

Abstract
Influenza patients frequently display increased susceptibility to Streptococcus pneumoniae co-infection and sepsis, the prevalent cause of mortality during influenza pandemics, but the detailed mechanisms by which an influenza infection predisposes patients to suffer pneumococcal pneumonia is not fully understood. A murine model for influenza infection closely reflects the observations in human patients, since if the animals that have recovered from influenza A virus (IAV) sublethal infection are challenged with S. pneumoniae, they undergo a usually fatal uncontrolled cytokine response. We have previously demonstrated both in vitro and in vivo that the expression and secretion of galectin-1 (Gal1) and galectin-3 (Gal3) are modulated during IAV infection, and that the viral neuraminidase unmasks galactosyl moieties in the airway epithelia. In this study we demonstrate in vitro that the binding of secreted Gal1 and Gal3 to the epithelial cell surface modulates the expression of SOCS1 and RIG1, and activation of ERK, AKT or JAK/STAT1 signaling pathways, leading to a disregulated expression and release of pro-inflammatory cytokines. Our results suggest that the activity of the viral and pneumococcal neuraminidases on the surface of the airway epithelial cells function as a “danger signal” that leads to rapid upregulation of SOCS1 expression to prevent an uncontrolled inflammatory response. The binding of extracellular Gal1 or Gal3 to the galactosyl moieties unmasked on the surface of airway epithelial cells can either “fine-tune” or severely disregulate this process, respectively, the latter potentially leading to hypercytokinemia.
Keywords: galectin, neuraminidase, SOCS, cytokines, chemokines, RIG1, signaling pathway
INTRODUCTION
Influenza patients frequently display increased susceptibility to bacterial infections, such as streptococcal pneumonia. Influenza co-infection with one of the prevalent pathogens, Streptococcus pneumoniae can lead to sepsis, which is the major cause of mortality during influenza pandemics. The synergic effect of the influenza A virus (IAV) and S. pneumoniae has been responsible for nearly all influenza deaths in 1918 influenza pandemic and up to 34% of 2009 pandemic influenza A (Chertow and Memoli 2013). The detailed mechanisms by which an influenza infection predisposes patients to suffer pneumococcal pneumonia and progress to uncontrolled hypercytokinemia, however, are not fully understood. In a murine model, the animals that have recovered from IAV sublethal infection undergo a usually fatal uncontrolled cytokine response after challenged with S. pneumonia, which closely reflects the observations in human patients (Alonso et al. 2003; Seki et al. 2004; Speshock et al. 2007; Bermejo-Martin et al. 2010; Yang et al. 2010; Monsalvo et al. 2011; Zuniga et al. 2011; Damjanovic et al. 2013). The reasons are unknown, but upon IAV infection, an antiviral response is initiated by the retinoic acid inducible gene 1 (RIG1), a helicase of the RIG-1 like receptor (RLR) family, which recognizes the cytosolic viral RNA and signals via the interferon-alpha/beta receptor 1 (IFNAR1)-dependent pathway to induce the expression and release of interferons and other cytokines (Poltorak et al. 1998; Takeuchi and Akira 2008). In turn, the potentially excessive production of cytokines and activation of both innate and adaptive immune mechanisms is controlled by the upregulation of expression of suppressors of cytokine signaling (SOCS) particularly SOCS1, SOCS3, and SOCS4 (Pothlichet et al. 2008; Ramirez-Martinez et al. 2013; Kedzierski et al. 2014). SOCS are a multigene family of intracellular proteins, which have been shown to regulate the responses of immune cells to the cytokines by negative regulation of the cytokine-induced JAK-STAT pathway. Most SOCS proteins are induced by cytokines, and therefore participate in a classical negative feedback loop to inhibit cytokine signal transduction (Ilangumaran et al. 2004; Fletcher and Starr 2005; Naka et al. 2005; Davey et al. 2006). There are eight members of the SOCS family (SOCS1-8), each of which has a central SRC homology 2 (SH2) domain, an amino-terminal domain of variable length and a divergent sequence, and a carboxy-terminal 40-amino-acid domain that it is known as the SOCS box (Yoshimura et al. 2007). SOCS1 protein levels, directly induced by viable microbes, are important for host defense by inhibiting both type I and type II interferon (IFN) signaling (Zimmermann et al. 2006).
The cell surface glycocalyx, comprising glycoproteins, glycolipids and other glycans, plays a major role in modulation of host-pathogen interactions by encoding key information necessary for microbial adhesion and entry as well as for the induction of the ensuing immune responses by the host. The structural changes in cell surface glycans resulting from the enzymatic activity (glycosidases and glycosyltransferases) from both host and pathogen, as well as the reciprocal recognition of these carbohydrate moieties by their glycan-binding proteins, represent a dynamic interplay that can define the outcome of the host-pathogen interaction. For example, the glycoproteins hemagglutinin (HA) and neuraminidase (NA) displayed on the IAV surface play critical roles in viral adhesion and infection, as well as the release of the newly assembled virions (Fukuyama and Kawaoka 2011; Luo 2012). HA is a major viral surface glycoprotein, which initiates the infection by binding to sialic acid (SA) moieties present on the cell surface. Once the virus is internalized by endocytosis the HA is cleaved into HA1 and HA2 subunits. When the replication process is terminated, the NA facilitates the release of progeny virions that infect neighboring cells (Fukuyama and Kawaoka 2011; Luo 2012). Moreover, viral neuraminidase contributes to the increase of bacterial adhesion and dissemination by cleaving the respiratory epithelial cell sialic acids (Feng et al. 2013; Nita-Lazar et al. 2015).
Among the host glycan-binding proteins, the critical roles of C-type lectins in pathogen recognition have been described in considerable detail, as well as the ensuing effector functions such as complement activation (Vasta and Ahmed 2008). In recent years, however, evidence has accumulated in support of key roles of galectins in defining the outcome of microbial infection (Vasta 2009). Galectins are a family of soluble β-galactoside-binding proteins that are synthesized in the cytosol and may carry out their biological roles in the nuclear compartment, cytoplasm, cell surface, and extracellular space. They are classified into three major structural types: proto-, chimera-, and tandem-repeat. Galectins are expressed and secreted to the extracellular space by various cell types, including epithelial cells, neutrophils, macrophages, dendritic cells, B and T cells. Further, a substantial body of evidence supports their critical roles both in the regulation of immune homeostasis as well as pattern recognition receptors in the recognition of glycans on the viral or bacterial surfaces. Recent evidence suggests their roles in the lung innate immune response to pneumococcal infection (Sato et al. 2002), although the mechanisms remain largely unknown.
