

Abstract
A series of 3-(benzilidine)indolin-2-one derivatives were synthesized and evaluated for their in vitro binding to alpha synuclein (α-syn), beta amyloid (Aβ), and tau fibrils. Compounds with a single double bond in the 3-position had only a modest affinity for α-syn and no selectivity for α-syn versus Aβ or tau fibrils. Homologation to the corresponding diene analogs yielded a mixture of Z,E and E,E isomers; substitution of the indoline nitrogen with an N-benzyl group resulted in increased binding to α-syn and reasonable selectivity for α-syn versus Aβ and tau. Introduction of a para-nitro group into the benzene ring of the diene enabled separation of the Z,E and E,E isomers and led to the identification of the Z,E configuration as the more active regioisomer. The data described here provide key structural information in the design of probes which bind preferentially to α-syn versus Aβ or tau fibrils.
INTRODUCTION
It is generally accepted that many neurodegenerative disorders are characterized by the accumulation of misfolded proteins which lead to the formation of insoluble protein aggregates in the CNS.1, 2 For example, Alzheimer’s disease (AD) is characterized by the formation of two different insoluble protein aggregates: 1) beta amyloid (Aβ plaques, which consists of aggregated beta amyloid protein, Aβ1-42; and 2) neurofibrillary tangles (NFTs), which consist of aggregates of hyperphosphorylated tau protein. The diagnosis of AD historically relied on the cognitive assessment of patients with moderate to severe memory deficits. Although a progressive cognitive decline resulting in a severe impairment of daily function was considered consistent with “Alzheimer-like dementia”, the clinical diagnosis of AD could not be confirmed until postmortem analysis demonstrated the presence of Aβ plaques and NFTs in the temporal and parietal cortex. A major breakthrough in the study of patients presenting with AD-like symptoms emerged with the development of [11C]PiB, a positron-labeled analog of thioflavin-T (Thio-T), the fluorescent dye used to visualize Aβ plaques in postmortem samples of AD brain.3, 4 Initial PET studies in AD patients and healthy controls demonstrated increased [11C]PiB retention in the frontal cortex in the patients with a clinical diagnosis of a progressive loss of cognitive function in comparison with normal controls.5 More recent studies in patients with familial AD have clearly shown that PiB(+) Aβ plaque formation occurs early in the disease process, and suggest amyloid plaques may represent an antecedent biomarker of AD.6, 7 The subsequent emergence of 18F-labeled Aβ imaging agents including [18F]fluorbetapir,8, 9 [18F]florbetaben,10 and [18F]flumetamol11 has enabled the expansion of PET studies in AD patients to imaging centers without an on-site cyclotron facility. Recently, a number of 18F- and 11C-labeled agents for imaging tau deposits in NFTs have been reported.12–14 These agents are expected to provide valuable information on the temporal separation between the formation of Aβ plaques (a possible antecedent biomarker) and NFT formation (thought to reflect neuronal loss in AD).6 This ability to measure the neuropathological time course of Aβ plaque and NFT formation in patients will be important in the evaluation of new disease-modifying therapeutics aimed at slowing the clinical progression of AD.
Another protein capable of forming insoluble aggregates in brain is alpha synuclein (α-syn), which is the principal species found in Lewy bodies (LBs) and Lewy neurites (LNs).15, 16 LBs are a dense core of insoluble aggregates of α-syn found in the cell bodies of neurons, principally located in the brain stem and subcortical regions of the CNS. LNs are abnormal axons or dendrites that contain insoluble aggregates of α-syn and have a more diffuse morphology than LBs. The formation of LBs and LNs are the primary pathological features of a collection of neurological disorders referred to as “synucleinopathies”, which include Parkinson’s disease (PD), Dementia with Lewy Bodies (DLB), multiple system atrophy (MSA), and Picks’ disease.17 In addition, 50% of familial and sporadic AD patients show evidence of LBs at autopsy.18 Furthermore, AD patients with concomitant LB pathology demonstrate an accelerated cognitive decline and may represent a subset of AD patients.18 These data suggest that a PET radiotracer for imaging insoluble α-syn aggregates should be useful in the study of the formation of LBs and LNs in a diverse panel of CNS disorders.
Recently, a number of phenothiazine analogs including the tricyclic ligands SIL23, SIL5, and SIL26, the fluorescent dyes LDS 798 and LDS 730 and the indolinone 5 ((Figure 1) have shown reasonable in vitro selectivity for α-syn versus Aβ and tau fibrils.19–24 Although these compounds are noteworthy as the first analogs shown to label insoluble α-syn aggregates, their low in vitro stability and moderate affinity for α-syn aggregates limits their utility as PET imaging agents. Therefore, there is a need to identify new chemical scaffolds which can serve as lead compounds for PET radiotracer development. In this study, we describe the synthesis and in vitro evaluation of a novel class of compounds, the indolinone-diene analogs, having high affinity and selectivity for α-syn aggregates which could serve as a second generation lead compound for PET radiotracer development.
Figure 1.
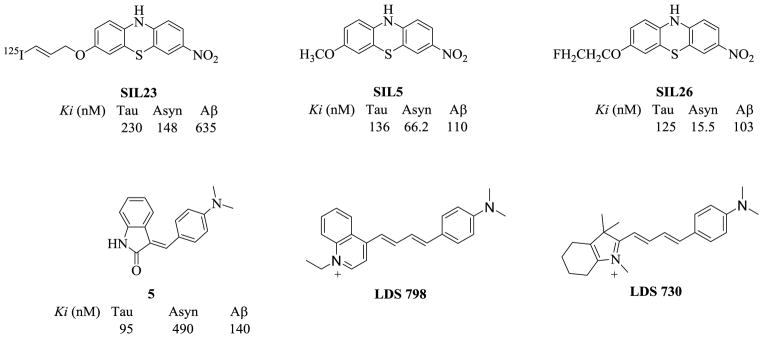
Reported ligands for tau, α-synuclein, and Aβ.
RESULTS
Chemistry
The first step in the design of a probe for imaging α-syn aggregates involved identifying a suitable lead compound for our structure-activity relationship (SAR) studies. An earlier publication reported that compound 5 (Figure 1) had a modest affinity for α-syn fibrils but low selectivity versus Aβ and tau fibrils.20 We synthesized a number of indolinone analogs to determine if it would be possible to improve the affinity of compound 5 for α-syn and reduce its affinity for Aβ and tau fibrils by exploring substituents in the benzylidene aromatic ring and alkylation the indolin-2-one nitrogen atom. A recent publication additionally demonstrated that the fluorescent dyes LDS 798 and LDS 730 labeled Lewy bodies in postmortem PD cases.21 Therefore, the diene fragment of LDS 798 and LDS 730 was incorporated into compound 5 to determine if these groups would improve the affinity and selectivity of the new analogs for α-syn fibrils versus Aβ and tau fibrils.
The synthesis of the 3-benzylidene-indolin-2-one analogs is shown in Scheme 1. 3-(4-(dimethylamino)benzylidene)indolin-2-one (5), 3-(4-(dimethylamino)benzylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (6), and 3-(4-(dimethylamino)benzylidene)-5-hydroxyindolin-2-one (7) were prepared by condensing indolin-2-one (1), 1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one (2), 5-hydroxyindolin-2-one (3) and dimethylaminobenzaldehyde (4) in acetic acid with refluxing for 3 h. The nitrogen of 5 and 6 was alkylated by treatment of 5 or 6 with sodium hydride in DMF at 0 °C, followed by addition of select alkyl halides to give compounds 8–14. Similarly, indolin-2-one (1), 1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one (2) or 5-hydroxyindolin-2-one (3) was condensed with a 4-substituted cinnamaldehyde (15–17) in acetic acid with refluxing to give 3-(4-substituted-phenyl)allylidene)indolin-2-one (18–20), 3-(3-(4-substituted phenyl)allylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (21) or 3-(3-(4-(dimethylamino)phenyl)allylidene)-5-hydroxyindolin-2-one (22) respectively. The nitrogen of compound 18–21 were alkylated by treatment of 18, 19, 20 or 21 with sodium hydride in DMF at 0 °C, followed by the addition of select alkyl halides to give compounds 23–32. The nitro group of 28 was reduced to an amino group with iron powder and HCl in methanol to afford compound 33. 1-Phenylindolin-2-one (34) was condensed with 4 or 17 in in acetic acid with refluxing to afford 35 or 36, respectively (Scheme 1).
Scheme 1.

Synthesis of 3-(Benzylidene)indolin-2-one Derivativesa
aReagents: (a) CH3COOH, reflux 3 h; (b) 1) NaH, DMF, 2) CH3I, R-C6H4CH2X (X = Cl, Br), or ClCO2C2H5 (c) 28, Fe, CH3OH, HCl, reflux 3 h.
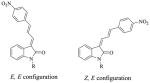
Compounds 5, 6, 7 and their nitrogen alkylated analogs (8–14, and 35) exist as either Z or E geometric isomers depending on the characteristics of the substituent at the C-3 position of the 3-substituted indolin-2-one or 3-substituted 1H-pyrrolo[2,3-b]pyridin-2(3H)-one. The 3-(4-substituted-phenyl)allylidene)indolin-2-one (18–20), 3-(3-(4-substituted phenyl)allylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (21) and 3-(3-(4-(dimethylamino)phenyl)allylidene)-5-hydroxyindolin-2-one (22) and their analogs (23–33, and 36), should theoretically have 4 geometric isomers for the double bond of each compound with Z,Z, E,E, Z,E, and E,Z configuration. Because only the trans (E isomer) of 4-substituded cinnamaldehyde was used for the synthesis, most of the compounds have only two geometric isomers: E,E or Z,E. We first attempted to separate the E,E and Z,E isomers by HPLC for binding assays. As an example, although the two isomers 20 and 27 could be separated by HPLC, the isolated pure Z,E and E,E isomers each isomerized within 20 min to a mixture of Z,E and E,E isomers. Because we were unable to obtain pure Z,E and E,E isomers for the binding assay, a mixture of the Z and E isomers for compounds 5–7, and their analogs (8–14 and 35), and mixtures of Z,E and E,E isomers of 18–22, and their analogs (23–32 and 36) were used to measure binding affinities; the ratio of the Z and E, or Z,E and E,E isomers was measured with HPLC and determined by 1H NMR spectra.
Similarly, compounds 39 and 42–46 were synthesized from condensation of 1 with 4-nitrobenzaldehyde (37) and (E)-3-(4-nitrophenyl)acrylaldehyde (40) to give 3-(4-nitrobenzylidene)indolin-2-one (38) and 3-(3-(4-nitrophenyl)allylidene)indolin-2-one (41) followed by alkylation of 38 and 41 with various alkyl halides (Scheme 2). Unlike other 4-substituted-benzaldehyde and 4-substituded cinnamaldehyde condensed analogs, the isomers of condensed products of 4-nitrobenzaldehyde (37) and (E)-3-(4-nitrophenyl)acrylaldehyde (40) and their derivatives could be separated by flash chromatography with CH2Cl2-ethyl ether (3:1) to afford 38a–39a and 38b–39b, and 41a–46a and 41b–46b, respectively. The stability of compounds 41a and 41b in solution was measured by HPLC. In a solvent mixture of CH2Cl2-MeOH (1:1), 41a was stable for 20 h as evidenced by the HPLC peak and retention time. In contrast, 41b was unstable under the same conditions, showing 33% isomerization to an un-identified isomer different from 41a (Supporting Information, Figure 4–7).
Scheme 2.

Synthesis of 3-(Benzylidene)indolin-2-one Derivativesa
aReagents: (a) CH3COOH, reflux 3 h; (b) 1) NaH, DMF, 2) CH3I, or R-C6H4CH2X (X = Cl, Br); (c) Fe, CH3OH, HCl, reflux 3 h.
The two isomers, forms Z,E and E,E of compound 41–46 could be distinguished by 2D NMR spectra analysis. Compounds 41a and 41b were used to determine configuration structure by 2D NMR. The E,E configured compounds should show a nuclear Overhauser effect (NOE) between the proton at the C-4 position and the vinyl proton, like H4 and Hb, whereas the Z,E configured compounds should not show a NOE effect between the proton at the C-4 position and the vinyl proton(s) in the C-3 substitution of the 3-substituted indolin-2-ones (Figure 2). The proton 1H shift of compound 41b was assigned by 1H NMR. In DMSO solution, the peak of H4 in C-4 position of indole overlapped with the peak of Hα in the nitrophenyl ring. After adding 10% D6-benzene, the two peaks is separated very well (H4, 8.15 ppm and Hα, 8.07 ppm, respectively). The H4 and Hb show a strong NOE effect in the 2D NMR spectra of 41b. Therefore, compound 41b is E,E configure, and 41a is Z,E configure (supporting information, Figure 1–3). The configurations of the remaining compounds 42a–46a and 42b–46b were assigned by comparison of their 1H NMR spectrum with 41a and 41b. The chemical shifts of its particular proton Hb (dd couple) is 8.81ppm for Z,E configuration and 7.78 ppm for E,E configure. Similarly, configurations of the compounds 5–14, and 35 were assigned based on the chemical shifts of particular protons, Ha, H-2 and/or H-6, by comparing the reported chemical shifts for compound 5;25 configurations of compounds 18–33 and 36 were assigned based on the chemical shifts of particular proton, Hb.
