

Abstract
G-protein-coupled receptors are eukaryotic membrane proteins with broad biological and pharmacological relevance. Like all membrane-embedded proteins, their location and orientation are influenced by lipids, which can also impact protein function via specific interactions. Extensive simulations totaling 0.25 ms reveal a process in which phospholipids from the membrane’s cytosolic leaflet enter the empty G-protein binding site of an activated β2 adrenergic receptor and form salt-bridge interactions that inhibit ionic lock formation and prolong active-state residency. Simulations of the receptor embedded in an anionic membrane show increased lipid binding, providing a molecular mechanism for the experimental observation that anionic lipids can enhance receptor activity. Conservation of the arginine component of the ionic lock among Rhodopsin-like G-protein-coupled receptors suggests that intracellular lipid ingression between receptor helices H6 and H7 may be a general mechanism for active-state stabilization.
Introduction
Intercellular communication is essential for many facets of multicellular life (1, 2, 3, 4). At a conceptual midpoint of all intercellular communication pathways lies the cell membrane, whose combination of low and high dielectric lamellae provides a physical barrier to solute passage (5, 6, 7, 8). Proteins embedded within these membranes enable homeostasis and communication by providing selective permeability to both matter and information (9, 10, 11, 12, 13, 14, 15).
The functional dynamics of membrane proteins are influenced by lipid composition, which controls membrane bulk properties such as thickness, fluidity, and surface potential, and provisions lipid species that can engage in specific and functionally relevant lipid-protein interactions (16, 17, 18, 19, 20). These interactions are mediated by membrane protein residues that are often conserved and, at the cytosolic interface, most commonly involve arginine (21).
G-protein-coupled receptors (GPCRs) are a class of transmembrane protein that relay extracellular information into eukaryotic cells (9, 22, 23, 24, 25). The human genome contains over 800 different GPCRs, which have evolved to bind thousands of different chemicals and evoke varied cellular responses (26). As such, GPCRs play crucial roles in olfaction, vision, appetite, mood, heart rate, blood pressure, and pain control (27), and are targeted by 30% of drugs (28).
In a simplified representation, a GPCR exists in either an active or inactive state. Extracellular ligands called agonists stabilize or induce the receptor’s active states, which have guanine nucleotide exchange factor activity toward a cytosolically bound, inactive, guanosine-diphosphate-containing Gαβγ protein heterotrimer (29). This guanine nucleotide exchange factor activity causes the release of guanosine-diphosphate from the Gα subunit (29), which then tends to bind guanosine-triphosphate. Ensuing conformational changes in the G-protein lead to dissociation of the Gα subunit from the Gβγ heterodimer, both of which are then free to initiate downstream signaling pathways (30). Cytosolic phosphorylation of the receptor by kinases (31, 32) enables arrestin recruitment, additional signaling, and cellular desensitization to agonist-mediated stimulation via receptor internalization (33).
The well-studied beta 2 adrenergic receptor (β2AR) provides an excellent model system for defining the activation mechanisms available to GPCRs. Sixteen crystal structures of the β2AR are currently available. Ten of these structures are inactive and tightly packed at the cytosolic surface (34, 35, 36, 37, 38, 39, 40), with receptor helix 6 (H6) approaching H3; in the six apparently active structures (41, 42, 43, 44), the cytosolic end of H6 is splayed outward, facilitating receptor engagement with a guanine nucleotide-free heterotrimeric G-protein (41) or a mimetic nanobody (42, 43, 44) (see Fig. 1).
Figure 1.

Cytosolic view of H6 splaying upon receptor activation. Receptor helices are shown as cylinders colored by PDB ID. H6 splaying, quantified by cytosolic H3–H6 separation, is larger for (blue and cyan) active than (red and orange) inactive crystal structures. To see this figure in color, go online.
Surprisingly, agonist binding does not appear to be sufficient to fully stabilize active states of the β2AR (45, 46). All existing crystallographic structures of this receptor’s active states are bound to a protein that engages and presumably stabilizes the cleft between receptor helices H3 and H6 (41, 42, 43, 44). Even covalent, biochemically active agonist-receptor complexes crystallize in an inactive state (40) unless cocrystallized with a nanobody that binds at the receptor’s cytosolic face (44). Molecular simulations corroborate the fragile nature of the receptor’s active states in the absence of an intracellular binding partner. Simulations starting from active states exhibit spontaneous receptor deactivation on the low microsecond timescale (47, 48), even in the presence of an extracellular agonist (40, 48). This deactivation often involves the formation of a salt bridge between an arginine in H3 and a glutamic acid in H6 (40, 48, 49), an ionic lock that is thought to stabilize inactive states (50). Consistently, inactive conformations of the receptor do not spontaneously activate on achievable timescales during unbiased simulations with (51) or without (49, 52, 53) agonist. It is currently unclear how ligand binding generates efficient signal transduction and whether the binding of downstream signaling proteins involves induced fit, conformational selection, or both. Nevertheless, recent double electron-electron resonance and 19F NMR spectroscopy suggest that, even in the absence of a cytosolic binding protein, agonist binding induces 15–60% of the receptor’s conformational ensemble to adopt an active-like state with extensive H6 splaying (46).
Here, we investigate specific interactions of membrane lipids with the β2AR and provide a mechanism that explains how phospholipids may act as facultative cofactors to prolong the receptor’s residency in active states or activation intermediates. Specifically, a phospholipid from the membrane’s cytosolic leaflet can insert between receptor helices H6 and H7 and form a salt bridge with the arginine component of the ionic lock. This lipid binding occurs much more frequently in active than inactive receptor states, suggesting that lipid binding can stabilize active states via conformational selection. Of importance, we show that anionic lipids outcompete zwitterionic lipids for arginine binding and may therefore be even more disruptive to ionic lock formation and receptor deactivation, consistent with available experimental data.
Materials and Methods
The simulation system consists of a truncated form of the human β2AR embedded in a hydrated lipid bilayer comprising zwitterionic POPC or anionic POPG (Fig. S1 in the Supporting Material). Simulations use a wild-type receptor sequence that is based on the resolved residues in the crystal structure of a mutant construct that maintains the wild-type profile of guanosine-triphosphateγS binding in apo, agonist-(isoproterenol), and inverse-agonist-(ICI-118551) bound states (Protein Data Bank (PDB) ID: 4GBR) (35). This sequence is truncated at the N- and C-termini and in intracellular loop 3. Specifically, in comparison to the receptor’s wild-type sequence (UniProt ID: P07550), the sequence in our simulations is Δ(M1-D29), Δ(S236-K263), and Δ(L342-L412). Residues D2.50 and E3.41 are protonated and the C341 palmitoyl group (54) is not included. Residue identifiers follow the Ballesteros and Weinstein format: N.M, where N is the transmembrane helix number and M equals 50 plus the offset from the most conserved residue in that helix (55).
