

Abstract
Cigarette smoking is common despite its adverse effects on health, such as cardiovascular disease and stroke. Understanding the mechanisms that contribute to the addictive properties of nicotine makes it possible to target them to prevent the initiation of smoking behavior and/or increase the chance of successful quit attempts. While highly addictive, nicotine is not generally considered to be as reinforcing as other drugs of abuse. There are likely other mechanisms at work that contribute to the addictive liability of nicotine. Nicotine modulates aspects of the endocrine system, including the thyroid, which is critical for normal cognitive functioning. It is possible that nicotine’s effects on thyroid function may alter learning and memory, and this may underlie some of its addictive potential. Here, we review the literature on thyroid function and cognition, with a focus on how nicotine alters thyroid hormone signaling and the potential impact on cognition. Changes in cognition are a major symptom of nicotine addiction. Current anti-smoking therapies have modest success at best. If some of the cognitive effects of nicotine are mediated through the thyroid hormone system, then thyroid hormone agonists may be novel treatments for smoking cessation therapies. The content of this review is important because it clarifies the relationship between smoking and thyroid function, which has been ill-defined in the past. This review is timely because the reduction in smoking rates we have seen in recent decades, due to public awareness campaigns and public smoking bans, has leveled off in recent years. Therefore, novel treatment approaches are needed to help reduce smoking rates further.
Keywords: Nicotine, Thyroid, Acetylcholine, Cognition, Learning and memory
1. Introduction
A large portion of the population is affected by some form of thyroid disorder, and this can have effects on cognitive function. Some estimates place the percentage of the population with a thyroid disorder at 10% (Muller et al., 1995). The thyroid hormone signaling system is required for proper neural formation (Iglesias et al., 1996; Thompson and Potter, 2000) and low thyroid hormone levels during gestation can contribute to impaired cognitive development (Pop et al., 1999, 2003). Additionally, thyroid dysfunction during adulthood is associated with altered cognitive processes and mental disorders such as major depressive disorder (Mistry et al., 2009). Thyroid function is often implicated in developmental disorders such as cretinism (Mistry et al., 2009; Cheng et al., 2010) and metabolic disorders (Smith et al., 2002). In addition to thyroid function being critical for development, thyroid hormone receptors (TRs) continue to be expressed in the adult brain and have an array of effects that could contribute to adult neural functioning, synaptic plasticity, and cognition.
Thyroid function can be modulated by multiple factors (Koibuchi and Iwasaki, 2006) and an increasing amount of evidence suggests that cigarette smoking can alter thyroid function (Kapoor and Jones, 2005). Currently, little is known about how cognition is affected by cigarette smoking/nicotine-induced thyroid alterations. Nicotine is an addictive drug that has been used throughout history and across the world. It has low reinforcing properties (Henningfield and Goldberg, 1983), suggesting that nicotine possesses other unique qualities that contribute to its high rate of use. Some examples may include the effects of nicotine on attention, learning and memory, and/or anxiety (Gould, 2010; Gould and Leach, 2013). For instance, the ability of nicotine to facilitate maladaptive associations could contribute to context-evoked cigarette cravings (Gould, 2006). In contrast, chronic cigarette smoking/nicotine use is associated with deficits in cognitive function (Durazzo et al., 2010). Specifically, longitudinal studies have identified negative consequences of chronic cigarette smoking on cognition and memory (Richards et al., 2003; Sabia et al., 2008). Further, research suggests there is an association between cigarette smoking and impaired cognitive control later in life (for review, see Poorthuis et al., 2009). In support of a causative role of nicotine in these long-lasting cognitive deficits, adolescent nicotine exposure in rodents caused long-lasting disruptions in attention and memory (Counotte et al., 2009; Mateos et al., 2010; Portugal et al., 2012). Deficits in cognitive processes brought on by chronic nicotine and withdrawal from smoking and nicotine use may contribute to continued use as an attempt by negatively affected individuals to return to baseline levels of cognition (Gould and Leach, 2013). It is possible that nicotine alters thyroid function, and this may underlie some of its cognitive effects.
In this review, we examine how nicotine may interact with thyroid hormone signaling and if it is likely to alter cognitive processes through this interaction. First, background information on thyroid signaling is included in order to understand potential mechanisms whereby nicotine may affect thyroid signaling. To this end, we include a thorough characterization of the thyroid hormone signaling system and how thyroid signaling contributes to synaptic plasticity, learning, and memory so that we may explore how nicotine affects these processes. Then, we review the literature on the relationship between cigarette smoking and thyroid function from both an epidemiological (human) and experimental (rodent) perspective (Table 1).
Table 1.
Glossary of key terms.
| Term | Definition |
|---|---|
| Choline acetyltransferase (ChAT) | Enzyme critical for the synthesis of acetylcholine from acetyl coenzyme A and choline |
| TRα, TRα1 | Thyroid receptor alpha, Thyroid receptor alpha1 |
| TRβ | Thyroid receptor beta |
| Thyroxine, T4 | Primary synthesis product of the thyroid, inactive thyroid hormone |
| Tri-iodothyronine, T3 | Metabolically active form of thyroid hormone |
| Thyroid stimulating hormone, TSH | Regulatory hormone responsible for stimulating thyroid hormone synthesis and release |
| Thyroid binding globulin, TBG | Main thyroid hormone carrier molecule |
| Propylthiouracil, PTU | Hypothyroid-inducing compound that works through the inhibition of thyroxine synthesis and deiodinase 1 activity |
| Bromodeoxyuracil, BrdU | Modified form of thymidine that is incorporated into the DNA of proliferating cells during mitosis |
| Deiodinase 1, 2, 3 | Enzymes that remove iodine molecules from thyroid hormone to activate/inactivate them |
2. Thyroid hormone
2.1. Hormone activation and transport
Thyroid hormones are synthesized in the thyroid gland and circulate throughout the bloodstream (Schussler, 2000). Thyroid hormones are thought to enter the brain through the end feet of astrocytes (Schroeder and Privalsky, 2014). Deiodinase enzymes activate and deactivate thyroid hormones by removing iodine molecules, which alters their metabolic activity. Deiodinases 1 and 2 activate thyroid hormone by converting thyroxine (T4) to tri-iodothyronine (T3), while deiodinase 3 deactivates thyroid hormone by converting T3 to di-iodothyronine (T2) (Gereben et al., 2008). Approximately half of the T4 secreted by the thyroid gland is deiodinated to T3 by deiodinase 1 in the kidney and liver (St Germain et al., 2009). The remainder of T4 to T3 conversion occurs via the activity of deiodinase 2, which has a very specific tissue and sub-cellular localization. In most tissues, deiodinase 2 is located in the endoplasmic reticulum, providing a long-lasting and proximal T3 presence within the nucleus (Gereben et al., 2008). Neurons, however, lack deiodinase 2 activity but contain thyroid hormone receptors (TRs), which can regulate neuronal gene transcription (Cheng et al., 2010). Glial cells, on the other hand, contain deiodinase 2, thus providing the necessary T3 for thyroid regulation of neuronal function (Guadano-Ferraz et al., 1997; Asteria, 1998). Glial cells have traditionally been thought of as playing a supporting role in neural function, but recent evidence suggests glial cells are critically involved in cognitive processes (Ben Achour and Pascual, 2010). It is becoming well accepted that glial cells contain receptors and can release chemical signals, or “gliotransmitters” (for review see Carmignoto, 2000). Glial-derived T3 may be another important contributor to neural function.
Recently, an elegant set of studies showed the thyroid hormone signaling capabilities of glial cells and their relationship to neuronally expressed genes under TR control. Freitas et al. (2010) showed that administration of T4 to a co-culture of neurons and glia enhanced T3-mediated neuronal genes, but had no effect on neuronal gene expression when glial cells were absent. They also found that the glia effect was due to deiodinase 2 activity. These findings show that deiodinase 2 activity in astrocytes converts T4 to T3 to alter transcription of genes in nearby neurons. Alterations in neuronal levels of T3 may affect learning and memory through its effects on gene transcription. Another way to regulate thyroid activity in addition to deiodinase activity is through changes in thyroid hormone transporters, which are important for the proper localization of thyroid hormones. Major thyroid hormone transporters include monocarboxylate transporter 8 (MCT8), which is critical for T3 transport, and the organic anion transporting polypeptides (OATPs), which are critical for T4 transport (Fig. 1). Morte et al. (2010) showed that deiodinase 2 and MCT8 are both required to provide enough T3 for normal transcriptional activity in the cortex of mice.
Fig. 1.
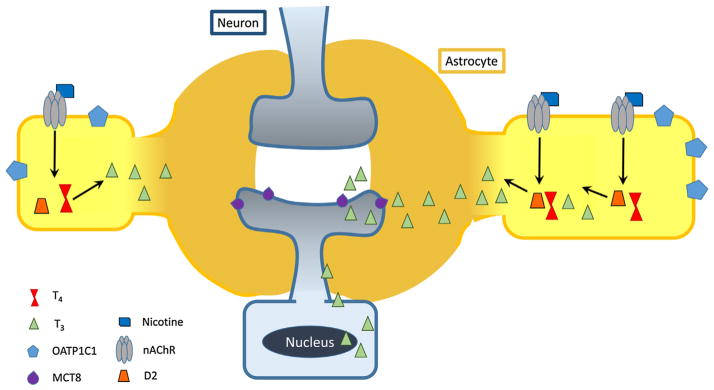
Putative effects of nicotine on thyroid signaling through astrocytes. Nicotine binds to nAChRs on astrocytes to stimulate deiodinase 2 (D2) activity. Deiodinase 2 converts T4 to T3 in astrocytes and active thyroid hormone passively diffuses to nearby neurons. T3 binds to receptors in the nucleus of target neurons where it acts to regulate gene transcription activity. Thyroid hormone transporters with higher affinity for T4 (OATP1C1) and T3 (MCT8) are included on astrocytes and neurons, respectively.
Thyroid-specific enhancer-binding protein (T/EBP), or thyroid transcription factor 1 (TTF-1) is critical for normal thyroid development and is also expressed in the brain (Lazzaro et al., 1991). Overall, thyroid hormones are controlled by a complex hormone signaling system commonly referred to as the hypothalamic–pituitary–thyroid axis. Thyroid hormones are secreted by the thyroid gland. Subsequently, they are activated and transported throughout the body and brain where they alter gene product levels in various tissues. The following sections will describe thyroid hormone mechanism of action in order to better understand its role in neural function.