Figure 2.
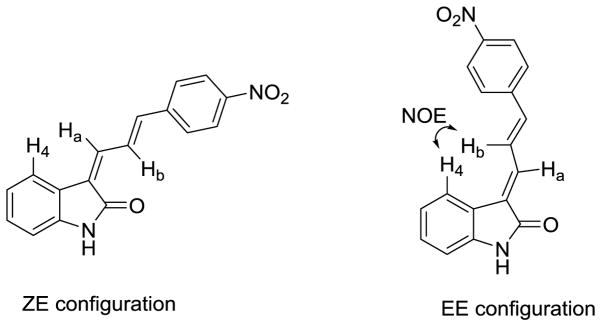
Nuclear Overhauser effect in E,E configuration of compound 41.
Radiosynthesis
The synthesis of the mesylate precursor 49 and the 18F-labeling are shown in Scheme 3. Radiosynthesis of [18F]46a was carried out manually starting from 49 under conventional conditions using tert-amyl alcohol as the solvent to afford [18F]46a with a yield of 19.3 ± 6.3 mCi (n = 6) (decay corrected) with specific activity ranging from 800 to 5000 mCi/μmol at the end of synthesis. The radiochemical purity was > 99%.
Scheme 3.

Synthesis of Labeling Precursor 48 and Radiosynthesis of [18F]46a
Reagents: (a) 1) NaH, DMF, 2) BrCH2C6H4OCH2CH2Br; (b) AgOMs, CH3CN; (c) [18F]KF, K2CO3, K222, tert-amyl alcohol.
Fibril Binding Assays
Thio-T competitive binding assays were utilized to measure affinity and selectivity for α-syn fibrils. Thio-T is a weakly fluorescent dye that displays a dramatic increase in fluorescence when bound to amyloid fibrils. Thio-T assays for α-syn fibrils were modified from our previous studies22 to improve the consistency of binding results for different classes of compounds. Furthermore, we developed additional competitive binding assays with Aβ and tau fibrils in order to evaluate selectivity. Under the conditions used for these studies, we determined that the affinity constants (Kd) of Thio-T for α-syn, Aβ and tau fibrils were 1850 nM, 32 nM, and 2700 nM respectively (Supplemental Figure S5). The competitive binding assays measured displacement of Thio-T binding by increasing concentrations of each indolinone compound, using fixed concentrations of fibrils and Thio-T. The Thio-T concentration was set at 150% of the Kd value determined for each fibril species).
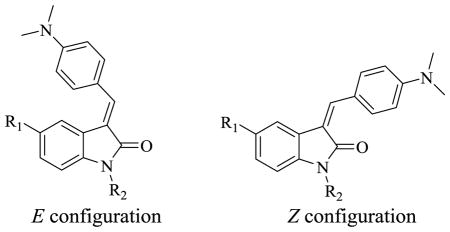
All final compounds in the manuscript possess a purity of at least 95% as determined by elemental analysis or HPLC analysis. The competitive binding results for indolinone and aza-indolinone analogs having a single double bond are shown in Table 1. In our assay, lead compound 5, with an E:Z ratio of 75:25, had a similar affinity for α-syn and Aβ fibrils (Ki ~ 85 nM) and a 3-fold lower affinity for tau fibrils (Ki = 262 nM). The corresponding aza-indolinone analog 6 had a reversed E:Z ratio and a higher affinity for Aβ (Ki = 40 nM) versus α-syn and tau fibrils (Ki = 125 – 190 nM). Substitution of the 5-position of 5 with a hydroxyl group 7 resulted in a reduction in affinity for α-syn fibrils (Ki = 407 nM) and no change in affinity for Aβ and tau fibrils. Substitution of the indolinone nitrogen atom (8–14, 35) had only a minor effect on the affinity of this series for all three fibrils. Replacement of the dimethyl amino group of 5 and 9 with a nitro group permitted the isolation of Z and E regioisomers. The Z isomer 38a had a higher affinity for α-syn (Ki = 347 nM) than the corresponding E isomer 38b (Ki = 620 nM), but the affinity was too low to be useful as a PET probe for imaging α-syn, a feature observed in all of the analogs having a single double bond in the indolinone ring system (Tables 1 and 3).
Table 1.
Ki Values of 3-(Benzylidene)indolin-2-one Derivatives for α-Syn, Aβ and Tau
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R1 | X | R | E:Z | α-Syn (nM) | Aβ (nM) | Tau (nM) | Log Pa |
|
| ||||||||
| 5 | H | CH | H | 75:25 | 84.4±13.5 | 91.6±18.5 | 261.5±4.9 | 3.0 |
| 6 | H | N | H | 26:74 | 124.3±46.1 | 40.0±22.3 | 191.3±39.5 | 2.4 |
| 7 | OH | CH | H | 67:33 | 407.1±98.6 | 94.8±22.4 | 212.4±110.9 | 2.6 |
| 8 | H | CH | CH3 | 84:16 | 507.6±10.0 | 155.1±8.8 | 617.6±182.0 | 3.2 |
| 9 | H | CH | PhCH2 | 83:17 | 144.4±53.2 | 260.4±78.5 | 797.8±70.2 | 4.9 |
| 10 | H | CH | p-CH3OC6H4CH2 | 52:48 | 114.5±53.9 | 251.5±52.0 | 431.7±123.3 | 4.8 |
| 11 | H | CH | p-PyridylCH2 | 77:23 | 149.8±45.7 | 87.1±34.1 | 364.0±121.1 | 3.6 |
| 12 | H | CH | CO2C2H5 | 62:38 | 342.6±114.4 | 139.9±70.1 | 1116.2±69.1 | 3.8 |
| 13 | H | N | PhCH2 | 66:34 | 350.2±201.3 | 190.2±84.8 | 858.8±172.0 | 4.3 |
| 14 | H | N | p-CH3OC6H4CH2 | 69:31 | 79.5±22.1 | 113.3±2.6 | 853.5±158.6 | 4.2 |
| 35 | H | CH | Ph | 94:6 | 290.2±120.2 | 179.2±71.3 | 789.8±153.7 | 4.9 |
Calculated by ChemBioDraw Ultra 13.0.
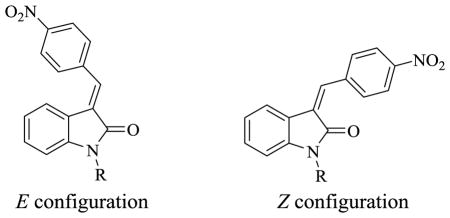
Table 3.
Ki Values of 3-(Benzylidene)indolin-2-one Derivatives for α-Syn, Aβ and Tau
| ||||||
|---|---|---|---|---|---|---|
| Compd | R | E or Z | α-Syn (nM) | Aβ (nM) | Tau (nM) | Log Pa |
| 38a | H | Z | 347.2±88.9 | 325.2±73.5 | 362.5±21.4 | 2.7 |
|
| ||||||
| 38b | H | E | 621.8±149.0 | 439.2±162.8 | 528.5±77.0 | 2.7 |
|
| ||||||
| 39a | PhCH2 | Z | 482.9±139.8 | 459.5±49.9 | 1756.7±104.5 | 3.8 |
|
| ||||||
| 39b | PhCH2 | E | 959.3±376.5 | 694.7±72.0 | 1410.0±298.4 | 3.8 |
|
| ||||||
| ||||||
|
| ||||||
| Compd | R | E,E or Z,E | α-Syn (nM) | Aβ (nM) | Tau (nM) | Log Pa |
|
| ||||||
| 41a | H | Z,E | 14.6±7.5 | 36.2±12.9 | 27.1±3.7 | 3.4 |
| 41b | H | E,E | 211.4±22.7 | 60.0±19.1 | 208.7±110.3 | 3.4 |
| 42a | C6H5CH2 | Z,E | 25.0±12.7 | 214.2±52.1 | 121.5±18.8 | 4.5 |
| 42b | C6H5CH2 | E,E | 192.7±57.4 | 235.4±143.0 | 590.5±74.7 | 4.5 |
| 43a | p-CH3OC6H4CH2 | Z,E | 12.9±4.9 | 130.8±64.4 | 72.4±24.1 | 4.7 |
| 43b | p-CH3OC6H4CH2 | E,E | 81.9±31.3 | 138.5±40.2 | 318.1±104.3 | 4.7 |
| 44a | m-CH3OC6H4CH2 | Z,E | 3.8±0.6 | 109.7±0.4 | 228.6±113.7 | 4.7 |
| 44b | m-CH3OC6H4CH2 | E,E | 64.7±14.6 | 264.8±64.4 | 354.6±211.7 | 4.7 |
| 45a | p,m-di-CH3OC6H4CH2 | Z,E | 3.5±0.2 | 73.6±27.3 | 151.7±64.7 | 4.8 |
| 45b | p,m-di-CH3OC6H4CH2 | E,E | 53.3±15.0 | 129.5±16.0 | 630.3±250.7 | 4.8 |
| 46a | p-FCH2CH2OC6H4CH2 | Z,E | 2.1±0.3 | 142.4±36.9 | 80.1±12.0 | 4.7b |
| 46b | p-FCH2CH2OC6H4CH2 | E,E | 76.1±41.2 | 125.3±40.1 | 455.7±66.1 | 4.7 |
Calculated by ChemBioDraw Ultra 13.0.
Measured: 4.18 ± 0.02 (n = 3).
The results of the in vitro binding data for the indolinone-diene analogs are shown in Tables 2 and 3. As with the analogs of 5, indolinone-diene analogs having either a hydrogen, methoxy, or dimethylamino group in the pendant aromatic ring consisted of a mixture of regioisomers, in this case E,E and Z,E isomers. Substitution of the para-position of the pendant phenyl group of 18 with a methoxy group (19) resulted in an increase in affinity for α-syn (Ki = 62 nM versus 206 nM), Aβ (Ki = 125 nM versus 267 nM), and tau fibrils (Ki = 169 nM versus 522 nM), with a slight preference in affinity for α-syn. However, a similar substitution with a dimethylamino group resulted in a compound (20) having a higher affinity for Aβ (Ki = 27 nM) versus α-syn (Ki = 41 nM) and tau (Ki = 54 nM). The corresponding aza-indolinone-diene analog of 20 (i.e., 21) had high affinity (Ki ~12 – 35 nM), for all three fibrils. Substitution of the indolinone nitrogen atom of 18 with a benzyl group resulted in 23, which was primarily in the Z,E configuration, and did not lead to an improvement in affinity for α-syn (Ki = 227 nM), Aβ (Ki = 700 nM) or tau (Ki = 680 nM) fibrils. However, substitution of the para position of 24 with either a methoxy group (27) or nitro group (28) resulted in an improvement in affinity for α-syn fibrils versus Aβ and tau fibrils, but it is not clear if this improvement in affinity was due to the substituent effect or the preference of these analogs for the E,E isomer. The other analogs in Table 2, which consisted of a mixture of E,E and Z,E isomers had modest affinity for α-syn, Aβ and tau fibrils. The most interesting compound in this series was 28, which has a 4 nM affinity for α-syn fibrils and 20-fold lower affinity for Aβ and tau fibrils.
Table 2.