Molecular dynamics simulations are conducted with a single-precision compilation of version 4.6.1 of the GROMACS simulation package (56). Macromolecules are modeled by the CHARMM 22/27 protein force field (57) with grid-based energy correction maps (58), and the CHARMM 36 lipid force field (59) as implemented in GROMACS (60, 61). This lipid force field reproduces experimental bulk properties of POPG (62). The water model is TIP3P (63) with CHARMM modifications (57). Water molecules are rigidified with SETTLE (64) and bond lengths in protein and lipid are constrained with P-LINCS (65) using sixth-order coupling and a single iteration. Lennard-Jones interactions are evaluated using an atom-based cutoff, gradually switching off the potential energies of interactions between 0.8 and 1.2 nm. Coulomb interactions are calculated using the smooth particle-mesh Ewald method (66, 67) with a Fourier grid spacing of 0.12 nm. Simulation in the NpT ensemble is achieved by semiisotropic coupling to Berendsen barostats (68) at 1 bar with coupling constants of 4 ps and temperature coupling the simulation system using velocity Langevin dynamics (69) at 310 K with a coupling constant of 1 ps. The integration time step is 2 fs. The nonbonded pairlist is updated every 20 fs. Further details are provided in the Supporting Material.
Results
We conduct replicate simulations of active conformations of the apo form of the human β2AR embedded in a hydrated zwitterionic lipid bilayer (Fig. S1). Initial receptor conformations are drawn from crystal structures in which the β2AR is bound to an agonist and either a G-protein (41) or a G-protein-mimicking nanobody (42), which are hereafter referred to as G-protein- and nanobody-derived structures, respectively. To obtain a statistical measure of receptor dynamics, we conduct 52 1-μs simulations of each receptor conformation.
Receptor deactivation
We use three metrics to quantify the extent of receptor deactivation in our simulations: collapse of the cytosolic binding pocket, inward swinging of H6 toward H3, and formation of the ionic lock (Fig. 2, A–C). These measures are strongly correlated, with absolute values of pairwise Pearson correlation coefficients ≥0.8 (Fig S2). Interestingly, the nanobody-derived structure deactivates 5–10 times more often than the G-protein-derived structure (Table 1 and Fig. 2, D and E). During this deactivation process, ionic lock formation and occlusion of the receptor’s cytosolic binding pocket are in some cases reversible (Fig. S3, A and C). However, once H6 swings inward to sample dH3–H6 values <0.8 nm the receptor remains in the inactive conformational basin for the remainder of the simulation (Fig. S3 B).
Figure 2.

Receptor deactivation. (A–C) Cytosolic view of representative structures from simulations depicting (left) active and (right) inactive states of the receptor for each of the following deactivation metrics: (A) volumetric clash between the (orange) simulated apo receptor and the (black) C-terminal helix of a Gα protein based on postsimulation modeling, Vclash (see Supporting Materials and Methods); (B) approach of the cytosolic ends of H3 and H6 measured by the distance between (yellow spheres) Cα atoms of R3.50 and L6.34, dH3–H6; and (C) formation of the ionic lock as measured by the minimum Nη-Oε distance between (sticks) R3.50 and E6.30, dlock (also depicted in Movie S1). (D–F) Probability histograms of deactivation metrics in simulations of (solid red line) G-protein- and (dashed blue line) nanobody-derived structures. Dotted gray lines in parts (D) and (F) represent the data from G-protein-derived simulations when dH3–H6 ≥ 1.54 nm, the crystallographic active-state displacement. Vertical bars represent standard error. Arrows and PDB identifiers denote abscissa values for selected crystal structures. To see this figure in color, go online.
Table 1.
Receptor deactivation
| Crystallized with | % of Simulations Exhibiting |
% of Time |
Active State Half-Life (μs) | ||
|---|---|---|---|---|---|
| H3–H6 Closurea | Ionic Lockb | Closeda | Lockedb | ||
| G-protein | 25 ± 2 | 12 ± 0 | 7 ± 1 | 2 ± 1 | 2.9 ± 0.3 |
| Nanobody | 83 ± 2 | 81 ± 4 | 61 ± 3 | 46 ± 1 | 0.6 ± 0.2 |
Cα distance between R3.50 and L6.34, dH3–H6 < 0.94 nm.
Minimum Nη-Oε distance between R3.50 and E6.30, dlock < 0.35 nm.
Even when the apo receptor is in an active state with H6 splayed outward at least as much as it is in the G-protein-derived structure (dH3–H6≥1.54 nm; Fig. 1), protein breathing motions including side-chain flexibility, loop rearrangement, and movement of H5 toward H3 lead the receptor to take up some of the space occupied by the G-protein in its crystallographic pose (41), hereafter referred to as the G-protein volume. In simulations of the G-protein-derived structure, the average G-protein volume occluded by the receptor when dH3–H6 ≥ 1.54 nm is 0.19 ± 0.01 nm3, or about eight heavy atoms (Fig. 2 D). Therefore, our simulations indicate that G-protein binding requires some form of induced fit or conformational selection even when H6 is splayed outward.
To assess the generality of these results, the entire set of simulations is repeated with the AMBER99SB-ILDN protein force field (70) and the Stockholm lipid parameters (71). These AMBER simulations corroborate our conclusion that the nanobody-derived structure deactivates more rapidly than the G-protein-derived structure (Supporting Results, Table S2, and Fig. S7, B–D).
Lipid binding to the receptor’s cytosolic pocket
The side chain of the R3.50 component of the ionic lock is located in the core of the receptor’s binding pocket in all available crystal structures of the β2AR. Surprisingly, our simulations indicate that a phospholipid can insert between the cytosolic ends of receptor helices H6 and H7 and form a salt bridge with the guanidino moiety of this arginine residue when the receptor is in an active state (Fig. 3 A). Conversely, receptor deactivation tends to bring H6 toward H7 and exclude lipids from the G-protein binding pocket (Fig. 3 B). Lipid binding is quantified in Table 2.
