biol Chem. 2007;282:7893–7902. doi: 10.1074/jbc.M610107200. [DOI] [PubMed] [Google Scholar]
- Onishi T, Hayashi N, Theriault RL, Hortobagyi GN, Ueno NT. Future directions of bone-targeted therapy for metastatic breast cancer. Nat Rev Clin Oncol. 2010;7:641–651. doi: 10.1038/nrclinonc.2010.134. [DOI] [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29:22–28. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Park IC, Lee SY, Jeon DG, Lee JS, Hwang CS, Hwang BG, Lee SH, Hong WS, Hong SI. Enhanced expression of cathepsin L in metastatic bone tumors. J Korean Med Sci. 1996;11:144–148. doi: 10.3346/jkms.1996.11.2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulick MG, Bogyo M. Application of activity-based probes to the study of enzymes involved in cancer progression. Curr Opin Genet Dev. 2008;18:97–106. doi: 10.1016/j.gde.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann I, Mayer C, Stypmann J, Biniossek ML, Tobin DJ, Engelen MA, Dandekar T, Grune T, Schild L, Peters C, Reinheckel T. Lysosomal, cytoskeletal, and metabolic alterations in cardiomyopathy of cathepsin L knockout mice. FASEB J. 2006;20:1266–1268. doi: 10.1096/fj.05-5517fje. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Diel IJ. Osteoporosis due to cancer treatment: pathogenesis and management. J Clin Oncol. 2000;18:1570–1593. doi: 10.1200/JCO.2000.18.7.1570. [DOI] [PubMed] [Google Scholar]
- Potts W, Bowyer J, Jones H, Tucker D, Freemont AJ, Millest A, Martin C, Vernon W, Neerunjun D, Slynn G, Harper F, Maciewicz R. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int J Exp Pathol. 2004;85:85–96. doi: 10.1111/j.0959-9673.2004.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prence EM, Dong JM, Sahagian GG. Modulation of the transport of a lysosomal enzyme by PDGF. J Cell Biol. 1990;110:319–326. doi: 10.1083/jcb.110.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin MS, Doherty PJ, Gottesman MM. The tumor promoter phorbol 12-myristate 13-acetate induces a program of altered gene expression similar to that induced by platelet-derived growth factor and transforming oncogenes. Proc Natl Acad Sci U S A. 1986;83:357–360. doi: 10.1073/pnas.83.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachner TD, Hadji P, Hofbauer LC. Novel therapies in benign and malignant bone diseases. Pharmacol Ther. 2012;134:338–344. doi: 10.1016/j.pharmthera.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103–25108. doi: 10.1074/jbc.R109.041087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid IR. Glucocorticoid osteoporosis--mechanisms and management. Eur J Endocrinol. 1997;137:209–217. doi: 10.1530/eje.0.1370209. [DOI] [PubMed] [Google Scholar]
- Roato I, D'Amelio P, Gorassini E, Grimaldi A, Bonello L, Fiori C, Delsedime L, Tizzani A, De Libero A, Isaia G, Ferracini R. Osteoclasts are active in bone forming metastases of prostate cancer patients. PLoS One. 2008;3:e3627. doi: 10.1371/journal.pone.0003627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22:271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- Rozhin J, Wade RL, Honn KV, Sloane BF. Membrane-associated cathepsin L: a role in metastasis of melanomas. Biochem Biophys Res Commun. 1989;164:556–561. doi: 10.1016/0006-291x(89)91755-5. [DOI] [PubMed] [Google Scholar]
- Sadaghiani AM, Verhelst SH, Gocheva V, Hill K, Majerova E, Stinson S, Joyce JA, Bogyo M. Design, synthesis, and evaluation of in vivo potency and selectivity of epoxysuccinyl-based inhibitors of papain-family cysteine proteases. Chem Biol. 2007;14:499–511. doi: 10.1016/j.chembiol.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Saleh Y, Siewinski M, Kielan W, Ziolkowski P, Grybos M, Rybka J. Regulation of cathepsin B and L expression in vitro in gastric cancer tissues by egg cystatin. J Exp Ther Oncol. 2003;3:319–324. doi: 10.1111/j.1533-869x.2003.01105.x. [DOI] [PubMed] [Google Scholar]