Ki Values of 3-(Benzylidene)indolin-2-one Derivatives for α-Syn, Aβ and Tau

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # | R1 | X | R2 | R3 | EE:ZE | α-Syn (nM) | Aβ (nM) | Tau (nM) | Log Pa |
|
| |||||||||
| 18 | H | CH | H | H | 77:23 | 206.7±8.7 | 266.8±136.5 | 522.1±152.6 | 3.2 |
| 19 | H | CH | OCH3 | H | 78:22 | 61.1±9.6 | 125.8±42.6 | 169.0±22.3 | 3.1 |
| 20 | H | CH | N(CH3)2 | H | 41:59 | 40.7±8.7 | 27.6±4.8 | 53.7±9.7 | 3.5 |
| 21 | H | N | N(CH3)2 | H | 38:62 | 11.5±2.0 | 15.3±5.5 | 35.1±12.3 | 2.9 |
| 22 | OH | CH | N(CH3)2 | H | 47:53 | 54.0±17.9 | 37.1±13.6 | 48.0±17.0 | 3.1 |
| 23 | H | CH | H | CH2Ph | 0:100 | 226.9±64.0 | 703.3±214.7 | 682.4±120.2 | 5.2 |
| 24 | H | CH | OCH3 | CH2Ph | 84:16 | 72.5±22.6 | 741.0±254.6 | 717.6±295.3 | 5.0 |
| 25 | H | CH | N(CH3)2 | CH3 | 63:37 | 63.7±29.6 | 56.0±14.8 | 229.1±121.4 | 3.7 |
| 26 | H | CH | N(CH3)2 | CH2Ph | 65:35 | 40.9±25.2 | 357.9±161.7 | 374.0±209.3 | 5.5 |
| 27 | H | CH | N(CH3)2 | p-CH3OC6H4CH2 | 52:48 | 54.6±11.4 | 129.6±41.4 | 239.7±157.6 | 5.3 |
| 28 | H | CH | N(CH3)2 | p-NO2C6H4CH2 | 65.35 | 4.2±0.8 | 92.4±21.9 | 90.0±48.8 | 5.5 |
| 29 | H | CH | N(CH3)2 | p-PyridylCH2 | 50:50 | 92.0±11.2 | 45.9±19.4 | 188.0±68.0 | 4.1 |
| 30 | H | CH | N(CH3)2 | CO2C2H5 | 51:49 | 45.3±9.0 | 84.0±35.0 | 263.2±110.5 | 4.4 |
| 31 | H | N | N(CH3)2 | CH2Ph | 80:20 | 68.3±31.2 | 85.1±23.0 | 257.0±116.7 | 4.8 |
| 32 | H | N | N(CH3)2 | p-CH3OC6H4CH2 | 87:13 | 29.8±6.6 | 107.0±18.9 | 204.3±26.7 | 4.7 |
| 33 | H | CH | N(CH3)2 | p-NH2C6H4CH2 | 55:45 | 43.6±20.0 | 98.9±12.1 | 131.1±51.5 | 4.7 |
| 36 | H | CH | N(CH3)2 | Ph | 29:71 | 68.1±33.0 | 286.8±142.2 | 465.1±94.6 | 5.4 |
| 47 | H | H | NH2 | H | 44.8±15.6 | 50.8±18.5 | 38.3±28.7 | 2.4 | |
Calculated by ChemBioDraw Ultra 13.0.
Introduction of a para nitro group into the benzene ring of the diene moiety enabled the isolation of both the E,E and Z,E regioisomers. In general, the Z,E regioisomers were more potent than the corresponding E,E configuration (Table 3). Although compound 41a had a similar affinity for α-syn, Aβ and tau fibrils (Ki ~ 15 – 35 nM), substitution of the indolinone nitrogen with a benzyl group 42a resulted in increase in selectivity for α-syn (Ki = 40 nM) versus Aβ (Ki = 214 nM) and tau (Ki = 121 nM) fibrils. The most potent and selective compound in the series was 46a (Ki = 2 nM) and a 70-fold selectivity versus Ab and 40-fold selectivity versus tau fibrils. The 2-fluoroethoxy-group in the benzyl moiety enabled 18F-labeling for direct binding assays with α-syn, Aβ and tau fibrils.
Fluorescent tissue staining
The fluorescent properties of compound 20, and its high affinity for α-syn, Aβ, and tau fibrils (Figure 3) encouraged us to investigate in vitro fluorescent staining of postmortem brain samples from PD/DLB and PD cases. Sections from midbrain tissue of PD/DLB and temporal lobe tissue sections from neuropathologically verified AD cases were incubated with 5 μM of compound 20 and imaged using fluorescence microscopy. Antibody labeling was performed on adjacent sections to determine the protein species labeled by the fluorescent probe. Compound 20 was found to label both Lewy bodies and Aβ plaques, which was anticipated given the high affinity of this compound for α-syn and Aβ fibrils. In addition, the clear staining of LBs and Aβ plaques showed good agreement between the in vitro binding studies using the fibrillary protein and insoluble protein aggregates found in postmortem samples of PD/DLB and AD brain samples.
Figure 3.

Fluorescent microscopy studies of compound 20 in postmortem samples of PD/DLB and AD brain. Note the high labeling of both Lewy Bodies and Aβ plaques, which was confirmed with appropriate immunohistochemistry labeling.
Saturation binding assays to measure the affinity of 46a for α-syn fibrils
To further confirm the binding affinity determined in Thio-T assays, we synthesized [18F]46a and directly measured its binding affinity in saturation binding assays with α-syn fibrils. We observed specific binding of [18F]46a to α-syn fibrils with an average Kd of 8.9 nM (Figure 4a). We observed consistent binding values for three independently prepared fibril batches. Scatchard analysis indicates that the binding fits a one-site model (Figure 4b). We also determined binding affinity of 46a for Aβ and tau fibrils. The average Kd values for Aβ and tau fibrils were 271 nM and 50 nM, respectively, in two independent assays (Figure 5).
Figure 4. [18F]46a binds recombinant α-syn fibrils with an average Kd of 8.9 nM.

Fibrils prepared from recombinant α-syn were incubated with increasing concentrations of [18F]46a. Nonspecific binding was determined in parallel reactions utilizing 20 as the competitor or in reactions containing no fibrils. A representative plot of specific binding versus [18F]46a concentration is shown in A. Data points represent mean ± s.d. (n = 3). The data were analyzed by curve fitting to a one-site binding model using nonlinear regression. Kd values were determined by fitting the data to the equation Y=Bmax*X/(X+Kd). Scatchard analysis of binding is shown in B. Similar results were obtained in more than three independent experiments.
Figure 5. Radioligand binding studies demonstrate that [18F]46a binds with lower affinity to synthetic Aβ1–42 fibrils or recombinant tau fibrils compared to α-syn fibrils.

Binding affinities of [18F]46a to Aβ (A) and tau (C) fibrils were determined in saturation binding studies. The average Kd values of Aβ and tau fibrils were 271 nM and 50 nM, respectively. Data points represent mean ± s.d. (n = 3). Scatchard plots of binding values are shown in (B) for Aβ and (D) for tau. Similar results were obtained in two independent experiments.
DISCUSSION
The past decade has witnessed significant advances in the field of the development of PET-based radiotracers for imaging insoluble protein aggregates which occur in many neurodegenerative disorders. Radiotracers such as [11C]PiB, [18F]fluorbetapir, [18F]florbetaben and [18F]flutemetamol are widely used in clinical PET studies, and it is currently believed that imaging Aβ plaques with these radiotracers is capable of identifying patients for risk for developing AD.6, 7 More recent studies have focused on imaging tau; clinical PET studies conducted with [18F]T807 (7-(6-18F-fluoropyridin-3-yl)-5H-pyrido[4,3-b]indole), [18F]THK-5105 (6-[(3-18F-fluoro-2-hydroxy)propoxy]-2-(4-dimethylaminophenyl)quinoline), [18F]THK-5117 (6-[(3-18F-fluoro-2-hydroxy)propoxy]-2-(4-methylaminophenyl)quinoline), and [11C]PBB3 (2-((1E,3E)-4-(6-(11C-methylamino)pyridin-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol) have shown promise in imaging NFTs in AD patients.26 A logical next step in the evolution of PET radiotracers for imaging neurodegeneration is the development of a probe capable of imaging α-syn aggregates in Lewy bodies and Lewy neurites in PD.
A key step in the process of developing PET radioligands is the identification of a suitable lead compound for structure-activity relationship studies. The lead compound for the Aβ imaging agent [11C]PiB, was Thio-T, a fluorescent dye used for staining Aβ plaques in postmortem samples of AD brain tissue.3, 27, 28 The 18F-labeled compounds [18F]fluorbetapir and [18F]fluorbetapen were developed using X-34, a structural analog of the fluorescent dye Congo Red as the lead compound.29, 30 Thio-T also labels α-syn aggregates in Lewy bodies and Lewy neurites, but its low fluorescent staining of α-syn and higher affinity for Aβ plaques (which led to the development of [11C]PiB) suggest that it would not be a good lead compound for a PET radiotracer to image insoluble α-syn aggregates in PD. Although several tricyclic analogs (SIL23, SIL26, SIL5) were recently reported as ligands that bind aggregated α-syn in postmortem PD brain tissue assays,22, 23 their affinity and selectivity is not optimal for imaging α-syn aggregates in vivo.
The indolinone analog 5 (Figure 1) was previously identified as having modest binding affinity to α-syn in a rapid throughput screening assay potential lead compounds as a tau imaging agent for PET.20 We used this compound as the basis of the SAR studies described in this study. The first series of compounds focused on direct analogs of 5 and explored the effect of different substitutions in the indolinone ring system. Most of the compounds within this series had only modest affinity for α-syn and no selectivity towards α-syn, Aβ or tau fibrils. Although most of the compounds in this series consisted of a mixture of E and Z regioisomers, it was possible to separate compounds 38 and 39 into their E and Z isomers; the Z configuration was found to have a higher affinity for α-syn, Aβ and tau fibrils than the corresponding E isomer.
The presence of a diene moiety in the structure of the dyes LDS 798 and LDS 730 encouraged us to add a second double bond into the structure of 5. This change in structure resulted in improved affinity for α-syn fibrils; this indolinone-diene compound (20), which was also a mixture of E,E and Z,E regioisomers, had a high affinity for both Aβ and tau fibrils. Fortunately the strong fluorescence of compound 20 enabled us to confirm that the indolinone-dienes could label both Lewy bodies and Aβ plaques in postmortem samples of PD and AD brain. It was difficult to identify clear structure-activity relationships because of the EE:ZE mixture for each compound, which varied depending on the nature of the substitution of the indolinone nitrogen. However, it was possible to reach some conclusions by comparing structural congeners having a similar EE:ZE ratio. Substitution of the indolinone nitrogen with bulky substituents such as a benzyl or substituted benzyl group versus an N-methyl group appeared to improve affinity and selectivity for α-syn versus Aβ and tau. The 4-nitrobenzyl analog 28 was the most potent compound in this series, having an affinity for α-syn of 4 nM and 20-fold selectivity for α-syn versus Aβ and tau.
Introduction of the 4-nitrobenzene ring into the diene moiety enabled the separation of stable Z,E and E,E regioisomers, and led to the identification of the Z,E configuration as being the more active of the regioisomers. A number of compounds were identified having a high affinity for α-syn, and good selectivity for α-syn versus Aβ and tau fibrils. The most noteworthy compound in this series was 46a, which had a high affinity (2 nM) and excellent selectivity for α-syn versus Aβ and tau fibrils. This was also confirmed via direct binding studies with the corresponding 18F-labeled compound in α-syn, Aβ and tau fibrils. Unfortunately, the high log P value of this compound (4.18) made it difficult to obtain reliable and reproducible binding data from insoluble α-syn obtained from PD brain (results not shown). The high log P and potential of the nitro group to be reduced to the corresponding amino group in vivo indicates that [18F]46a will not be a useful in vivo PET tracer for imaging Lewy bodies and Lewy neurites in PD brain. However, our SAR results are noteworthy since they identified structural features leading to preferential binding to α-syn versus Aβ and tau fibrils. Consequently, compound 46a will serve as a secondary lead compound for further SAR studies. Other analogs within this series can serve as potential radioligands for in vitro binding assays for α-syn and represent an improvement to the current assay which uses Thio-T as a screening ligand, which binds more potently to Aβ versus α-syn, and tau fibrils (Supplemental Figure S5).
In summary, a series of indolinone and indolinone-diene analogs were synthesized and in vitro binding assays were conducted for α-syn, Aβ and tau fibrils. In general, the indolinone-diene analogs had a higher affinity for all three fibrils versus the corresponding indolinone congeners. The presence of two different regioisomers made it difficult to identify clear structure-activity relationships for the two series of compounds, but this was overcome by introduction of a para-nitro substituent into the benzene ring of the ene and diene fragment. Within the indolinone-diene series, the Z,E configuration was the more stable of the two regioisomers; substitution of the indolinone nitrogen with a benzyl moiety improved both the α-syn affinity and selectivity versus Aβ and tau fibrils. The results of this study have identified clear structural requirements for preparing compounds having a preferential affinity for α-syn versus Aβ and tau fibrils.