We have previously shown both in vitro and in vivo that the expression and secretion of galectins, particularly galectin-1 (Gal1) and galectin-3 (Gal3), are modulated during IAV infection, and that the viral neuraminidase unmasks galactosy moieties in the airway epithelia (Nita-Lazar et al. 2015). The increased levels of galectins remaining in the bronchoalveolar space upon recovery from influenza further contribute to the increased binding of Gal3 and enhancement of pneumococcal adhesion by cross-linking the bacteria to the cell surface (Nita-Lazar et al. 2015). Thus, we proposed that the activity of the influenza and pneumococcal neuraminidases, which unmask galactosyl moieties on the airway epithelial cell surface, and act synergistically with the increased Gal3 and Gal1 concentrations in the bronchoalveolar space to promote galectin binding and additional pneumococcal adhesion as shown in our previous study, could also regulate the cytokine/chemokine expression and release by the airway epithelial cells and lead to the typical cytokine storm. In this study, we demonstrate in vitro that the combined activity of microbial neuraminidases and the secreted Gal1 and Gal3 at the epithelial cell surface modulate the expression of SOCS1 and RIG1 and activation of ERK, AKT or JAK/STAT1 signaling pathways, leading to a disregulated expression and release of pro-inflammatory cytokines.
MATERIALS AND METHODS
Reagents
Phenylmethanesulfonyl fluoride (PMSF), trypsin, and bacterial neuraminidases α (2⇒3,6,8,9) Arthrobacter ureafaciens, Clostridium perfringens were purchased from Sigma (St. Louis, MO), or QA-bio (Palm Desert, CA), respectively. Antibodies against NF-κB p65 and phospho-NF-κB p65 (Ser 311) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against integrin β3, phospho-integrin β3, ERK, phospho-ERK, AKT, phospho-AKT, p38 MAPK, phospho-p38 MAPK, STAT1 and phospho-STAT1 were purchased from Sigma (Saint Louis, MO). Protease inhibitor cocktail set I, β-mercaptoethanol, and 100x penicillin/streptomycin were obtained from Calbiochem (La Jolla, CA). Dialysis tubing (MW:6-8000) was purchased from Spectrum Laboratories (Rancho Dominguez, CA). TRIzol Reagent was obtained from Invitrogen (Camarillo, CA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Cellgro (Manassas, VA). Prestained broad range protein marker was obtained from Cell Signaling Technology (Danvers, MA). PD Mini Trap G10 was purchased from GE Healthcare (Pittsburgh, PA). Mini Protean TGX precast gels, phosphate buffered saline (PBS), resolving buffer, and stacking buffer were all purchased from Bio-Rad (Hercules, CA). One minute western blot stripping buffer was obtained from GM Biosciences (Frederick, MD). Polyvinylidene fluoride (PVDF) membrane was purchased from Millipore Thermo Scientific (Rockford, IL). Western Lightning Plus-ECL was purchased from PerkinElmer Inc (Waltham, MA). Molecular biology grade agarose was purchased from Denville Scientific Inc (Metuchen, NJ). Dream Taq PCR master mix (2X) and Revertaid first strand cDNA synthesis kit was purchased from Thermo Scientific (Pittsburgh, PA), Fetal bovine serum (FBS) was obtained from Quality Biological Inc (Gaithersburg, MD). Oligonucleotide primers for RT-PCR were synthesized by Sigma-Aldrich, Human proinflamatory 9-Plex ultra-sensitive kit and human IL-15 ultra-sensitive kit were purchased from Meso Scale Discovery (Gaithersburg, MD).
Expression and purification of recombinant human galectin-1 (rhGal1) and human galectin-3 (rhGal3)
Expression of rhGal1 and rhGal3 were performed using the pT7 (ML-1) and pET30 Ek/Lic vectors in the Escherichia coli BL21 (DE3) (Novagen; Billerica, MA) cells and induced by 0.1 mM isopropyl D-thiogalactoside (Sigma-Alderich) at 23 °C for 16 h in 3 liters of LB medium containing 100 μg/ml amphicilin and 30 μg/ml kanamycin. The soluble proteins extracted with Bugbuster (Novagen) containing 1mM PMSF and 0.07% β-mercaptoethanol (2-ME), contained most of the recombinant proteins: rhGal1 and rhGal3 (approximately 80%). These fractions were loaded onto a column packed with 4 ml of lactose-Sepharose. After washing the column thoroughly with 0.07% 2-ME in 1:10 PBS [PBS (1:10)/2-ME] for rhGal1 and 0.07% 2-ME in PBS (1X) for rhGal3, the rhGal1 was eluted with 0.1M lactose in PBS (1:10)/2-ME and rhGal3 with 0.1M lactose in PBS (1X). From a 3-liter E.coli culture, approximately 17 mg of rhGal1 and 30 mg of rhGal3 were purified. Carbamidomethylation of rhGal1 was performed as reported earlier (Feng et al. 2013). Purified rhGal1 (17 mg) was absorbed on a 1 ml of DEAE-Sepharose pre-equilibrated with PBS (1:10)/2-ME, and incubated for 1 h at 4 °C with slow agitation. The resin was poured into a column and after extensive washing with PBS (1:10), the column was overlaid with 3 ml of 0.1M iododacetamide/0.1 M lactose and incubated for 1h at 4 °C in the dark. After washing the column with 50 mM lactose in PBS (1:10), the bound protein (crhGal1) was eluted with PBS (1:10)/0.5 M NaCl/0.1M lactose.
Airway epithelial cell primary cultures and cell line
A549 cells (human alveolar type II epithelial cell line derived from a lung adenocarcinoma; ATCC: CCL-185, Manassas, VA) were cultured in DMEM containing 10% FBS suplemented with 50 units/ml penicillin and 50 μg/ml streptomycin as described (Lillehoj et al. 2012).