Figure 3.

Zwitterionic lipid binding to the R3.50 component of the ionic lock. (A and B) Representative conformations of (A) active, lipid bound and (B) inactive, lipid unbound states. Cα atoms used in evaluation of dH3–H6 are shown as yellow spheres. (C and D) Probability histograms of (C) dH3–H6 and (D) dlock for (solid black line) lipid-unbound and (dashed gray line) lipid-bound states. (E) Probability histograms of the minimum Nη-O distance between R3.50 and a lipid, dR3.50-lipid, for simulations of (solid red line) G-protein- and (dashed blue line) nanobody-derived structures. Probability density maps depicting dR3.50-lipid vs. dH3–H6 and dlock are presented in Fig. S4. To see this figure in color, go online.
Table 2.
Zwitterionic lipid binding to R3.50
| Crystallized with | No. of Distinct Binding Eventsa,b | % of Lipid-Bound R3.50b |
|---|---|---|
| G-protein | 37 | 3 ± 1 |
| Nanobody | 10 | 0.2 ± 0.02 |
Not counting rebinding of a previously bound lipid.
Minimum Nη-O distance between R3.50 and a lipid, dR3.50-lipid < 0.35 nm.
The specific binding of membrane lipids to R3.50 occurs more often when H6 is splayed outward (Figs. 3 C and S4, A and B) and when the ionic lock is broken (Figs. 3 D and S4, C and D). The incompatibility of lipid binding with H6 closure and ionic lock formation suggests that the ingression of a lipid from the bilayer’s cytosolic leaflet may stabilize the receptor’s active state and compete with E6.30 for interaction with R3.50. This specific lipid binding is also more common in simulations of G-protein- than nanobody-derived structures (Table 2 and Fig. 3 E), pointing to a possible molecular basis for the different stability and lipid binding properties of these two structures in our simulations. Specifically, in the nanobody-derived structure, R3.50 extends toward the cytosol. Conversely, in the G-protein-derived structure, R3.50 extends toward H6/H7 where this cation may act to preorganize annular lipids for entry into the G-protein binding pocket.
In five of our simulations, a lipid from the cytosolic leaflet moves entirely into the G-protein binding pocket, sporadically maintaining salt-bridge interactions with R3.50 and remaining in the binding pocket until the simulations are terminated (Fig. S5). This complete lipid entry is only observed in simulations of the G-protein-derived structure. Even though a lipid remains in the receptor’s binding pocket for a total of 2.96 μs in these five simulations, the R3.50-lipid oxygen binding condition (dR3.50-lipid < 0.35 nm) is only met for 1.12 μs due to the high degree of mobility experienced by a bound zwitterionic lipid (Movie S3). Taking the full duration of these lipid invasion events into consideration, zwitterionic lipids bind R3.50 and/or enter the receptor’s binding pocket 6% of the time across all 52 simulations initiated from this receptor structure. Extension of two of these simulations to 14 and 3 μs clarifies that this is a stable or metastable interaction in which the lipid remains in the receptor’s G-protein binding pocket and the receptor remains active (Figs. 4 and S6).
Figure 4.

Complete invasion of the receptor’s cytosolic binding pocket by a zwitterionic lipid. (A) Probability density map of (black, labeled P) the phosphorus atom of the bound phospholipid and (cyan, labeled 1–7) transmembrane helices viewed from the cytosol in a single 14-μs simulation initiated from the G-protein-derived structure. Each helix is represented by four Cα atoms near the headgroup region of the bilayer’s cytosolic leaflet (see Supporting Materials and Methods). (B) Time trajectories of H6 splaying, dH3–H6, ionic lock formation, dlock, and R3.50 binding for the single identified lipid, dR3.50-lipid. Ordinate units are nm. (C–F) Cytosolic view of representative conformations from time points noted in (B). This simulation is depicted in Movie S3. Corresponding data for another 3-μs simulation in which a lipid dissociated from the receptor’s annular lipids and moved completely into the receptor’s cytosolic binding pocket are provided in Fig. S6. To see this figure in color, go online.
AMBER simulations corroborate the specific binding of a lipid to the R3.50 component of the ionic lock (Table S3 and Fig. S7 A). They also support our conclusions that lipids bind more extensively to the G-protein- than to the nanobody-derived structure (Table S3 and Fig. S7 E) and that lipid entry to the receptor’s cytosolic binding occurs more often when H6 is splayed outward (Fig. S7 F) and when the ionic lock is broken (Fig. S7 G). However, compared to the CHARMM simulations, lipid binding to R3.50 is at least an order of magnitude less stable in AMBER simulations (Tables 2 and S3 and Figs. 3 E and S7 E), which also do not feature any of the complete lipid insertion events that we observe in five of the CHARMM simulations (Figs. 4, S5, and S6). The source of this discrepancy is unclear.
Anionic lipids bind R3.50 more frequently
The specific interaction between a lipid and R3.50 suggests that a cytosolically bound phospholipid may be able to transiently stabilize active states of the receptor. If this is the case, charge complementarity may lead anionic lipids to occupy the receptor’s cytosolic binding pocket and stabilize active receptor states with greater frequency than the zwitterionic lipids used in the aforementioned simulations. Indeed, free energy profiles characterizing the interaction between analogs of amino acid side chains and lipid headgroups indicate that arginine binding strength follows the progression glutamate/aspartate > anionic lipids > zwitterionic lipids (Fig. S8). Therefore, we conduct 52 1-μs simulations of the apo β2AR embedded in an anionic phosphatidylglycerol bilayer using the G-protein-derived receptor structure.
As hypothesized, salt-bridge interactions between lipids and R3.50 are more common with anionic lipids (Table 3 and Fig. S9 D) than with zwitterionic lipids (Table 2 and Fig. 3 E). Anionic lipid binding is also largely incompatible with complete H6 closure (Fig. S9 E) or ionic lock formation (Fig. S9 F). Moreover, anionic lipids enhance the stability of the receptor’s active state, increasing its half-life by a factor of three (Tables 1 and 3), although it is unclear if this active-state stabilization is entirely due to R3.50-lipid interactions. Complete lipid entry to the receptor’s binding pocket occurs in two of these simulations (not shown). Anionic lipid binding is depicted in Movie S4.
Table 3.