EXPERIMENTAL SECTION
General methods and materials
All chemicals were obtained from standard commercial sources and used without further purification. All reactions were carried out using standard air-free and moisture-free techniques under an inert nitrogen atmosphere with dry solvents unless otherwise stated. Flash column chromatography was conducted using Scientific Adsorbents, Inc. silica gel, 60A, “40 Micron Flash” (32–63 μm). Melting points were determined by using MEL-TEMP 3.0 apparatus and are uncorrected. Routine 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz on Agilent Technologies spectrometers. All chemical shifts were reported as a part per million (ppm) downfield from tetramethylsilane (TMS). All coupling constants (J) are given in Hertz (Hz). Splitting patterns are typically described as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Elemental analysis (C, H, N) was determined by Atlantic Microlab, Inc., Norcross, GA. Purities of final products were determined to be >95% by elemental analysis. [18F]Fluoride was produced at Washington University by the 18O(p,n)18F reaction through proton irradiation of enriched (95%) [18O] water in the RDS111 cyclotron. HPLC analysis was performed with an UV detector and a scintillation NaI (Tl) detector and associated electronics for radioactivity detection. An Agilent SB-C18 250×9.4 mm 5 μm semi-preparative column and an Agilent SB-C18 250×4.6 mm 5 μm analytical column were used for preparative purification and for post-synthesis analysis of chemical and radiochemical purity and specific activity, respectively. The semi-preparative HPLC conditions were: 62% acetonitrile and 38% ammonium formate buffer (0.1 M, pH 4.5) with a flow rate of 4 mL/min and UV at 264 nM for purification; the mobile phase for QC analysis was 80% acetonitrile and 20% ammonium formate buffer with a flow rate of 1.5 mL/min. Radio-TLC was accomplished using a Bioscan AR-2000 imaging scanner (Bioscan, Inc. Washington DC).
Synthesis method 1
The 2-oxindole (20 mmol) and aldehyde (20 mmol) in acetic acid (50 mL) and 37% HCl (1 mL) were heated at reflux for 3 h, then water (500 mL) was added at rt. The solids were filtered out and recrystallized with methanol to afford the 3-(substituted) indolin-2-one condensation product.
Synthesis method 2
The 3-(substituted) indolin-2-one (1.0 mmol) in THF 5 mL) was added to 60% NaH (1.5 mmol) at 0 °C. After 15 min, the alkyl halide (3 mmol) was added. The reaction mixture was stirred for 8 h, ethyl acetate (75 mL) was added, washed with water (50 mL x 2), saturated NaCl (50 mL) and dried over Na2SO4. After evaporation of the ethyl acetate, the crude product was purified by silica gel column chromatography eluting with hexane-CH2Cl2-EtOAc (10:10:3, v/v/v) to afford the nitrogen alkylated compound.
3-(4-(Dimethylamino)benzylidene)indolin-2-one (5) was prepared by method 1 with compound 1 and 4 to afford the desired product, (5), as a red solid (90%), mp 190.5–191.9 °C. 1H NMR (400 MHz, CDCl3) δ 9.20 and 8.74 (s, 1H), 8.40, and 7.69 (d, J = 8.8 Hz, 2H), 7.90 and 7.48 (d, J = 7.6 Hz, 1H), 7.79 and 7.47 (s, 1H), 7.18 and 7.00 (t, J = 7.6 Hz, 1H), 7.15 and 6.92 (t, J = 8.0 Hz, 1H), 6.94 and 6.87 (d, J = 7.6 Hz, 2H), 3.07 (s, 6H). (13C NMR (100 MHz, CDCl3) δ 171.4, 168.6, 151.4, 141.0,139.1, 138.9, 138.7, 134.9, 132.2, 128.5, 127.0, 126.6, 122.6, 122.4, 122.3, 122.1, 121.4, 121.2, 120.4, 118.1, 111.4, 111.2, 110.1, 109.3, 40.1, 40.0. Anal. C17H16N2O Calcd. C: 77.25, H: 6.10, N: 10.60. Found C: 77.24, H: 6.29, N: 10.60.
3-(4-(Dimethylamino)benzylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (6) was prepared by method 1 with compound 2 and 4 to afford the desired product, (6), as a red solid (47%), mp 247.4–248.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.40 and 7.64 (d, J = 8.8 Hz, 2H), 8.10 and 7.67 (d, J = 6.4 Hz, 2H), 7.87 and 7.42 (s, 1H), 6.95 and 6.88 (t, J = 7.2 Hz, 1H), 6.76 (d, J = 8.4 Hz, 2H), 3.10 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.5, 167.7, 156.6, 154.2, 152.6, 152.2, 147.0, 145.8, 140.6, 139.8, 135.6, 132.9, 128.8, 125.6, 122.3, 121.2, 121.1, 120.4, 118.4, 117.6, 117.4, 116.7, 112.1, 111.6, 40.2. Anal. C16H15N3O.0.25H2O Calcd. C: 71.82, H: 5.75, N: 15.70. Found C: 71.89, H: 5.83, N: 15.58.
3-(4-(Dimethylamino)benzylidene)-5-hydroxyindolin-2-one (7) was prepared by method 1 with compound 3 and 4 to afford the desired product, (7), as a red solid (77%), mp 221.1–222.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 8.89 (s, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.43 (s, 1H), 7.26 (s, 1H), 6.78 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.57 (d, J = 7.6 Hz, 1H), 2.99 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.9, 152.1, 151.7, 137.4, 135.0, 132.2, 123.6, 122.9, 121.6, 115.7, 111.9, 110.4, 109.8, 40.1. Anal. C17H16N2O2.0.125H2O Calcd. C: 72.26, H: 5.80, N: 9.91. Found C: 72.31, H: 5.80, N: 9.80.
3-(4-(Dimethylamino)benzylidene)-1-methylindolin-2-one (8) was prepared by method 2 with compound 5 and CH3I to afford the desired product, (8), as a red solid (55%), mp 100.3–101.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.42 and 7.67 (d, J = 8.0 Hz, 2H), 7.91 and 7.49 (d, J = 7.6 Hz, 1H), 7.80 and 7.46 (s, 1H), 7.24 (t, J = 7.2 Hz, 1H), 7.03 and 6.94 (t, J = 7.2 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 2H), 3.29 (s, 3H), 3.07 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.4, 151.3, 143.5, 138.8, 138.3, 134.8, 132.0, 128.4, 122.3, 122.2, 122.1, 122.0, 121.4, 121.3, 111.4, 111.1, 107.9, 107.5, 40.1, 26.1. Anal. C18H18N2O Calcd. C: 77.67, H: 6.52, N: 10.06. Found C: 77.45, H: 6.77, N: 9.83.
1-Benzyl-3-(4-(dimethylamino)benzylidene)indolin-2-one (9) was prepared by method 2 with compound 5 and benzyl bromide to afford the desired product, (9), as an orange solid (82%), mp 111.0–112.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.44 and 7.69 (d, J = 8.8 Hz, 2H), 7.92 and 7.50 (d, J = 7.6Hz, 1H), 7.87 and 7.51 (s, 1H), 7.35-7.22 (m, 5H), 7.11 (t, J = 7.6 Hz, 1H), 6.90 (t, J = 7.2 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 6.71 (m, 1H), 5.01 (s, 2H), 3.06 (s, 6H). (13C NMR (100 MHz, CDCl3) δ 169.4, 152.0, 151.4, 142.6, 140.3, 139.2, 138.6, 136.7, 136.4, 134.9, 132.1, 128.7, 128.6, 128.3, 127.4, 127.3, 126.9, 122.5, 122.2, 122.1, 122.0, 121.5, 121.4, 117.8, 111.4, 111.2, 108.9, 108.4, 43.7, 40.1, 40.0. Anal. C24H22N2O Calcd. C: 81.33, H: 6.26, N: 7.90. Found C: 81.22, H: 6.34, N: 7.96.
3-(4-(Dimethylamino)benzylidene)-1-(4-methoxybenzyl)indolin-2-one (10) was prepared by method 2 with compound 5 and 4-methoxybenzyl chloride to afford the desired product, (10), as an orange solid (61%), mp 154.8–155.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.44 and 7.68 (d, J = 8.8 Hz, 2H), 7.91(d, J = 7.6 Hz, 1H), 7.86 and 7.49 (s, 1H), 7.48 and 7.26 (d, J = 5.2 Hz, 1H), 7.28 (d, J = 8.8 Hz, 2H), 7.10 (t, J = 7.6 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.82 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 169.3, 166.6, 158.8, 151.9, 151.4, 142.6, 140.4, 139.1, 138.5, 134.9, 132.1, 128.9, 128.7, 128.5, 128.3, 125.7, 122.5, 122.1, 121.4, 121.3, 112.0, 117.8, 114.1, 114.0, 111.4, 111.2, 108.9, 108.4, 55.2, 43.1, 42.9, 40.1, 40.0. Anal. C25H24N2O2 Calcd. C: 78.10, H: 6.29, N: 7.29. Found C: 77.85, H: 6.47, N: 7.24.
3-(4-(Dimethylamino)benzylidene)-1-(pyridin-4-ylmethyl)indolin-2-one (11) was prepared by method 2 with compound 5 and 4-(bromomethyl)pyridine to afford the desired product, (11), as a red solid (56%), mp 153.0–153.7 °C. 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 4.8 Hz, 2H), 8.43 and 7.97 (d, J = 8.0 Hz, 1H), 7.89 and 7.55 (s, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 5.2 Hz, 2H), 7.14 (t, J = 7.6 Hz, 1H), 6.95 (t, J = 7.6 Hz, 1H), 6.76 (d, J = 9.2 Hz, 2H), 6.63 (d, J = 8.0 Hz, 1H), 5.02 (s, 2H), 3.09 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.4, 151.5, 150.2, 150.1, 145.5, 141.8, 139.9, 135.0, 132.3, 128.3, 122.3, 122.2, 122.0, 121.9, 121.8, 121.3, 118.0, 111.4, 111.2, 108.5, 42.6, 40.1, 40.0. Anal. C23H21N3O Calcd. C: 77.72, H: 5.96, N: 11.82. Found C: 77.64, H: 6.12, N: 11.74.
Ethyl 3-(4-(dimethylamino)benzylidene)-2-oxoindoline-1-carboxylate (12) was prepared by method 2 with compound 5 and ethyl chloroformate to afford the desired product, (12), as a red solid (59%), mp 121.7–123.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 and 7.67 (d, J = 8.4 Hz, 2H), 7.98 and 7.89 (d, J = 7.6 Hz, 2H), 7.82 and 7.50 (s, 1H), 7.52 and 7.29 (t, J = 7.6 Hz, 1H), 7.17 and 7.07 (t, J = 7.6 Hz, 1H), 6.74 (d, J = 8.4 Hz, 2H), 4.51 (q, J = 7.2 Hz, 2H), 3.09 (s, 6H), 1.48 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.3 151.7, 140.5, 140.1, 135.5, 132.3, 128.6, 123.7, 122.8, 121.7, 121.4, 117.5, 115.0, 114.8, 111.3, 111.1, 63.1, 40.1, 14.3. Anal. C20H20N2O3 Calcd. C: 71.41, H: 5.99, N: 8.33. Found C: 71.57, H: 6.15, N: 8.33.
1-Benzyl-3-(4-(dimethylamino)benzylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (13) was prepared by method 2 with compound 6 and benzyl bromide to afford the desired product, (13), as a red solid (70%), mp 144.7–145.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.41 and 7.61 (d, J = 8.8 Hz, 2H), 8.10 and 8.05 (d, J = 7.6 Hz, 1H), 7.90 and 7.41 (s, 1H), 7.49 (d, J = 7.6 Hz, 2H), 7.31-7.21 (m, 4H), 6.91 and 6.83 (dd, J = 7.6 Hz, 5.6 Hz, 1H), 6.73 and 6.70 (d, J = 8.8 Hz, 2H), 5.13 and 5.11 (s, 2H), 3.06 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.9, 166.2, 155.7, 153.6, 152.3, 151.7, 146.5, 145.5, 140.7, 140.3, 137.4, 137.2, 135.3, 132.4, 128.5, 128.4, 128.3, 127.3, 127.2, 124.3, 122.1, 121.7, 120.4, 112.0, 117.5, 117.3, 116.9, 111.5, 111.2, 42.5, 42.3, 40.1, 40.0. Anal. C23H21N3O Calcd. C: 77.72, H: 5.96, N: 11.82. Found C: 77.86, H: 6.05, N: 11.81.