RT-PCR
Total RNA from cultured A549 cells was extracted with TRIzol Reagent. RNA was quantified in a Nanodrop Bioanalyzer at 260/280 nm. Complementary DNA (cDNA) was synthesized using Revertaid first cDNA synthesis kit from 1μg of total RNA according to the manufacturer’s instructions. cDNAs were amplified using Dream Taq PCR Master Mix (2X) and the following primers: human SOCS1, forward, 5′-TTTTCGCCCTTAGCGTGAA-3′; reverse, 5′-GCGGCGCGGCGCCGCCACG-3′; human RIG1, forward, 5′-ACCAGACCTCCTCTTGGC-3′ ; reverse, 5′-GAAGGGGCAGATGGCTGT-3′; human β-actin, forward, 5′-CCGCGCTCGTCGTCGACAAC-3′; reverse, 5′-GCTCTGGGCCTCGTCGCCC-3′. The PCR products were fractionated on 1% agarose gels and visualized by ethidium bromide staining.
Real-time PCR
Total RNA from cultured A549 cells was extracted and cDNA was synthesized as described above. cDNA transcribed from 10 ng of total RNA was amplified at 7500 Fast Real-Time PCR System (Applied Biosystems) using Fast SYBR Green Master Mix according to the manufacture’s recommendation and the following primers: human SOCS1, forward, 5′-GACGCCTGCGGATTCTACTG -3′; reverse, 5′-CACGCTAAGGGCGAAAAAGC -3′; human RPS13 5′-CGAAAGCATCTTGAGAGGAACA -3′; reverse, 5′-TCGAGCCAAACGGTGAATC -3′. The relative gene expression of SOCS1 was calculated using the ΔCt method, and normalized to the reference gene, PRS13, as an internal control (Cross et al. 2012).
Enzyme treatments
Cultured A549 cells were subject to neuraminidase treatment (300 mU of neuraminidase per 1 × 106 cells in 200 μl) at 37 °C for 1 h in serum-free DMEM. After incubation, the cells were washed three times in PBS and resuspended in DMEM without FBS for detection of galectins and cytokines.
Western Blot
Cells were lysed with ice cold 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1%(v/v) Triton X, protease inhibitor, 0.1mM PMSF. The cell lysates were assayed for protein concentration at 280 nm using Nanodrop Bioanalyzer. Equal amounts of protein were resolved by electrophoresis on commercial gradient SDS-polyacrylamide gels and transferred to PVDF membrane. The membranes were blocked in 5% nonfat milk and then probed with primary antibodies at 1:1000 dilutions. Next, membranes were washed in PBS-T followed by incubation with HRP-linked secondary antibody (1:3000) and the results were visualized with Western Lightning Plus-ECL reagents.
Expression of SOCS1 and RIG1, and secretion of cytokines in A549 cells exposed to neuraminidase and galectins
A549 cells grown on 6 well plates were treated with NeuK (combination of A. ureafaciens and C. perfringens neuraminidases, 300mU per 1 × 106 cells in 200 μl) at 37 °C for 1 h. After the treatment, cells were washed three times with PBS then incubated in FBS-free DMEM with rhGal1 or rhGal3 (15 μg/ml, final concentration) for 1 h. Cells were collected and total RNA obtained was used for detection of SOCS1 and RIG1 transcripts. The culture medium was collected and added to plated pre-coated with cytokine-specific antibodies for assessment of cytokine levels using an MSD human proinflamatory 9-Plex Assay Ultra-Sensitive Kit to measure IFN-γ, TNF-α, IL-1β, IL-6, IL-8, IL-10 and IL-12, and MSD human IL-15 Assay Ultra-Sensitive Kit to determine levels of IL-15, following MSD manufacturer’s instructions.
Statistical analyses
The protein or RT-PCR amplicon bands were quantified using Image J software. Comparison of two groups was performed by Student’s t-test for the comparison of non-paired samples. All results with p<0.05 were considered statistically significant.
RESULTS AND DISCUSSION
We previously reported that the expression and secretion of galectins, particularly galectin-1 (Gal1) and galectin-3 (Gal3), are modulated during IAV infection, and that their levels in the broncholalveolar space remain high upon recovery from influenza (Nita-Lazar et al. 2015). This observation led us to test in an in vitro system whether microbial neuraminidases and galectins could contribute to the dysregulation of the lung immune response with the ensuing cytokine storm. In a prior study (Nita-Lazar et al. 2015) we comparatively examined in primary small airway epithelial cells (SAEC) and the airway epithelial cell line A549 cells the expression of galectins and their secretion to the extracellular environment (Nita-Lazar et al. 2015). Moreover, the viral neuraminidase unmasks galactosyl moieties in the airway epithelia, which promotes binding of the abovementioned galectins and contributes to enhance pneumococcal adhesion by cross-linking the bacteria to the cell surface. Like in the lung tissues, both the SAEC and the A549 cells express galectins 1 and 3, and secrete them to the extracellular space. Thus, we considered it a suitable in vitro model to further characterize the potential role(s) of microbial sialidases and host galectins in the regulation of the immune response to influenza and a subsequent pneumococcal infection, with a particular focus on their effects on the expression of SOCS and cytokine expression and secretion profiles.
Expression of SOCS and RIG1 in epithelial lung carcinoma cells (A549) exposed to microbial neuraminidase and galectins
First, we examined by real-time PCR the potential variability of SOCS1 expression in A549 cells during the culture period for which the experiments were designed. Our results revealed that SOCS1 expression levels in A549 cells remained constant over the 4-day growth period, under standard culture conditions (data not shown). Next, we tested the potential effects of microbial neuraminidase treatment and exposure to extracellular Gal1 and Gal3 on the expression of SOCS1 and RIG1. In a previous study (Nita-Lazar et al, 2015), we had validated the use of a combination of A. ureafaciens and C. perfringens neuraminidases (NeuK) as an enzymatic treatment that would reflect the activities of the influenza and pneumococcal neuraminidases on the airway epithelial surface. Activity of the exogenous NeuK on the A549 cells increased SOCS1 expression levels in about 2-fold (Fig 1A), which is consistent with the upregulation of SOCS1 expression observed upon in vivo influenza infection (Pothlichet et al. 2008), Exposure of untreated cells to rhGal1, significantly decreased SOCS1 expression in 20–30%, but when the cells had been pre-treated with NeuK, the SOCS1 expression returned to the control levels. Exposure of the untreated or NeuK-treated A549 cells to rhGal3, however, downregulated SOCS1 expression in about 60–70% of the control levels.
Fig 1. Exposure of lung carcinoma A549 cells to microbial neuraminidase and extracellular Gal1 and Gal3 modulates expression of SOCS1 and RIG1.