Lipid binding and receptor deactivation in simulations with anionic lipids
| No. of Distinct Lipid Binding Eventsa,b | % of Lipid-Bound R3.50b (%) | % of Simulations Exhibiting |
% of Time |
Active State Half-Life (μs) | ||
|---|---|---|---|---|---|---|
| H3–H6 Closurec | Ionic Lockd | Closedc | Lockedd | |||
| 83 | 14 ± 1 | 12 ± 0 | 21 ± 2 | 4 ± 2 | 2 ± 1 | 9.4 ± 1.6 |
Not counting rebinding of a previously bound lipid.
Minimum Nη-O distance between R3.50 and a lipid, dR3.50-lipid < 0.35 nm.
Cα distance between R3.50 and L6.34, dH3–H6 < 0.94 nm.
Minimum Nη-Oε distance between R3.50 and E6.30, dlock < 0.35 nm.
Both lipid types most frequently interact with R3.50 by way of a salt bridge between the side chain of this receptor arginine and the free oxygen atoms in the lipid’s phosphate group, although other lipid oxygen atoms also contribute to this interaction, especially those in the headgroup glycerol moiety of phosphatidylglycerol lipids (Fig. 5).
Figure 5.
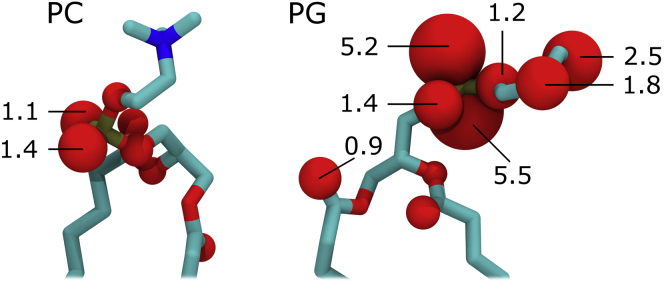
Lipidic oxygen atoms involved in R3.50 binding. (Left) zwitterionic PC and (right) anionic PG lipids are shown with oxygen atom volume proportional to time spent interacting with an Nη atom of R3.50 (d < 0.35 nm) in simulations of the G-protein-derived crystal structure. Numbers represent the percentage of time for which this binding criterion is met (oxygen atoms without numbers are <0.5%). To see this figure in color, go online.
Of importance, lipid binding at R3.50 does not necessarily preclude G-protein binding. Bound zwitterionic and anionic lipids occupy, on average, 0.11 ± 0.02 and 0.055 ± 0.002 nm3 of the G-protein volume, respectively (Fig. 6 A). These volumes correspond to four and two heavy atoms, respectively, and are substantially less than the G-protein volume occupied by the receptor itself when dH3–H6 ≥ 1.54 nm (Fig. 2 D). Protrusion of receptor and lipid into the G-protein binding volume is depicted in Fig. 6, B and C. Just as the receptor’s conformation likely responds to the presence of a G-protein, a receptor-bound lipid may reorganize or be extruded back into the membrane upon receptor-G-protein interaction, especially since the bound lipid can be quite dynamic (Figs. 4 and S4 and Movies S2 and S3). However, if the bound lipid interferes with G-protein binding, the net effect of lipid binding on receptor activity is then given by the relationship between stabilization of the receptor’s active state and G-protein interference.
Figure 6.

Lipid and G-protein binding. (A) Probability histogram of volume overlap between a receptor-bound lipid and the C-terminal helix of a Gα protein based on postsimulation modeling, Vclash-lipid. Data are obtained from simulations of the G-protein-derived structure with (solid black line) zwitterionic, PC, and (dashed red line) anionic, PG, lipids. The dotted gray line represents the PC data after omitting the five simulations in which the bound lipid completely invaded the receptor’s binding pocket (Fig. S5). Distributions of Vclash vs. Vclash-lipid are provided in Fig. S10. (B and C) Two cytosolic depictions of the same representative snapshot from simulations in which an anionic lipid is bound at R3.50 and the values of Vclash and Vclash-lipid are similar to the mean values for lipid-bound states in these simulations (values for this snapshot are 0.2 and 0.05 nm3, respectively). To see this figure in color, go online.
Discussion
Based on a vast number of simulations (N = 260 totaling 254 μs), we consistently identify spontaneous invasion of the β2AR’s cytosolic binding pocket by membrane lipids, which bind the R3.50 component of the ionic lock by inserting their headgroup between receptor helices H6 and H7 (Figs. 3, 4, and 6, C and D) and stabilize the receptor’s active states (Figs. 3, C and D, S4, and S9, E and F). This specific lipid binding is reminiscent of the pathway by which the sn-2-arachidonoylglycerol ligand has been predicted to bind the cannabinoid type 2 (CB2) receptor (72), though in this case occurring in the cytosolic membrane leaflet. Similarly, experimental studies highlight the differential influence of lipid- and detergent-based environments on the equilibrium between open and closed states of potassium channel KscA (73), the stability of CC-chemokine receptor 5 (74), the ligand binding strength (75) and activation dynamics (76) of the β2AR, and the activity of rhodopsin (77, 78, 79, 80).
Additional simulations show that anionic lipids enter the receptor’s binding pocket and interact with R3.50 more readily than zwitterionic lipids (Tables 2 and 3 and Fig. 5), providing a detailed molecular mechanism for the experimental finding that anionic lipids can enhance receptor activity. Specifically, Inagaki et al. (81) showed that the addition of phosphatidylglycerol to nanodisks containing rat neurotensin receptor 1 increases the rate of nucleotide exchange at Gqα much more dramatically than it increases the receptor’s affinity for the peptide agonist neurotensin, even in nanodisks with purely anionic lipids. Likewise, Kimura et al. (82) showed that phosphatidylserine enhances the activity of the human CB2 receptor in reconstituted liposomes, although in this case receptor activity is reduced at very high concentrations of anionic lipids (1:4 zwitterionic/anionic). Rhodopsin activation is also enhanced in the presence of phosphatidylserine (83), though the pH dependence of rhodopsin’s activation (84) suggests that anionic lipids may be acting indirectly via proton enrichment. Finally, G-protein-coupled receptor kinases, which may occupy the receptor’s G-protein binding pocket (85), phosphorylate activated receptors with greater efficacy in the presence of anionic lipids (86, 87).