2.2. Thyroid hormones’ role in gene regulation
Thyroid hormones and their receptors constitute a mechanism by which gene transcription can be altered in a highly dynamic way. Thyroid hormone receptors are members of the nuclear receptor superfamily that includes the retinoic acid receptors, vitamin D3 receptor, and steroid receptors (Gronemeyer et al., 2004). Thyroid hormone receptors bind to specific DNA sequences called thyroid hormone response elements (T3REs) consisting of the sequence (A/G)GGT(C/A/G)A. Thyroid hormone response elements occur most often as direct repeats (i.e., two iterations of the same sequence) separated by 4 nucleotides. Thyroid hormone receptors exist in homo- or heterodimeric complexes. Often, retinoic acid receptors will form a complex with TRs to form these heterodimers. Various genes may be differentially controlled by homodimeric TR complexes compared to heterodimeric receptor complexes (Wu et al., 2001). Unlike some of the other hormone receptors, TRs are found constitutively bound to DNA with or without the hormone present and they recruit different complexes depending on whether they are liganded or unliganded. Thyroid hormone receptors are encoded by two genes (α and β) and alternative splicing creates several isoforms of these receptors. The most abundant isoforms of TRs in the brain are TRα1, TRα2, TRβ1 and TRβ2 (Cheng et al., 2010). Currently, little is known about the specific TR isoforms responsible for control of specific gene regulation, although recent studies have made progress in this regard (Gil-Ibanez et al., 2013). Gene regulation effects of thyroid hormone are carried out through epigenetic mechanisms that include the recruitment of co-activators and co-repressors that function as histone acetyltransferases and histone deacetylation enzymes (Cheng et al., 2010). These histone modifications alter the physical structure of DNA making it more or less accessible for the transcriptional machinery. Thyroid-associated epigenetic mechanisms regulate N-methyl-D-aspartate (NMDA) receptor signaling. Lee et al. (2003) found that changes in thyroid status affects hippocampal NR1 and NR2b expression. These findings underscore the importance of thyroid functioning to intact neuronal signaling, as NMDA receptors are a key component of synapses that participate in synaptic plasticity and other learning and memory processes (Gould et al., 2002).
Furthermore, thyroid signaling regulates brain development through changes in expression of genes such as reelin (Pathak et al., 2010; Sui and Li, 2010; Sui et al., 2010). Reelin is a protein critical for proper neuronal migration and cortical layering (D’Arcangelo et al., 1995; Curran and D’Arcangelo, 1998; Pathak et al., 2010; Tan et al., 2010). TRs control reelin gene expression through epigenetic mechanisms (Sui and Li, 2010). Reelin is typically associated with disorders of development, but the protein is also present in adult neuronal tissue (Garcia-Ayllon et al., 2003; Beffert et al., 2005; Chen et al., 2005; Qiu et al., 2006; Pujadas et al., 2010) and is implicated in many functions related to synaptic plasticity. Reelin gene expression remains under TR control during adulthood, as it was during development. Reelin mRNA and protein were upregulated in the hippocampus after systemic or direct hippocampal infusions of T3 (Sui et al., 2010). Reelin has been implicated in both glutamate signaling in the adult brain (Qiu et al., 2006) and normal cholinergic signaling (Sigala et al., 2007). Therefore, the effects of thyroid signaling on neurotransmission may be mediated in part by reelin.
2.3. Thyroid hormones’ effects on cognition
Thyroid hormones have long been implicated in learning and memory processes. As will be reviewed, changes in thyroid hormone signaling both during development and adulthood can disrupt learning and memory. Further, supplemental thyroid hormone treatment can, in certain situations, improve cognition. Various rodent models indicate that a lack of adequate thyroid hormone during development disrupts learning and memory processes. Studies with a genetic mouse (hyt/hyt) model of hypothyroidism revealed that congenital hypothyroidism caused deficits in spatial and non-spatial navigation (Anthony et al., 1993). The work of Provost et al. (1999) demonstrated polychlorinated bisphenol treatment during development significantly reduced T3 and T4 levels after treatment ended, which also led to spatial navigation deficits in adulthood. Even transient periods of reduced exposure to adequate thyroid hormones during development can lead to long-lasting effects on cognition (Opazo et al., 2008). It has also been suggested that brief periods of lower thyroid hormone levels in pregnant mothers, especially prior to fetal production of thyroid hormones, can produce an autism-like phenotype in children (Roman, 2007; Sullivan, 2009; Hoshiko et al., 2011; de Cock et al., 2012; Roman et al., 2013).
In addition to thyroid hormone being important during development, thyroidectomy-induced hypothyroidism occurring in adulthood produces learning and memory impairment as well. Briefly, both short and long-term spatial working memory are disrupted in this “adult-onset” model of hypothyroidism (Alzoubi et al., 2006b, 2009). Additionally, supplemental thyroid hormone abolished the hypothyroidism-induced deficits (Alzoubi et al., 2009). Thyroid disruption, either in adulthood or during development, can produce profound neurocognitive symptoms, and environmental factors, such as polychlorinated bisphenols and nicotine (as will be described later), that contribute to thyroid dysfunction should be identified and avoided.
Further support of a role for thyroid signaling in learning and memory processes comes from studies that administered supplemental thyroid hormone to rodents. Smith et al. (2002) found enhanced Morris water maze performance in rats administered levothyroxine. Similarly, Leach et al. (2014) demonstrated that levothyroxine administered to mice prior to contextual and delay-cued fear conditioning enhanced performance in both learning paradigms. Further, infusion of T3 directly into the hippocampus also enhanced contextual and trace-cued fear conditioning (Sui et al., 2006). Taken together, these studies reveal that thyroid hormone can directly enhance cognitive performance in a variety of hippocampus-dependent (and some hippocampus-independent) tasks.
Supplemental thyroid hormone is also effective at reversing deficits in various pharmacological and disease models. Specifically, levothyroxine reversed scopolamine-induced deficits in the Morris water maze (Smith et al. 2002), suggesting an interaction between acetylcholine and thyroid hormone signaling. Levothyroxine has also demonstrated efficacy in reversing amyloid pathology in a mouse model of Alzheimer’s disease, a disease characterized by loss of cholinergic function (Fu et al., 2010). Finally, levothyroxine administered to mice experiencing nicotine withdrawal completely abolished deficits in hippocampal learning (Leach et al., 2014). To determine a potential mechanism for the effects of thyroid hormone on learning and memory, Smith et al. (2002) demonstrated that administration of levothyroxine to rats led to an increase in cholinergic activity in the hippocampus, as assessed by cholinesterase levels. This finding suggests a direct link between these two signaling systems. Cholinergic signaling in the central nervous system is important for many cognitive processes (Hasselmo and Bower, 1993; Sarter and Bruno, 1997; Hasselmo, 2006) and nicotine signaling through hippocampal nAChRs are important for its effects on learning, memory, and synaptic plasticity (for review, see Gould and Leach, 2013).
Genetic alterations of thyroid receptors (TRs) also affect cognition. TRα1 mutant mice, harboring a mutation rendering TRα1 highly resistant to thyroid hormone (i.e., 10-fold lower affinity for the hormone), showed a deficit in novel object recognition (Venero et al., 2005) and supplemental thyroid hormone ameliorated the deficit by overcoming the low-affinity receptor binding and returning the system to a functionally euthyroid state (i.e., normal thyroid function). Similarly, Wilcoxon et al. (2007) showed that by reducing thyroid hormone synthesis and activation in TRα KO mice, a striking deficit in Morris water maze performance was observed compared to wildtype mice exposed to the same treatments (Wilcoxon et al., 2007), indicating that the knockouts were more susceptible to thyroid disruption. Further, TRα1 knockout mice demonstrated normal delay-cued and contextual fear conditioning at 24 h, but showed a deficit in extinction when tested for contextual freezing one week later (Guadano-Ferraz et al., 2003), indicating there may be subtle alterations in cognition regardless of thyroid hormone status in TRα1 knockout mice. TRα, and TRα1 in particular, are clearly important for thyroid’s effects on cognition, particularly when thyroid function is disrupted.
A mouse strain that expresses low-affinity TRβ(TRβ1 and TRβ2) also displayed learning deficits in an operant lever pressing task (McDonald et al., 1998). Similar to the results described above in low-affinity TRα1 mutants, a functionally hypothyroid condition is produced by the low-affinity receptor mutant model that likely drives the observed cognitive deficits. When tested under euthyroid conditions, complete TRβ knockouts (TRβ1 and TRβ2) also showed normal performance in several hippocampus-dependent memory tasks (Forrest et al., 1996b; Leach et al., 2015). TRβ KO mice did exhibit reductions in auditory cued fear conditioning (Leach et al., 2015), but this was presumably driven by deficient auditory processing observed in these mice (Forrest et al., 1996a). No studies to date have evaluated the effect of thyroid disruption on cognition in TRβ knockout mice. It is possible that inducing thyroid dysfunction would reveal deficits in cognitive processes in these mice, similar to those observed in TRα1 knockouts. As with TRα1, TRβ function is important for cognition, especially when thyroid function at the receptor is functionally disrupted. Various rodent models have been used to study the specific contributions thyroid signaling makes to neural development, neural functioning, and cognitive processing (see Table 2 for summary). Thyroid hormone signaling has far reaching effects on cognition. Thyroid signaling affects learning and memory in both hippocampus-dependent and hippocampus-independent paradigms, indicating the potential for broad involvement in cognitive processes. Therefore, the effects of nicotine on thyroid function could perturb cognition.
Table 2.
Behavioral effects observed in mice harboring mutations related to thyroid function or wildtype rats with thyroid hormone status manipulation.
| Gene | Mutation | Manipulation | Effect on cognition | Task used | Neural effects | Effect on transcription | Citation |
|---|---|---|---|---|---|---|---|
| Deiodinase 2 | Knockout | Euthyroid and hypothyroid (PTU) | N/A | N/A | Minimal | Abnormal regulation of negatively regulated genes | Morte et al. (2010) |
| MCT8 | Knockout | Euthyroid and hypothyroid (PTU) | N/A | N/A | N/A | None | Morte et al. (2010) |
| TRα1 | Resistance | N/A | Deficit | NOR | Decrease in neurogenesis | Repression | Venero et al. (2005) |
| TRα1 | Knockout | N/A | No effect Deficit |
CFC Extinction |
Increase in neurogenesis | No longer regulated by TRα1 | Guadano-Ferraz et al. (2003) and Kapoor et al. (2010) |
| TRα | Knockout | Hypothyroid (PTU) | Deficit | MWM | N/A | No longer regulated by TRα | Wilcoxon et al. (2007) |
| hyt/hyt | Hypothyroid | N/A | Deficit | MWM | N/A | Repression | Anthony et al. (1993) |
| TRβ | Resistance | N/A | N/A | N/A | Repression | Portella et al. (2010) | |
| TRβ1 | Resistance | N/A | Deficit | Operant | N/A | Repression | McDonald et al. (1998) |
| TRβ1 | Knockout | N/A | None | MWM CFC |
Minimal | No longer regulated by TRβ1 | Forrest et al. (1996) and Leach et al. (2015) |
| Wildtype rat | None | Hypothyroid (thyroidectomy) | Deficit | WRAM | Deficits in LTP | Repression | Alzoubi et al. (2006) and Alzoubi et al. (2009) |
| Wildtype rat | None | Hyperthyroid (T3 or T4 administration) | Enhancement | CFC, MWM | Increased Reelin and cholinergic activity | Activation | Sui et al. (2006) |
| Wildtype rat | None | Subclinical hypothyroid (hemi-thyroidectomy) | Deficit | MWM | Increased ERK activity | Localized effects | Ge et al. (2012) |
↓ = decrease, ↑ = increase, ↔ = no change, (n.d.) = not determined, fT3 = free T3, and fT4 = free T4.