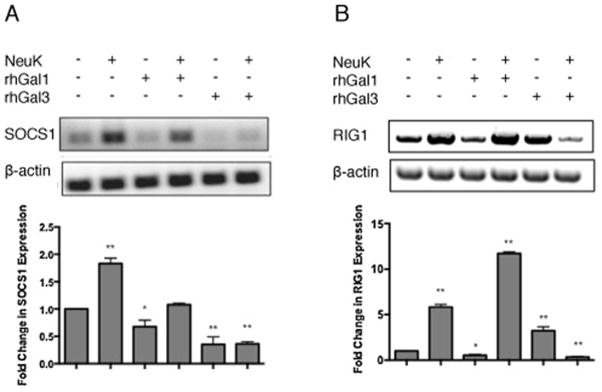
Total RNA was extracted from A549 control (Ctrl) or neuraminidase (Arthrobacter ureafaciens and Clostridium perfringens) treated cells (NeuK) incubated in presence or absence of 15 μg/ml exogenous rhGal1 or rhGal3 for 1 h. (A) SOCS1 transcript level was analyzed by RT-PCR. (B) RIG1 transcript levels were analyzed by RT-PCR. Bar graphs show the fold change in mRNA expression levels in neuraminidase treated cells as well as galectin treated cells in comparison with control cell without neuraminidase and galectin treatment (Ctrl) after normalized to β-actin. All studies represent one of three independent experiments. *p <0.05; **p<0.001, non paired Student’s t test.
Exposure of the airway epithelial cells to microbial neuraminidase and galectins had opposite effects on SOCS1 expression. While the microbial neuraminidase significantly increased SOCS1 expression levels (consistent with the increased pro-inflammatory cytokine expression), the subsequent exposure to Gal1 compensated this effect, retracting SOCS1 expression to the control levels, suggesting that Gal1 would have a homeostatic effect that would enable a controlled inflammatory response aimed a clearing the potential threat. The regulatory roles of Gal1 in immune homeostasis have been well established (Camby et al. 2006; Rabinovich and Ilarregui 2009). Gal1 displays anti-inflammatory activity (Rabinovich et al. 1999; Santucci et al. 2003; Toscano et al. 2006), mediates apoptosis of activated T-cells in peripheral tissues (Koh et al. 2008), and exerts antiproliferative effects on epithelial tumor cell lines by inhibition of the Ras-MEK-ERK pathway (Fischer et al. 2005). The anti-inflammatory activity of Gal1 takes place through the inhibition of IκB-α degradation that is induced by pro-inflammatory stimuli, the increase of cytoplasmic retention of p65, and a decrease of DNA binding activity of NF-κB, that attenuates NF-κB activation and impairs the transcriptional activation of immune genes (Toscano et al. 2011). Our results indicate that at the same time, Gal1 downregulates SOCS1 expression. In both the unexposed and neuraminidase-exposed cells, the extracellular Gal3 also had an effect opposite to the upregulatory effect of neuraminidase on SOCS1 expression, since it significantly decreased the SOCS1 transcript levels. Like we observed for Gal1, downregulation of SOCS1 expression by Gal3 would enable the robust cytokine response directed to induce the effective anti-microbial response. This is consistent with the protective role of Gal3 reported for pneumococcal infection in a murine model (Sato et al. 2002). In experimental pneumococcal infections in the mouse, Gal3 functions as a soluble adhesion molecule to mediate the integrin-independent recruitment of neutrophils to S. pneumonia-infected lungs (Sato et al. 2002). Furthermore, in contrast with the anti-inflammatory activity of Gal1, Gal3 displays pro-inflammatory activity. For example, Gal3 can directly activate neutrophils, augment neutrophil phagocytosis of bacteria, delay neutrophil apoptosis, facilitate phagocytosis of apoptotic neutrophils by macrophages, and exert bacteriostatic in vitro activity against S. pneumoniae (Farnworth et al. 2008). Our results revealed that upon a prior influenza infection, the high levels of Gal1 and Gal3 remaining in the bronchoalveolar space during the recovery period are further increased immediately after a pneumococcal challenge (Nita-Lazar et al. 2015). Thus, the synergistic effect of increased Gal3 levels in the extracellular space together with the availability of additional galactosyl moieties on the epithelial cell surface resulting from the influenza neuraminidase, would downregulate SOCS1 expression far below the unexposed control levels, leading to an uncontrolled hypercytokinemia (Pothlichet et al. 2008). SOCS1, a JAK binding protein, belongs to the SOCS family that is responsible for key negative feedback mechanisms to prevent uncontrolled inflammation (Croker et al. 2008). Upon IAV challenge, SOCS1 expression is upregulated through a RIG1/MAVS/IFNAR1-dependent pathway (Pothlichet et al. 2008), which suppresses expression of inflammatory cytokines, e.g. IL-6, TNF-α, IL-10, CCL3, CCL5, CCL4 and CXCL8, to prevent the cytokine storm (Ramirez-Martinez et al. 2013). Thus, upregulation of SOCS1 provides a negative feedback mechanism to set inflammation under control after pathogen clearance. In this context, the galectin-mediated downregulation of SOCS1 expression would upset this balance, leading to a cytokine storm, especially on tissues exposed to the viral neuraminidases. It is possible that exposure of the epithelial cell surface to the pneumococcal neuraminidase would synergistically enhance this effect upon the pneumococcal challenge (Nita-Lazar et al. 2015).
Expression of RIG1 was significantly increased by NeuK treatment alone (6-fold) whereas exposure to rhGal1 alone decreased RIG1 expression to about 50% of the unexposed control (Fig 1B). Exposure of NeuK-pretreated A549 cells to rhGal1 increased RIG1 expression in about 12-fold. The effects of rhGal3 on RIG1 expression were opposite to rhGal1: exposure to rhGal3 alone upregulated RIG1 expression in about 4-fold, whereas the NeuK treatment followed by exposure to rhGal3 downregulated RIG1 expression to about 20% of the unexposed controls. RIG1 is a member of the RIG-I-like receptor family, and functions as a pattern recognition receptor (PRR) and sensor for viruses such as influenza A, Sendai virus, and flavivirus (Yoneyama and Fujita 2009; Yoo et al. 2014). In addition to all-trans retinoic acid (ATRA), LPS (TLR4-mediated), type I interferon, and other agonists up-regulate RIG1 expression (Matsumiya and Stafforini 2010). RIG1 activates two distinct downstream TRAF-mediated signaling pathways: the IRF3/7 and NFkB pathways (Matsumiya and Stafforini 2010). Together with SOCS1, upregulation of mRNA levels of RIG1 in IAV infection suppresses expression of inflammatory cytokines, and thus prevents hypercytokinemia (Ramirez-Martinez et al. 2013). Furthermore, upon IAV infection the upregulation of RIG1 induces upregulation of SOCS1 expression (Pothlichet et al. 2008), and our results on the effects of neuraminidases and galectins on epithelial cells are consistent with the association of RIG1 and SOCS1 expression.
