

Abstract
In recent years, in vitro transcribed messenger RNA (mRNA) has emerged as a potential therapeutic platform. To fulfill its promise, effective delivery of mRNA to specific cell types and tissues needs to be achieved. Lipid nanoparticles (LNPs) are efficient carriers for short-interfering RNAs and have entered clinical trials. However, little is known about the potential of LNPs to deliver mRNA. Here, we generated mRNA-LNPs by incorporating HPLC purified, 1-methylpseudouridine-containing mRNA comprising codon-optimized firefly luciferase into stable LNPs. Mice were injected with 0.005–0.250 mg/kg doses of mRNA-LNPs by 6 different routes and high levels of protein translation could be measured using in vivo imaging. Subcutaneous, intramuscular and intradermal injection of the LNP-encapsulated mRNA translated locally at the site of injection for up to 10 days. For several days, high levels of protein production could be achieved in the lung from the intratracheal administration of mRNA. Intravenous and intraperitoneal and to a lesser extent intramuscular and intratracheal deliveries led to trafficking of mRNA-LNPs systemically resulting in active translation of the mRNA in the liver for 1–4 days. Our results demonstrate that LNPs are appropriate carriers for mRNA in vivo and have the potential to become valuable tools for delivering mRNA encoding therapeutic proteins.
Keywords: mRNA, pseudouridine, luciferase, nanoparticle, non-viral gene delivery
Graphical abstract
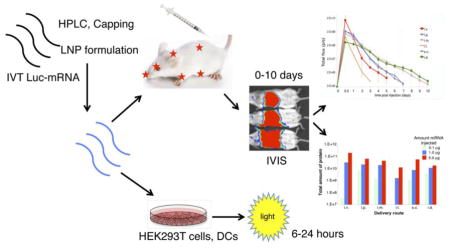
Graphical representation of experimental approaches and data acquisition. Methods for the delivery and analysis of data are described in the Materials and Methods and Results sections.
Introduction
mRNA-based therapy was first described in 1992 with the injection of vasopressin-encoding mRNA into rats with diabetes insipidus1, but underwent little development other than as an immunogen delivery vector until its immunogenicity and stability were addressed2. mRNA-based therapy has several conceptual advantages over other nucleic acid-based approaches. First, mRNA cannot integrate into the host genome, so there is no potential danger for insertional mutagenesis. Second, mRNA only requires cytosolic delivery in the host cell where it is translated into functional protein. Third, mRNA is translated transiently in cells and is degraded in a relatively short but controllable amount of time.
In order to use mRNA therapeutically, four main hurdles need to be overcome, 1) poor translatability, 2) lack of RNA stability, 3) inefficient in vivo delivery, and 4) its activation of innate immune sensors. To increase mRNA translatability and stability, 5′ cap, optimized 5′- and 3′-UTRs, and coding sequence and poly(A)-tail modifications can be introduced into the molecule3,4. Incorporation of modified nucleosides, such as pseudouridine or 1-methylpseudouridine and HPLC purification of the in vitro transcribed mRNA further increase protein translation and make mRNA immunologically silent2. The use of non-immunogenic mRNA is crucial, because a series of innate immune receptors (TLR3, TLR7, TLR8, RIG-I, MDA5, NOD2, PKR and others) recognize RNA resulting in the release of type I interferons and activation of interferon-inducible genes and inhibition of translation5.
Efficient delivery of mRNA into target cells in vivo is a major challenge. A variety of formulations have been developed to protect the nucleic acid from RNases and facilitate its uptake into cells. Positively charged lipids, cationic polypeptides, polymers, micelles or dendrimers have been used for in vivo RNA delivery, some formulations have already entered clinical trials in the field of cancer immunotherapy6.
There has been significant progress in overcoming many of the difficulties associated with in vivo delivery of mRNA and lipid nanoparticles (LNPs), containing ionizable cationic lipids, represent one of the most advanced technological platforms. The LNPs used in this study are 70 to 100 nm particles, prepared using an ionizable amino lipid, phospholipid, cholesterol and a PEGylated lipid, similar in composition to the LNPs that have recently proven to be safe and efficient tools for siRNA delivery7,8. Robust gene silencing has been demonstrated in several species including non-human primates9,10 and LNPs with encapsulated siRNAs have been successfully tested in human clinical trials. However, our knowledge is limited concerning mRNA delivery by LNPs. Geall and colleagues have recently developed an RNA self-replicating vaccine formulated in LNPs or a nanoemulsion and found that an intramuscular injection with very low doses of RNA induced protective immune responses against respiratory syncytial and influenza virus in mice11,12 and against respiratory syncytial virus and CMV in non-human primates13.