2.4. Thyroid hormones’ effects on synaptic plasticity
Thyroid hormone signaling contributes to short- and long-term synaptic plasticity in normal individuals and any alterations by nicotine would perturb this functioning. Thyroidectomies led to profound deficits in paired pulse facilitation (a model of short-term synaptic plasticity) and increased neurotransmitter release, both of which were reversed by thyroid hormone administration (Vara et al., 2002). This indicates that normal thyroid hormone signaling is critical to intact short-term synaptic plasticity. Thyroid hormone is critical for the development of hippocampal (CA1) LTP (Niemi et al., 1996). The work of Gerges and colleagues have demonstrated that thyroid hormone is important for both the early and late phases of LTP (Gerges et al., 2001; Gerges and Alkadhi, 2004), which are protein synthesis-independent and dependent, respectively (Frey et al., 1988). Further, Gerges, Alkadhi and colleagues provided molecular evidence that a reduction in thyroid hormone leads to a reduction in phosphorylated levels of hippocampal extracellular-signal regulated kinase (ERK1/2), which is critical for hippocampus-dependent learning and memory (Atkins et al., 1998; Selcher et al., 1999; Adams et al., 2000; Szapiro et al., 2003). Alzoubi and Alkadhi (2007) also determined that thyroidectomized rats demonstrated reduced levels of adenylyl cyclase I, calcium calmodulin kinase IV (CaMKIV), and phosphorylated and total cyclic adenosine monophosphate response element binding protein (CREB) and that hippocampal slices subjected to LTP protocols did not show activity-dependent changes in hypothyroid rats. These studies provide further evidence that intact thyroid hormone signaling is critical to synaptic plasticity that underlies the molecular basis of learning and memory. Another set of studies revealed a significant disruption of LTP in hippocampal tissue from hypothyroid rat pups (Taylor et al., 2008). Overall, deficient thyroid hormone signaling leads to disrupted synaptic plasticity.
2.5. Thyroid hormones’ effect on neural development and adult neurogenesis
Thyroid hormone is critical for successful brain development, and therefore it is of interest to determine what bi-directional effects environmental exposures, such as cigarette smoking, may have on this system. Specifically, there is strong evidence that TRβ signaling is required for normal cerebellar development (Portella et al., 2010). In mice lacking TRβ completely, the majority of thyroid hormone responsive genes were normally expressed (Gil-Ibanez et al., 2013), indicating that other transcription factors maintain control over these genes even in the absence of regulation by TRβ. In addition to being implicated in cerebellar development, it has been demonstrated that thyroid signaling has a critical role in hippocampal development as well (Madeira et al., 1992; Gong et al., 2010a,b). Briefly, hypothyroidism led to decreased cell survival in the hippocampus (Gong et al., 2010a) and decreased nerve fiber numbers (Gong et al., 2010b). Further, hippocampal sections from hypothyroid rats showed reduced pyramidal layer volume (CA1 and CA3) and reduced number of pyramidal cells in CA1.
There is also evidence that TRα regulates adult neurogenesis (Kapoor et al., 2010). These studies provide evidence that TRs regulate adult hippocampal neurogenesis and the levels of thyroid hormone play a critical role in this process. It has been suggested that adult neurogenesis in the hippocampus is critical for some forms of learning and memory (Shors et al., 2002). This is a potential mechanism whereby intact thyroid signaling plays a crucial role in cognitive processes. Conversely, TRβ (TRβ1 and TRβ2) knockout mice exhibited increased progenitor activity, but did not show increased neurogenesis after 4 weeks (Kapoor et al., 2011). This indicates a potential contribution to adult neurogenesis, but the relationship is unclear and requires further study.
3. Interaction between nicotine and thyroid
3.1. Nicotine’s effects on thyroid function
There has been a long standing interest in the effect of cigarette smoking on endocrine function and this interest includes putative effects of nicotine on the thyroid hormone system (Kapoor and Jones, 2005). As cigarette smoke contains many chemicals, an important issue is which components have the greatest effect on endocrine function. Nicotine is the main addictive component of cigarette smoke (USDHHS, 1988), but there are also other toxic compounds that are absorbed through smoking. Non-nicotine toxins in cigarettes, such as thiocyanate and 2,3-hydroxypyridine, may cause disruption in thyroid function (Kapoor and Jones, 2005). Thiocyanate decreases iodide absorption and causes goiter in the absence of sufficient iodine. Decreased iodide absorption directly reduces the capacity of the thyroid to synthesize T4. 2,3-Hydroxypyridine prevents de-iodization and activation of T4, indicating that there may be a reduced supply of the active thyroid hormone, T3. Nicotine seems to have effects on thyroid function, but these other components of cigarette smoke may also contribute to changes in thyroid function. The vast number of chemicals found in cigarette smoke makes it difficult to determine the specific components responsible for any alterations in thyroid activity in human cigarette smokers.
There is a strong association between smoking and Grave’s disease, a disorder of severe hyperthyroidism (Bartalena et al., 1989; Prummel and Wiersinga, 1993; Bertelsen and Hegedus, 1994; Utiger, 1998; Vestergaard, 2002). One interpretation of this association is that smoking may increase thyroid secretion, directly contributing to the development of a hyperthyroid state; another is that smoking decreases thyroid secretion and that smokers with Grave’s disease smoke in an effort to self-medicate. In support of the former, several studies demonstrated that smokers had higher thyroid hormone levels than non-smokers. One study demonstrated that smokers had higher serum T3 levels, with no difference in serum T4 or thyroid stimulating hormone (TSH) (Christensen et al., 1984), indicating potential hyperthyroidism. The thyroid and pituitary contain a negative feedback loop, such that higher levels of thyroid hormones act to reduce the release of TSH from the pituitary. Therefore, TSH is often used as a measure of thyroid function (although TSH levels alone do not describe the full range of thyroid function). A second study demonstrated that smokers had higher T4 and lower TSH than non-smokers (Fisher et al., 1997), also suggesting hyperthyroidism. Another study demonstrated no difference in serum T3, but significantly lower TSH levels in smokers (Ericsson and Lindgarde, 1991), suggesting subclinical hyperthyroidism. A fourth study, only examining TSH levels, found lower TSH in current and former smokers compared to never smokers (Asvold et al., 2007), which may indicate subclinical hyperthyroidism. Additionally, one study identified changes in thyroid function including higher free T3 (i.e., hormone cleaved from thyroxine-binding globulin, TBG), higher free T4, and lower TSH (Vejbjerg et al., 2008). Further, even brief passive/secondhand smoke exposure significantly increased total T3 and free T4 levels (Metsios et al., 2007; Flouris et al., 2008). It is important to note that some studies looked specifically at free levels of hormone, while others looked at overall (bound and free) levels. Measurements of free hormone levels are often described as an assessment of the readily available hormone; however, total levels of hormone, including those bound to TBG, may represent hormone reserves that would be available for substantial localized use should the system require it (Schussler, 2000).
It is also possible that smoking decreases thyroid secretion, leading to attempts of self-medication in patients with Grave’s disease. One study showed that smoking cigarettes decreased T4 levels (Banerjee and Muthu, 1994). Another study found that heavy smokers had reduced levels of T4 and T3 (Sepkovic et al., 1984). Smoking also increased the incidence of hypothyroidism in subjects with Hashimoto’s thyroiditis (Fukata et al., 1996), an autoimmune disease characterized by antibodies that develop against thyroglobulin and thyroid peroxidase, two molecules critical for thyroid hormone synthesis (Pearce et al., 2003). Similarly, Muller et al. (1995) compared smokers versus non-smokers in several categories of thyroid function: women with normal thyroid function (euthyroid), women who were subclinically hypothyroid, and women with overt hypothyroidism. The authors found that subclinical hypothyroid women who smoked had significantly higher T3 to free T4 ratios than non-smoking subclinical hypothyroid women, driven by a significant decrease in free T4 levels (p < 0.05). In addition, hypothyroid women who smoked exhibited more severe clinical symptoms of hypothyroidism than non-smoking hypothyroid women (Muller et al., 1995). Further, studies have found that smoking increased the rate of hypothyroxinemia (low T4) in women of reproductive age (Vanderver et al., 2007), which may adversely affect their children (this will be discussed in more detail later in this review). Maternal smoking during pregnancy may also induce a mild iodine deficiency, which indicates a disruption in thyroid function (Hiéronimus et al., 2012). Mild iodine deficiency during pregnancy contributed to long-lasting cognitive deficits in children (Bath et al., 2013), presumably due to iodine’s role in thyroid hormone synthesis.
Finally, a recent report found that smoking cessation was associated with a dramatic increase in the risk (i.e., odds ratio of 7.36 and 6.34 for those who quit smoking less than 1 year ago or 1–2 years ago, respectively) of developing autoimmune hypothyroidism (Carle et al., 2012), suggesting that withdrawal from chronic nicotine may also disrupt thyroid function. It is possible that smoking cigarettes has differential effects based on the population studied, such that individuals who smoke may be predisposed to thyroid disruption if they have pre-existing thyroid disease, are women of reproductive age, or are heavy smokers. The positive and negative correlations between smoking and thyroid function are summarized in Table 3.
Table 3.