The exposure of epithelial cells to extracellular Gal1 or Gal3 had opposite effects on RIG1 expression, and either excacerbated or reversed dramatically the effects of prior exposure to microbial neuraminidase. Gal1 alone downregulated the expression of RIG1, whereas if the cells had been pretreated with neuraminidase, the effect on RIG1 expression was dramatically reversed. In contrast, the presence of extracellular Gal3 alone significantly upregulated RIG1, while it had the opposite effect on the neuraminidase exposed cells. The results strongly suggest that cleavage of terminal sialic acids by the microbial neuraminidase exposed on the cell surface additional or novel galactosyl moieties that could activate different signaling pathways leading to either upregulation or downregulation of RIG1, and in turn, of SOCS1. While a cytokine-mediated signaling pathway can upregulate SOCS1 expression, viral infection can also do so through various mechanisms. For example, upon IAV infection SOCS1 expression is mediated by RIG1 (Pothlichet et al. 2008), but if the cells are exposed to the nonstructural protein1 (NS1) of the respiratory syncytial virus (RSV), SOCS1 is upregulated in a RIG1- or TLR3-independent manner (Xu et al. 2014). Therefore, it becomes clear that the associated expression of RIG1 and SOCS1 depends on the external stimuli, and in this context, the extracellular Gal1 and Gal3 may play different roles depending of the prior exposure of the cells to viral or bacterial neuraminidase.
Activation of signaling pathways in A549 cells exposed to neuraminidase and galectins
Based on the modulation of SOCS and RIG1 expression by the synergic activities of microbial neuraminidase and galectins, we examined the possible signaling pathways that may be activated in the process. Phosphorylation levels of integrin β3 (INTβ3), ERK, AKT, p38 MAPK, STAT1 and NFκB were assessed by western blot in untreated (control; Ctrl) or NeuK-treated A549 cells either in the presence or absence of 15 μg/ml rhGal1 or rhGal3 (Fig 2). The phosphorylation level of p38, a MAPK component, increased 2.5 fold in NeuK-treated A549 cells, but ERK, AKT and STAT1, as a JAK/STAT pathway, remained unchanged. The treatment of A549 cells with exogenous rhGal1 increased the phosphorylation levels of ERK, p38 and STAT1 between 2- and 3-fold, but decreased AKT to 50% of the control level. The NeuK treatment of A549 changed the effect of exogenous rhGal1 on AKT, increasing it up to 2-fold. The other proteins, ERK and STAT1, were also increased, but p38 remained at the level of the NeuK-treated A549 control. Exogenous rhGal3 induced an opposite effect for AKT phosphorylation than rhGal1 in A549 cells, as AKT phosphorylation increased up to 4-fold (Fig 2). The phosphorylation levels induced by exogenous rhGal3 in A549 also increased for ERK and STAT1, but remained unchanged for p38. The NeuK treatment of A549 cells changed the effect of exogenous rhGal3 for p38 (decreasing the phophorylation more than 50%) compared with rhGal3 effect on untreated cells. rhGal3 induced a 3-fold increase for ERK, 1.5-fold for AKT and up to 2-fold for STAT1. The phosphorylation of NF-κB was enhanced by addition of rhGal1 (up to 3-fold) or rhGal3 (up to 2-fold). The NeuK treatment of A549 cells alone did not modify the NF-κB phosphorylation level, but the further exposure to rhGal3 enhanced phosphorylation of NF-κB up to 3-fold (Fig 2).
Fig 2. Activation of regulatory signaling pathways in A549 cells is modulated by exposure to microbial neuraminidase and extracellular Gal1 and Gal3.

Total cell lysates were extracted from A549 control (Ctrl) or neuraminidase-treated cells (NeuK) incubated in the presence or absence of 15 μg/ml rhGal1 and rhGal3, and subject to 4 to 20% gradient SDS-PAGE. Total and phosphorylated protein levels of Integrin beta3 (INTb3), ERK, AKT, p38 MAPK, STAT1 and NF-κB were assessed by western blot. Bar graphs show the fold changes of phosphorylation levels in treated cells in comparison with control cells after normalization to total proteins. Shown images and the bar graphs are representative of data from at least two independent experiments. *p <0.05; **p<0.001, non paired Student’s t test.
The modulation of SOCS and RIG1 expression by the synergic activities of microbial neuraminidase and galectins at the airway cell surface, suggested that selected signaling pathways may be activated in the process leading to regulatory effects on expression and release of pro- and anti-inflammatory cytokines and chemokines. Our results revealed that the exposure of the epithelial cells to microbial neuraminidase and Gal1 or Gal3 led to the selective activation of different key regulatory signaling pathways. The activity of the microbial neuraminidase on the surface of A549 cells only activated the p38 MAPK signaling pathway, and enhanced INTβ3 phosphorylation, while all other pathways examined showed no phosphorylation changes in their selected components tested. This finding is not only consistent with the observation that infection by IAV inhibits expression of type I IFN and IL-6 by increasing SOCS expression via p38 MAPK activation (Bode et al. 2001; Pauli et al. 2008), but further suggests that the activity of influenza neuraminidase on the airway epithelial cell surface may be responsible for inducing the anti-inflammatory response resulting from enhancing SOCS expression via RIG1 and p38 MAPK activation. It is noteworthy that type I IFN display a pro-inflammatory role by activating NK cells (Nguyen et al. 2002), but it can also exert anti-inflammatory activity by inhibiting IL1, IL18 and IL12, or by enhancing IL10 production (Billiau 2006; Guarda et al. 2011; Gonzalez-Navajas et al. 2012; Arimori et al. 2013). In this regard, our study showed that the exposure of A549 cells to Gal1 maintained the MAPK activation levels, but significantly increased the phosphorylation levels of ERK, p38, STAT1 and NF-κB, while if the cells were previously exposed to neuraminidase the phosphorylation levels were increased further. Exposure of A549 cells to exogenous NeuK greatly increases INTβ3 phosphorylation. Further, desialylation increases LPS-mediated TLR4 activation (Feng et al. 2012), and β2 integrin interacts with TLR4 and modulate the NF-κB and MAPK activation (Perera et al. 2001; Yee and Hamerman 2013). Because INTβ3 may serve as coreceptor of TLR2 and boost the TLR2 signaling in response to viral or bacterial stimuli (Gianni et al. 2012; Gianni and Campadelli-Fiume 2014), its activation/phosphorylation could facilitate the interaction with TLR and modulate the downstream signaling or meditae the NF-κB activation directly. Effects of the exposure of the untreated cells to Gal3 were opposite to neuraminidase since except for p38, all components tested were significantly phosphorylated, particularly ERK and AKT. However, if the cells had been previously exposed to neuraminidase, exposure to Gal3 reduced p38 phosphorylation below the control levels, while phosphorylation of AKT was reduced to half, while phosphorylation levels of INTβ3 and NF-κB were maximally increased. The effect of Gal3 towards activation of AKT was opposite to that observed for Gal1.