In the present study, LNP-encapsulated, HPLC-purified, 1-methylpseudouridine-containing mRNA encoding firefly luciferase was delivered into cultured cells and mice. Mice were injected with 0.1 μg (0.005 mg/kg), 1 μg (0.050 mg/kg), and 5 μg (0.250 mg/kg) of mRNA-LNP formulations by different routes, including intravenous, intraperitoneal, subcutaneous, intramuscular, intradermal, and intratracheal. A series of comparative studies were carried out investigating the levels and duration of mRNA translation into the encoded protein. We have found that administration of mRNA-LNP complexes results in large amounts of protein production in vivo for varying lengths of time demonstrating that LNPs are suitable tools for highly efficient mRNA delivery.
2. Materials and methods
2.1. Ethics statement
Humans
Primary human cells utilized in these experiments were isolated from leukopheresis samples obtained from healthy volunteers. Written informed consent using a protocol approved by the University of Pennsylvania Institutional Review Board was used.
Mice
The investigators faithfully adhered to the “Guide for the Care and Use of Laboratory Animals” by the Committee on Care of Laboratory Animal Resources Commission on Life Sciences, National Research Council. The animal facilities at the University of Pennsylvania are fully accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC). All studies were conducted using protocols approved by the University of Pennsylvania IACUC.
2.2. Cells
Human embryonic kidney (HEK) 293T cells (American Type Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM L–glutamine (Life Technologies) and 10% fetal calf serum (FCS) (HyClone) (complete medium). Immature human dendritic cells (DCs) were generated from elutriated monocytes cultured in RPMI (Life Technologies), containing glutamine (2 mM), 10% FCS, human IL-4 (100 ng/ml) and human GM-CSF (50 ng/ml) (R&D). Cells were maintained with fresh medium containing IL-4 and GM-CSF every 3 days and used on day 7.
2.3. mRNA production
mRNAs were produced as previously described14 using linearized plasmids encoding codon-optimized firefly luciferase (pLuc19) and T7 RNA polymerase (Megascript, Ambion). mRNAs were transcribed to contain 130 nucleotide-long poly(A) tails. 1-methylpseudouridine-5′-triphosphate (TriLink) instead of UTP was used to generate modified nucleoside-containing mRNA. RNAs were capped using the m7G capping kit with 2′-O-methyltransferase (ScriptCap, CellScript) to obtain cap1. mRNA was purified by Fast Protein Liquid Chromatography (FPLC) (Akta Purifier, GE Healthcare) as described earlier15. All RNAs were analyzed by denaturing or native agarose gel electrophoresis and were stored frozen at −20°C.
2.4. Formulation of the mRNA
Lipofectin (Invitrogen) complexing was performed, as described previously2 using 0.8 μl of Lipofectin and 0.1 to 1.0 μg of mRNA per well of a 96-well plate. Complexing of mRNA to TransIT mRNA (TransIT) (Mirus Bio) was performed according to the manufacturer’s instructions combining 0.1 or 0.3 μg mRNA with TransIT reagents, TransIT mRNA (0.34 μl) and Boost (0.22 μl) in a final volume of 18 μl DMEM, which was added to a single 96-well of cells.
HPLC-purified 1-methylpseudouridine-containing firefly luciferase-encoding mRNA was encapsulated in LNPs using a self-assembly process in which an aqueous solution of mRNA at pH 4.0 is rapidly mixed with a solution of lipids dissolved in ethanol8. LNPs used in this study were similar in composition to those described previously7,8, which contain an ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG-lipid (50:10:38.5:1.5 mol/mol), encapsulated RNA-to-total lipid ratio of ~0.05 (wt/wt) and a diameter of ~80nm. mRNA-LNP formulations were stored at −80°C at a concentration of mRNA of ~1 μg/μl.
2.5. Cell transfections
For Lipofectin complexed mRNA, medium was removed and 47 μl of complexed mRNA was added to 5 × 104 HEK293T or DCs per well. Cells were incubated for 1 h and the Lipofectin-mRNA mixture was replaced with 200 μl complete medium. For TransIT complexed mRNA, 17 μl of complex was added to cells cultured in 183 μl complete medium. mRNA-LNPs were preincubated with 0.1 μg human recombinant ApoE3 (Sigma) protein in 6 μl AIM V medium (Life Technologies) for 5 minutes at 37 °C or not. After preincubation, mRNA-LNPs were added to the cells cultured in 194 μl AIM V medium. Cells were lysed in firefly-specific lysis reagent (Promega) at 18 h or at the indicated times post mRNA addition. Aliquots were assayed for enzyme activity using the firefly luciferase assay system (Promega) and a MiniLumat LB 9506 luminometer (Berthold/EG&G; Wallac).