Overview of human cigarette smoking and thyroid function studies.
| Population | Smoking status | Main findings | Citation | Interpretation |
|---|---|---|---|---|
| Grave’s hyperthyroidism | Chronic smokers | 47.9% of Grave’s disease | Bartalena et al. (1989) | Grave’s patients smoke at extremely high rates compared to the general population |
| Chronic smokers | 64.2% of Grave’s opthalmopathy | |||
| Grave’s hyperthyroidism | Chronic smokers | 1.9× ↑ risk of Grave’s disease | Prummel and Wiersinga (1993) | Smoking increases the risk of developing Grave’s disease and Grave’s opthalmopathy |
| Chronic smokers | 7.7× ↑ risk of Grave’s opthalmopathy | |||
| Euthyroid | Chronic smokers | ↑T3, ↔T4, ↔TSH | Christensen et al. (1984) | Smoking increases one measure of thyroid function (T3) |
| Former smokers | ↔T3, ↔T4, ↔TSH | |||
| Euthyroid | Chronic smokers | ↔T3, (n.d.)T4, ↓TSH | Ericsson and Lindgarde (1991) | Smoking increases thyroid function (only in indirect measurement) |
| Former smokers | ↔T3, (n.d.)T4, ↓TSH | |||
| Euthyroid | Chronic smokers | (n.d.)T3, ↑T4, ↓TSH | Fisher et al. (1997) | Smoking increases thyroid secretion |
| Euthyroid | Chronic smokers | (n.d.)T3, (n.d.)T4, ↓TSH | Asvold et al. (2007) | Smoking increases thyroid function (only indirectly assessed) |
| Former smokers | (n.d.)T3, (n.d.)T4, ↓TSH | |||
| Euthyroid | Chronic smokers | ↑fT3, ↑fT4, ↓TSH | Vejbjerg et al. (2008) | Smoking increases thyroid hormone function |
| Euthyroid | Secondhand smoke | ↑T3, ↑fT4, (n.d.)TSH | Metsios et al. (2007) and Flouris et al. (2008) | Smoking increases thyroid hormone function |
| Euthyroid | Chronic smokers | ↓fT3, ↓fT4, ↑TSH | Soldin et al. (2009) | Smoking decreases thyroid hormone function |
| Secondhand smoke | ↓fT3, ↓fT4, ↑TSH | |||
| Euthyroid | Chronic smokers | ↑fT3, ↑fT4, ↓TSH | Jorde and Sundsfjord (2006) | Smoking increases thyroid hormone function |
| Euthyroid | Filtered cigarette smokers | ↔T3, ↓T4, ↑TSH | Banerjee and Muthu (1994) | Smoking reduces thyroid function |
| Non-filtered cigarette smokers | ↔T3, ↓T4, ↑SH | |||
| Euthyroid | Light and moderate smokers | ↔T3, ↔T4, ↔TSH | Sepkovic et al. (1984) | Heavy smoking reduces thyroid function |
| Heavy smokers | ↓T3, ↓T4, ↔TSH | |||
| Hashimoto’s thyroiditis | Chronic smokers | 76.4% hypothyroid | Fukata et al. (1996) | Smoking increases the rates of hypothyroidism in subjects with Hashimoto’s thyroiditis |
| Former smokers | 61.9% hypothyroid | |||
| Women 15–44 years old | Chronic smokers (≥31/day) | ↑ rate of hypothyroxinemia normal rate of hypothyroxinemia | Vanderver et al. (2007) | Heavy smoking increases rate of severe low T4 syndrome |
| Chronic smokers (≤30/day) | ||||
| Euthyroid | Chronic smokers | ↔T3, ↔ T4, ↔TSH | Muller et al. (1995) | Smoking reduces thyroid function in subclinical hypothyroid subjects |
| Subclinical hypothyroid | Chronic smokers | ↔T3, ↓T4, ↑TSH | ||
| Hypothyroid | Chronic smokers | ↔T3, ↔T4, ↔TSH | ||
| Hypothyroid | Former smokers (<1 year) | 7.36× ↑ risk of hypothyroidism | Carle et al. (2012) | Smoking cessation increases the risk of developing overt hypothyroidism |
| Hypothyroid | Former smokers (1–2 years) | 6.34× ↑ risk of hypothyroidism |
↓ = decrease, ↑ = increase, ↔ = no change, (n.d.) = not determined, fT3 = free T3, and fT4 = free T4.
Studies utilizing rodent models are better able to elucidate the cause-effect relationship between nicotine and thyroid signaling due to the experimental control these models provide. Few rodent studies have evaluated the effects of nicotine on adult thyroid hormone function. First, acute nicotine administered to adult male rats did not affect T3 or T4 levels at any point during a 24-h period (Cam and Bassett, 1983). Because nicotine may differentially affect subjects with various predispositions (as described above), nicotine was evaluated in rodent models of hypothyroidism (thyroidectomy-induced). Twice daily injections of nicotine for 4–6 weeks did not affect T4 or TSH levels in either euthyroid or hypothyroid adult male rats (Alzoubi et al., 2006b). Similarly, Colzani et al. (1998) carried out a series of experiments to determine the effects of chronic nicotine on thyroid function in male euthyroid, subclinical hypothyroid, and hypothyroid rats. There was no effect of nicotine on T3, T4, or TSH in any of the groups tested.
Null effects described above may be due to several possibilities. First, based on human association studies, women (particularly those at reproductive age or who have subclinical thyroid dysfunction) are at the highest risk for smoking-induced thyroid disruption (Muller et al., 1995; Vanderver et al., 2007), whereas men were less likely to be affected. In support of this, de Oliveira et al. (2011) found that exposure of chronic nicotine to lactating dams significantly lowered maternal free T4, suggesting that chronic nicotine may produce a hypothyroid state in these rats. A second possibility for the null effects described above is that the doses of nicotine tested were too low to affect thyroid function, as human studies suggest that heavy smokers may be at increased risk for thyroid dysfunction (Sepkovic et al., 1984). To address this other possibility, and because it is also possible that withdrawal from chronic nicotine may affect thyroid function (Carle et al., 2012), Leach et al. (2014) tested the effects of chronic and withdrawal from chronic nicotine on thyroid hormone levels at a dose of nicotine that more closely approximates the plasma-nicotine levels observed in heavy smokers (Turner et al., 2011, 2013; Wilkinson and Gould, 2013). The results indicated that nicotine withdrawal significantly reduced serum T4 levels, and that both chronic and withdrawal from chronic nicotine treatment increased the ratio of T3 to T4, indicating potential alterations in thyroid hormone function.
Thorough analysis of the effects of cigarette smoking and nicotine on thyroid function reveal some key findings. Several factors likely contribute to whether cigarette smoking alters thyroid function and in what direction. Individuals with Grave’s disease and Grave’s opthalmopathy are more likely to smoke and this may represent an attempt at self-medication to reduce high levels of thyroid hormone or it may represent cigarette smoking-induced thyroid hyperfunctionality. Other factors appear to increase the likelihood that smoking will disrupt thyroid function. These factors include sex, particularly women at reproductive age or who have subclinical thyroid dysfunction (Muller et al., 1995; Vanderver et al., 2007), heavy smoking (Sepkovic et al., 1984), and autoimmune thyroid disease (Fukata et al., 1996). Additionally, both smoking cessation in humans (Carle et al., 2012) and withdrawal from chronic nicotine in mice (Leach et al., 2014) disrupt thyroid function. Individuals who smoke, or are attempting to quit smoking, with any of these potential risk factors would likely benefit from careful thyroid monitoring by a health professional.
3.2. Thyroid effects on cholinergic function
Just as nicotine may alter thyroid function, thyroid hormone may directly regulate cholinergic signaling by controlling the expression of the gene responsible for the synthesis of acetylcholine, ChAT. The ChAT gene is regulated by TRs (Quirin-Stricker et al., 1994). Many in vitro and in vivo studies have correlated thyroid hormone signaling with ChAT protein expression, especially during development (Juarez de Ku et al., 1994; Quirin-Stricker et al., 1994; Sawin et al., 1998; Provost et al., 1999). Thyroid hormone control of gene and subsequent protein expression are important for many metabolic processes including signaling systems critical for learning, memory, and synaptic plasticity (i.e., NMDA receptors, reelin, and ChAT). Thus, the ability of thyroid to control gene expression could contribute to cell signaling cascades critical for learning and memory.
The authors were unable to find evidence of thyroid control of nicotinic acetylcholine receptor expression or trafficking. There have, however, been observed links between the thyroid-specific enhancer-binding protein (T/EBP), also known as thyroid transcription factor-1 (TTF-1) or Nkx2.1, and the nicotinic cholinergic system. Specifically, TTF-1 was shown to regulate the α5 subunit of the nAChR in lung epithelial cells (Reynolds et al., 2010) and α7 subunits in the lung (Reynolds and Hoidal, 2005). Other neural activities of TTF-1 are facilitating cerebrospinal fluid development (Kim et al., 2007) and successful development of the ventral fore-brain and the pituitary (Kimura et al., 1996).
3.3. Developmental effects of cigarette smoking and nicotine on thyroid function
Smoking has been implicated in altered thyroid function in adult smokers, yet the effects of exposure to cigarette smoking on development may be even more profound. Parents who smoke may risk long term neurodevelopmental harm to their children, which could translate into deficits in learning, memory, and the synaptic plasticity underlying these processes. While it is important to understand the risk factors and extent to which cigarette smoking affects thyroid function in the adult, it is even more critical to understand the adverse effects of cigarette smoke on children of smokers because it may have long-lasting effects on their quality of life.
As mentioned previously, smoking during pregnancy induced a mild iodine deficiency (Hiéronimus et al., 2012), which may contribute to long-lasting cognitive deficits in children (Bath et al., 2013). Prospective iodine treatment of smoking-induced mild iodine deficiency ameliorated negative effects on infant thyroid function (Hiéronimus et al., 2012), which may reduce the risk of adverse cognitive effects in these children. Similarly, a study conducted by Chanoine et al. (1991) observed an increase in the size of the thyroid in infants born to smoking mothers. This occurred with a corresponding increase in thyroglobulin concentrations in the same infants (Chanoine et al., 1991), which has been proposed as a marker of iodine deficiency (Pacini et al., 1984; Sava et al., 1986). Recently, animal studies have begun to elucidate the long-term consequences on offspring of mothers exposed to chronic nicotine (Oliveira et al., 2009; de Oliveira et al., 2011) and chronic cigarette smoke (Santos-Silva et al., 2013).
Rodent models have demonstrated that prenatal and perinatal nicotine administration disrupted thyroid function. Oliveira et al. (2009) demonstrated that nicotine administration to a lactating rat dam produced lower serum free T3 and free T4 with higher TSH levels when the pups were examined at 15 days of age. Recently, de Oliveira et al. (2011) revealed that exposure of lactating dams to chronic nicotine reduced free T3 and free T4 levels in offspring. The findings from these studies revealed short- and long-term detrimental effects of maternal nicotine exposure to thyroid function in rats, without the confound of other toxic compounds such as thiocyanate or 2,3-hydroxypyridine.
Findings describing the developmental effects of nicotine on thyroid function are not entirely consistent. For instance, one study revealed decreased TSH levels in infants whose parents smoke (Meberg and Marstein, 1986), which would suggest a hyperthyroid state. It is unclear what may underlie these discrepant findings. Further, rodent studies have revealed similar contrary findings to those described above. Specifically, some experiments that administered nicotine to pregnant dams identified no differences in thyroid function in the pups (Chen and Kelly, 2005). These results did, however, reveal an increase in the offspring body weights when they were evaluated in adulthood, indicating a likely change in offspring metabolism. However, Chen and Kelly (2005) only looked at thyroid hormone levels at one developmental time point (10 day old pups) and therefore, long-term effects on thyroid function after maternal nicotine exposure cannot be ruled out. Finally, contrary to what would be expected from the human literature, offspring of mice chronically exposed to tobacco smoke showed no abnormalities in thyroid hormone function (Santos-Silva et al., 2013). Despite these discrepant findings, it seems likely that parental nicotine/smoking has severe long-term effects on thyroid hormone function of the offspring.