Cytokine secretion from A549 cells exposed to microbial neuraminidase and galectins
Finally, we examined the potential impact of the downregulation of SOCS and RIG1 by the combination of microbial neuraminidases and galectins on the expression and secretion of cytokines. For this, we quantitatively assessed the cytokine released to the supernatant by the galectin and NeuK-treated cells. Treatment of the cells with microbial neuraminidase alone enhanced to various degrees the extracellular levels of most cytokines tested [interferon-γ (IFNγ); tumor necrosis factor-α (TNFα); interleukin-1β (IL-1β); IL-6; IL-8; and IL-12] whereas IL-10 and IL-15 remained unchanged as compared to the untreated A549 cells (Fig. 3). When the untreated A549 cells were exposed to 15 μg/ml of exogenous rhGal1 the levels of IFNγ, IL-6, IL-8, and IL-10 increased, while TNFα, IL-1β, and IL-12 remained unaffected. Gal3 exposure (15 μg/ml) increased levels of IFNγ, TNFα, IL-1β, IL-6, and IL-8, whereas levels of IL-10 and IL-12 were unchanged. Pre-exposure of the A549 cells to NeuK further enhanced the rhGal1-mediated upregulation of all cytokines tested, particularly noticeable for the pro-inflammatory cytokines IL-6 (4-fold) and IL-15 (25- fold) and the chemokine IL-8 (5-fold). Similarly to rhGal3, the NeuK treatment of the A549 cells further upregulated most cytokine and chemokine levels in the culture supernatant, with most significant increases for IL-6 (8-fold), IL-8 (15-fold), and IL-15 (20-fold), but had no effect on IL-1β, IL-10 and IL-12.
Fig 3. Cytokines release by A549 cells.

A549 control (Ctrl) or neuraminidase treated (NeuK) cells were stimulated in presence or absence of 15 μg/ml exogenous rhGal1 and rhGal3. The cytokines (IFN-γ, TNF-α, IL-1β, IL-6, IL-8, IL-10, IL-12 and IL-15) released to the culture medium was measured by MSD multi-spot assay. *p <0.05; **p<0.001, non paired Student’s t test.
The exposure of A549 cells that had been subjected to prior neuraminidase exposure to exogenous Gal1 reversed the anti-inflammatory response of neuraminidase toward a pro-inflammatory response by moderately downregulating SOCS expression to reach the control levels. This is supported by the observation that in addition to enhancing the release of pro-inflammatory cytokines such as IFNγ, TNFα, and IL-6, and the chemokine IL-8, exposure of the cells to extracellular Gal1 also increased the release of the anti-inflammatory cytokine IL-10, suggesting a balance between pro- and anti-inflammatory activities. This would take place by the activation of the ERK and AKT pathways, which involved in type I IFN production that in turn, induce pro-inflammatory responses by activating the JAK/STAT pathway. As a result, a controlled antimicrobial innate immune response aimed at clearing the microbial challenge would be fully enabled. The binding of Gal3 to the airway epithelial surface exposed to viral and bacterial neuraminidase, however, dramatically downregulates the expression of SOCS1 and RIG1 in neuraminidase-exposed epithelial cells further enhancing the pro-inflammatory responses resulting from the Gal1-mediated mechanism described above. The maximal levels of NF-κB phosphorylation levels observed in cells exposed to both neuraminidase and Gal3 suggest that expression and release of pro-inflammatory cytokines are significantly enhanced. Consistent with this observation are the increases of all pro-inflammatory cytokines and chemokines tested, whereas release of IL-10 remained unchanged. Proinflammatory cytokine signaling at the cell surface induces expression of SOCS proteins through the STAT1 pathway. In turn, the upregulated SOCS inhibit the JAK/STAT signaling, which negatively affects the downstream expression of many inflammatory cytokines, thereby providing a feedback loop to prevent the overwhelming inflammatory responses. More specifically, SOCS1 blocks p38, c-Jun N-terminal kinase (JNK), and nuclear factor κB (NF-κB) activation, and downregulates the expression of transforming growth factor-β-activated kinase 1 (TAK1) (Choi et al. 2013). Our results revealed that exogenous Gal3 downregulated expression of SOCS1 expression most likely through the p38 pathway, whose phosphoration was significantly inhibited by Gal3. Among the multiple layers of negative feedback regulation, the p38 MAPK pathway plays a central role to many of the feedback loops to prevent unprovoked, excessive, or unduly prolonged expression of pro-inflammatory genes (Clark and Dean 2012). For example, p38 MAPK upregulates expression of SOCS3, which negatively regulates IL-6 expression (Ehlting et al. 2007; Kiu et al. 2007). Therefore, inhibition of the p38 MAPK pathway signaling by Gal3, would ultimately lead to an uncontrolled upregulation of the expression of inflammatory cytokines.
