

Abstract
Maturity onset diabetes of the young (MODY) is a rare form of diabetes mellitus typically seen in young adults that results from pancreatic beta-cell dysfunction. MODY4 is a rare subtype caused by a PDX1 mutation. In this case, we present a nonobese 26-year-old male with polyuria and polydipsia. Lab work showed a blood glucose of 511 mg/dL, no ketones or antibodies (insulin, islet cell, and glutamic acid decarboxylase [GAD]), C-peptide of 1.6 ng/mL, and A1c 9.3%. Genetic analysis revealed a novel nonsense mutation in the PDX1 gene, consistent with MODY type 4. Given this patient’s particular genetic mutation affecting the incretin pathway, sitagliptin was substituted for glyburide, which led to significant improvement in glycemic control. Our case report identifies a unique mutation in a rare form of MODY and outlines management of ensuing diabetes through targeting its inherent genetic mutation.
Keywords: MODY, MODY4, diabetes, genetics, dipeptidyl peptidase-4 inhibitor
Introduction
Maturity onset diabetes of the young (MODY) is a monogenic form of diabetes characterized by absence of ketosis, autosomal dominant inheritance, primary defect in pancreatic beta-cell function, and onset before the age of 25.1,2 MODY type 3 is the most common form and is caused by mutations in the HNF4A gene.1 MODY type 1 is caused by mutations in the related HNF1A gene.1 MODY type 2 is also common and is caused by a mutation in the glucokinase (GCK) gene, leading to hyperglycemia, which is usually treated with diet alone.1 MODY type 5 is caused by mutations in the HNF1B gene and is associated with diabetes mellitus and pancreatic atrophy with renal and genital defects.1
MODY type 4 is caused by mutations in the PDX1 gene, which encodes the insulin promoter factor-1 (IPF1), a homeodomain-containing transcription factor.3 It is a rare form of MODY, with collective literature stemming from a few families known with this gene mutation.3,4 Management of MODY4 in a nonobese adult has not been previously described, and the particular genetic mutation in this case report appears novel.
This case offers insight into a rare disease entity with few case reports and very little known therapeutics. While MODY4 itself is a rare disease entity, our MODY4 patient is particularly unique in that he displays a novel PDX1 mutation not cited in previous literature, does not display obesity typically seen in MODY4, presented at an earlier age with diabetes than typical MODY4 cases, and required treatment for diabetes at the time of diagnosis. Our case report is also particularly significant in that the therapeutics prescribed herein were based on the genetic mutations inherent in this rare form of diabetes; which has not been previously postulated in other case reports of MODY4. As this patient responded favor-ably to a therapeutic regimen based on this particular genetic mutation, this case proposes a therapeutic modality for a disease that has minimal recommended therapeutics.
Case Presentation
A 26-year-old nonobese (BMI 23 kg/m2) male presented with symptoms of polyuria, fatigue, and visual changes over several months. Past medical history included hypertension. In addition to his antihypertensive medications (lisinopril and metoprolol), he was taking multivitamin and loratadine–pseudophedrine (PRN) on a daily basis. Basic metabolic profile showed nonfasting glucose of 511 mg/dL, c-peptide of 1.6 ng/mL (1.0–7.6 ng/mL), HbA1c of 9.3%, negative urine ketones, and negative screening antibodies (islet cell and glutamic acid decarboxylase antibodies). Family history revealed a paternal uncle diagnosed with diabetes in his 20s who died of a diabetic coma in his 40s as well as multiple obese relatives in the patient’s paternal grandmother’s family with type 2 diabetes. The patient’s father has had episodic hyperglycemia although does not carry a diagnosis of diabetes. The patient was started on metformin and pioglitazone with improvement in his glycemic control after one month of therapy with twice daily blood sugar readings ranging between 120 mg/dL and 220 mg/dL.
Given the patient’s age, habitus, and presentation, there was high suspicion of MODY as the underlying etiology for his diabetes; however, genetic testing took several months for insurance approval. After optimizing metformin and pioglitazone, his A1c improved rapidly to 5.9% within 3 months of therapy and the patient decided to follow up with his primary care physician for further management. However, the patient eventually became intolerant to metformin because of nausea and diarrhea and was switched to glyburide 5 mg while continuing pioglitazone by his primary physician. It is unclear as to when he developed the intolerance to metformin as this was noted by his primary care physician outside of our hospital system. This new regimen led to worsened glycemic control, with blood sugars ranging between 200 mg/dL and 300 mg/dL. Further therapy was discussed with the patient pending genetic testing.
Once authorized, genetic testing was undertaken and showed a heterozygous mutation in the PDX1 gene (c.694_697delGGCGinsAGCT p.Gly232Serfsx2), consistent with MODY type 4. This mutation caused a deletion of four nucleotides and insertion of four different nucleotides, causing the wildtype glycine to be substituted by serine at position 232 and introducing a novel stop codon at position 233 (Fig. 1).5
Figure 1.