The simulations outlined in this article are initiated from active conformations of the receptor. Therefore, these simulations assess the ability of lipids to stabilize, but not induce, active states, and the proposed mechanism requires that active-like splaying of H6 occurs in agonist-bound receptors before their interaction with a downstream signaling protein or mimetic nanobody. Crystal structures of this receptor are very similar in complex with agonists (40) and inverse agonists (34, 36) and to date only G-protein- and mimetic nanobody-stabilized crystals capture the receptor’s active state (41, 42, 43, 44). Nevertheless, in solution, agonist binding alone (in the absence of a G-protein or nanobody) appears to increase the mobility of the intracellular end of H6 (45, 88). This is the same region of the receptor that undergoes a large-scale conformational change upon activation and protein binding at the receptor’s cytosolic face (Fig. 1) and that must separate from H7 to permit lipid entry (Figs. 3, A and B, and 6, C and D). Specifically, the agonist BI-167107 shifts the receptor’s conformational ensemble toward a presumed activation intermediate in which the dynamics of a cytosolic region of H6 including C6.27 is increased (45). Likewise, the agonist isoproterenol causes L6.34-T6.45 to become less protected from deuterium/hydrogen exchange, consistent with increased structural plasticity (88). Moreover, both of these agonists alone lead the β2AR to sample conformations in which the distance between L6.28 and N4.40 reported by double electron-electron resonance spectroscopy increases by ≥0.8 nm (46). Although 19F NMR spectroscopy indicates that this H6 splaying may represent an on-pathway activation intermediate with distinct conformational features from fully active G-protein- or nanobody-bound receptor states (46), the agonist-driven increase in mobility (45, 88) and splaying (46) of the cytosolic end of H6 supports the possibility that the specific lipid binding described in this article may occur before receptor interaction with a G-protein. The hypothesized mechanism by which a lipid may stabilize a receptor’s active state via conformational selection is depicted schematically in Fig. 7.
Figure 7.

Proposed role of lipid binding in putative GPCR activation pathways. Extracellular agonist binding enhances the mobility of the cytosolic end of receptor helix H6. Transient outward splaying of this helix permits lipid binding to the R3.50 component of the ionic lock, stabilizing the receptor’s active state before its interaction with a downstream signaling protein. This lipid binding is more common with anionic lipids, which provide further stabilization. Receptor helices H6 and H7 are shown in (I) inactive, (A) active, or (I∗) inactive intermediate states as they interact with (yellow star) agonist, (red and blue circle and sticks) lipid, and (green blob) a cytosolic binding protein such as a G-protein. To see this figure in color, go online.
There is a precedent for the specific formation of a salt bridge between a lipid headgroup and an arginine residue in a GPCR, albeit at the receptor’s extracellular face. In the orthosteric site of the sphingosine 1-phosphate 1 receptor, the phosphate group of an antagonist sphingolipid mimic is coordinated by an arginine residue (89), whose mutation reduces the potency of a variety of agonists (90). To our knowledge, reports of specific nonligand lipid/fatty acid binding sites on GPCRs have thus far been restricted to the receptor’s periphery (34, 37, 91, 92, 93, 94, 95, 96), with the exception of extracellular invasion of an oleic acid acyl chain between H1 and H7 of the adenosine A2A receptor (95). In simulations, the cytosolic H6/H7 interface of inactive rhodopsin forms tight interactions with docosahexanoic acid, stearic acid, and cholesterol (96) and docosahexanoic penetrates rhodopsin’s helical interfaces nonspecifically (97).
Localization of R3.50 in the membrane’s cytosolic headgroup plane favorably situates this residue to alternately bind a lipid or form the ionic lock. The specific binding between a lipid from the membrane’s cytosolic leaflet and R3.50 appears to be incompatible with the receptor’s completely inactive states (Figs. 3, C and D, and S9, E and F), and may act as a metaphorical foot in the door of the activated receptor as it awaits favorable interaction with downstream signaling proteins including G-proteins, G-protein-coupled receptor kinases, and arrestins (98). This lipid-protein binding may also affect or underlie a receptor’s ability to differentially couple with various G-protein families by regulating the size of the receptor’s cytosolic binding pocket (99). Furthermore, the likelihood of this specific protein-lipid interaction depends on membrane composition (Fig. 5), which varies with cell type (100, 101), cell-cycle stage (102), disease (101, 103), diet (104), acquired drug resistance (105), and localization in membrane microdomains (106), thus providing novel avenues for receptor regulation as generally proposed by Inagaki et al. (81) and defined mechanistically in this work. Finally, the overwhelming conservation of R3.50 across type-A GPCRs (107) suggests that the hypothesis developed in this article for the β2AR may be broadly relevant across this pharmaceutically important class of membrane proteins.
Author Contributions
C.N., R.P., and A.E.G. designed research, C.N. performed research, C.N. and H.D.H. analyzed data, C.N. and A.E.G. wrote the article.
Acknowledgments
We thank R. Scott Prosser for useful discussion. Computations are performed at 1) Orcinus at the WestGrid high performance computing (HPC) consortium (www.westgrid.ca), 2) Guillimin and Colosse at the CLUMEQ HPC consortium of Calcul Québec (www.calculquebec.ca), 3) GPC at the SciNet HPC consortium (108), all of which are resources of Compute Canada (www.computecanada.ca), and 4) Stampede at the Texas Advanced Computing Center at the University of Texas at Austin (www.tacc.utexas.edu), to which access is provided by the Extreme Science and Engineering Discovery Environment (XSEDE grant TG-MCB130178), which is supported by National Science Foundation (NSF) grant No. ACI-1053575.
SciNet is funded by the Canada Foundation for Innovation (CFI) under the auspices of Compute Canada; the Government of Ontario; Ontario Research Fund - Research Excellence; and the University of Toronto. CLUMEQ is funded by CFI, the Natural Sciences and Engineering Research Council of Canada (NSERC), and Fonds de Recherche Nature et Technologies Québec. C.N. is funded by a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR). R.P. is funded by CIHR Operating Grant MOP-43998 and NSERC Discovery grant 418679. This work is funded in part by NSF grant MCB-1050966.
Editor: Emad Tajkhorshid.
Footnotes
Angel E. García’s present address is CNLS MSB258, Los Alamos National Laboratory, Los Alamos, New Mexico.
Supporting Materials and Methods, Supporting Results, ten figures, three tables, and four movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(15)00863-2.
Supporting Citations
References (109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128) appear in the Supporting Material.