- Samaiya M, Bakhshi S, Shukla AA, Kumar L, Chauhan SS. Epigenetic regulation of cathepsin L expression in chronic myeloid leukaemia. J Cell Mol Med. 2011;15:2189–2199. doi: 10.1111/j.1582-4934.2010.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedel J, Seemayer CA, Pap T, Neidhart M, Kuchen S, Michel BA, Gay RE, Muller-Ladner U, Gay S, Zacharias W. Targeting cathepsin L (CL) by specific ribozymes decreases CL protein synthesis and cartilage destruction in rheumatoid arthritis. Gene Ther. 2004;11:1040–1047. doi: 10.1038/sj.gt.3302265. [DOI] [PubMed] [Google Scholar]
- Scher CD, Dick RL, Whipple AP, Locatell KL. Identification of a BALB/c-3T3 cell protein modulated by platelet-derived growth factor. Mol Cell Biol. 1983;3:70–81. doi: 10.1128/mcb.3.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert G, Walenta S, Sundfor K, Rofstad EK, Mueller-Klieser W. Correlation of high lactate levels in human cervical cancer with incidence of metastasis. Cancer Res. 1995;55:4757–4759. [PubMed] [Google Scholar]
- Sheahan K, Shuja S, Murnane MJ. Cysteine protease activities and tumor development in human colorectal carcinoma. Cancer Res. 1989;49:3809–3814. [PubMed] [Google Scholar]
- Shridhar R, Zhang J, Song J, Booth BA, Kevil CG, Sotiropoulou G, Sloane BF, Keppler D. Cystatin M suppresses the malignant phenotype of human MDA-MB-435S cells. Oncogene. 2004;23:2206–2215. doi: 10.1038/sj.onc.1207340. [DOI] [PubMed] [Google Scholar]
- Siemann DW, H M. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacology and Therapeutics. 2015 doi: 10.1016/j.pharmthera.2015.06.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siewinski M, Saleh Y, Gryboc M, Murawski M, Ekonjo GB, Ziolkowski P, Janocha A, Symonowicz K. Determination of cysteine peptidases-like activity and their inhibitors in the serum of patients with ovarian cancer treated by conventional chemotherapy and vitamin E. J Exp Ther Oncol. 2004;4:189–193. [PubMed] [Google Scholar]
- Singh N, Das P, Datta Gupta S, Sahni P, Pandey RM, Gupta S, Chauhan SS, Saraya A. Prognostic significance of extracellular matrix degrading enzymes-cathepsin L and matrix metalloproteases-2 [MMP-2] in human pancreatic cancer. Cancer Invest. 31:461–471. doi: 10.3109/07357907.2013.820318. [DOI] [PubMed] [Google Scholar]
- Singh N, Das P, Datta Gupta S, Sahni P, Pandey RM, Gupta S, Chauhan SS, Saraya A. Prognostic significance of extracellular matrix degrading enzymes-cathepsin L and matrix metalloproteases-2 [MMP-2] in human pancreatic cancer. Cancer Invest. 2013;31:461–471. doi: 10.3109/07357907.2013.820318. [DOI] [PubMed] [Google Scholar]
- Sivaparvathi M, Yamamoto M, Nicolson GL, Gokaslan ZL, Fuller GN, Liotta LA, Sawaya R, Rao JS. Expression and immunohistochemical localization of cathepsin L during the progression of human gliomas. Clin Exp Metastasis. 1996;14:27–34. doi: 10.1007/BF00157683. [DOI] [PubMed] [Google Scholar]
- Skrzypczak M, Springwald A, Lattrich C, Haring J, Schuler S, Ortmann O, Treeck O. Expression of cysteine protease cathepsin L is increased in endometrial cancer and correlates with expression of growth regulatory genes. Cancer Invest. 2012;30:398–403. doi: 10.3109/07357907.2012.672608. [DOI] [PubMed] [Google Scholar]
- Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, Koller BH. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- Solau-Gervais E, Zerimech F, Lemaire R, Fontaine C, Huet G, Flipo RM. Cysteine and serine proteases of synovial tissue in rheumatoid arthritis and osteoarthritis. Scand J Rheumatol. 2007;36:373–377. doi: 10.1080/03009740701340172. [DOI] [PubMed] [Google Scholar]
- Song J, Jones LM, Chavarria GE, Charlton-Sevcik AK, Jantz A, Johansen A, Bayeh L, Soeung V, Snyder LK, Lade SD, Jr, Chaplin DJ, Trawick ML, Pinney KG. Small-molecule inhibitors of cathepsin L incorporating functionalized ring-fused molecular frameworks. Bioorg Med Chem Lett. 2013;23:2801–2807. doi: 10.1016/j.bmcl.2012.12.025. [DOI] [PubMed] [Google Scholar]
- Stearns NA, Dong JM, Pan JX, Brenner DA, Sahagian GG. Comparison of cathepsin L synthesized by normal and transformed cells at the gene, message, protein, and oligosaccharide levels. Arch Biochem Biophys. 1990;283:447–457. doi: 10.1016/0003-9861(90)90666-m. [DOI] [PubMed] [Google Scholar]