3-(4-(Dimethylamino)benzylidene)-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (14) was prepared by method 2 with compound 6 and 4-methoxybenzyl chloride to afford the desired product, (14), as a red solid (70%), mp 123.5–124.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.41 and 8.04 (d, J = 8.4 Hz, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.89 and 7.41 (s, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 6.93-6.80 (m, 3H), 6.73 and 6.71 (d, J = 9.2 Hz, 2H), 5.06 and 5.04 (s, 2H), 3.75 (s, 3H), 3.07 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.8, 166.1, 158.9, 158.8, 155.7, 153.6, 152.3, 151.7, 146.4, 145.4, 140.5, 140.1, 135.2, 132.3, 129.8, 129.7, 129.5, 128.3, 124.2, 122.1, 121.7, 120.4, 120.0, 117.6, 117.2, 116.8, 113.8, 111.4, 111.2, 55.2, 41.9, 41.7, 40.1, 40.0. Anal. C24H23N3O2 Calcd. C: 74.78, H: 6.01, N: 10.90. Found C: 75.04, H: 6.10, N: 10.86.
3-(3-Phenylallylidene)indolin-2-one (18) was prepared by method 1 with compound 1 and 15 to afford the desired product, (18), as an orange solid (91%), mp 203.9–205.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.60-8.53 (m, 1H), 8.30 (s, 1H), 7.73-7.58 (m, 3H), 7.51-7.30 (m, 4H), 7.26-7.15 (m, 2H), 7.09-6.85 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 170.3, 169.1, 144.5, 143.1, 141.0, 139.8, 136.5, 136.4, 136.0, 129.7, 129.3, 129.0, 128.8, 128.7, 127.8, 127.7, 125.7, 124.8, 124.4, 124.2, 123.7, 123.4, 123.0, 122.1, 121.8, 119.5, 110.2, 109.7. Anal. C17H13NO.0.125H2O Calcd. C: 81.82, H: 5.33, N: 5.61. Found C: 81.73, H: 5.47, N: 5.48.
3-(3-(4-Methoxyphenyl)allylidene)indolin-2-one (19) was prepared by method 1 with compound 1 and 16 to afford the desired product, (19), as an orange solid (85%), mp 226.1–226.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.46 and 10.41 (s, 1H), 7.93 (d, J = 7.2 Hz, 1H), 7.72 (d, J = 7.6 Hz, 1H), 7.60-7.50 (m, 2H), 7.29-7.07 (m, 3H), 6.97 (m, 3H), 6.81 (d, J = 7.2 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.4, 168.8, 161.1, 160.8, 145.0, 142.7, 142.3, 141.2, 136.5, 135.6, 130.3, 129.5, 129.3, 129.2, 129.1, 129.0, 124.9, 124.5, 124.4, 124.3, 122.7, 122.3, 121.8, 121.5, 121.4, 120.1, 115.1, 114.8, 110.0, 109.8, 55.8. Anal. C18H15NO2 Calcd. C: 77.96, H: 5.45, N: 5.05. Found C: 77.66, H: 5.60, N: 5.02.
3-(3-(4-(Dimethylamino)phenyl)allylidene)indolin-2-one (20) was prepared by method 1 with compound 1 and 17 to afford the desired product, (20), as an orange solid (82%), mp 227.8–229.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.37 and 10.33 (s, 1H), 8.18 and 7.45 (dd, J = 15.6 Hz, 11.6 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.60 and 7.39 (d, J = 8.8 Hz, 2H), 7.47 and 7.24 (d, J = 12.0 Hz, 1H), 7.20 and 7.04 (d, J = 15.6 Hz, 1H), 7.14 and 7.09 (t, J = 7.6 Hz, 1H), 6.96 and 6.89 (t, J = 7.6 Hz, 1H), 6.80 and 6.75 (d, J = 7.2 Hz, 1H), 6.72 and 6.71 (d, J = 8.8 Hz, 2H), 2.97 and 2.95 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.5, 168.9, 151.8, 151.6, 146.4, 144.3, 141.9, 140.7, 136.6, 130.3, 129.5, 128.5, 124.7, 124.2, 124.0, 123.2, 122.8, 122.4, 121.6, 121.6, 121.2, 119.8, 112.5, 112.3, 109.9, 109.7, 40.2. Anal. C19H18N2O.0.125H2O Calcd. C: 77.99, H: 6.29, N: 9.57. Found C: 77.95, H: 6.32, N: 9.56.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (21) was prepared by method 1 with compound 2 and 17 to afford the desired product, (21), as a purple solid (74%), mp 273.6–274.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.96 and 10.92 (s, 1H), 8.22 and 7.79 (d, J = 7.2 Hz, 1H), 7.99 and 7.96 (d, J = 4.8 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.44-7.34 (m, 2H), 7.26 and 7.08 (d, J = 15.2 Hz, 1H), 6.95 and 6.91 (d, J = 7.4 Hz, 5.2 Hz, 1H), 6.70 (d, J = 8.8 Hz, 2H), 2.97 and 2.96 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.1, 156.0, 155.1, 152.0, 151.8, 147.9, 146.4, 145.8, 139.7, 138.6, 130.7, 130.4, 129.9, 126.3, 123.9, 123.8, 120.8, 119.7, 119.0, 118.5, 117.7, 117.5, 117.4, 112.5, 112.2, 40.1. Anal. C18H17N3O Calcd. C: 74.20, H: 5.88, N: 14.42. Found C: 73.92, H: 5.84, N: 14.32.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-5-hydroxyindolin-2-one (22) was prepared by method 1 with compound 1 and 15 to afford the desired product, (22), as a purple solid (81%), mp >280 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.06 and 10.02 (s, 1H), 8.93 and 8.91 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.38-6.88 (m, 5H), 6.71 (d, J = 7.2 Hz, 2H), 6.59-6.54 (m, 2H), 2.96 and 2.94 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.5, 169.1, 152.5, 152.4, 151.7, 151.5, 145.7, 144.0, 137.0, 136.0, 134.5, 133.4, 129.9, 129.4, 125.5, 124.3, 124.0, 123.8, 123.7, 123.2, 119.8, 118.5, 115.0, 112.6, 112.4, 111.1, 110.3, 110.1, 110.0, 106.9, 40.1. Anal. C19H18N2O2.0.25H2O Calcd. C: 73.41, H: 6.00, N: 9.01. Found C: 73.50, H: 6.09, N: 8.72.
1-Benzyl-3-(3-phenylallylidene)indolin-2-one (23) was prepared by method 2 with compound 18 and benzyl bromide to afford the desired product, (23), as an orange solid (90%), mp 124.6–125.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.67 (dd, J = 15.8 Hz, 11.6 Hz, 1H), 7.62 (d, J = 7.2 Hz, 2H), 7.46 (d, J = 7.6 Hz, 1H), 7.39-7.25 (m, 9H), 7.14 (t, J = 7.6 Hz, 1H), 7.05 (d, J = 16.0 Hz, 1H), 6.99 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 4.97 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 167.4, 143.0, 141.6, 136.4, 136.3, 129.3, 128.8, 128.7, 128.6, 127.8, 127.5, 127.3, 124.5, 124.4, 123.5, 121.9, 119.3, 108.9, 43.3. Anal. C24H19NO Calcd. C: 85.43, H: 5.68, N: 4.15. Found C: 85.22, H: 5.88, N: 4.13.
1-Benzyl-3-(3-(4-methoxyphenyl)allylidene)indolin-2-one (24) was prepared by method 2 with compound 19 and benzyl bromide to afford the desired product, (24), as an orange solid (78%), mp 167.3–169.3 °C. 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 7.2 Hz, 1H), 7.59-7.51 (m, 3H), 7.35-7.25 (m, 5H), 7.17-7.11 (m, 2H), 7.04 and 6.99 (t, J = 7.6 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 9.2Hz, 1H), 6.72 and 6.70 (d, J = 8.0 Hz, 1H), 4.99 (s, 2H), 3.86 and 3.84 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 168.8, 161.0, 160.8, 144.3, 143.1, 142.5, 136.9, 136.7, 136.4, 136.2, 129.5, 129.3, 128.9, 128.7, 128.4, 128.2, 127.4, 127.3, 127.2, 123.9, 123.2, 122.7, 122.6, 122.0, 121.7, 121.4, 119.0, 114.5, 114.3, 109.1, 108.8, 55.4, 43.6. Anal. C25H21NO2 Calcd. C: 81.72, H: 5.76, N: 3.81. Found C: 81.57, H: 5.93, N: 3.82.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-methylindolin-2-one (25) was prepared by method 2 with compound 20 and CH3I to afford the desired product, (25), as an orange solid (67%), mp 170.5–171.5 °C. 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 7.2 Hz, 1H), 7.53-7.41 (m, 3H), 7.30 (d, J = 11.6 Hz, 1H), 7.24 and 7.21 (t, J = 7.6 Hz, 1H), 7.09 and 6.97 (d, J = 14.8 Hz, 1H), 7.07 and 7.00 (t, J = 8.0 Hz, 1H), 6.83 and 6.79 (d, J = 8.0 Hz, 1H), 6.70 and 6.68 (d, J = 8.8 Hz, 2H), 3.27 (s, 3H), 3.02 and 3.02 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.9, 167.7, 151.4, 151.2, 145.5, 144.3, 143.0, 141.8, 137.6, 137.4, 129.6, 129.4, 127.8, 127.6, 124.6, 124.1, 124.0, 122.9, 122.8, 122.2, 121.7, 121.5, 120.3, 119.0, 118.5, 112.0, 111.9, 107.8, 107.6, 40.2, 26.0, 25.6. Anal. C20H20N2O Calcd. C: 78.92, H: 6.62, N: 9.20. Found C: 78.73, H: 6.72, N: 9.29.
1-Benzyl-3-(3-(4-(dimethylamino)phenyl)allylidene)indolin-2-one (26) was prepared by method 2 with compound 20 and benzyl bromide to afford the desired product, (26), as an orange solid (85%), mp 131.5–133.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 5.6 Hz, 1H), 7.84 and 7.58 (d, J = 7.6 Hz, 1H), 7.62 (d, J = 12.4 Hz, 1H), 7.53-7.64 (m, 4H), 7.36-7.20 (m, 5H), 7.12 and 7.01 (d, J = 14.8 Hz, 1H), 6.95 and 6.90 (t, J = 5.6 Hz, 1H), 6.69 (d, J = 8.8 Hz, 2H), 5.08 (s, 2H), 3.05 and 3.03 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.4, 155.3, 151.6, 151.5, 146.9, 146.0, 139.7, 139.3, 137.3, 137.2, 130.0, 129.7, 129.0, 128.5, 128.4, 128.2, 127.3, 123.7, 120.3, 120.0, 118.3, 117.6, 117.5, 111.9, 42.4, 40.1. Anal. C26H24N2O Calcd. C: 78.71, H: 6.08, N: 11.02. Found C: 78.27, H: 6.16, N: 10.87.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-(4-methoxybenzyl)indolin-2-one (27) was prepared by method 2 with compound 20 and 4-methoxybenzyl chloride to afford the desired product, (27), as an orange solid (47%), mp 169.5–170.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.46 and 7.46 (dd, J = 15.4 Hz, 11.6 Hz, 1H), 7.70 and 7.41 (d, J = 7.6 Hz, 1H), 7.57 and 7.33 (d, J = 12.0 Hz, 1H), 7.52-7.46 (m, 2H), 7.28-7.24 (m, 2H), 7.14-6.94 (m, 3H), 6.84-6.81 (m, 2H), 6.74-6.65 (m, 3H), 4.91 (s, 2H), 3.75 (s, 3H), 3.02 and 3.01 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.0, 167.7, 158.9, 151.4, 151.2, 145.7, 144.6, 142.1, 140.9, 137.9, 137.8, 129.7, 129.5, 128.7, 128.6, 128.5, 127.7, 127.5, 124.6, 124.1, 124.0, 123.1, 122.9, 122.0, 121.7, 121.5, 121.2, 120.3, 118.9, 118.6, 114.0, 112.0, 111.9, 108.9, 108.6, 55.2, 43.0, 42.7, 40.2, 40.1. Anal. C27H26N2O2 Calcd. C: 79.00, H: 6.38, N: 6.82. Found C: 79.08, H: 6.51, N: 6.75.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-(4-nitrobenzyl)indolin-2-one (28) was prepared by method 2 with compound 20 and 4-nitrobenzyl bromide to afford the desired product, (28), as an orange solid (83%), mp 204.2–205.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 7.2 Hz, 1H), 7.61 and 7.40 (d, J = 12.4 Hz, 1H), 7.54-7.45 (m, 5H), 7.17-7.00 (m, 3H), 6.71-6.61 (m, 3H), 5.08 (s, 2H), 3.06 and 3.04 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.0, 167.6, 151.6, 147.4, 146.6, 145.5, 144.0, 138.8, 138.6, 137.7, 129.8, 129.7, 128.0, 127.9, 127.7, 127.5, 124.0, 123.9, 123.1, 122.3, 122.1, 121.1, 120.1, 118.8, 118.7, 112.0, 111.9, 108.4, 108.1, 43.0, 42.7, 40.2. Anal. C26H23N3O3 Calcd. C: 73.39, H: 5.45, N: 9.88. Found C: 73.48, H: 5.63, N: 9.81.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-(pyridin-4-ylmethyl)indolin-2-one (29) was prepared by method 2 with compound 20 and 4-(bromomethyl)pyridine to afford the desired product, (29), as a purple solid (51%), mp 194.1–195.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 4.4 Hz, 2H), 7.76 (d, J = 7.2 Hz, 1H), 7.61 (d, J = 12.4 Hz, 1H), 7.54-7.39 (m, 3H), 7.20-7.02 (m, 5H), 6.71 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 7.6 Hz, 1H), 5.00 (s, 2H), 3.06 and 3.03 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.0, 151.5, 150.1, 146.4, 145.5, 145.3, 141.4, 138.5, 129.8, 129.6, 127.8, 123.9, 123.1, 122.2, 122.0, 121.3, 118.7, 112.0, 111.9, 108.5, 42.6, 40.2. Anal. C25H23N3O Calcd. C: 78.71, H: 6.08, N: 11.02. Found C: 78.76, H: 6.27, N: 10.95.