Supporting Material
Side and cytosolic views. Side chains of ionic lock components R3.50 and E6.30 are shown as sticks.
{kind=link}
Side and cytosolic views. Side chains of ionic lock components R3.50 and E6.30 are shown as surfaces. Lipid carbon atoms are yellow.
{kind=link}
Cytosolic view. Ionic lock components are colored for (blue) R3.50 and (red) E6.30. Lipid carbon atoms are yellow.
{kind=link}
Side and cytosolic views. The side chain of ionic lock component R3.50 and E6.30 is shown as a surface. Lipid carbon atoms are cyan.
{kind=link}
References
- 1.Singer S.J. Intercellular communication and cell-cell adhesion. Science. 1992;255:1671–1677. doi: 10.1126/science.1313187. [DOI] [PubMed] [Google Scholar]
- 2.Denef C. Paracrinicity: the story of 30 years of cellular pituitary crosstalk. J. Neuroendocrinol. 2008;20:1–70. doi: 10.1111/j.1365-2826.2007.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santner A., Estelle M. Recent advances and emerging trends in plant hormone signalling. Nature. 2009;459:1071–1078. doi: 10.1038/nature08122. [DOI] [PubMed] [Google Scholar]
- 4.Singh A.B., Harris R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell. Signal. 2005;17:1183–1193. doi: 10.1016/j.cellsig.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 5.Abbott N.J., Patabendige A.A.K., Begley D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Stevens T., Garcia J.G.N., Malik A.B. Mechanisms regulating endothelial cell barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279:L419–L422. doi: 10.1152/ajplung.2000.279.3.L419. [DOI] [PubMed] [Google Scholar]
- 7.Marguet D., Lenne P.-F., He H.-T. Dynamics in the plasma membrane: how to combine fluidity and order. EMBO J. 2006;25:3446–3457. doi: 10.1038/sj.emboj.7601204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsegian A. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 1969;221:844–846. doi: 10.1038/221844a0. [DOI] [PubMed] [Google Scholar]
- 9.De Lean A., Stadel J.M., Lefkowitz R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 10.Heidel A.J., Lawal H.M., Glöckner G. Phylogeny-wide analysis of social amoeba genomes highlights ancient origins for complex intercellular communication. Genome Res. 2011;21:1882–1891. doi: 10.1101/gr.121137.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marban E., Yamagishi T., Tomaselli G.F. Structure and function of voltage-gated sodium channels. J. Physiol. 1998;508:647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro J.A. Thinking about bacterial populations as multicellular organisms. Annu. Rev. Microbiol. 1998;52:81–104. doi: 10.1146/annurev.micro.52.1.81. [DOI] [PubMed] [Google Scholar]
- 13.Bonner J.T. Induction of stalk cell differentiation by cyclic AMP in the cellular slime mold Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA. 1970;65:110–113. doi: 10.1073/pnas.65.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar N.M., Gilula N.B. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 15.Langley J.N. On the reaction of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and to curari. J. Physiol. 1905;33:374–413. doi: 10.1113/jphysiol.1905.sp001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras F.-X., Ernst A.M., Brügger B. Specificity of intramembrane protein-lipid interactions. Cold Spring Harb. Perspect. Biol. 2011;3:a004705. doi: 10.1101/cshperspect.a004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunte C. Specific protein-lipid interactions in membrane proteins. Biochem. Soc. Trans. 2005;33:938–942. doi: 10.1042/BST20050938. [DOI] [PubMed] [Google Scholar]
- 18.Ernst A.M., Contreras F.X., Wieland F. Determinants of specificity at the protein-lipid interface in membranes. FEBS Lett. 2010;584:1713–1720. doi: 10.1016/j.febslet.2009.12.060. [DOI] [PubMed] [Google Scholar]
- 19.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 20.Laganowsky A., Reading E., Robinson C.V. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palsdottir H., Hunte C. Lipids in membrane protein structures. Biochim. Biophys. Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Kobilka B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerione R.A., Sibley D.R., Lefkowitz R.J. Reconstitution of a hormone-sensitive adenylate cyclase system. The pure beta-adrenergic receptor and guanine nucleotide regulatory protein confer hormone responsiveness on the resolved catalytic unit. J. Biol. Chem. 1984;259:9979–9982. [PubMed] [Google Scholar]
- 24.Katritch V., Cherezov V., Stevens R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefkowitz R.J. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 26.Fredriksson R., Lagerström M.C., Schiöth H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 27.Bockaert J., Pin J.P. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rask-Andersen M., Almén M.S., Schiöth H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 29.Oldham W.M., Hamm H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 30.Rosenbaum D.M., Rasmussen S.G.F., Kobilka B.K. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitcher J.A., Freedman N.J., Lefkowitz R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 32.Ribas C., Penela P., Mayor F., Jr. The G-protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 33.Reiter E., Ahn S., Lefkowitz R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cherezov V., Rosenbaum D.M., Stevens R.C. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou Y., Weis W.I., Kobilka B.K. N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One. 2012;7:e46039. doi: 10.1371/journal.pone.0046039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasmussen S.G.F., Choi H.-J., Kobilka B.K. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 37.Hanson M.A., Cherezov V., Stevens R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wacker D., Fenalti G., Stevens R.C. Conserved binding mode of human β2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bokoch M.P., Zou Y., Kobilka B.K. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenbaum D.M., Zhang C., Kobilka B.K. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen S.G.F., DeVree B.T., Kobilka B.K. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasmussen S.G.F., Choi H.-J., Kobilka B.K. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ring A.M., Manglik A., Kobilka B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weichert D., Kruse A.C., Gmeiner P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA. 2014;111:10744–10748. doi: 10.1073/pnas.1410415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim T.H., Chung K.Y., Prosser R.S. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J. Am. Chem. Soc. 2013;135:9465–9474. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manglik A., Kim T.H., Kobilka B.K. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell. 2015;161:1101–1111. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nygaard R., Zou Y., Kobilka B.K. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dror R.O., Arlow D.H., Shaw D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA. 2011;108:18684–18689. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dror R.O., Arlow D.H., Shaw D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ballesteros J.A., Jensen A.D., Javitch J.A. Activation of the β 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 51.Dror R.O., Pan A.C., Shaw D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozcan O., Uyar A., Akten E.D. Effect of intracellular loop 3 on intrinsic dynamics of human β2-adrenergic receptor. BMC Struct. Biol. 2013;13:29. doi: 10.1186/1472-6807-13-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanni S., Neri M., Rothlisberger U. Observation of “ionic lock” formation in molecular dynamics simulations of wild-type β 1 and β 2 adrenergic receptors. Biochemistry. 2009;48:4789–4797. doi: 10.1021/bi900299f. [DOI] [PubMed] [Google Scholar]