Maternal cigarette smoking likely affects fetal thyroid function and this may have long-term detrimental outcomes to the individual. Even subtle alterations in thyroid hormone function during development can lead to long-lasting deficits in cognition (Bath et al., 2013). Since exposure to adequate levels of thyroid hormones throughout development (gestation and beyond) are critical for normal cognitive development (Roman, 2007; Opazo et al., 2008; Sullivan, 2009; Hoshiko et al., 2011; de Cock et al., 2012; Roman et al., 2013), environmental compounds that disrupt thyroid function, like nicotine, should be avoided.
4. Nicotine, thyroid, and cognition
Thyroid signaling has an established role in learning, memory, and cognition, as does nicotine (Gould and Leach, 2013). If nicotine alters thyroid function, then this may contribute to nicotine’s effects on learning and memory. The interaction between nicotine and thyroid signaling in various rodent models have recently been investigated (Table 4). Twice daily nicotine for 4–6 weeks reversed a deficit in water radial arm maze performance in thyroidectomy-induced hypothyroid rats (Alzoubi et al., 2006b). In the same rat model, thyroidectomies caused impairments in LTP and ERK1/2 and CREB phosphorylation (Alzoubi and Alkadhi, 2007) and nicotine reversed these deficits as well (Alzoubi et al., 2006a,b, 2007). Therefore, nicotine may be acting through the ERK1/2 signaling pathway to ameliorate hypothyroidism-induced deficits in LTP, similar to the way nicotine enhances contextual conditioning through ERK1/2 signaling (Raybuck and Gould, 2007). Further, Leach et al. (2014) demonstrated that levothyroxine (L-T4) treatment ameliorated nicotine withdrawal-induced deficits in learning, presumably by normalizing the thyroid dysfunction described in the previous section. Overall, nicotine not only reverses hypothyroidism-induced deficits in learning and synaptic plasticity, but supplemental thyroid hormone reverses nicotine withdrawal-related cognitive deficits as well.
Table 4.
Overview of studies regarding nicotine exposure and thyroid function.
| Population | Experimental conditions | Main findings | Citation | Interpretation |
|---|---|---|---|---|
| Euthyroid | Acute nicotine exposure | ↔T3, ↔T4, (n.d.)TSH | Leach et al. (2015) | Acute nicotine does not affect thyroid function, but thyroid receptors are selectively activated by learning in the presence of nicotine and TRβ is critical for acute nicotine’s effects on learning |
| Euthyroid | 12 day nicotine exposure (24 h withdrawal) | ↔T3, ↓T4, (n.d.)TSH | Leach et al. (2014) | Nicotine withdrawal decreases thyroid function, but chronic nicotine does not affect thyroid function |
| Euthyroid | 13 day nicotine exposure | ↔T3, ↔T3, (n.d.)TSH | ||
| Euthyroid | 7 day nicotine exposure | ↔T3, ↔T3, ↔T3 | Colzani et al. (1998) | Short-term nicotine exposure does not affect thyroid function |
| Hemithyroidectomized | 7 day nicotine exposure | ↔T3, ↔T3, ↔T3 | ||
| Thyroidectomized | 7 day nicotine exposure | ↔T3, ↔T3, ↔T3 | ||
| Euthyroid | 4–6 weeks twice daily nicotine | (n.d.)T3, ↔T4, ↔TSH | Alzoubi et al. (2006) | Nicotine reversed behavioral and electrophysiological deficits of hypothyroidism, but did not affect thyroid function |
| Thyroidectomized | 4–6 weeks twice daily nicotine | (n.d.)T3, ↔T4, ↔TSH | ||
| 15 Day old pups | Nicotine during lactation | ↓fT3, ↓fT4, ↑TSH | Oliveira et al. (2009) | Nicotine decreases thyroid function of offspring (short- and long-term) |
| 180 Day old pups | Nicotine during lactation | ↓fT3, ↓fT4, ↓TSH | ||
| 15 Day old pups | Nicotine during lactation | ↓fT3, ↓fT4, ↑TSH | de Oliveira et al. (2011) | Nicotine decreases thyroid function of mother and offspring (short-term) |
| 21 Day old pups | Nicotine during lactation | ↔fT3, ↔fT4, ↔T3, | ||
| 15 Day maternal exposure (Dams) | Nicotine during lactation | ↔fT3, ↓fT4, ↑TSH | ||
| 21 Day old pups | Smoke exposure during lactation | ↔fT3, ↔fT4, (n.d.)TSH | Santos-Silva et al. (2013) | Smoke exposure for this duration does not affect offspring thyroid hormones |
| 10 Day old pups | Prenatal nicotine exposure | (n.d.)T3, ↔T4, (n.d.)TSH | Chen and Kelly (2005) | Prenatal nicotine does not affect thyroid function at this timepoint |
NOR = novel object recognition, CFC = contextual fear conditioning, MWM = Morris water maze, WRAM = water radial arm maze, and Operant = lever pressing acquisition.
Recent research has implicated thyroid signaling in the acute effects of nicotine on learning. While acute nicotine and/or learning did not affect serum levels of T3, T4, or the T3/T4 ratio, hippocampus-dependent learning in the presence of acute nicotine activated hippocampal TRs (Leach et al., 2015). Leach et al. (2015) tested 319 unique transcription factors, using transcription factor array technology, and 2 out of 3 “hits” for selective effects of nicotine on learning consisted of TR activity (direct repeat 4 and T3RE half-site activity, respectively). Further, the ability of nicotine to enhance contextual learning was abolished in mice lacking TRβ, but was intact in wildtype mice and knockout mice lacking TRα1, indicating a selective role of TRβ in nicotine’s effects on learning (Leach et al., 2015).
Recently, through the use of animal models, a direct link between nicotine and thyroid signaling has been established and this effect seems to modulate learning. Nicotine reversed hypothyroidism-induced learning-related deficits at the molecular, cellular, and behavioral level (Alzoubi et al., 2006a,b, 2007). Further, thyroid hormone normalized nicotine withdrawal-induced behavioral deficits (Leach et al., 2014). Finally, learning in the presence of acute nicotine recruited TR activity, and nicotine-augmented learning required intact TRβ signaling (Leach et al., 2015). An interesting possibility is that acute nicotine increases the supply of neuronal T3 by stimulating deiodinase 2 activity in nearby glial cells (Gondou et al., 1999), which may contribute to observed behavioral effects. In support of this putative relationship, hippocampal astrocytes contain nicotinic acetylcholine receptors (Gahring et al., 2004), and thyroid signaling represents a mechanism whereby glial cells may contribute substantially to neural function. This putative model is described in Fig. 1. Modern immunohistochemical and microscopy techniques may now be capable of assessing nicotine’s transient effects on neural T3 and T4, and their specific localization (i.e., glial vs neuronal and nuclear vs cytosolic), but these studies have not been conducted.
5. Conclusion
Cigarette smoking dynamically alters thyroid function, and the direction of the relationship may relate to the underlying susceptibilities of the individuals involved. For instance, women of reproductive age, women with subclinical hypothyroidism, heavy smokers, or individuals attempting to quit smoking may be at increased risk for hypothyroidism-like symptoms. In contrast, the general population and those with hyperthyroidism may be affected by nicotine in the opposite direction (i.e., produce hyperthyroidism-like effects). It is also possible that subjects with high basal levels of thyroid hormone, including subjects with Grave’s disease, smoke cigarettes to self-medicate, but this supposition requires further testing. Further, cigarette smoking/nicotine administration during pregnancy/lactation alters thyroid hormone status of the offspring, which could have long-lasting effects on cognition, learning, and memory.
The hypothalamic–pituitary–thyroid axis regulates thyroid signaling that can control gene expression in a highly temporal and tissue-specific manner. In the central nervous system, glial-derived deiodinase activity provides active thyroid hormone to neurons and controls transcription of developmentally important and plasticity-related genes such as NMDA receptor subunits, ChAT, critical for cholinergic function, and reelin. Further, thyroid hormone is critical not only during development, but also during adulthood for efficient neural signaling and cognition.
Nicotine’s interaction with thyroid signaling also has consequences for cognitive processes in adulthood. Specifically, learning in the presence of acute nicotine increased TR activity, and intact TRβ was critical for these effects. Further, nicotine withdrawal-induced deficits in learning were associated with disrupted thyroid hormone levels and deficits were reversed/ameliorated by thyroid hormone supplementation. In total, nicotine reversed/ameliorated hypothyroidism-induced deficits in learning and memory at the molecular, cellular, and behavioral level.
In summary, thyroid function should be clinically monitored in cigarette smokers and those interested in quitting smoking, in order to avoid adverse effects on thyroid function and cognition. Successfully monitoring and treating thyroid status may result in significantly higher rates of abstinence, which has remained stubbornly low despite pharmacological interventions. In addition, during pregnancy, thyroid hormone status should be closely monitored in current or former smokers to allow for successful normalization of thyroid function to eliminate the possibility of detrimental effects on the cognitive development of their children.
Acknowledgments
This work was funded with grant support from the National Institute on Drug Abuse (T.J.G., DA017949, DA024747). PTL was supported by NIH-NIDA Training Grant (DA007237). The authors would like to thank Tatiana Kazdoba for her assistance in creating the figure.