Ethyl 3-(3-(4-(dimethylamino)phenyl)allylidene)-2-oxoindoline-1-carboxylate (30) was prepared by method 2 with compound 20 and ethyl chloroformate to afford the desired product, (30), as a red solid (57%), mp 169.9–170.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.39 and 7.53 (dd, J = 15.2 Hz, 12.0 Hz, 1H), 7.98 and 7.91 (d, J = 8.0 Hz, 1H), 7.77 and 7.46 (d, J = 7.2 Hz, 1H), 7.52 and 7.50 (d, J = 9.2 Hz, 2H), 7.37-7.02 (m, 4H), 6.70 and 6.67 (d, J = 8.8 Hz, 2H), 4.51 (q, J = 7.2 Hz, 2H), 3.05 and 3.04 (s, 6H), 1.49 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.5, 151.5, 147.3, 146.4, 139.3, 139.1, 137.3, 130.0, 129.8, 128.0, 127.9, 124.7, 124.3, 124.1, 124.0, 123.8, 122.4, 120.2, 119.2, 118.5, 118.1, 115.1, 115.0, 112.0, 111.9, 63.1, 40.1, 14.4. Anal. C22H22N2O3 Calcd. C: 72.91, H: 6.12, N: 7.73. Found C: 73.18, H: 6.04, N: 7.80.
1-Benzyl-3-(3-(4-(dimethylamino)phenyl)allylidene)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (31) was prepared by method 2 with compound 21 and benzyl bromide to afford the desired product, (31), as a purple solid (72%), mp 203.1–204.0 °C. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 5.2 Hz, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.61 (d, J = 12.0 Hz, 1H), 7.52-7.34 (m, 4H), 7.31-7.20 (m, 4H), 7.11 (d, J = 15.2Hz, 1H), 6.94 (dd, J = 7.4 Hz, 5.2 Hz, 1H), 6.68 (d, J = 9.2 Hz, 2H), 5.01 (s, 2H), 3.03 and 3.02 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.4, 155.3, 151.6, 151.5, 146.9, 146.0, 145.9, 139.6, 139.3, 137.3, 137.2, 130.0, 129.7, 129.0, 128.5, 128.4, 128.2, 127.3, 125.0, 124.2, 123.7, 120.3, 120.0, 119.0, 118.7, 118.3, 117.6, 117.4, 111.9, 42.4, 42.1, 40.1. Anal. C25H23N3O Calcd. C: 78.71, H: 6.08, N: 11.02. Found C: 78.88, H: 6.21, N: 10.93.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridin-2(3H)-one (32) was prepared by method 2 with compound 21 and 4-methoxybenzyl chloride to afford the desired product, (32), as a purple solid (81%), mp 189.7–191.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 5.6 Hz, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.60 (d, J = 12.8 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.30 (dd, J = 14.8 Hz, 12.4 Hz, 1H), 7.11 (d, J = 14.8 Hz, 1H), 6.93 (dd, J = 7.2 Hz, 5.6 Hz, 1H), 6.81 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 5.01 (s, 2H), 3.75 (s, 3H), 3.03(s, 6H). (13C NMR (100 MHz, CDCl3) δ 168.3, 158.8, 155.3, 151.6, 146.8, 145.9, 139.2, 129.8, 129.7, 129.5, 129.0, 123.7, 120.1, 118.4, 117.6, 117.4, 113.8, 111.9, 55.2, 41.8, 40.1. Anal. C26H25N3O2 Calcd. C: 75.89, H: 6.12, N: 10.21. Found C: 75.97, H: 6.13, N: 10.18.
1-(4-Aminobenzyl)-3-(3-(4-(dimethylamino)phenyl)allylidene)indolin-2-one (33) A mixture of 28 (425 mg, 1 mmol) and Fe powder (560 mg, 10 mmol) in methanol (30 mL) and 37% HCl (1 mL) was heated to reflux for 3 h. The solid was filtered out, the filtrate was evaporated, ethyl acetate (75 mL) was added, washed with saturated Na2CO3 (50 mL), water (50 mL), NaCl (50 mL), and dried over Na2SO4. After removed of the solvent, the crude product was purified by silica gel column chromatography eluting with EtOAc-MeOH (10:1, v/v) to afford 263 mg (66%) of 33 as a red solid, mp 221.0–222.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, J = 7.6 Hz, 1H), 7.61 (d, J = 8.8 Hz, 2H), 7.51-7.36 (m, 2H), 7.26 (d, J = 14.4 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 7.6 Hz, 1H), 6.70 (d, J = 8.4 Hz, 2H), 6.46 (d, J = 8.4 Hz, 2H), 4.98 (s, 2H), 4.71 (s, 2H), 2.96 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 168.1, 151.9, 148.4, 147.2, 142.2, 137.5, 130.4, 128.8, 128.3, 124.0, 123.9, 123.6, 122.6, 122.1, 121.5, 118.6, 114.3, 112.3, 109.4, 42.7, 40.1. Anal. C26H25N3O.0.5H2O Calcd. C: 77.20, H: 6.48, N: 10.39. Found C: 77.74, H: 6.34, N: 10.31.
3-(4-(Dimethylamino)benzylidene)-1-phenylindolin-2-one (35) was prepared by method 1 with compound 4 and 34 to afford the desired product, (35), as a yellow solid (88%), mp 171.1–172.8 °C. 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 6.8 Hz, 1H), 7.89 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.53-7.40 (m, 5H), 7.16 (t, J = 7.2 Hz, 1H), 6.97 (t, J = 7.2 Hz, 1H), 6.84 (d, J = 6.8 Hz, 1H), 6.77 (d, J = 8.8 Hz, 2H), 3.01 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.7, 151.4, 143.3, 139.6, 135.0, 132.2, 129.5, 128.3, 127.7, 126.9, 122.2, 122.1, 121.9, 111.4, 109.2, 40.1. Anal. C23H20N2O Calcd. C: 81.15, H: 5.92, N: 8.23. Found C: 81.01, H: 6.04, N: 8.18.
3-(3-(4-(Dimethylamino)phenyl)allylidene)-1-phenylindolin-2-one (36) was prepared by method 1 with compound 17 and 34 to afford the desired product, (36), as a purple solid (77%), mp 206.8–207.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.48 (dd, J = 15.2 Hz, 11.6 Hz, 1H), 7.54-7.45 (m, 7H), 7.39 (d, J = 12.0 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 15.6 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.64 (d, J = 8.8 Hz, 2H), 2.99 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 167.1, 151.4, 145.0, 141.6, 138.5, 135.0, 129.8, 129.5, 127.6, 127.5, 126.7, 124.6, 124.2, 122.1, 121.0, 120.5, 110.8, 112.0, 109.1, 40.2. Anal. C25H22N2O Calcd. C: 81.94, H: 6.05, N: 7.74. Found C: 81.92, H: 6.25, N: 7.59.
(Z)-3-(4-Nitrobenzylidene)indolin-2-one (38a) and (E)-3-(4-nitrobenzylidene)indolin-2-one (38b) were prepared by method 1 with compound 1 and 37. The crude product was purified by silica gel column chromatography eluting with CH2Cl2-EtOAc (100:5, v/v), compound 38a eluted first and was afforded as a red solid (41%), mp 247.5–248.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 8.8 Hz, 2H), 8.28 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.53 (s, 1H), 7.42 (s, 1H), 7.29 (t J = 7.2 Hz, 1H), 7.08 (t, J = 7.2 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.2, 147.8, 142.0, 140.7, 133.9, 132.9, 131.5, 130.9, 130.6, 124.6, 124.4, 123.6, 121.8, 121.1, 110.1. Anal. C15H10N2O3.0.125H2O Calcd. C: 67.10, H: 3.85, N: 10.43. Found C: 67.08, H: 3.81, N: 10.47. Compound 38b eluted second as a red solid (48%), mp 252.6–252.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.31 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.63 (s, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 6.81 (t, J = 7.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 168.6, 147.9, 143.9, 141.9, 133.4, 131.5, 130.9, 130.5, 124.4, 123.3, 121.8, 120.7, 110.8. Anal. C15H10N2O3 Calcd. C: 67.67, H: 3.79, N: 10.52. Found C: 67.54, H: 3.71, N: 10.60.
(Z)-1-Benzyl-3-(4-nitrobenzylidene)indolin-2-one (39a) was prepared by method 2 with compound 38a and benzyl bromide to afford the desired product, (39a), as a red solid (76%), mp 200.4–201.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 9.2 Hz, 2H), 8.28 (d, J = 8.4 Hz, 2H), 7.57 (s, 1H), 7.56 (d, J = 6.8 Hz, 1H), 7.32-7.22 (m, 6H), 7.06 (t, J = 7.2 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 4.98 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 165.7, 148.0, 142.2, 139.8, 135.7, 133.5, 132.3, 130.2, 129.4, 128.8, 127.7, 127.3, 123.5, 123.4, 122.3, 119.8, 109.2, 43.7. Anal. C22H16N2O3.0.25H2O Calcd. C: 73.22, H: 4.61, N: 7.76. Found C: 72.93, H: 4.50, N: 7.75.
(E)-1-Benzyl-3-(4-nitrobenzylidene)indolin-2-one (39b) was prepared by method 2 with compound 38b and benzyl bromide to afford the desired product, (39b), as red solid (66%), mp 161.9-162.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 8.4 Hz, 2H), 7.87 (s, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 7.2 Hz, 1H), 7.35-7.26 (m, 5H), 7.20 (t, J = 7.6 Hz, 1H), 6.86 (t, J = 7.6 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 5.00 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 167.9, 147.9, 143.9, 141.7, 135.6, 133.7, 130.9, 130.0, 129.7, 128.8, 127.7, 127.3, 124.0, 123.0, 122.2, 120.4, 109.6, 43.9. Anal. C22H16N2O3 Calcd. C: 74.15, H: 4.53, N: 7.86. Found C: 74.14, H: 4.61, N: 7.84.
(Z)-3-((E)-3-(4-Nitrophenyl)allylidene)indolin-2-one (41a) and (E)-3-((E)-3-(4-Nitrophenyl)allylidene)indolin-2-one (41b) were prepared by method 1 with compound 1 and 40, the crude product was purified by silica gel column chromatography eluting with CH2Cl2-EtOAc (100:5, v/v). Compound 41a eluted first and was afforded as a red solid (41%), mp 256.2–257.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.58 (dd, J = 15.6 Hz, 11.2 Hz, 1H), 8.22 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 7.6 Hz, 1H), 7.54 (d, J = 11.2 Hz, 1H), 7.22 (d, J = 15.6 Hz, 1H), 7.17 (t, J = 7.2 Hz, 1H), 6.93 (t, J = 7.6 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 168.6, 147.5, 143.1, 142.0, 139.5, 134.6, 130.1, 128.6, 128.3, 128.0, 124.7, 123.7, 121.7, 120.9, 110.1. Anal. C17H12N2O3 Calcd. C: 69.86, H: 4.14, N: 9.58. Found C: 69.59, H: 4.03, N: 9.51. Compound 41b eluted second and was afforded as a red solid (46%), mp 263.8-264.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.19 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 8.4 Hz, 3H), 7.87 (dd, J = 15.0 Hz, 12.8 Hz, 1H), 7.42 (d, J = 14.8 Hz, 1H), 7.24 (d, J = 12.4 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 6.98 (t, J = 7.6 Hz, 1H), 6.2 (d, J = 8.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.1, 147.6, 143.1, 142.8, 141.8, 133.6, 130.2, 129.3, 128.4, 127.8, 125.1, 124.4, 122.2, 121.9, 110.3. Anal. C17H12N2O3.0.50H2O Calcd. C: 67.77, H: 4.35, N: 9.30. Found C: 67.94, H: 4.73, N: 8.87.