- 54.O’Dowd B.F., Hnatowich M., Bouvier M. Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys-341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J. Biol. Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 55.Ballesteros J.A., Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 56.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 58.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 59.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bjelkmar P., Larsson P., Lindahl E. Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010;6:459–466. doi: 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- 61.Piggot T.J., Piñeiro Á., Khalid S. Molecular dynamics simulations of phosphatidylcholine membranes: a comparative force field study. J. Chem. Theory Comput. 2012;8:4593–4609. doi: 10.1021/ct3003157. [DOI] [PubMed] [Google Scholar]
- 62.Kang H., Klauda J.B. Molecular dynamics simulations of palmitoyloleoylphosphatidylglycerol bilayers. Mol. Simulat. 2014;41:948–954. [Google Scholar]
- 63.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 64.Miyamoto S., Kollman P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 65.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 66.Darden T., York D., Pedersen L. Particle mesh Ewald: An N•log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 67.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 68.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 69.van Gunsteren W.F., Berendsen H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1988;1:173–185. [Google Scholar]
- 70.Lindorff-Larsen K., Piana S., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jämbeck J.P.M., Lyubartsev A.P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 2012;8:2938–2948. doi: 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- 72.Hurst D.P., Grossfield A., Reggio P.H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010;285:17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Imai S., Osawa M., Shimada I. Functional equilibrium of the KcsA structure revealed by NMR. J. Biol. Chem. 2012;287:39634–39641. doi: 10.1074/jbc.M112.401265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshiura C., Kofuku Y., Shimada I. NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers. J. Am. Chem. Soc. 2010;132:6768–6777. doi: 10.1021/ja100830f. [DOI] [PubMed] [Google Scholar]
- 75.Whorton M.R., Bokoch M.P., Sunahara R.K. A monomeric G-protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. USA. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kofuku Y., Ueda T., Shimada I. Functional dynamics of deuterated β2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2014;53:13376–13379. doi: 10.1002/anie.201406603. [DOI] [PubMed] [Google Scholar]
- 77.Brown M.F. Influence of nonlamellar-forming lipids on rhodopsin. Curr. Top. Membr. 1997;44:285–356. [Google Scholar]
- 78.Botelho A.V., Gibson N.J., Brown M.F. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 2002;41:6354–6368. doi: 10.1021/bi011995g. [DOI] [PubMed] [Google Scholar]
- 79.Soubias O., Teague W.E., Gawrisch K. Evidence for specificity in lipid-rhodopsin interactions. J. Biol. Chem. 2006;281:33233–33241. doi: 10.1074/jbc.M603059200. [DOI] [PubMed] [Google Scholar]
- 80.Soubias O., Gawrisch K. The role of the lipid matrix for structure and function of the GPCR rhodopsin. Biochim. Biophys. Acta. 2012;1818:234–240. doi: 10.1016/j.bbamem.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Inagaki S., Ghirlando R., Grisshammer R. Modulation of the interaction between neurotensin receptor NTS1 and Gq protein by lipid. J. Mol. Biol. 2012;417:95–111. doi: 10.1016/j.jmb.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kimura T., Yeliseev A.A., Gawrisch K. Recombinant cannabinoid type 2 receptor in liposome model activates g protein in response to anionic lipid constituents. J. Biol. Chem. 2012;287:4076–4087. doi: 10.1074/jbc.M111.268425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gibson N.J., Brown M.F. Role of phosphatidylserine in the MI-MII equilibrium of rhodopsin. Biochem. Biophys. Res. Commun. 1991;176:915–921. doi: 10.1016/s0006-291x(05)80273-6. [DOI] [PubMed] [Google Scholar]
- 84.Wong J.K., Ostroy S.E. Hydrogen ion changes of rhodopsin I. Proton uptake during the metarhodopsin I 478 metarhodopsin II 308 reaction. Arch. Biochem. Biophys. 1973;154:1–7. doi: 10.1016/0003-9861(73)90028-3. [DOI] [PubMed] [Google Scholar]
- 85.Boguth C.A., Singh P., Tesmer J.J.G. Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 2010;29:3249–3259. doi: 10.1038/emboj.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Onorato J.J., Gillis M.E., Ruoho A.E. The β-adrenergic receptor kinase (GRK2) is regulated by phospholipids. J. Biol. Chem. 1995;270:21346–21353. doi: 10.1074/jbc.270.36.21346. [DOI] [PubMed] [Google Scholar]
- 87.Homan K.T., Glukhova A., Tesmer J.J.G. Regulation of G protein-coupled receptor kinases by phospholipids. Curr. Med. Chem. 2013;20:39–46. [PubMed] [Google Scholar]
- 88.West G.M., Chien E.Y., Griffin P.R. Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. Structure. 2011;19:1424–1432. doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanson M.A., Roth C.B., Stevens R.C. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Loenen P.B., de Graaf C., Alewijnse A.E. Agonist-dependent effects of mutations in the sphingosine-1-phosphate type 1 receptor. Eur. J. Pharmacol. 2011;667:105–112. doi: 10.1016/j.ejphar.2011.05.071. [DOI] [PubMed] [Google Scholar]
- 91.Paila Y.D., Tiwari S., Chattopadhyay A. Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta. 2009;1788:295–302. doi: 10.1016/j.bbamem.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 92.Sengupta D., Chattopadhyay A. Identification of cholesterol binding sites in the serotonin1A receptor. J. Phys. Chem. B. 2012;116:12991–12996. doi: 10.1021/jp309888u. [DOI] [PubMed] [Google Scholar]