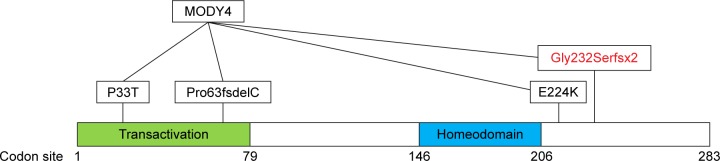
Schematic of PDX1 transcription factor showing known MODY4 mutations (black) and novel MODY4 gene mutation (red) at respective codon sites (5). P: proline; E: glutamic acid; K: lysine; G: glycine; T: threonine.
Given this subtype of MODY with its potential to impair incretin pathway,6 as well as the patient’s desire to avoid injectable therapy, the patient was recommended to start 100 mg sitagliptin, discontinue the sulfonylurea, and continue the thiazolidinedione. Following initiation of sitagliptin, the patient had significantly improved glycemic control, particularly with improved postprandial blood sugar readings. The patient reported frequent postprandial blood glucoses well into the 300s prior to the initiation of sitagliptin (approximate A1c 10–11%), which improved to postprandial glucoses in the 150–180 mg/dL range, within a month of sitagliptin initiation. A1c was down to 8.5% three months after initiation of sitagliptin.
The patient has remained on sitagliptin since its initiation and has had no side effects on therapy. Despite the improvement in postprandial hyperglycemia, the patient’s fasting glucoses eventually began to rise (three months after sitagliptin and pioglitazone combination), necessitating initiation of basal insulin with 10 units of glargine each morning. We had anticipated that the sitagliptin would have had more of an effect on fasting glucose as well, but considering that direct transcription of insulin was likely impaired by IPF1 dysfunction,7 it was decided that basal insulin glargine would be optimal for maintenance of euglycemia.
Discussion
Owing to the rarity of MODY4, a significant portion of medical knowledge of its diagnostics and therapeutics reside in several large kindreds. Both obese and nonobese patients with MODY4 have been described at a variety of ages. The relatively small number of patients who have been fully characterized does not allow the authors to definitively describe phenotype of age and body habitus in this condition. Two families have been reported to have similar PDX1 mutations involving deletion of a single nucleotide causing frameshift and downstream truncation (Pro63fsX60; c.188delC).3,4 One of the familial cohorts with MODY4 showed obesity, which was not observed in the other family of MODY4 patients.4 Another PDX1 mutation (p.Pro33Thr;c.97C>A) has also been isolated in an Italian family with predominance of type 2 DM, MODY4, or gestational diabetes mellitus.8 There is a paucity of literature concerning the correlation of therapeutics with MODY4, though unspecified oral hypoglycemic agents and insulin have both been cited.4
Our MODY4 patient is unique in that he displays a novel PDX1 mutation, does not display obesity, presented at an early age with diabetes, and required treatment for diabetes. Mutations in the PDX1 gene cause impaired IPF1 protein function with resultant globally impaired insulin production due to direct effects on gene transcription.7 The IPF1 protein plays a central role in pancreatic development and beta-cell function and interacts with multiple genes as a transcriptional activator binding to the promoter region of genes encoding glucokinase, islet amyloid polypeptide, and glucose transporter.9 The indirect effects of IPF1 appear to be crucial for glucose-stimulated insulin release, which appears to have major implications for postprandial hyperglycemia that our patient experienced.6 Importantly, the incretin pathway appears to be indirectly impaired in PDX1 mutations (Fig. 2).
Figure 2.

Effects of DPP4 inhibition and IPF1 within the incretin pathway.
Abbreviations: DPP4, dipeptidyl peptidase-4; GLP-1, glucagon-like peptide-1; IPF1, insulin promoter factor-1.
PDX has a direct activating effect on the transcription of insulin by binding to AT-rich regions, which are located upstream of the insulin and somatostatin genes.7 Interestingly, IPF1 also appears to exert indirect influence on insulin secretion through other pathways. The FGFR1 pathway in beta islet cells is important for proper sensing of blood glucose level and would appear to be important in maintenance of postprandial glucose levels. IPF1 is required for maintenance of this pathway as an upstream regulator.10 Most importantly, it has also been shown that IPF1 also exerts indirect control over insulin secretion by binding to an enhancer site of the G-protein-coupled protein Gpr40. Gpr40 itself responds to the presence of free fatty acids and contributes to insulin secretion after glucose load both directly and indirectly through the mediation of incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP).6
Incretin hormones are important regulators of postprandial glucose homeostasis as they increase insulin secretion and reduce glucagon secretion.11 Pharmacologic incretin therapy could be initiated through one of two methods: either directly through synthetic analogs such as GLP-1 via injection or through inhibition of endogenous DPP4 through DPP4 inhibitors. While either therapy might be efficacious for this genetic mutation pathway, GLP-1 therapy usually involves less desirable side effects such as nausea, vomiting, and is available only via injectable therapy. DPP4 inhibitors on the other hand are given via oral administration and do not have side effects of nausea, vomiting, and potential weight loss.