(Z)-1-Benzyl-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (42a) was prepared by method 2 with compound 41a and benzyl bromide to afford the desired product, (42a), as a red solid (76%), mp 224.5–225.8 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (dd, J = 15.6 Hz, 11.6 Hz, 1H), 8.25 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 12.0 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.33-7.18 (m, 7H), 7.00 (t, J = 7.6 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 4.93 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.8, 143.0, 140.3, 137.1, 135.5, 131.3, 130.1, 129.1, 128.7, 128.2, 127.8, 126.7, 124.7, 122.5, 120.8, 109.6, 40.6. Anal. C24H18N2O3.0.125H2O Calcd. C: 74.94, H: 4.78, N: 7.28. Found C: 74.83, H: 4.85, N: 7.18.
(E)-1-Benzyl-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (42b) was prepared by method 2 with compound 41b and benzyl bromide to afford the desired product, (42b), as a red solid (67%), mp 237.1–237.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 8.4 Hz, 2H), 7.79 (dd, J = 15.2 Hz, 12.4 Hz, 1H), 7.72 (d, J = 8.4 Hz, 3H), 7.54 (d, J = 12.0 Hz, 1H), 7.323-7.17 (m, 7H), 7.07 (t, J = 7.6 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 4.99 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 168.3, 147.8, 143.3, 142.2, 140.6, 135.9, 134.1, 129.7, 128.7, 128.0, 127.7, 127.6, 127.3, 127.2, 124.3, 123.8, 122.3, 122.0, 109.5, 43.7. Anal. C24H18N2O3 Calcd. C: 75.38, H: 4.74, N: 7.33. Found C: 75.30, H: 6.78, N: 7.28.
(Z)-1-(4-Methoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (43a) was prepared by method 2 with compound 41a and 4-methoxybenzyl chloride to afford the desired product, (43a), as a red solid (55%), mp 182.6–183.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.82 (dd, J = 15.6 Hz, 11.6 Hz, 1H), 8.21 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 12.8 Hz, 1H), 7.28 (d, J = 9.2 Hz, 2H), 7.19 (t, J = 8.0 Hz, 1H), 7.03 (d, J = 16.8 Hz, 1H), 7.01 (t, J = 7.8 Hz, 1H), 6.85 (d, J = 8.0 Hz, 2H), 6.75 (d, J = 7.6 Hz, 1H), 4.90 (s, 2H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.1, 159.1, 147.5, 142.7, 142.2, 139.0, 134.2, 129.6, 128.7, 128.5, 128.1, 128.0, 127.1, 124.1, 123.0, 122.1, 119.8, 114.2, 109.1, 55.2, 42.9. Anal. C25H20N2O4 Calcd. C: 72.80, H: 4.89, N: 6.79. Found C: 72.52, H: 5.03, N: 6.67.
(E)-1-(4-Methoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (43b) was prepared by method 2 with compound 41b and 4-methoxybenzyl chloride to afford the desired product, (43b), as a red solid (47%), mp 237.7–238.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 8.0 Hz, 2H), 7.78 (dd, J = 15.0 Hz, 12.0 Hz, 1H), 7.71 (d, J = 8.0 Hz, 2H), 7.53 (d, J = 12.0 Hz, 1H), 7.26-7.21 (m, 4H), 7.18 (d, J = 14.8 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.84 (d, J = 8.0 Hz, 2H), 6.77 (d, J = 8.4 Hz, 1H), 4.91 (s, 2H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 168.2, 159.1, 153.8, 147.8, 143.3, 142.2, 140.5, 134.0, 129.7, 128.7, 128.0, 127.8, 127.4, 124.3, 123.7, 122.2, 122.0, 114.1, 109.5, 55.2, 43.2. Anal. C25H20N2O4 Calcd. C: 72.80, H: 4.89, N: 6.79. Found C: 72.75, H: 5.01, N: 6.73.
(Z)-1-(3-Methoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (44a) was prepared by method 2 with compound 41a and 3-methoxybenzyl bromide to afford the desired product, (44a), as a red solid (52%), mp 193.3–194.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 16.0 Hz, 11.6 Hz, 1H), 8.21 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 12.0 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.03 (d, J = 17.2 Hz, 1H), 7.01 (t, J = 7.2 Hz, 1H), 6.91 (d, J = 7.6 Hz, 1H), 6.86 (s, 1H), 6.80 (d, J = 8.8 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 4.94 (s, 2H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.2, 159.9, 147.5, 142.7, 142.2, 139.1, 137.6, 134.3, 129.9, 129.6, 128.5, 128.0, 127.0, 124.1, 123.0, 122.1, 119.8, 119.5, 113.2, 112.7, 109.1, 55.2, 43.3. Anal. C25H20N2O4 Calcd. C: 72.80, H: 4.89, N: 6.79. Found C: 72.80, H: 5.06, N: 6.66.
(E)-1-(3-Methoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (44b) was prepared by method 2 with compound 41b and 3-methoxybenzyl bromide to afford the desired product, (44b), as a red solid (59%), mp 237.7–239.0 °C. 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 8.0 Hz, 2H), 7.82-7.67 (m, 3H), 7.62-7.45 (m, 1H), 7.33-7.18 (m, 3H), 7.08-7.04 (m, 2H), 6.91-6.75 (m, 4H), 4.95 (s, 2H), 3.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 140.6, 134.1, 129.8, 128.0, 127.3, 124.3, 124.2, 123.7, 122.3, 119.5, 113.1, 112.8, 109.5, 55.2, 43.7. Anal. C25H20N2O4 Calcd. C: 72.80, H: 4.89, N: 6.79. Found C: 72.87, H: 4.91, N: 6.72.
(Z)-1-(3,4-Dimethoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (45a) was prepared by method 2 with compound 41a and 3,4-dimethoxybenzyl bromide to afford the desired product, (45a), as a red solid (55%), mp 199.6–200.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 15.4 Hz, 11.6 Hz, 1H), 8.21 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 7.2 Hz, 1H), 7.32 (d, J = 12.0 Hz, 1H), 7.19 (t, J = 7.2 Hz, 1H), 7.05-7.01 (m, 2H), 6.87 (m, 2H), 6.80 (d, J = 7.6 Hz, 1H), 6.76 (d, J = 7.6 Hz, 1H), 4.90 (s, 2H), 3.85 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 167.2, 149.3, 148.5, 147.5, 142.7, 142.2, 139.1, 134.3, 129.6, 128.5, 128.0, 127.1, 124.1, 123.0, 122.1, 119.8, 119.7, 111.1, 110.6, 109.1, 56.0, 55.9, 43.3. Anal. C26H22N2O5 Calcd. C: 70.58, H: 5.01, N: 6.33. Found C: 70.70, H: 5.14, N: 6.26.
(E)-1-(3,4-Dimethoxybenzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (45b) was prepared by method 2 with compound 41b and 3,4-methoxybenzyl bromide to afford the desired product, (45b), as a red solid (51%), mp 201.3–201.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 7.2 Hz, 2H), 7.82-7.71 (m, 2H), 7.72 (d, J = 6.8 Hz, 2H), 7.44 (d, J = 12.0 Hz, 1H), 7.26-7.17 (m, 2H), 7.08 (t, J = 7.2 Hz, 1H), 6.86 (m, 2H), 6.80 (d, J = 7.2 Hz, 2H), 4.92 (s, 2H), 3.84 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 168.3, 149.3, 148.5, 147.8, 143.3, 142.2, 140.6, 134.1, 129.7, 128.4, 128.0, 127.7, 127.3, 124.3, 123.8, 122.3, 122.0, 119.7, 111.1, 110.6, 109.5, 55.9, 55.8, 43.6. Anal. C26H22N2O5 Calcd. C: 70.58, H: 5.01, N: 6.33. Found C: 70.76, H: 5.26, N: 6.26.
(Z)-1-(4-(2-Fluoroethoxy)benzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (46a) was prepared by method 2 with compound 41a and 4-(2-fluoroethoxy)benzyl bromide to afford the desired product, (46a), as a red solid (61%), mp 187.8–188.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 16.0 Hz, 11.6 Hz, 1H), 8.21 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 11.6 Hz, 1H), 7.28 (d, J = 8.8 Hz, 2H), 7.19 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 16.0 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 7.6 Hz, 1H), 4.91 (s, 2H), 4.73 (dt, J = 47.2 Hz, 4.0 Hz, 2H), 4.17 (dt, J = 28.4 Hz, 4.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.2, 157.9, 147.5, 142.7, 142.1, 139.1, 134.3, 129.6, 128.8, 128.7, 128.5, 128.0, 127.1, 124.1, 123.0, 122.1, 119.8, 114.9, 109.1, 81.8 (d, J = 169.7 Hz), 67.1 (d, J = 20.1 Hz), 42.8. Anal. C26H21FN2O4 Calcd. C: 70.26, H: 4.76, N: 6.30. Found C: 70.46, H: 4.91, N: 6.25.
(E)-1-(4-(2-Fluoroethoxy)benzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (46b) was prepared by method 2 with compound 41b and 4-(2-fluoroethoxy)benzyl bromide to afford the desired product, (46b), as a red solid (55%), mp 225.4–226.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 8.4 Hz, 2H), 7.78 (dd, J = 15.0 Hz, 12.0 Hz, 1H), 7.72 (d, J = 8.4 Hz, 3H), 7.53 (d, J = 12.0 Hz, 1H), 7.27-7.17 (m, 4H), 7.07 (t, J = 7.6 Hz, 1H), 6.87 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.0 Hz, 1H), 4.92 (s, 2H), 4.73 (dt, J = 47.6 Hz, 4.0 Hz, 2H), 4.18 (dt, J = 28.0 Hz, 4.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 168.2, 157.9, 147.8, 143.3, 142.2, 140.6, 134.1, 129.7, 128.7, 128.6, 128.0, 127.8, 127.3, 124.3, 123.8, 122.3, 122.0, 114.9, 109.4, 81.8 (d, J = 169.7 Hz), 67.1 (d, J = 20.1 Hz), 43.2. Anal. C26H21FN2O4 Calcd. C: 70.26, H: 4.76, N: 6.30. Found C: 70.22, H: 4.68, N: 6.31.
3-(3-(4-Aminophenyl)allylidene)indolin-2-one (47) A mixture of 41a (146 mg, 0.5 mmol) and Fe powder (280 mg, 5 mmol) in methanol (15 mL) and 37% HCl (0.5 mL) was heated to reflux for 3 h. The solid was filtered out, the filtrate was evaporated, ethyl acetate (75 mL) was added, washed with saturated Na2CO3 (50 mL), water (50 mL), NaCl (50 mL), and dried over Na2SO4. After removed of the solvent, the crude product was purified by silica gel column chromatography eluting with EtOAc-MeOH (10:1, v/v) to afford 98 mg (67%) of 47 as a red solid, mp 263.3–264.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 and 10.31 (s, 1H), 8.14 and 7.39 (dd, J = 15.6 Hz, 12.0 Hz, 1H), 7.86 and 7.44 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.26 and 7.15 (d, J = 11.2 Hz, and 12.8 Hz, 1H), 7.13 and 7.08 (t, J = 8.0 Hz, 1H), 6.94 and 6.88 (t, J = 8.0 Hz, 1H), 6.80 and 6.75 (d, J = 7.6 Hz, 1H), 6.57 (d, J = 8.0 Hz, 2H), 5.79 and 5.75 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.6, 169.0, 151.7, 151.4, 147.0, 144.8, 141.8, 140.6, 137.7, 136.8, 130.6, 129.9, 128.4, 128.1, 124.7, 124.1, 123.9, 123.8, 123.2, 122.4, 122.0, 121.6, 121.2, 119.5, 119.1, 118.0, 114.4, 114.2, 109.8, 109.6. Anal. C17H14N2O.0.125H2O Calcd. C: 77.18, H: 5.43, N: 10.59. Found C: 77.34, H: 5.58, N: 10.37.