- 93.Cang X., Yang L., Jiang H. Cholesterol-β1 AR interaction versus cholesterol-β2 AR interaction. Proteins. 2014;82:760–770. doi: 10.1002/prot.24456. [DOI] [PubMed] [Google Scholar]
- 94.Lee J.Y., Lyman E. Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc. 2012;134:16512–16515. doi: 10.1021/ja307532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu W., Chun E., Stevens R.C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grossfield A., Feller S.E., Pitman M.C. A role for direct interactions in the modulation of rhodopsin by ω-3 polyunsaturated lipids. Proc. Natl. Acad. Sci. USA. 2006;103:4888–4893. doi: 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pitman M.C., Grossfield A., Feller S.E. Role of cholesterol and polyunsaturated chains in lipid-protein interactions: molecular dynamics simulation of rhodopsin in a realistic membrane environment. J. Am. Chem. Soc. 2005;127:4576–4577. doi: 10.1021/ja042715y. [DOI] [PubMed] [Google Scholar]
- 98.Szczepek M., Beyrière F., Scheerer P. Crystal structure of a common GPCR-binding interface for G-protein and arrestin. Nat. Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rose A.S., Elgeti M., Hildebrand P.W. Position of transmembrane helix 6 determines receptor G protein coupling specificity. J. Am. Chem. Soc. 2014;136:11244–11247. doi: 10.1021/ja5055109. [DOI] [PubMed] [Google Scholar]
- 100.Sessions A., Horwitz A.F. Differentiation-related differences in the plasma membrane phospholipid asymmetry of myogenic and fibrogenic cells. Biochim. Biophys. Acta. 1983;728:103–111. doi: 10.1016/0005-2736(83)90442-x. [DOI] [PubMed] [Google Scholar]
- 101.Pratt H.P.M., Saxon A., Graham M.L. Membrane lipid changes associated with malignant transformation and normal maturation of human lymphocytes. Leuk. Res. 1978;2:1–10. [Google Scholar]
- 102.Atilla-Gokcumen G.E., Muro E., Eggert U.S. Dividing cells regulate their lipid composition and localization. Cell. 2014;156:428–439. doi: 10.1016/j.cell.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Escribá P.V. Membrane-lipid therapy: a new approach in molecular medicine. Trends Mol. Med. 2006;12:34–43. doi: 10.1016/j.molmed.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 104.Wince L.C., Rutledge C.O. The effect of dietary lipid on The binding of [3H] dihydroalprenolol and adenylate cyclase activity in rat atria. J. Pharmacol. Exp. Ther. 1981;219:625–631. [PubMed] [Google Scholar]
- 105.May G.L., Wright L.C., Mountford C.E. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int. J. Cancer. 1988;42:728–733. doi: 10.1002/ijc.2910420517. [DOI] [PubMed] [Google Scholar]
- 106.Sonnino S., Prinetti A. Membrane domains and the “lipid raft” concept. Curr. Med. Chem. 2013;20:4–21. [PubMed] [Google Scholar]
- 107.Mirzadegan T., Benkö G., Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loken C., Gruner D., Van Zon R. SciNet: lessons learned from building a power-efficient top-20 system and data centre. J. Phys. Conf. Ser. 2010;256:012026. [Google Scholar]
- 109.Jo S., Kim T., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 110.Jo S., Lim J.B., Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiang Z., Soto C.S., Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. USA. 2002;99:7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soto C.S., Fasnacht M., Honig B. Loop modeling: sampling, filtering, and scoring. Proteins. 2008;70:834–843. doi: 10.1002/prot.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krivov G.G., Shapovalov M.V., Dunbrack R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rath P., DeCaluwé L.L.J., Rothschild K.J. Fourier transform infrared difference spectroscopy of rhodopsin mutants: light activation of rhodopsin causes hydrogen-bonding change in residue aspartic acid-83 during meta II formation. Biochemistry. 1993;32:10277–10282. doi: 10.1021/bi00090a001. [DOI] [PubMed] [Google Scholar]
- 115.Fahmy K., Jäger F., Siebert F. Protonation states of membrane-embedded carboxylic acid groups in rhodopsin and metarhodopsin II: a Fourier-transform infrared spectroscopy study of site-directed mutants. Proc. Natl. Acad. Sci. USA. 1993;90:10206–10210. doi: 10.1073/pnas.90.21.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nygaard R., Valentin-Hansen L., Schwartz T.W. Conserved water-mediated hydrogen bond network between TM-I, -II, -VI, and -VII in 7TM receptor activation. J. Biol. Chem. 2010;285:19625–19636. doi: 10.1074/jbc.M110.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kohlhoff K.J., Shukla D., Pande V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014;6:15–21. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Romo T.D., Grossfield A., Pitman M.C. Concerted interconversion between ionic lock substates of the β(2) adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys. J. 2010;98:76–84. doi: 10.1016/j.bpj.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schmidt T.H., Kandt C. LAMBADA and InflateGRO2: efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J. Chem. Inf. Model. 2012;52:2657–2669. doi: 10.1021/ci3000453. [DOI] [PubMed] [Google Scholar]
- 120.Rowland R.S., Taylor R. Intermolecular nonbonded contact distances in organic crystal structures: comparison with distances expected from van der Waals radii. J. Phys. Chem. 1996;100:7384–7391. [Google Scholar]
- 121.Williams, T., and C. Kelly. 2010. Gnuplot 4.4: an interactive plotting program.
- 122.Torrie G.M., Valleau J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 123.Neale C., Madill C., Pomès R. Accelerating convergence in molecular dynamics simulations of solutes in lipid membranes by conducting a random walk along the bilayer normal. J. Chem. Theory Comput. 2013;9:3686–3703. doi: 10.1021/ct301005b. [DOI] [PubMed] [Google Scholar]
- 124.Grossfield, A. WHAM: the weighted histogram analysis method. http://www.membrane.urmc.rochester.edu/content/wham. Accessed September 15, 2014.
- 125.Kumar S., Rosenberg J.M., Kollman P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 126.Neumann R.M. Entropic approach to Brownian movement. Am. J. Phys. 1980;48:354–357. [Google Scholar]
- 127.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 128.Dahl A.C.E., Chavent M., Sansom M.S.P. Bendix: intuitive helix geometry analysis and abstraction. Bioinformatics. 2012;28:2193–2194. doi: 10.1093/bioinformatics/bts357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Side and cytosolic views. Side chains of ionic lock components R3.50 and E6.30 are shown as sticks.
Side and cytosolic views. Side chains of ionic lock components R3.50 and E6.30 are shown as surfaces. Lipid carbon atoms are yellow.
Cytosolic view. Ionic lock components are colored for (blue) R3.50 and (red) E6.30. Lipid carbon atoms are yellow.
Side and cytosolic views. The side chain of ionic lock component R3.50 and E6.30 is shown as a surface. Lipid carbon atoms are cyan.