References
- Adams JP, Roberson ED, English JD, Selcher JC, Sweatt JD. MAPK regulation of gene expression in the central nervous system. Acta Neurobiol Exp (Warsz) 2000;60:377–394. doi: 10.55782/ane-2000-1357. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Aleisa AM, Alkadhi KA. Molecular studies on the protective effect of nicotine in adult-onset hypothyroidism-induced impairment of long-term potentiation. Hippocampus. 2006a;16:861–874. doi: 10.1002/hipo.20217. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Aleisa AM, Alkadhi KA. Nicotine prevents disruption of the late phase LTP-related molecular cascade in adult-onset hypothyroidism. Hippocampus. 2007;17:654–664. doi: 10.1002/hipo.20306. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Aleisa AM, Gerges NZ, Alkadhi KA. Nicotine reverses adult-onset hypothyroidism-induced impairment of learning and memory: behavioral and electrophysiological studies. J Neurosci Res. 2006b;84:944–953. doi: 10.1002/jnr.21014. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Alkadhi KA. A critical role of CREB in the impairment of late-phase LTP by adult onset hypothyroidism. Exp Neurol. 2007;203:63–71. doi: 10.1016/j.expneurol.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Alzoubi KH, Gerges NZ, Aleisa AM, Alkadhi KA. Levothyroxin restores hypothyroidism-induced impairment of hippocampus-dependent learning and memory: behavioral, electrophysiological, and molecular studies. Hippocampus. 2009;19:66–78. doi: 10.1002/hipo.20476. [DOI] [PubMed] [Google Scholar]
- Anthony A, Adams PM, Stein SA. The effects of congenital hypothyroidism using the hyt/hyt mouse on locomotor activity and learned behavior. Horm Behav. 1993;27:418–433. doi: 10.1006/hbeh.1993.1031. [DOI] [PubMed] [Google Scholar]
- Asteria C. Crucial role for type II iodothyronine deiodinase in the metabolic coupling between glial cells and neurons during brain development. Eur J Endocrinol. 1998;138:370–371. doi: 10.1530/eje.0.1380370. [DOI] [PubMed] [Google Scholar]
- Asvold BO, Bjoro T, Nilsen TI, Vatten LJ. Tobacco smoking and thyroid function: a population-based study. Arch Intern Med. 2007;167:1428–1432. doi: 10.1001/archinte.167.13.1428. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Banerjee KK, Muthu PM. Effect of cigarette smoking on thyroid hormone homeostasis. Ind J Med Res. 1994;99:74–76. [PubMed] [Google Scholar]
- Bartalena L, Martino E, Marcocci C, Bogazzi F, Panicucci M, Velluzzi F, Loviselli A, Pinchera A. More on smoking habits and Graves’ ophthalmopathy. J Endocrinol Invest. 1989;12:733–737. doi: 10.1007/BF03350047. [DOI] [PubMed] [Google Scholar]
- Bath SC, Steer CD, Golding J, Emmett P, Rayman MP. Effect of inadequate iodine status in UK pregnant women on cognitive outcomes in their children: results from the Avon Longitudinal Study of Parents and Children (ALSPAC) Lancet. 2013;382:331–337. doi: 10.1016/S0140-6736(13)60436-5. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Ben Achour S, Pascual O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int. 2010;57:440–445. doi: 10.1016/j.neuint.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Bertelsen JB, Hegedus L. Cigarette smoking and the thyroid. Thyroid. 1994;4:327–331. doi: 10.1089/thy.1994.4.327. [DOI] [PubMed] [Google Scholar]
- Cam GR, Bassett JR. The effect of acute nicotine administration on plasma levels of the thyroid hormones and corticosterone in the rat. Pharmacol Biochem Behav. 1983;19:559–561. doi: 10.1016/0091-3057(83)90135-1. [DOI] [PubMed] [Google Scholar]
- Carle A, Bulow Pedersen I, Knudsen N, Perrild H, Ovesen L, Banke Rasmussen L, Jorgensen T, Laurberg P. Smoking cessation is followed by a sharp but transient rise in the incidence of overt autoimmune hypothyroidism – a population-based, case–control study. Clin Endocrinol (Oxf) 2012;77:764–772. doi: 10.1111/j.1365-2265.2012.04455.x. [DOI] [PubMed] [Google Scholar]
- Carmignoto G. Reciprocal communication systems between astrocytes and neurones. Prog Neurobiol. 2000;62:561–581. doi: 10.1016/s0301-0082(00)00029-0. [DOI] [PubMed] [Google Scholar]
- Chanoine JP, Toppet V, Bourdoux P, Spehl M, Delange F. Smoking during pregnancy: a significant cause of neonatal thyroid enlargement. Br J Obstet Gynaecol. 1991;98:65–68. doi: 10.1111/j.1471-0528.1991.tb10313.x. [DOI] [PubMed] [Google Scholar]
- Chen WJ, Kelly RB. Effect of prenatal or perinatal nicotine exposure on neonatal thyroid status and offspring growth in rats. Life Sci. 2005;76:1249–1258. doi: 10.1016/j.lfs.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I, Herz J. Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SB, Ericsson UB, Janzon L, Tibblin S, Melander A. Influence of cigarette smoking on goiter formation, thyroglobulin, and thyroid hormone levels in women. J Clin Endocrinol Metab. 1984;58:615–618. doi: 10.1210/jcem-58-4-615. [DOI] [PubMed] [Google Scholar]
- Colzani R, Fang SL, Alex S, Braverman LE. The effect of nicotine on thyroid function in rats. Metabolism. 1998;47:154–157. doi: 10.1016/s0026-0495(98)90212-8. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Spijker S, Van de Burgwal LH, Hogenboom F, Schoffelmeer AN, De Vries TJ, Smit AB, Pattij T. Long-lasting cognitive deficits resulting from adolescent nicotine exposure in rats. Neuropsychopharmacology. 2009;34:299–306. doi: 10.1038/npp.2008.96. [DOI] [PubMed] [Google Scholar]
- Curran T, D’Arcangelo G. Role of reelin in the control of brain development. Brain Res Brain Res Rev. 1998;26:285–294. doi: 10.1016/s0165-0173(97)00035-0. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- de Cock M, Maas YG, van de Bor M. Does perinatal exposure to endocrine disruptors induce autism spectrum and attention deficit hyperactivity disorders? Review Acta Paediatr. 2012;101:811–818. doi: 10.1111/j.1651-2227.2012.02693.x. [DOI] [PubMed] [Google Scholar]
- de Oliveira E, de Moura EG, Santos-Silva AP, Pinheiro CR, Claudio-Neto S, Christian Manhaes A, Passos MC, Lisboa PC. Neonatal hypothyroidism caused by maternal nicotine exposure is reversed by higher T3 transfer by milk after nicotine withdraw. Food Chem Toxicol. 2011;49:2068–2073. doi: 10.1016/j.fct.2011.04.040. [DOI] [PubMed] [Google Scholar]
- Durazzo TC, Meyerhoff DJ, Nixon SJ. Chronic cigarette smoking: implications for neurocognition and brain neurobiology. Int J Environ Res Public Health. 2010;7:3760–3791. doi: 10.3390/ijerph7103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson UB, Lindgarde F. Effects of cigarette smoking on thyroid function and the prevalence of goitre, thyrotoxicosis and autoimmune thyroiditis. J Intern Med. 1991;229:67–71. doi: 10.1111/j.1365-2796.1991.tb00308.x. [DOI] [PubMed] [Google Scholar]
- Fisher CL, Mannino DM, Herman WH, Frumkin H. Cigarette smoking and thyroid hormone levels in males. Int J Epidemiol. 1997;26:972–977. doi: 10.1093/ije/26.5.972. [DOI] [PubMed] [Google Scholar]
- Flouris AD, Metsios GS, Jamurtas AZ, Koutedakis Y. Sexual dimorphism in the acute effects of secondhand smoke on thyroid hormone secretion, inflammatory markers and vascular function. Am J Physiol Endocrinol Metab. 2008;294:E456–E462. doi: 10.1152/ajpendo.00699.2007. [DOI] [PubMed] [Google Scholar]
- Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet. 1996a;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. EMBO J. 1996b;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- Freitas BC, Gereben B, Castillo M, Kallo I, Zeold A, Egri P, Liposits Z, Zavacki AM, Maciel RM, Jo S, Singru P, Sanchez E, Lechan RM, Bianco AC. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120:2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Fu AL, Zhou CY, Chen X. Thyroid hormone prevents cognitive deficit in a mouse model of Alzheimer’s disease. Neuropharmacology. 2010;58:722–729. doi: 10.1016/j.neuropharm.2009.12.020. [DOI] [PubMed] [Google Scholar]
- Fukata S, Kuma K, Sugawara M. Relationship between cigarette smoking and hypothyroidism in patients with Hashimoto’s thyroiditis. J Endocrinol Invest. 1996;19:607–612. doi: 10.1007/BF03349026. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Persiyanov K, Dunn D, Weiss R, Meyer EL, Rogers SW. Mouse strain-specific nicotinic acetylcholine receptor expression by inhibitory interneurons and astrocytes in the dorsal hippocampus. J Comp Neurol. 2004;468:334–346. doi: 10.1002/cne.10943. [DOI] [PubMed] [Google Scholar]
- Garcia-Ayllon MS, Segui D, Perales M, Lopez-Hurtado E, Prieto JJ, Saez-Valero J. Acetylcholinesterase level and molecular isoforms are altered in brain of Reelin Orleans mutant mice. J Neurochem. 2003;87:773–779. doi: 10.1046/j.1471-4159.2003.02052.x. [DOI] [PubMed] [Google Scholar]
- Ge JF, Peng L, Hu CM, Wu TN. Impaired learning and memory performance in a sub clinical hypothyroidism rat model induced by hemi-thyroid electrocauterisation. J Neuroendocrinol. 2012;24:953–961. doi: 10.1111/j.1365-2826.2012.02297.x. [DOI] [PubMed] [Google Scholar]
- Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges NZ, Alkadhi KA. Hypothyroidism impairs late LTP in CA1 region but not in dentate gyrus of the intact rat hippocampus: MAPK involvement. Hippocampus. 2004;14:40–45. doi: 10.1002/hipo.10165. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Stringer JL, Alkadhi KA. Combination of hypothyroidism and stress abolishes early LTP in the CA1 but not dentate gyrus of hippocampus of adult rats. Brain Res. 2001;922:250–260. doi: 10.1016/s0006-8993(01)03181-x. [DOI] [PubMed] [Google Scholar]
- Gil-Ibanez P, Morte B, Bernal J. Role of thyroid hormone receptor subtypes alpha and beta on gene expression in the cerebral cortex and striatum of postnatal mice. Endocrinology. 2013;154:1940–1947. doi: 10.1210/en.2012-2189. [DOI] [PubMed] [Google Scholar]
- Gondou A, Toyoda N, Nishikawa M, Yonemoto T, Sakaguchi N, Tokoro T, Inada M. Effect of nicotine on type 2 deiodinase activity in cultured rat glial cells. Endocr J. 1999;46:107–112. doi: 10.1507/endocrj.46.107. [DOI] [PubMed] [Google Scholar]
- Gong J, Dong J, Wang Y, Xu H, Wei W, Zhong J, Liu W, Xi Q, Chen J. Developmental iodine deficiency and hypothyroidism impair neural development, up-regulate caveolin-1 and down-regulate synaptophysin in rat hippocampus. J Neuroendocrinol. 2010a;22:129–139. doi: 10.1111/j.1365-2826.2009.01943.x. [DOI] [PubMed] [Google Scholar]