Conclusions
Our results suggest that the activity of the viral and pneumococcal neuraminidase on the surface of the airway epithelial cells function as a “danger signal” that alerts the system and rapidly induces expression of SOCS to prevent an uncontrolled inflammatory response, which is “fine-tuned” by the presence of galectins in the extracellular space and their binding to galactosyl moieties unmasked on the surface of airway epithelial cells. Thus, the high levels of Gal1 in the bronchoalveolar space during the influenza infection and recovery counteract the effect of the viral neuraminidase on the airway epithelium, and enables an effective response of both pro- (IFNγ, TNFα, IL-6, IL-8) and anti-inflammatory cytokines and chemokines that “fine-tunes” the inflammatory response and the controlled clearance of the viral infection. During the recovery from influenza infection, however, the levels of Gal3 in the bronchoalveolar space increase dramatically as early as 1h upon the pneumococcal challenge, and as a consequence, the expression of SOCS1 via RIG1 and p38 MAPK is further downregulated, multiple proinflammatory pathways are activated, particularly ERK and JAK/STAT, and NF-κB is maximally phosporylated. These results in significant increases in the expression and release of pro-inflammatory cytokines while the anti-inflammatory cytokines, such as IL-10 remain unaffected. As a consequence, the pro-and anti-inflammatory cytokine balance would be grossly tilted towards an exaggerated response leading to a hypercytokinemia and septic shock.
Highlights.
Microbial neuraminidases unmask galectin ligands at the airway epithelial surface
Galectin 1 and galectin 3 modulate the expression of SOCS1 and RIG1
Galectins modulate the activation of ERK, AKT or JAK/STAT1 signaling pathways
Leads to a disregulated expression and release of pro-inflammatory cytokines
Acknowledgments
This work was financially supported by grant 5R01GM070589-06 from the National Institutes of Health to GRV.
Footnotes
Supported by grant 5R01GM070589-06 from the National Institutes of Health to GRV
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso JM, Guiyoule A, et al. A model of meningococcal bacteremia after respiratory superinfection in influenza A virus-infected mice. FEMS Microbiol Lett. 2003;222(1):99–106. doi: 10.1016/S0378-1097(03)00252-0. [DOI] [PubMed] [Google Scholar]
- Arimori Y, Nakamura R, et al. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res. 2013;99(3):230–237. doi: 10.1016/j.antiviral.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Bermejo-Martin JF, Martin-Loeches I, et al. Host adaptive immunity deficiency in severe pandemic influenza. Crit Care. 2010;14(5):R167. doi: 10.1186/cc9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiau A. Interferon: the pathways of discovery I. Molecular and cellular aspects. Cytokine Growth Factor Rev. 2006;17(5):381–409. doi: 10.1016/j.cytogfr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Bode JG, Ludwig S, et al. The MKK6/p38 mitogen-activated protein kinase pathway is capable of inducing SOCS3 gene expression and inhibits IL-6-induced transcription. Biol Chem. 2001;382(10):1447–1453. doi: 10.1515/BC.2001.178. [DOI] [PubMed] [Google Scholar]
- Camby I, Le Mercier M, et al. Galectin-1: a small protein with major functions. Glycobiology. 2006;16(16840800):157. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. JAMA. 2013;309(3):275–282. doi: 10.1001/jama.2012.194139. [DOI] [PubMed] [Google Scholar]
- Choi YS, Park JK, et al. Cytokine signaling-1 suppressor is inducible by IL-1beta and inhibits the catabolic effects of IL-1beta in chondrocytes: its implication in the paradoxical joint-protective role of IL-1beta. Arthritis Res Ther. 2013;15(6):R191. doi: 10.1186/ar4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Dean JL. The p38 MAPK Pathway in Rheumatoid Arthritis: A Sideways Look. Open Rheumatol J. 2012;6:209–219. doi: 10.2174/1874312901206010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Kiu H, et al. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19(4):414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AS, Hyun SW, et al. NEU1 and NEU3 sialidase activity expressed in human lung microvascular endothelia: NEU1 restrains endothelial cell migration, whereas NEU3 does not. J Biol Chem. 2012;287(19):15966–15980. doi: 10.1074/jbc.M112.346817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanovic D, Lai R, et al. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am J Pathol. 2013;183(3):868–880. doi: 10.1016/j.ajpath.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Davey GM, Heath WR, et al. SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens. 2006;67(1):1–9. doi: 10.1111/j.1399-0039.2005.00532.x. [DOI] [PubMed] [Google Scholar]
- Ehlting C, Lai WS, et al. Regulation of suppressor of cytokine signaling 3 (SOCS3) mRNA stability by TNF-alpha involves activation of the MKK6/p38MAPK/MK2 cascade. J Immunol. 2007;178(5):2813–2826. doi: 10.4049/jimmunol.178.5.2813. [DOI] [PubMed] [Google Scholar]
- Farnworth SL, Henderson NC, et al. Galectin-3 reduces the severity of pneumococcal pneumonia by augmenting neutrophil function. Am J Pathol. 2008;172(2):395–405. doi: 10.2353/ajpath.2008.070870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Ghosh A, et al. The galectin CvGal1 from the eastern oyster (Crassostrea virginica) binds to blood group A oligosaccharides on the hemocyte surface. J Biol Chem. 2013;288(34):24394–24409. doi: 10.1074/jbc.M113.476531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Stamatos NM, et al. Sialyl residues modulate LPS-mediated signaling through the Toll-like receptor 4 complex. PLoS One. 2012;7(4):e32359. doi: 10.1371/journal.pone.0032359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Zhang L, et al. Neuraminidase reprograms lung tissue and potentiates lipopolysaccharide-induced acute lung injury in mice. J Immunol. 2013;191(9):4828–4837. doi: 10.4049/jimmunol.1202673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer C, Sanchez-Ruderisch H, et al. Galectin-1 interacts with the {alpha}5{beta}1 fibronectin receptor to restrict carcinoma cell growth via induction of p21 and p27. J Biol Chem. 2005;280(44):37266–37277. doi: 10.1074/jbc.M411580200. [DOI] [PubMed] [Google Scholar]