- Stypmann J, Glaser K, Roth W, Tobin DJ, Petermann I, Matthias R, Monnig G, Haverkamp W, Breithardt G, Schmahl W, Peters C, Reinheckel T. Dilated cardiomyopathy in mice deficient for the lysosomal cysteine peptidase cathepsin L. Proc Natl Acad Sci U S A. 2002;99:6234–6239. doi: 10.1073/pnas.092637699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhan DR, Siemann DW. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin Exp Metastasis. 2013;30:891–902. doi: 10.1007/s10585-013-9590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpizio S, Franceschini N, Piattelli A, Di Sebastiano P, Innocenti P, Selvaggi F. Cathepsins and pancreatic cancer:the 2012 update. Pancreatology. 12:395–401. doi: 10.1016/j.pan.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Sulpizio S, Franceschini N, Piattelli A, Di Sebastiano P, Innocenti P, Selvaggi F. Cathepsins and pancreatic cancer: the 2012 update. Pancreatology. 2012;12:395–401. doi: 10.1016/j.pan.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Svatek RS, Karam J, Karakiewicz PI, Gallina A, Casella R, Roehrborn CG, Shariat SF. Role of urinary cathepsin B and L in the detection of bladder urothelial cell carcinoma. J Urol. 2008;179:478–484. doi: 10.1016/j.juro.2007.09.037. discussion 484. [DOI] [PubMed] [Google Scholar]
- Tagami K, Kakegawa H, Kamioka H, Sumitani K, Kawata T, Lenarcic B, Turk V, Katunuma N. The mechanisms and regulation of procathepsin L secretion from osteoclasts in bone resorption. FEBS Lett. 1994;342:308–312. doi: 10.1016/0014-5793(94)80522-9. [DOI] [PubMed] [Google Scholar]
- Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins mediate tumor metastasis. World J Biol Chem. 2013;4:91–101. doi: 10.4331/wjbc.v4.i4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Cai J, Shen D, Bian Z, Yan L, Wang YX, Lan J, Zhuang GQ, Ma WZ, Wang W. Lysosomal cysteine peptidase cathepsin L protects against cardiac hypertrophy through blocking AKT/GSK3beta signaling. J Mol Med (Berl) 2009;87:249–260. doi: 10.1007/s00109-008-0423-2. [DOI] [PubMed] [Google Scholar]
- Thomssen C, Oppelt P, Janicke F, Ulm K, Harbeck N, Hofler H, Kuhn W, Graeff H, Schmitt M. Identification of low-risk node-negative breast cancer patients by tumor biological factors PAI-1 and cathepsin L. Anticancer Res. 1998;18:2173–2180. [PubMed] [Google Scholar]
- Thomssen C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P, Janicke F, Graeff H. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clin Cancer Res. 1995;1:741–746. [PubMed] [Google Scholar]
- Tomoo K. Development of cathepsin inhibitors and structure-based design of cathepsin B-specific inhibitor. Curr Top Med Chem. 2010;10:696–707. doi: 10.2174/156802610791113441. [DOI] [PubMed] [Google Scholar]
- Torkar A, Lenarcic B, Lah T, Dive V, Devel L. Identification of new peptide amides as selective cathepsin L inhibitors: the first step towards selective irreversible inhibitors? Bioorg Med Chem Lett. 2013;23:2968–2973. doi: 10.1016/j.bmcl.2013.03.041. [DOI] [PubMed] [Google Scholar]
- Troen BR, Gal S, Gottesman MM. Sequence and expression of the cDNA for MEP (major excreted protein), a transformation-regulated secreted cathepsin. Biochem J. 1987;246:731–735. doi: 10.1042/bj2460731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy AM, Sheahan K, Mulcahy HE, Duffy MJ, Hyland JM, O'Donoghue DP. Expression of Cathepsin B and L antigen and activity is associated with early colorectal cancer progression. Eur J Cancer. 2004;40:1610–1616. doi: 10.1016/j.ejca.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Tumminello FM, Leto G, Pizzolanti G, Candiloro V, Crescimanno M, Crosta L, Flandina C, Montalto G, Soresi M, Carroccio A, Bascone F, Ruggeri I, Ippolito S, Gebbia N. Cathepsin D, B and L circulating levels as prognostic markers of malignant progression. Anticancer Res. 1996;16:2315–2319. [PubMed] [Google Scholar]
- Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- Turk V, Brzin J, Kotnik M, Lenarcic B, Popovic T, Ritonja A, Trstenjak M, Begic-Odobasic L, Machleidt W. Human cysteine proteinases and their protein inhibitors stefins, cystatins and kininogens. Biomed Biochim Acta. 1986;45:1375–1384. [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWalde A, Hurria A. Aging and osteoporosis in breast and prostate cancer. CA Cancer J Clin. 2011;61:139–156. doi: 10.3322/caac.20103. [DOI] [PubMed] [Google Scholar]