2.6. Administration of LNPs to mice
Female BALB/c mice aged 6 weeks were purchased from Harlan Laboratories. Increasing amounts (0.1 μg, 1.0 μg or 5.0 μg) of mRNA-LNPs in Dulbecco’s Phosphate Buffered Saline (PBS) were injected into animals intradermally (30 μl), intraperitoneally (200 μl), subcutaneously (200 μl), intramuscularly (30 μl) and intravenously (100 μl) with 3/10cc insulin syringes (BD Biosciences) using standard techniques16. A Penn-Century Microsprayer Aerosolizer (Model IA-1C) and FMJ-250 high pressure syringe was used for intratracheal delivery of mRNA-LNPs via intubation17.
2.7. Bioluminescence imaging studies
Bioluminescence imaging was performed with an IVIS Spectrum imaging system (Caliper Life Sciences). Mice were administered D-luciferin (Regis Technologies) at a dose of 150 mg/kg intraperitoneally. Mice were anesthetized after receiving D-luciferin in a chamber with 3% isoflurane (Piramal Healthcare Limited) and placed on the imaging platform while being maintained on 2% isoflurane via a nose cone. Mice were imaged at 5 minutes post administration of D-luciferin using an exposure time of 5 seconds or longer to ensure that the signal acquired was within effective detection range (above noise levels and below CCD saturation limit). Bioluminescence values were quantified by measuring photon flux (photons/second) in the region of interest where bioluminescence signal emanated using the Living IMAGE Software provided by Caliper.
2.8. Mathematical and statistical analyses
The half-life of protein translation by the delivered mRNA was calculated using the equation: t1/2=Δt X ln(2)/ln(N0/Nt), where Δt is the time between measurements, N0 is the starting value and Nt is the value at the end of the time being evaluated. Time intervals where saturation was evident and where translation was ending, low values were not calculated to exclude the effect of protein half-life. Area under the curve calculations were performed using the equation: AUC=Σ ti+1 − ti)/2 X (Ci + Ci+1, where ti is the starting time point, ti+1 is the finishing time point, Ci is the starting value and Ci+1 is the finishing value for each measurement over time. Means, standard error of the means, and student’s paired t-tests were determined using Microsoft Excel Software.
3. Results
3.1. Transfection of HEK293T cells and primary human DCs with luciferase mRNA formulated with lipofectin, TransIT or LNPs
Immortalized cell lines and primary cells are barely transfectable with naked mRNA but 60 to 95 percent transfection efficiencies can be obtained when mRNA is complexed with cationic polymer or lipid-based reagents, such as TransIT or Lipofectin18. In the first set of experiments, HEK293T cells and human monocyte-derived dendritic cells (hDCs) were transfected with luciferase encoding nucleoside-modified mRNA complexed with TransIT and Lipofectin or encapsulated into LNPs (Fig. 1A). Notably, cells were exposed to Lipofectin-mRNA complexes for an hour in serum-free medium whereas cells were incubated with TransIT-mRNA and LNP-mRNA complexes in complete medium. In HEK293T cells, translation levels were slightly higher when LNPs delivered the mRNA compared to cationic polymer/lipid or lipid (Fig. 1A). On the contrary, in DCs, the level of luciferase was lower when LNPs were used for formulation of the mRNA as compared to using the commercially available reagents. Unlike the standard transfection reagents named above, the LNPs exhibit a net neutral surface charge at physiological pH, which results in maximal delivery to hepatocytes in vivo7, but the absence of a positive surface charge reduces cell uptake in vitro. In vivo, ionizable LNP containing nucleic acid therapeutics bind ApoE and are taken up by hepatocytes through receptor mediated endocytosis19,20. Incubation of mRNA-LNPs with recombinant ApoE protein prior to transfection of DCs significantly increased uptake and subsequent translation of the luciferase encoding mRNA (Fig. 1A). HEK293T cells are decorated with ApoE receptor and produce its ligand as well21, hence, not surprisingly, addition of exogenous ApoE did not result in elevated luciferase production in this cell line (Fig. 1A).
Fig. 1.