- Gong J, Liu W, Dong J, Wang Y, Xu H, Wei W, Zhong J, Xi Q, Chen J. Developmental iodine deficiency and hypothyroidism impair neural development in rat hippocampus: involvement of doublecortin and NCAM-180. BMC Neurosci. 2010b;11:50. doi: 10.1186/1471-2202-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ. Nicotine and hippocampus-dependent learning: implications for addiction. Mol Neurobiol. 2006;34:93–107. doi: 10.1385/MN:34:2:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ. Addiction and cognition. Addict Sci Clin Pract. 2010;5:4–14. [PMC free article] [PubMed] [Google Scholar]
- Gould TJ, Leach PT. Cellular, molecular, and genetic substrates underlying the impact of nicotine on learning. Neurobiol Learn Mem. 2013 doi: 10.1016/j.nlm.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ, McCarthy MM, Keith RA. MK-801 disrupts acquisition of contextual fear conditioning but enhances memory consolidation of cued fear conditioning. Behav Pharmacol. 2002;13:287–294. doi: 10.1097/00008877-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- Guadano-Ferraz A, Benavides-Piccione R, Venero C, Lancha C, Vennstrom B, Sandi C, DeFelipe J, Bernal J. Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol Psychiatry. 2003;8:30–38. doi: 10.1038/sj.mp.4001196. [DOI] [PubMed] [Google Scholar]
- Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A. 1997;94:10391–10396. doi: 10.1073/pnas.94.19.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–715. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Bower JM. Acetylcholine and memory. Trends Neurosci. 1993;16:218–222. doi: 10.1016/0166-2236(93)90159-j. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Goldberg SR. Nicotine as a reinforcer in human subjects and laboratory animals. Pharmacol Biochem Behav. 1983;19:989–992. doi: 10.1016/0091-3057(83)90405-7. [DOI] [PubMed] [Google Scholar]
- Hiéronimus S, Ferrari P, Gal J, Berthier F, Azoulay S, Bongain A, Fénichel P, Brucker-Davis F. Relative impact of iodine supplementation and maternal smoking on cord blood thyroglobulin in pregnant women with normal thyroid function. Eur Thyroid J. 2012;1:264–273. doi: 10.1159/000342915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko S, Grether JK, Windham GC, Smith D, Fessel K. Are thyroid hormone concentrations at birth associated with subsequent autism diagnosis? Autism Res. 2011;4:456–463. doi: 10.1002/aur.219. [DOI] [PubMed] [Google Scholar]
- Iglesias T, Caubin J, Stunnenberg HG, Zaballos A, Bernal J, Munoz A. Thyroid hormone-dependent transcriptional repression of neural cell adhesion molecule during brain maturation. EMBO J. 1996;15:4307–4316. [PMC free article] [PubMed] [Google Scholar]
- Jorde R, Sundsfjord J. Serum TSH levels in smokers and non-smokers. The 5th Tromso study. Exp Clin Endocrinol Diabetes. 2006;114:343–347. doi: 10.1055/s-2006-924264. [DOI] [PubMed] [Google Scholar]
- Juarez de Ku LM, Sharma-Stokkermans M, Meserve LA. Thyroxine normalizes polychlorinated biphenyl (PCB) dose-related depression of choline acetyltransferase (ChAT) activity in hippocampus and basal forebrain of 15-day-old rats. Toxicology. 1994;94:19–30. doi: 10.1016/0300-483x(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Kapoor D, Jones TH. Smoking and hormones in health and endocrine disorders. Eur J Endocrinol. 2005;152:491–499. doi: 10.1530/eje.1.01867. [DOI] [PubMed] [Google Scholar]
- Kapoor R, Ghosh H, Nordstrom K, Vennstrom B, Vaidya VA. Loss of thyroid hormone receptor beta is associated with increased progenitor proliferation and NeuroD positive cell number in the adult hippocampus. Neurosci Lett. 2011;487:199–203. doi: 10.1016/j.neulet.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor R, van Hogerlinden M, Wallis K, Ghosh H, Nordstrom K, Vennstrom B, Vaidya VA. Unliganded thyroid hormone receptor {alpha}1 impairs adult hippocampal neurogenesis. FASEB J. 2010 doi: 10.1096/fj.10-161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JG, Son YJ, Yun CH, Kim YI, Nam-Goong IS, Park JH, Park SK, Ojeda SR, D’Elia AV, Damante G, Lee BJ. Thyroid transcription factor-1 facilitates cerebrospinal fluid formation by regulating aquaporin-1 synthesis in the brain. J Biol Chem. 2007;282:14923–14931. doi: 10.1074/jbc.M701411200. [DOI] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Koibuchi N, Iwasaki T. Regulation of brain development by thyroid hormone and its modulation by environmental chemicals. Endocr J. 2006;53:295–303. doi: 10.1507/endocrj.kr-69. [DOI] [PubMed] [Google Scholar]
- Lazzaro D, Price M, de Felice M, Di Lauro R. The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development. 1991;113:1093–1104. doi: 10.1242/dev.113.4.1093. [DOI] [PubMed] [Google Scholar]
- Leach PT, Holliday Ma E, Kutlu MG, Gould TJ. Withdrawal from chronic nicotine reduces thyroid hormone levels and levothyroxine treatment ameliorates nicotine withdrawal-induced deficits in hippocampus-dependent learning in C57BL/6J mice. Nicotine Tob Res. 2014 doi: 10.1093/ntr/ntu229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach PT, Kenney JW, Connor DA, Gould TJ. Thyroid receptor beta involvement in the effects of acute nicotine on hippocampus-dependent memory. Neuropharmacology. 2015;93C:155–163. doi: 10.1016/j.neuropharm.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PR, Brady D, Koenig JI. Thyroid hormone regulation of N-methyl-D-aspartic acid receptor subunit mRNA expression in adult brain. J Neuroendocrinol. 2003;15:87–92. doi: 10.1046/j.1365-2826.2003.00959.x. [DOI] [PubMed] [Google Scholar]
- Madeira MD, Sousa N, Lima-Andrade MT, Calheiros F, Cadete-Leite A, Paula-Barbosa MM. Selective vulnerability of the hippocampal pyramidal neurons to hypothyroidism in male and female rats. J Comp Neurol. 1992;322:501–518. doi: 10.1002/cne.903220405. [DOI] [PubMed] [Google Scholar]
- Mateos B, Borcel E, Loriga R, Luesu W, Bini V, Llorente R, Castelli M, Viveros MP. Adolescent exposure to nicotine and/or the cannabinoid agonist CP 55, 940 induces gender-dependent long-lasting memory impairments and changes in brain nicotinic and CB1 cannabinoid receptors. J Psychopharmacol. 2010 doi: 10.1177/0269881110370503. [DOI] [PubMed] [Google Scholar]
- McDonald MP, Wong R, Goldstein G, Weintraub B, Cheng SY, Crawley JN. Hyperactivity and learning deficits in transgenic mice bearing a human mutant thyroid hormone beta1 receptor gene. Learn Mem. 1998;5:289–301. [PMC free article] [PubMed] [Google Scholar]
- Meberg A, Marstein S. Smoking during pregnancy – effects on the fetal thyroid function. Acta Paediatr Scand. 1986;75:762–766. doi: 10.1111/j.1651-2227.1986.tb10287.x. [DOI] [PubMed] [Google Scholar]
- Metsios GS, Flouris AD, Jamurtas AZ, Carrillo AE, Kouretas D, Germenis AE, Gourgoulianis K, Kiropoulos T, Tzatzarakis MN, Tsatsakis AM, Koutedakis Y. A brief exposure to moderate passive smoke increases metabolism and thyroid hormone secretion. J Clin Endocrinol Metab. 2007;92:208–211. doi: 10.1210/jc.2006-0762. [DOI] [PubMed] [Google Scholar]
- Mistry N, Wass J, Turner MR. When to consider thyroid dysfunction in the neurology clinic. Pract Neurol. 2009;9:145–156. doi: 10.1136/jnnp.2008.167163. [DOI] [PubMed] [Google Scholar]
- Morte B, Ceballos A, Diez D, Grijota-Martinez C, Dumitrescu AM, Di Cosmo C, Galton VA, Refetoff S, Bernal J. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology. 2010;151:2381–2387. doi: 10.1210/en.2009-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B, Zulewski H, Huber P, Ratcliffe JG, Staub JJ. Impaired action of thyroid hormone associated with smoking in women with hypothyroidism. N Engl J Med. 1995;333:964–969. doi: 10.1056/NEJM199510123331503. [DOI] [PubMed] [Google Scholar]
- Niemi WD, Slivinski K, Audi J, Rej R, Carpenter DO. Propylthiouracil treatment reduces long-term potentiation in area CA1 of neonatal rat hippocampus. Neurosci Lett. 1996;210:127–129. doi: 10.1016/0304-3940(96)12676-8. [DOI] [PubMed] [Google Scholar]
- Oliveira E, Moura EG, Santos-Silva AP, Fagundes AT, Rios AS, Abreu-Villaca Y, Nogueira Neto JF, Passos MC, Lisboa PC. Short- and long-term effects of maternal nicotine exposure during lactation on body adiposity, lipid profile, and thyroid function of rat offspring. J Endocrinol. 2009;202:397–405. doi: 10.1677/JOE-09-0020. [DOI] [PubMed] [Google Scholar]
- Opazo MC, Gianini A, Pancetti F, Azkcona G, Alarcon L, Lizana R, Noches V, Gonzalez PA, Marassi MP, Mora S, Rosenthal D, Eugenin E, Naranjo D, Bueno SM, Kalergis AM, Riedel CA. Maternal hypothyroxinemia impairs spatial learning and synaptic nature and function in the offspring. Endocrinology. 2008;149:5097–5106. doi: 10.1210/en.2008-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini F, Lari R, La Ricca P, Grasso L, Taddei D, Bardini N, Fenzi GF, Di Bartolo F, Baschieri L, Pinchera A. Serum thyroglobulin in newborns’ cord blood, in childhood and adolescence: a physiological indicator of thyroidal status. J Endocrinol Invest. 1984;7:467–471. doi: 10.1007/BF03348452. [DOI] [PubMed] [Google Scholar]
- Pathak A, Sinha RA, Mohan V, Mitra K, Godbole MM. Maternal thyroid hormone before the onset of fetal thyroid function regulates reelin and downstream signaling cascade affecting neocortical neuronal migration. Cereb Cortex. 2010 doi: 10.1093/cercor/bhq052. [DOI] [PubMed] [Google Scholar]
- Pearce EN, Farwell AP, Braverman LE. Thyroiditis. N Engl J Med. 2003;348:2646–2655. doi: 10.1056/NEJMra021194. [DOI] [PubMed] [Google Scholar]
- Poorthuis RB, Goriounova NA, Couey JJ, Mansvelder HD. Nicotinic actions on neuronal networks for cognition: general principles and long-term consequences. Biochem Pharmacol. 2009;78:668–676. doi: 10.1016/j.bcp.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Pop VJ, Brouwers EP, Vader HL, Vulsma T, van Baar AL, de Vijlder JJ. Maternal hypothyroxinaemia during early pregnancy and subsequent child development: a 3-year follow-up study. Clin Endocrinol (Oxf) 2003;59:282–288. doi: 10.1046/j.1365-2265.2003.01822.x. [DOI] [PubMed] [Google Scholar]
- Pop VJ, Kuijpens JL, van Baar AL, Verkerk G, van Son MM, de Vijlder JJ, Vulsma T, Wiersinga WM, Drexhage HA, Vader HL. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf) 1999;50:149–155. doi: 10.1046/j.1365-2265.1999.00639.x. [DOI] [PubMed] [Google Scholar]