- Verdoes M, Oresic Bender K, Segal E, van der Linden WA, Syed S, Withana NP, Sanman LE, Bogyo M. Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. J Am Chem Soc. 2013;135:14726–14730. doi: 10.1021/ja4056068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60:916–921. [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Higashi T, Hashimoto M, Tomoda I, Tominaga S, Hashimoto N, Morimoto S, Yamauchi Y, Nakatsukasa H, Kobayashi M, et al. Elevation of tissue cathepsin B and L activities in gastric cancer. Hepatogastroenterology. 1987;34:120–122. [PubMed] [Google Scholar]
- Watanabe M, Higashi T, Watanabe A, Osawa T, Sato Y, Kimura Y, Tominaga S, Hashimoto N, Yoshida Y, Morimoto S, et al. Cathepsin B and L activities in gastric cancer tissue: correlation with histological findings. Biochem Med Metab Biol. 1989;42:21–29. doi: 10.1016/0885-4505(89)90037-6. [DOI] [PubMed] [Google Scholar]
- Weber E, Gunther D, Laube F, Wiederanders B, Kirschke H. Hybridoma cells producing antibodies to cathepsin L have greatly reduced potential for tumour growth. J Cancer Res Clin Oncol. 1994;120:564–567. doi: 10.1007/BF01221037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yuan G, Liu W, Zhang Y, Chen W. Expression of cathepsin L in nasopharyngeal carcinoma and its clinical significance. Exp Oncol. 2009;31:102–105. [PubMed] [Google Scholar]
- Yamaguchi T, Naruishi K, Arai H, Nishimura F, Takashiba S. IL-6/sIL-6R enhances cathepsin B and L production via caveolin-1-mediated JNK-AP-1 pathway in human gingival fibroblasts. J Cell Physiol. 2008;217:423–432. doi: 10.1002/jcp.21517. [DOI] [PubMed] [Google Scholar]
- Yan JA, Xiao H, Ji HX, Shen WH, Zhou ZS, Song B, Chen ZW, Li WB. Cathepsin L is associated with proliferation and clinical outcome of urothelial carcinoma of the bladder. J Int Med Res. 2010;38:1913–1922. doi: 10.1177/147323001003800604. [DOI] [PubMed] [Google Scholar]
- Yang Z, Cox JL. Cathepsin L increases invasion and migration of B16 melanoma. Cancer Cell Int. 2007;7:8. doi: 10.1186/1475-2867-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasothornsrikul S, Greenbaum D, Medzihradszky KF, Toneff T, Bundey R, Miller R, Schilling B, Petermann I, Dehnert J, Logvinova A, Goldsmith P, Neveu JM, Lane WS, Gibson B, Reinheckel T, Peters C, Bogyo M, Hook V. Cathepsin L in secretory vesicles functions as a prohormone-processing enzyme for production of the enkephalin peptide neurotransmitter. Proc Natl Acad Sci U S A. 2003;100:9590–9595. doi: 10.1073/pnas.1531542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajc I, Sever N, Bervar A, Lah TT. Expression of cysteine peptidase cathepsin L and its inhibitors stefins A and B in relation to tumorigenicity of breast cancer cell lines. Cancer Lett. 2002;187:185–190. doi: 10.1016/s0304-3835(02)00452-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dai J, Yao Z, Lu Y, Dougall W, Keller ET. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res. 2003;63:7883–7890. [PubMed] [Google Scholar]
- Zhang W, Yang HC, Wang Q, Yang ZJ, Chen H, Wang SM, Pan ZM, Tang BJ, Li QQ, Li L. Clinical value of combined detection of serum matrix metalloproteinase-9, heparanase, and cathepsin for determining ovarian cancer invasion and metastasis. Anticancer Res. 2011;31:3423–3428. [PubMed] [Google Scholar]
- Zheng X, Chou PM, Mirkin BL, Rebbaa A. Senescence-initiated reversal of drug resistance: specific role of cathepsin L. Cancer Res. 2004;64:1773–1780. doi: 10.1158/0008-5472.can-03-0820. [DOI] [PubMed] [Google Scholar]
- Zheng X, Chu F, Chou PM, Gallati C, Dier U, Mirkin BL, Mousa SA, Rebbaa A. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: a putative mechanism. Am J Physiol Cell Physiol. 2009;296:C65–74. doi: 10.1152/ajpcell.00082.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker S, Cao J. Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: ready for prime time? Cancer Biol Ther. 2009;8:2371–2373. doi: 10.4161/cbt.8.24.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]