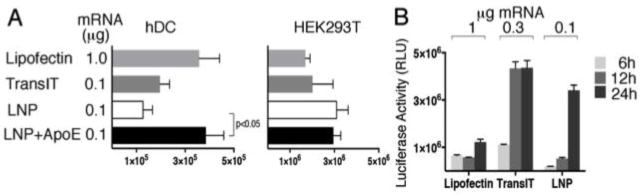
Efficiency of complexing reagents for in vitro luciferase mRNA transfection. Firefly luciferase expression following transfection of (A) HEK293T cells and human monocyte derived dendritic cells with Lipofectin (1 μg mRNA), TransIT (0.1 μg mRNA) or LNP-encapsulated luciferase mRNA (0.1 μg mRNA). ApoE recombinant protein was incubated with mRNA-LNPs where indicated prior to addition to the cells. (B) Firefly luciferase expression in HEK293T cells at 6, 12, and 24 hours following transfection of luciferase-encoding mRNA complexed with either Lipofectin, TransIT or encapsulated in LNPs. Error bars are standard error of the mean (SEM). p-value determined by paired student’s t-test.
The kinetics and dose response of protein translation were examined in HEK293T cells transfected with increasing amounts of Luc mRNA formulated with cationic polymer and/or lipid or LNPs. Transfecting higher amounts of cationic polymer and/or lipid-complexed Luc mRNA resulted in higher protein levels at 12 and 24 hours post transfection (Fig. 1B). Interestingly, LNP-formulated mRNA translated most efficiently at 24 hours post transfection regardless of the amount of mRNA transfected into cells (Fig. 1B). Most likely saturation of the translational or uptake capacity of the cells occurred at the lowest dose of delivered mRNA.
3.2. mRNA-LNP administration into mice
To examine the duration and distribution of protein production from mRNA-LNPs in vivo, 5.0 μg luciferase mRNA-LNPs were administered into mice using commonly applied therapeutic delivery routes and protein translation was followed over time. Intravenous (retro-orbital) and intraperitoneal delivery resulted in mRNA-LNP trafficking to the liver (Fig. 2), which is in accordance with previous observations for siRNA10,19. In the liver, mRNA was translated to functional protein and very strong bioluminescent signal was measured in the first 24 hours post injection. Translation abated relatively quickly in the liver and there was no measurable luciferase activity at 3 days after mRNA-LNP injection. A small fraction of activity remained at the site of the injections (around the eyes and in the abdomen) and continued translation was measured for up to 7 days demonstrating the rapid turnover of translatable mRNA in the liver.
Fig. 2.

Duration and translational pattern of mRNA-LNPs in mice injected by various routes. Representative IVIS images of groups of 3 BALB/c mice injected with 5.0 μg mRNA-LNP by the intradermal (i.d.), intramuscular (i.m.), subcutaneous (s.c.), intravenous (i.v.), intraperitoneal (i.p.) and intratracheal (i.t.) routes. Relative luminescence plot is shown and the scale of luminescence is indicated.
When mRNA-LNPs were injected intramuscularly and intratracheally, similar to intravenous and intraperitoneal deliveries, a large portion of the luciferase activity was detectable in the liver, demonstrating systemic spread of the nanoparticles. Also similar to intravenous and intraperitoneal deliveries, the high levels of protein produced in the liver occurred over a short duration with the majority of translation ceasing at day 2 post-injection (Fig. 2). Interestingly, significant bioluminescent signal could be measured in the lungs and muscles, as well, with the latter lasting for up to 8 days post injection.
Subcutaneous and intradermal mRNA-LNP deliveries only resulted in protein production at the site of injection (Fig. 2). The duration of active translation extended for 6 and 10 days in the subcutaneous and intradermal space, respectively (Fig. 2).
3.3. Kinetics of LNP-formulated mRNA translation in mice
The expression of a protein by delivering the encoding nucleoside-modified mRNA could be used for different therapies depending on the function of the protein. This includes rapid and high-level protein production, short or long durations of translation, or delivery to a specific site versus systemic delivery. Next, we investigated the kinetics and dose-response of protein production after injecting mice with 0.1 μg, 1.0 μg, 5.0 μg of mRNA-LNPs (Fig. 3). Intravenous delivery resulted in the highest amount of peak protein produced and the shortest duration of expression. The highest bioluminescent signal was measured at 4 hours followed by a sharp decrease in protein production. Two days after the mRNA-LNP delivery, translation levels were 2 orders of magnitude lower compared to those measured at 4 hours and baseline expression was observed at day 3 post transfection. The mRNA translation had similar kinetics following intratracheal administration, but with a lower peak level of expression for the same amount of delivered mRNA (Fig. 3) and similar to other non-liver tissues longer duration of translation could be detected in the lung versus the liver. No signal could be measured when mice were injected with 0.1 μg of mRNA-LNPs.
Fig. 3.

Translational kinetics of mRNA-LNP delivered by different routes in vivo. Quantification of the bioluminescent signal measured in BALB/c mice injected with (A) 0.1 μg, (B) 1.0 μg or (C) 5.0 μg mRNA-LNPs by intradermal (i.d.), intramuscular (i.m.), subcutaneous (s.c.), intravenous (i.v.), intraperitoneal (i.p.) and intratracheal (i.t.) routes. Error bars are standard error of the mean (SEM).