- Fletcher J, Starr R. The role of suppressors of cytokine signalling in thymopoiesis and T cell activation. Int J Biochem Cell Biol. 2005;37(9):1774–1786. doi: 10.1016/j.biocel.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol. 2011;23(4):481–486. doi: 10.1016/j.coi.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Campadelli-Fiume G. The epithelial alphavbeta3-integrin boosts the MYD88-dependent TLR2 signaling in response to viral and bacterial components. PLoS Pathog. 2014;10(11):e1004477. doi: 10.1371/journal.ppat.1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Leoni V, et al. alphavbeta3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc Natl Acad Sci U S A. 2012;109(48):19792–19797. doi: 10.1073/pnas.1212597109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, et al. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12(2):125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Braun M, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Ilangumaran S, Ramanathan S, et al. Regulation of the immune system by SOCS family adaptor proteins. Semin Immunol. 2004;16(6):351–365. doi: 10.1016/j.smim.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Kedzierski L, Linossi EM, et al. Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog. 2014;10(5):e1004134. doi: 10.1371/journal.ppat.1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiu H, Hilton DJ, et al. Mechanism of crosstalk inhibition of IL-6 signaling in response to LPS and TNFalpha. Growth Factors. 2007;25(5):319–328. doi: 10.1080/08977190701830151. [DOI] [PubMed] [Google Scholar]
- Koh HS, Lee C, et al. CD7 expression and galectin-1-induced apoptosis of immature thymocytes are directly regulated by NF-kappaB upon T-cell activation. Biochem Biophys Res Commun. 2008;370(1):149–153. doi: 10.1016/j.bbrc.2008.03.049. [DOI] [PubMed] [Google Scholar]
- Lillehoj EP, Hyun SW, et al. NEU1 sialidase expressed in human airway epithelia regulates epidermal growth factor receptor (EGFR) and MUC1 protein signaling. J Biol Chem. 2012;287(11):8214–8231. doi: 10.1074/jbc.M111.292888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M. Influenza virus entry. Adv Exp Med Biol. 2012;726:201–221. doi: 10.1007/978-1-4614-0980-9_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumiya T, Stafforini DM. Function and regulation of retinoic acid-inducible gene-I. Crit Rev Immunol. 2010;30(6):489–513. doi: 10.1615/critrevimmunol.v30.i6.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalvo AC, Batalle JP, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2011;17(2):195–199. doi: 10.1038/nm.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka T, Fujimoto M, et al. Negative regulation of cytokine and TLR signalings by SOCS and others. Adv Immunol. 2005;87:61–122. doi: 10.1016/S0065-2776(05)87003-8. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Salazar-Mather TP, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol. 2002;169(8):4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- Nita-Lazar M, Banerjee A, et al. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol Immunol. 2015;65(1):1–16. doi: 10.1016/j.molimm.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli EK, Schmolke M, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4(11):e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera PY, Mayadas TN, et al. CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J Immunol. 2001;166(1):574–581. doi: 10.4049/jimmunol.166.1.574. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Pothlichet J, Chignard M, et al. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol. 2008;180(4):2034–2038. doi: 10.4049/jimmunol.180.4.2034. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Daly G, et al. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J Exp Med. 1999;190(3):385–398. doi: 10.1084/jem.190.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Ilarregui JM. Conveying glycan information into T-cell homeostatic programs: a challenging role for galectin-1 in inflammatory and tumor microenvironments. Immunol Rev. 2009;230(1):144–159. doi: 10.1111/j.1600-065X.2009.00787.x. [DOI] [PubMed] [Google Scholar]
- Ramirez-Martinez G, Cruz-Lagunas A, et al. Seasonal and pandemic influenza H1N1 viruses induce differential expression of SOCS-1 and RIG-I genes and cytokine/chemokine production in macrophages. Cytokine. 2013;62(1):151–159. doi: 10.1016/j.cyto.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci L, Fiorucci S, et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology. 2003;124(5):1381–1394. doi: 10.1016/s0016-5085(03)00267-1. [DOI] [PubMed] [Google Scholar]
- Sato S, Ouellet N, et al. Role of galectin-3 as an adhesion molecule for neutrophil extravasation during streptococcal pneumonia. J Immunol. 2002;168(4):1813–1822. doi: 10.4049/jimmunol.168.4.1813. [DOI] [PubMed] [Google Scholar]
- Seki M, Higashiyama Y, et al. Acute infection with influenza virus enhances susceptibility to fatal pneumonia following Streptococcus pneumoniae infection in mice with chronic pulmonary colonization with Pseudomonas aeruginosa. Clin Exp Immunol. 2004;137(1):35–40. doi: 10.1111/j.1365-2249.2004.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speshock JL, Doyon-Reale N, et al. Filamentous influenza A virus infection predisposes mice to fatal septicemia following superinfection with Streptococcus pneumoniae serotype 3. Infect Immun. 2007;75(6):3102–3111. doi: 10.1128/IAI.01943-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(1):17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Toscano MA, Campagna L, et al. Nuclear factor (NF)-kappaB controls expression of the immunoregulatory glycan-binding protein galectin-1. Mol Immunol. 2011;48(15–16):1940–1949. doi: 10.1016/j.molimm.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Toscano MA, Commodaro AG, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. 2006;176(10):6323–6332. doi: 10.4049/jimmunol.176.10.6323. [DOI] [PubMed] [Google Scholar]
- Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. 2009;7(6):424–438. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta GR, Ahmed H. Animal Lectins: A Functional View. CRC Press; 2008. [Google Scholar]
- Xu X, Zheng J, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology. 2014;57(2):65–73. doi: 10.1159/000357327. [DOI] [PubMed] [Google Scholar]
- Yang XX, Du N, et al. Gene expression profiles comparison between 2009 pandemic and seasonal H1N1 influenza viruses in A549 cells. Biomed Environ Sci. 2010;23(4):259–266. doi: 10.1016/S0895-3988(10)60061-X. [DOI] [PubMed] [Google Scholar]
- Yee NK, Hamerman JA. beta(2) integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur J Immunol. 2013;43(3):779–792. doi: 10.1002/eji.201242550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227(1):54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- Yoo JS, Kato H, et al. Sensing viral invasion by RIG-I like receptors. Curr Opin Microbiol. 2014;20:131–138. doi: 10.1016/j.mib.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, et al. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Murray PJ, et al. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. J Immunol. 2006;176(3):1840–1847. doi: 10.4049/jimmunol.176.3.1840. [DOI] [PubMed] [Google Scholar]
- Zuniga J, Torres M, et al. Inflammatory profiles in severe pneumonia associated with the pandemic influenza A/H1N1 virus isolated in Mexico City. Autoimmunity. 2011;44(7):562–570. doi: 10.3109/08916934.2011.592885. [DOI] [PubMed] [Google Scholar]