- Portella AC, Carvalho F, Faustino L, Wondisford FE, Ortiga-Carvalho TM, Gomes FC. Thyroid hormone receptor beta mutation causes severe impairment of cerebellar development. Mol Cell Neurosci. 2010;44:68–77. doi: 10.1016/j.mcn.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Portugal GS, Wilkinson DS, Turner JR, Blendy JA, Gould TJ. Developmental effects of acute, chronic, and withdrawal from chronic nicotine on fear conditioning. Neurobiol Learn Mem. 2012;97:482–494. doi: 10.1016/j.nlm.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost TL, Juarez de Ku LM, Zender C, Meserve LA. Dose- and age-dependent alterations in choline acetyltransferase (ChAT) activity, learning and memory, and thyroid hormones in 15- and 30-day old rats exposed to 1.25 or 12.5 PPM polychlorinated biphenyl (PCB) beginning at conception. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:915–928. doi: 10.1016/s0278-5846(99)00035-4. [DOI] [PubMed] [Google Scholar]
- Prummel MF, Wiersinga WM. Smoking and risk of Graves’ disease. JAMA. 1993;269:479–482. [PubMed] [Google Scholar]
- Pujadas L, Gruart A, Bosch C, Delgado L, Teixeira CM, Rossi D, de Lecea L, Martinez A, Delgado-Garcia JM, Soriano E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J Neurosci. 2010;30:4636–4649. doi: 10.1523/JNEUROSCI.5284-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM, Weeber EJ. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirin-Stricker C, Nappey V, Simoni P, Toussaint JL, Schmitt M. Trans-activation by thyroid hormone receptors of the 53 flanking region of the human ChAT gene. Brain Res Mol Brain Res. 1994;23:253–265. doi: 10.1016/0169-328x(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Raybuck JD, Gould TJ. Extracellular signal-regulated kinase 1/2 involvement in the enhancement of contextual fear conditioning by nicotine. Behav Neurosci. 2007;121:1119–1124. doi: 10.1037/0735-7044.121.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds PR, Allison CH, Willnauer CP. TTF-1 regulates alpha5 nicotinic acetylcholine receptor (nAChR) subunits in proximal and distal lung epithelium. Respir Res. 2010;11:175. doi: 10.1186/1465-9921-11-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds PR, Hoidal JR. Temporal-spatial expression and transcriptional regulation of alpha7 nicotinic acetylcholine receptor by thyroid transcription factor-1 and early growth response factor-1 during murine lung development. J Biol Chem. 2005;280:32548–32554. doi: 10.1074/jbc.M502231200. [DOI] [PubMed] [Google Scholar]
- Richards M, Jarvis MJ, Thompson N, Wadsworth ME. Cigarette smoking and cognitive decline in midlife: evidence from a prospective birth cohort study. Am J Public Health. 2003;93:994–998. doi: 10.2105/ajph.93.6.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman GC. Autism: transient in utero hypothyroxinemia related to maternal flavonoid ingestion during pregnancy and to other environmental antithyroid agents. J Neurol Sci. 2007;262:15–26. doi: 10.1016/j.jns.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Roman GC, Ghassabian A, Bongers-Schokking JJ, Jaddoe VW, Hofman A, de Rijke YB, Verhulst FC, Tiemeier H. Association of gestational maternal hypothyroxinemia and increased autism risk. Ann Neurol. 2013 doi: 10.1002/ana.23976. [DOI] [PubMed] [Google Scholar]
- Sabia S, Marmot M, Dufouil C, Singh-Manoux A. Smoking history and cognitive function in middle age from the Whitehall II study. Arch Intern Med. 2008;168:1165–1173. doi: 10.1001/archinte.168.11.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Silva AP, Oliveira E, Pinheiro CR, Santana AC, Nascimento-Saba CC, Abreu-Villaca Y, Moura EG, Lisboa PC. Endocrine effects of tobacco smoke exposure during lactation in weaned and adult male offspring. J Endocrinol. 2013;218:13–24. doi: 10.1530/JOE-13-0003. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP. Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res Brain Res Rev. 1997;23:28–46. doi: 10.1016/s0165-0173(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Sava L, Tomaselli L, Runello F, Belfiore A, Vigneri R. Serum thyroglobulin levels are elevated in newborns from iodine-deficient areas. J Clin Endocrinol Metab. 1986;62:429–432. doi: 10.1210/jcem-62-2-429. [DOI] [PubMed] [Google Scholar]
- Sawin S, Brodish P, Carter CS, Stanton ME, Lau C. Development of cholinergic neurons in rat brain regions: dose-dependent effects of propylthiouracil-induced hypothyroidism. Neurotoxicol Teratol. 1998;20:627–635. doi: 10.1016/s0892-0362(98)00020-8. [DOI] [PubMed] [Google Scholar]
- Schroeder AC, Privalsky ML. Thyroid hormones, t3 and t4, in the brain. Front Endocrinol. 2014;5:40. doi: 10.3389/fendo.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schussler GC. The thyroxine-binding proteins. Thyroid. 2000;10:141–149. doi: 10.1089/thy.2000.10.141. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepkovic DW, Haley NJ, Wynder EL. Thyroid activity in cigarette smokers. Arch Intern Med. 1984;144:501–503. [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala S, Zoli M, Palazzolo F, Faccoli S, Zanardi A, Mercuri NB, Spano P. Selective disarrangement of the rostral telencephalic cholinergic system in heterozygous reeler mice. Neuroscience. 2007;144:834–844. doi: 10.1016/j.neuroscience.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Smith JW, Evans AT, Costall B, Smythe JW. Thyroid hormones, brain function and cognition: a brief review. Neurosci Biobehav Rev. 2002;26:45–60. doi: 10.1016/s0149-7634(01)00037-9. [DOI] [PubMed] [Google Scholar]
- Soldin OP, Goughenour BE, Gilbert SZ, Landy HJ, Soldin SJ. Thyroid hormone levels associated with active and passive cigarette smoking. Thyroid. 2009;19:817–823. doi: 10.1089/thy.2009.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Germain DL, Galton VA, Hernandez A. Minireview: defining the roles of the iodothyronine deiodinases: current concepts and challenges. Endocrinology. 2009;150:1097–1107. doi: 10.1210/en.2008-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui L, Li BM. Effects of perinatal hypothyroidism on regulation of reelin and brain-derived neurotrophic factor gene expression in rat hippocampus: Role of DNA methylation and histone acetylation. Steroids. 2010;75:988–997. doi: 10.1016/j.steroids.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Sui L, Ren WW, Li BM. Administration of thyroid hormone increases reelin and brain-derived neurotrophic factor expression in rat hippocampus in vivo. Brain Res. 2010;1313:9–24. doi: 10.1016/j.brainres.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Sui L, Wang F, Liu F, Wang J, Li BM. Dorsal hippocampal administration of triiodothyronine enhances long-term memory for trace cued and delay contextual fear conditioning in rats. J Neuroendocrinol. 2006;18:811–819. doi: 10.1111/j.1365-2826.2006.01480.x. [DOI] [PubMed] [Google Scholar]
- Sullivan KM. Iodine deficiency as a cause of autism. J Neurol Sci. 2009;276:202. doi: 10.1016/j.jns.2008.09.016. (author reply 203) [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Tan XJ, Fan XT, Kim HJ, Butler R, Webb P, Warner M, Gustafsson JA. Liver X, receptor beta and thyroid hormone receptor alpha in brain cortical layering. Proc Natl Acad Sci U S A. 2010;107:12305–12310. doi: 10.1073/pnas.1006162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MA, Swant J, Wagner JJ, Fisher JW, Ferguson DC. Lower thyroid compensatory reserve of rat pups after maternal hypothyroidism: correlation of thyroid, hepatic, and cerebrocortical biomarkers with hippocampal neurophysiology. Endocrinology. 2008;149:3521–3530. doi: 10.1210/en.2008-0020. [DOI] [PubMed] [Google Scholar]
- Thompson CC, Potter GB. Thyroid hormone action in neural development. Cereb Cortex. 2000;10:939–945. doi: 10.1093/cercor/10.10.939. [DOI] [PubMed] [Google Scholar]
- Turner JR, Castellano LM, Blendy JA. Parallel anxiolytic-like effects and upregulation of neuronal nicotinic acetylcholine receptors following chronic nicotine and varenicline. Nicotine Tob Res. 2011;13:41–46. doi: 10.1093/ntr/ntq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Wilkinson DS, Poole RL, Gould TJ, Carlson GC, Blendy JA. Divergent functional effects of sazetidine-a and varenicline during nicotine withdrawal. Neuropsychopharmacology. 2013;38:2035–2047. doi: 10.1038/npp.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USDHHS. A Report of the Surgeon General. U.D. o. H. a. H. Services; 1988. The Health Consequences of Smoking: Nicotine Addiction. [Google Scholar]
- Utiger RD. Effects of smoking on thyroid function. Eur J Endocrinol. 1998;138:368–369. doi: 10.1530/eje.0.1380368. [DOI] [PubMed] [Google Scholar]
- Vanderver GB, Engel A, Lamm S. Cigarette smoking and iodine as hypothyroxinemic stressors in U.S. women of childbearing age: a NHANES III analysis. Thyroid. 2007;17:741–746. doi: 10.1089/thy.2006.0332. [DOI] [PubMed] [Google Scholar]
- Vara H, Martinez B, Santos A, Colino A. Thyroid hormone regulates neurotransmitter release in neonatal rat hippocampus. Neuroscience. 2002;110:19–28. doi: 10.1016/s0306-4522(01)00541-3. [DOI] [PubMed] [Google Scholar]
- Vejbjerg P, Knudsen N, Perrild H, Carle A, Laurberg P, Pedersen IB, Rasmussen LB, Ovesen L, Jorgensen T. The impact of smoking on thyroid volume and function in relation to a shift towards iodine sufficiency. Eur J Epidemiol. 2008;23:423–429. doi: 10.1007/s10654-008-9255-1. [DOI] [PubMed] [Google Scholar]
- Venero C, Guadano-Ferraz A, Herrero AI, Nordstrom K, Manzano J, de Escobar GM, Bernal J, Vennstrom B. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev. 2005;19:2152–2163. doi: 10.1101/gad.346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard P. Smoking, thyroid disorders–a meta-analysis. Eur J Endocrinol. 2002;146:153–161. doi: 10.1530/eje.0.1460153. [DOI] [PubMed] [Google Scholar]
- Wilcoxon JS, Nadolski GJ, Samarut J, Chassande O, Redei EE. Behavioral inhibition and impaired spatial learning and memory in hypothyroid mice lacking thyroid hormone receptor alpha. Behav Brain Res. 2007;177:109–116. doi: 10.1016/j.bbr.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DS, Gould TJ. Withdrawal from chronic nicotine and subsequent sensitivity to nicotine challenge on contextual learning. Behav Brain Res. 2013;250:58–61. doi: 10.1016/j.bbr.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Xu B, Koenig RJ. Thyroid hormone response element sequence and the recruitment of retinoid X receptors for thyroid hormone responsiveness. J Biol Chem. 2001;276:3929–3936. doi: 10.1074/jbc.M006743200. [DOI] [PubMed] [Google Scholar]