Intraperitoneal and subcutaneous injection of mRNA-LNPs resulted in similar peak levels and duration of active translation at the site of injection (Fig. 3). Subcutaneous delivery did not result in measurable luciferase expression in the liver or other anatomical sites. The rate of decrease in luciferase expression at both sites of injection was slower than after intravenous injection with less than one half log decrease between 4 and 24 hours. Translation could still be measured at day 6 after injection in the peritoneal cavity but not in the liver for intraperitoneal injection, and was lost at day 7 for animals injected with 5.0 μg mRNA-LNP (Fig. 3).
Intramuscular and intradermal delivery of mRNA-LNPs resulted in the longest duration of translation. In both cases, translation levels decreased gradually at a much slower rate over time and the duration of protein production was extended, up to 10 days post injection. Interestingly, intradermal delivery of mRNA-LNP resulted in a lower initial amount of protein, which was produced for a longer period of time, suggesting saturation of the uptake or translational capacity of the intradermal space.
The dose response differed at each site. For intravenous injection, a linear dose response was observed (Fig. 3). The lowest 0.1 μg dose administered, resulted in disproportionally less protein than expected, likely because during injection a similar portion of the delivered nanoparticles remained and translated in the eye socket at each dose. Subcutaneous, intratracheal, and intramuscular injection had linear dose response characteristics (Fig. 3). Intraperitoneal injection of mRNA-LNPs demonstrated a decrease in the expected maximal level of translation at the 5.0 μg dose, suggesting saturation of the uptake and translational capacity of the cells in the peritoneal cavity (Fig. 3 and Fig. 1sup). This saturation was most evident with intradermal delivery. All 3 doses, 5.0, 1.0 and 0.1 μg led to similar levels of peak translation at 4 hours post injection (Fig. 3 and Fig. 1sup), but the increased doses extended the duration of translation from 5 (0.1 μg) to 10 (5.0 μg) days.
3.4. Total protein produced and half-life of protein production
The total amount of protein produced over the full duration of translation can be estimated using area under the curve analyses. As luciferase catalytic activity has a short half-life (3 hours) in vivo22, its measurement reflects active translation of protein from the delivered mRNA. Different doses of mRNA delivered also had different relative levels of protein produced. At 5.0 μg, intravenous delivery gave the highest amount of total protein produced, even though it had a relatively short duration of active translation (Fig. 4). Subcutaneous, intramuscular, and intraperitoneal had similar levels of total protein produced, intradermal was slightly lower and intratracheal was substantially decreased (Fig. 4). The effect of saturation was particularly evident for intradermal delivery, where a non-linear dose response was detected over an increasing number of days with dosage increases (Fig. 3 and Fig. 1sup).
Fig. 4.
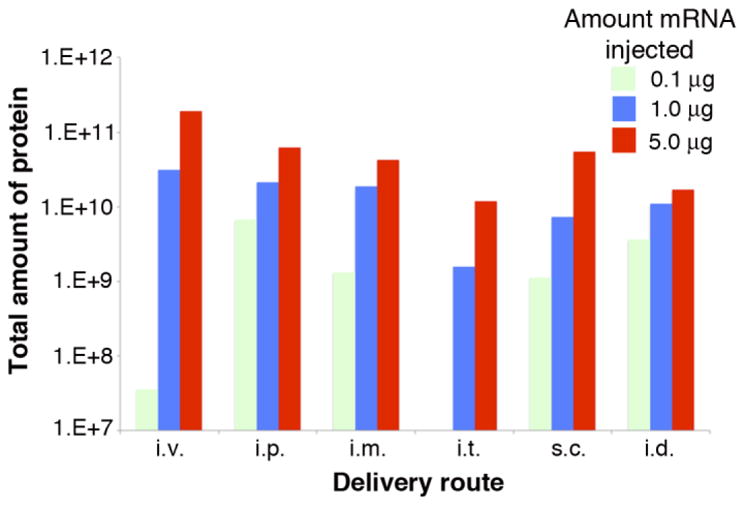
Total amount of protein produced for each amount of mRNA administered at each delivery site. Area under the curve was calculated as described in the Materials and Methods. The reduction beyond the expected linear dose response for 0.1 μg of mRNA delivered by the i.v. route was likely due to the observation that a similar amount of mRNA-LNPs remained in the eye socket for each dose delivered and this amount was a substantial fraction delivered for the 0.1 μg dose.