The therapeutic effect of DPP4 inhibition was demonstrated via the improvement of glycemic response. We presume that this effect was mediated by enhancement of GLP-1 levels. Owing to the identification of this mutation and what we know from literature, we initiated a DPP4-inhibitor. We did not assess the GLP-1/GIP levels, before or after treatment, as it is not part of routine management of patients with diabetes. GLP-1R signaling has been shown to modulate the endoplasmic reticulum (ER) stress response, which can promote beta-cell adaptation and survival. Since DPP4 inhibition increase GLP-1 levels, it is likely that modulation of ER stress response played a role in this patient’s therapeutic response to DPP4 inhibition.12 The rapid worsening of glycemic control in this patient, eventually requiring insulin therapy, was a puzzling phenomenon. Sulfonylureas have the potential to induce beta-cell apoptosis.13 This, along with the potential of this patient’s mutation to affect pancreatic development, may provide some explanation to the rapid worsening of glycemic control in this patient.
In conclusion, patients with MODY often present differently than patients with type 2 diabetes, have unique genetic mutations causative for their diabetes, and respond differently to treatment. MODY4 is a particularly rare form of MODY, with minimal therapeutic recommendations. Understanding the biochemical pathways inherent in various mutations of MODY is crucial for its optimal treatment. In this patient, we hypothesized a treatment protocol of sitagliptin, which was suggested by the patient’s molecular mutation with its defect in the incretin pathway. Dipeptidyl peptidase-4 inhibitors as a class of antihyperglycemics have not previously been recommended as a therapeutic agent in other literature sources about MODY4. This therapy resulted in significant improvement in glycemic control and may be a source of recommended therapeutics for other patients with MODY4.
Acknowledgments
This work was done through the VA Nebraska-Western Iowa Health Care System and is based upon work supported with resources and use of facilities at the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, and Biomedical Laboratory Research and Development.
Footnotes
ACADEMIC EDITOR: Nigel Irwin, Editor ion Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 515 words, excluding any confidential comments to the academic editor.
DISCLAIMER: The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Patient Consent
Written consent was obtained from the patient for publication of the submitted article.
Author Contributions
Conceived and designed the experiments: CM, ER, and VS. Analyzed the data: CM, ER, and VS. Wrote the first draft of the manuscript: CM. Contributed to the writing of the manuscript: CM, ER, and VS. Agree with manuscript results and conclusions: ER and VS. Jointly developed the structure and arguments for the paper: CM, ER, and VS. Made critical revisions and approved the final version: VS. All authors reviewed and approved the final manuscript.
REFERENCES
- 1.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 2.Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53(12):2504–2508. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 3.Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17(2):138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- 4.Fajans SS, Bell GI, Paz VP, et al. Obesity and hyperinsulinemia in a family with pancreatic agenesis and MODY caused by the IPF1 mutation Pro63fsX60. Transl Res. 2010;156(1):7–14. doi: 10.1016/j.trsl.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available at: http://AtlasGeneticsOncology.org.
- 6.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–2287. doi: 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma S, Jhala US, Johnson T, Ferreri K, Leonard J, Montminy M. Hormonal regulation of an islet-specific enhancer in the pancreatic homeobox gene STF-1. Mol Cell Biol. 1997;17(5):2598–2604. doi: 10.1128/mcb.17.5.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gragnoli C, Stanojevic V, Gorini A, Von Preussenthal GM, Thomas MK, Habener JF. IPF-1/MODY4 gene missense mutation in an Italian family with type 2 and gestational diabetes. Metabolism. 2005;54(8):983–988. doi: 10.1016/j.metabol.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 9.Edlund H. Factors controlling pancreatic cell differentiation and function. Diabetologia. 2001;44:1071–1079. doi: 10.1007/s001250100623. [DOI] [PubMed] [Google Scholar]
- 10.Hart AW, Baeza N, Apelqvist A, Edlund H. Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature. 2000;408(6814):864–868. doi: 10.1038/35048589. [DOI] [PubMed] [Google Scholar]
- 11.Lovshin JA, Zinman B. Blood pressure-lowering effects of incretin-based diabetes therapies. Can J Diabetes. 2014;38(5):364–371. doi: 10.1016/j.jcjd.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Yusta B, Baggio LL, Estall JL, et al. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4(5):391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J Clin Endocrinol Metab. 2005;90(1):501–506. doi: 10.1210/jc.2004-0699. [DOI] [PubMed] [Google Scholar]