We calculated the half-life of mRNA translation using the region of the activity versus time curve that excluded points associated with saturation and time points with low levels of luciferase activity, where previous high levels of luciferase activity would artificially increase the calculated half-life by introducing protein half-life effects. This demonstrated that liver translation by routes that result in primarily liver delivery of LNPs (intravenous and lung) resulted in the shortest half-life of mRNA translation, while the intradermal route had the longest (Table 1).
Table 1.
Half-life of protein production
| Delivery site | Half-life of mRNA translation (hours) |
|---|---|
|
| |
| intradermal | 29.6 |
| intramuscular | 20.6 |
| subcutaneous | 14.7 |
| intravenous | 6.8 |
| intraperitoneal | 14.8 |
| intratracheal | 7.5 |
4. Discussion
Nucleoside-modified mRNA has given new interest to the use of mRNA to express proteins for research and therapeutic purposes. mRNA does not require nuclear entry, it cannot induce insertional mutagenesis, and it results in both very rapid and highly controllable protein expression. Thus, the non-integrating and transient character of mRNA therapy is extremely well suited for using mRNA for expressing proteins and applying it for replacement therapies. To make mRNA a potent therapeutic, stable, highly translatable and non-immunogenic mRNA needs to be delivered into high numbers of target cells. To fulfill the first criteria, HPLC-purification and incorporation of modified nucleosides e.g. 1-methylpseudouridine and stabilizing elements, including 5′ cap, optimized 5′- and 3′-UTRs and poly(A)-tail into the mRNA sequence was necessary3,4. Over the past years, numerous reagents and methods have been developed for in vivo mRNA delivery but most of them have limited efficacy or serious side effects. A great variety of lipid-based formulations (lipoplexes, lipopolyplexes, cationic nanoemulsions) have been tested in mice and non-human primates with varying success (reviewed in 23). Commercially available cationic polymer and/or lipid-based reagents such as TransIT, Lipofectin and others can be efficiently used for in vitro mRNA delivery but have limited efficacy in vivo2,18. To selectively target DCs, mRNA complexes can be decorated with ligands for their lectin and scavenger receptors, such as mannose, dectins and langerin23. In addition to positively charged lipids, cationic polymers (dendrimers, poly-L-lysine, protamine, polyethylenimine) can also be potentially used for mRNA delivery24,25.
The development of LNPs containing ionizable amino lipids of the type described by Jayaraman et al. has enabled the clinical development of siRNA therapeutics for hepatic gene silencing9,10. Interestingly, there is almost no data on using this class of LNPs for mRNA delivery to date. Geall and colleagues have recently generated LNP-complexed self-replicating RNA vaccines against RSV and influenza virus in mice11,12 and found it to be very efficient even at low doses. We have developed methods for making stable, highly translatable and immunologically silent mRNA (reviewed in 14,15). This optimized, firefly luciferase-encoding mRNA was encapsulated into ionizable LNPs developed by Acuitas Therapeutics to deliver mRNA in vivo. In this study, we report on the high level and extended expression of nucleoside-modified mRNA complexed to LNPs delivered by intraperitoneal, intravenous, intradermal, subcutaneous, intramuscular and intratracheal routes.
We observed that in human dendritic cells but not in HEK293T cells efficient uptake of LNPs required exogenous ApoE. This result is in agreement with the finding that HEK293T cells express both ApoE and its receptor LRP121. We next investigated the mRNA-LNP expression kinetics in HEK293T cells and found that regardless of the amount of nanoparticles transfected into cells, the highest enzyme activity was measured at 24 hours post transfection and more mRNA-LNPs did not result in higher luciferase expression (Fig. 1B). This lack of dose response with LNPs in vitro could result from saturation of either the uptake pathway, which is likely different from that of the cationic transfection reagents, or translation capacity of the cells. To summarize the in vitro results, mRNA-LNPs are taken up by cultured cells with similar efficiencies to that of cationic polymer/lipid-based transfection reagents and saturation of uptake and/or translation occurs at low amounts of delivered mRNA.
Expression pattern, dosing, duration of protein translation and the amount of produced protein are the key determinants for in vivo use of mRNA encoding therapeutic proteins. Intradermally and intranodally administered naked and protamine complexed mRNA vaccines have entered clinical trials (ClinicalTrials.gov identifiers: NCT01915524, NCT01817738 and NCT01684241) but there have been no human trials on systemic delivery of mRNA to replace or deliver a therapeutic protein. Different applications have varying requirements for dosing, duration of expression, intracellular versus extracellular function and site of activity. Moderate protein expression from the mRNA that also stimulates the innate immune system for a few days might be sufficient to induce immune responses but will not be successful for supplementing therapeutic proteins. Here, we show that in vivo administration of mRNA-LNP complexes by various commonly used injection routes displays different expression patterns and kinetics. Depending on the site of injection, both local protein production and dissemination to the liver occurs. Interestingly, we observed that with intramuscular injection, the depth into the muscle also determined whether the majority of protein produced was in the muscle, for superficial injections, or in the liver, for deep muscular administration (unpublished observations). Importantly, very low doses (0.005 mg/kg) of mRNA-LNPs could be translated for several days following the tested delivery routes demonstrating the potential of these formulations for in vivo development.
BALB/c mice received intraperitoneal, intravenous, intradermal, subcutaneous, intramuscular and intratracheal injections of 0.1 μg, 1.0 μg, and 5.0 μg (0.005–0.250 mg/kg) mRNA-LNPs and the expression pattern and translational kinetics of the firefly luciferase was followed over time using in vivo imaging. It has been demonstrated that the depth of the luciferase activity has an impact on the strength of the luminescent signal. Far red and infrared light (between 600 and 900 nm) penetrates more efficiently, as tissues are more translucent, and firefly luciferase contains a significant near-IR component over 650 nm (reviewed in 26). Intraperitoneal, intravenous, and deep intramuscular injections of mRNA-LNPs resulted in strong protein production in the liver. The duration of protein expression in the liver differed from other tissues in that it was short with a rapid decrease in luciferase enzyme activity and return to baseline at day 3 post-injection (Fig. 2), i.e., a short half-life of translation (Table 1). This could indicate that the mRNA and/or the LNPs are turned over more rapidly in the liver. Thus, intravenous administration of mRNA-LNPs resulted in high translation rates obtained for a short amount of time and the highest total amounts of protein produced for the 1.0 and 5.0 μg doses (Fig. 4). Potent delivery of mRNA to liver following systemic administration of LNPs in vivo is in agreement with findings of other laboratories10,19 using siRNA-LNPs with similar compositions to those employed here, which target hepatocytes through ApoE-dependent, receptor-mediated endocytosis. Intramuscular (deep) and intratracheal delivery resulted in protein production both in the liver and at the injection sites (Fig. 2). Protein production for intramuscular delivery, especially superficial injection, in the muscle could be measured for up to 8 days and high amounts of protein was made (Fig. 3 and Fig. 4). Subcutaneous and intradermal delivery resulted in a more moderate peak protein translation at the injection sites with continued production for up to 10 days (Fig. 2–4). In the in vivo studies the strength of the luminescent signal is influenced by 1) the number of cells that are able to take up the mRNA-LNPs in the given tissue, 2) the amount of mRNA-LNPs that are taken up by cells 3) the translational capacity of a specific cell type, and 4) the duration of time that the mRNA-LNP is capable of being taken up by cells and translated (stability). Our data cannot differentiate between these elements. We believe that systemic delivery results in high luminescent signals due to the large number of potential target cells and fast turnover in the liver. Similarly, for i.m. and s.c. deliveries, the high luminescence is likely due to the large number of cells that take up the mRNA-LNPs and also the efficient uptake and translation of the particles. The kinetics of protein translation is different in the case of i.d. delivery. It is known that a relatively small number of cells reside in the intradermal space that can likely be quickly saturated with the nanoparticles that remain stable for an extended period of time, which could explain why weaker signals are measured for a prolonged period of time.
In a similar study, Phua et al. evaluated the in vivo properties of Stemfect-complexed unmodified luciferase encoding mRNAs administered to mice by intravenous, subcutaneous and intranasal routes27. They found that systemic delivery of the mRNA complexes resulted in the fast clearance of particles and exponential decrease in protein production over time in the spleen. Subcutaneously delivered mRNA formulations were expressed for several days at the site of injection. Notably, when they administered up to 26 μg of unmodified luciferase encoding mRNA, they measured 100–1000-fold weaker bioluminescent signals that clearly shows the superior properties of nucleoside-modified and HPLC-purified mRNA to standard mRNA combined with a potent LNP system. The half-life of the intravenously delivered mRNA was also substantially shorter than measured in this study, which can be partly due to the different turnover rates of mRNA-LNPs in different organs (spleen versus liver) but mainly attributable to the increased duration of translation of modified mRNA2,18.
We demonstrate the ability to deliver nucleoside-modified mRNA complexed to LNPs in a dose dependent manner at 6 different commonly used injection sites. Very high levels of protein for an extended duration of time are produced with variation between sites. This offers the additional flexibility, in addition to alterations in the mRNA that regulate peak and duration of translation, to tailor the delivery of encoded protein to match the required therapeutic kinetics. These studies will help to accelerate the transformation of mRNA therapeutics into clinical practice.
5. Conclusion
The expression profile of in vitro and in vivo delivered HPLC-purified, 1-methylpseudouridine, LNP-encapsulated firefly luciferase encoding mRNA was evaluated in this study. mRNA-LNP was efficiently taken up by immortalized, as well as, primary cells in vitro. Our major finding is the outstanding ability of mRNA-LNPs for in vivo mRNA delivery. To our knowledge, this is the first demonstration that a single injection of low dose (0.005–0.250 mg/kg) mRNA administered by various injection routes translated at high levels for up to 10 days depending on the dose and the site of the delivery. Our findings clearly show that mRNA is a promising non-viral delivery platform that has a bright future as a potent therapeutic and that delivery by LNPs that have performed well in clinical trials offers a clear step towards research application and clinical development.
Supplementary Material
Acknowledgments
Support from R01-AI-050484, R01-AI-090788, Bill and Melinda Gates Foundation CAVD grant is acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jirikowski GF, Sanna PP, Maciejewski-Lenoir D, Bloom FE. Reversal of diabetes insipidus in Brattleboro rats: intrahypothalamic injection of vasopressin mRNA. Science. 1992;255:996–998. doi: 10.1126/science.1546298. [DOI] [PubMed] [Google Scholar]
- 2.Kariko K, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nature reviews. Drug discovery. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 4.Weissman D. mRNA transcript therapy. Expert review of vaccines. 2015;14:265–281. doi: 10.1586/14760584.2015.973859. [DOI] [PubMed] [Google Scholar]
- 5.Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 6.Weide B, et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. Journal of immunotherapy. 2009;32:498–507. doi: 10.1097/CJI.0b013e3181a00068. [DOI] [PubMed] [Google Scholar]
- 7.Jayaraman M, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angewandte Chemie. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maier MA, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1570–1578. doi: 10.1038/mt.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akinc A, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nature biotechnology. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love KT, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geall AJ, et al. Nonviral delivery of self-amplifying RNA vaccines. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hekele A, et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerging Microbes & Infections. 2013;2:e52. doi: 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brito LA, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22:2118–2129. doi: 10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardi N, Muramatsu H, Weissman D, Kariko K. In vitro transcription of long RNA containing modified nucleosides. Methods in molecular biology. 2013;969:29–42. doi: 10.1007/978-1-62703-260-5_2. [DOI] [PubMed] [Google Scholar]
- 15.Weissman D, Pardi N, Muramatsu H, Kariko K. HPLC purification of in vitro transcribed long RNA. Methods in molecular biology. 2013;969:43–54. doi: 10.1007/978-1-62703-260-5_3. [DOI] [PubMed] [Google Scholar]
- 16.Machholz E, Mulder G, Ruiz C, Corning BF, Pritchett-Corning KR. Manual restraint and common compound administration routes in mice and rats. Journal of visualized experiments : JoVE. 2012 doi: 10.3791/2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das S, MacDonald K, Chang HY, Mitzner W. A simple method of mouse lung intubation. Journal of visualized experiments : JoVE. 2013:e50318. doi: 10.3791/50318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kariko K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic acids research. 2011;39:e142. doi: 10.1093/nar/gkr695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akinc A, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan X, et al. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Biochemical and biophysical research communications. 2005;328:57–62. doi: 10.1016/j.bbrc.2004.12.137. [DOI] [PubMed] [Google Scholar]
- 21.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Molecular & cellular proteomics : MCP. 2012;11:M111 014050. doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson JF, Hayes LS, Lloyd DB. Modulation of firefly luciferase stability and impact on studies of gene regulation. Gene. 1991;103:171–177. doi: 10.1016/0378-1119(91)90270-l. [DOI] [PubMed] [Google Scholar]
- 23.Midoux P, Pichon C. Lipid-based mRNA vaccine delivery systems. Expert review of vaccines. 2015;14:221–234. doi: 10.1586/14760584.2015.986104. [DOI] [PubMed] [Google Scholar]
- 24.Petsch B, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nature biotechnology. 2012;30:1210–1216. doi: 10.1038/nbt.2436. [DOI] [PubMed] [Google Scholar]
- 25.Fotin-Mleczek M, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. Journal of immunotherapy. 2011;34:1–15. doi: 10.1097/CJI.0b013e3181f7dbe8. [DOI] [PubMed] [Google Scholar]
- 26.Adams ST, Jr, Miller SC. Beyond D-luciferin: expanding the scope of bioluminescence imaging in vivo. Current opinion in chemical biology. 2014;21:112–120. doi: 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phua KK, Leong KW, Nair SK. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. Journal of controlled release : official journal of the Controlled Release Society. 2013;166:227–233. doi: 10.1016/j.jconrel.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.