(Z)-1-(4-(2-Bromoethoxy)benzyl)-3-((E)-3-(4-nitrophenyl)allylidene)indolin-2-one (48) was prepared by method 2 with compound 41a and 4-(2-bromoethoxy)benzyl bromide to afford the desired product, (48), as a red solid (78%), mp 192.3–193.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 15.8 Hz, 11.6Hz, 1H), 8.22 (d, J = 8.8 Hz, 2H), 7.73 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 12.0 Hz, 1H), 7.28 (d, J = 8.8 Hz, 2H), 7.194 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 17.2 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 6.75 (7.6 Hz, 1H), 4.91 (s, 2H), 4.25 (t, J = 6.2 Hz, 2H), 3.61 (t, J = 6.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.2, 157.6, 147.5, 142.7, 142.1, 139.1, 134.3, 129.6, 128.9, 128.8, 128.5, 128.0, 127.1, 124.1, 123.0, 122.1, 119.8, 115.0, 109.1, 67.8, 42.8, 29.0.
2-(4-(((Z)-3-((E)-3-(4-Nitrophenyl)allylidene)-2-oxoindolin-1-yl)methyl)phenoxy)ethyl methanesulfonate (49) A solution of 48 (127 mg, 0.5 mmol) and silver methanesulfonate (500 mg, 5 mmol) in acetonitrile (15 mL) was heated to reflux for 20 h. After evaporation of the solvent, the crude product was purified by silica gel column chromatography eluting with EtOAc: MeOH (10: 1, v/v) to afford 89 mg (68%) of 49 as red solid, mp 186.4–187.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.80 (dd, J = 15.4 Hz, 11.4 Hz, 1H), 8.21 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 11.6 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H), 7.19 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 16.4 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 6.73 (d, J = 7.6 Hz, 1H), 4.90 (s, 2H), 4.54 (t, J = 6.2 Hz, 2H), 4.20 (t, J = 6.2 Hz, 2H), 3.06 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.1, 157.5, 147.5, 142.7, 142.0, 139.2, 134.4, 129.6, 129.2, 128.9, 128.4, 128.0, 127.0, 124.1, 123.0, 122.2, 119.9, 114.8, 109.0, 67.9, 65.8, 42.8, 37.8.
Radiosynthesis of [18F]46a
[18F]Fluoride (50–200 mCi) in [18O]water (without treatment by trapping on quaternary methyl ammonia cartridge) was transferred into a Pyrex screw cap tube (10 mL) containing K2CO3 (0.3 mg, 2.2 μmol) and Kryptofix 222 (K222) (2.2 mg, 5.9 μmol); the activity was dried by azeotropic distillation at 105 °C using MeCN (3 × 1 mL) under a gentle flow of N2 gas. To the dried activity was added the mesylate precursor 49 (1.8 mg, 3.46 μmol) as solid with tert-amyl alcohol (300 μL) using a transfer pipette. The tube was capped and shaken in an oil bath of 105 °C to dissolve the precursor. The reaction mixture was heated at 105 °C for 10 min. After cooling to room temperature, the mixture was treated by passing through a silica column (5×20 mm); MeCN (6×0.5 mL) was used to rinse the tube and the column. The combined eluents were concentrated at 105 °C under a flow of N2 to almost dryness, and HPLC mobile phase (3 mL) was added to the above residue for HPLC purification. Under the specified conditions, [18F]46a eluted at 40 min. The HPLC fraction was collected and diluted with water (50 mL), the diluted solution was passed through a Waters Oasis HLB plus cartridge. The cartridge was rinsed with water (10 mL), and the final product was eluted with DMF for binding studies.
Binding Assay Methods
Preparation of recombinant α-syn and tau protein
Recombinant proteins were produced in E. coli and purified as previously described.23 Briefly, α-syn was extracted from E. coli using an osmotic shock method then purified by a combination of heat-precipitation and DEAE (diethylaminoethyl) ion exchange chromatography. Purified α-syn protein was dialyzed overnight in 10 mM Tris-HCl pH 7.6, 50 mM NaCl, 1 mM DTT. Preparations contained greater than 95% α-syn protein as determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, IL), with a typical yield of 30 mg protein per 250 ml culture.
Recombinant tau protein was produced in E. coli. BL21(DE3)RIL The bacterial cultures were transformed with a pRK172 bacterial expression plasmid encoding a human tau fragment containing the four microtubule binding repeats (amino acids 243–375), provided by Marc Diamond at Washington University. Cultures were inoculated and grown overnight as above for α-syn protein production. Purified tau protein was prepared using the previously described method and dialyzed overnight in 100 mM sodium acetate pH 7.0.31
Preparation of recombinant α-syn fibrils
Purified recombinant α-syn monomer (2 mg/ml) was incubated in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl for 72 h at 37 °C with shaking at 1000 rpm in an Eppendorf Thermomixer as previously described.23 Fibril concentration was determined by BCA protein assay to determine monomer concentration in the supernatant obtained by centrifuging the fibril reaction mix at 18,000 g for 15 min to separate fibrils from monomer. The measured decrease in α-syn monomer concentration was used to determine the concentration of fibrils in the 72 h fibril reaction mixture.
Preparation of Aβ1-42 fibrils
Synthetic Aβ1-42 peptide (1 mg) (Bachem, Torrance, CA) was first dissolved in 50 μl DMSO. An additional 925 μl of ultrapure water was added. Finally, 25 μl 1M Tris-HCl pH 7.6 was added to bring the final peptide concentration to 222 μM (1 mg/ml). The dissolved peptide was incubated for 30 h at 37 °C with shaking at 1000 rpm in an Eppendorf Thermomixer. Fibril formation was confirmed by Thio-T fluorescence. To determine the concentration of fibrils, the fibril reaction mix was centrifuged at 18,000 × g for 15 min to separate fibrils from monomer. The concentration of Aβ monomer in the supernatant was determined in a BCA protein assay using a BSA standard curve that contained DMSO at a percentage equivalent to the samples.
Preparation of recombinant tau fibrils
Purified recombinant tau monomer (300 μg/ml) was incubated in 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 25 μM low molecular weight heparin, 0.5 mM DTT for 48 h at 37 °C with shaking at 1000 rpm in an Eppendorf Thermomixer. To determine the concentration of fibrils, the fibril reaction mix was centrifuged at 18,000 × g for 15 min to separate fibrils from monomer. The concentration of tau monomer in the supernatant was determined in a BCA protein assay along with a BSA standard curve. The measured decrease in monomer concentration was used to determine the concentration of tau fibrils in the 48 h fibril reaction mixture.
Preparation of α-syn, Aβ1-42, and tau fibrils for binding and competition assays
The prepared fibril mixture was centrifuged at 18,000 × g for 15 min to prepare fibrils for binding assays. The supernatant was discarded and the fibril pellet was resuspended in 30 mM Tris-HCl pH 7.4, 0.1% BSA to achieve the desired concentration of fibrils for use in the assay.
In vitro saturation binding studies to determine binding affinity of Thio-T for fibrils
A fixed concentration (1 μM) of α-syn, Aβ, or tau fibrils were incubated for 2 h at 37 °C with increasing concentrations of Thio-T in 30 mM Tris-HCl pH 7.4, 0.1% BSA in a reaction volume of 150 μL. Nonspecific binding was determined in a duplicate set of binding reactions containing the same concentration of Thio-T with no fibrils. Fluorescence was determined in a Biotek plate reader using a 440/30 excitation filter and a 485/20 emission filter. All data points were performed in triplicate. The dissociation constant (Kd) and the maximal number of binding sites (Bmax) values were determined by fitting the data to the equation Y=Bmax*X/(X+Kd) by nonlinear regression using Graphpad Prism software (version 4.0).
Thio-T competition studies
Competition assays used a fixed concentration of α-syn, Aβ, or tau fibrils (1 μM), consisting of 3 μM, 50 nM and 4 μM of Thio-T respectively. The competitor reaction was diluted in 30 mM Tris-HCl pH 7.4, 0.1% BSA and added to the reactions in varying concentrations. To optimize the determination of selectivity, assays for all three fibril species were set up in parallel using a common set of intermediate compound dilutions for each analog. Reactions were incubated at room temperature for 1.5 h before quantifying bound ligand as described above for the saturation binding assay. Fluorescence was determined in a Biotek plate reader using a 440/30 excitation filter and a 485/20 emission filter. All data points were performed in triplicate. Nonspecific fluorescence was measured in parallel reactions containing Thio-T plus each concentration of competitor but no fibrils, and these measurements were subtracted from the reactions with fibrils to yield fibril-specific fluorescence. Data were analyzed using Graphpad Prism software (version 4.0) to obtain EC50 values by fitting the data to the equation Y=bottom+(top-bottom)/(1+10(x-logEC50)). Ki values were calculated from EC50 values using the equation Ki=EC50/(1+[radioligand]/Kd).
In vitro saturation binding studies of [18F]46a
α-Syn, Aβ, or tau fibrils (1 μM) were incubated for 1 h at 37 °C with increasing concentrations of [18F]46a in 30 mM Tris-HCl pH 7.4, 0.1% BSA in a reaction volume of 150 μL. A fixed ratio of labeled: unlabeled 46a was used for all radioligand concentrations. The exact labeled: unlabeled 46a ratio was measured in each experiment by counting a 2 μL sample of the radioligand preparation in a scintillation counter. Nonspecific binding was determined in the parallel binding reactions containing 20 as the competitor or in reactions containing no fibrils, both of which yielded similar nonspecific binding measurements. Bound and free radioligand were separated by vacuum filtration through 1.0 μm glass fiber filters in 96-well filter plates (Millipore), followed by three 200 μL washes with ice-cold assay buffer. Filters containing the bound ligand were mixed with 150 μL of Optiphase Supermix scintillation cocktail (PerkinElmer) and counted immediately. All data points were performed in triplicate. The dissociation constant (Kd) and the maximal number of binding sites (Bmax) values were determined by fitting the data to the equation Y = Bmax*X/(X+Kd) by nonlinear regression using Graphpad Prism software (version 4.0).
Human tissue acquisition
Post-mortem midbrain tissue from DLB and PD cases was obtained from the Harvard Brain Tissue Resource Center at McLean Hospital. Paraffin-embedded formalin-fixed tissue from the temporal lobe of AD cases was obtained through the Massachusetts Alzheimer’s Disease Research Center Brain Bank. The human brain tissue provided was neuropathologically defined as DLB tissue using standard criteria.
Labeling of synuclein pathology in human DLB and PD cases
Blocks of midbrain tissue from human PD/DLB cases were fixed with 4% paraformaldehyde and stored at −80°C. In preparation for immunohistochemistry, the tissue block was deparaffinized with xylene and rehydrated. Midbrain tissue was sliced into 40 μm thick sections with a freezing sledge Leica SM 2000R microtome. Free-floating tissue was permeabilized with 0.5% Triton-X 100, then incubated with normal goat serum (NGS) at room temperature for 1 h with shaking. Primary H3C antibody (1:5000 in 1.5% NGS) to α-syn was incubated overnight at 4 °C with shaking, and then washed with 1 × PBS three times. The tissue was then incubated for 2 h at room temperature with a fluorescent secondary antibody labeled with Alexa Fluor 488 (1:200 in PBS). Tissue was then treated with the compound to be tested. Each tissue section was incubated at room temperature for 30 min with 5 μM of test compound dissolved in PBS. The tissue was washed with 1 × PBS three times, and then mounted onto Adhesion SuperFrost Plus slide and cover slipped. The tissue was imaged with an Olympus BX51 upright microscope (Olympus America Inc., Melville, NY) using standard excitation/emission filters for Alexa Fluor 488 and Cy5.
Staining of plaques in human tissue from AD cases
Paraffin-embedded paraformaldehyde (4%) fixed sections of temporal lobe of neuropathologically verified human AD cases were deparaffinized. Tissue sections were stained with 0.05% Thioflavin S Thio-T (Sigma, St. Louis, MO) in 50% ethanol for 5 min to visualize senile plaques. Tissue was then incubated with 5 μM of compound in PBS for 20 min at room temperature. Slides were rinsed in PBS, coverslipped, and then imaged on an Olympus BX51 microscope using a 20x Plan Neofluar objective (NA=0.7) using standard excitation/emission filters for AlexaFluor488 and Cy5.
Supplementary Material
Acknowledgments
This study was funded in part by the Michael J. Fox Alpha Synuclein Imaging Consortium, Washington University Institute of Clinical and Translational Sciences Grant CTSA406, and the Charles and Joanne Knight Alzheimer’s Research Initiative of the Washington University Alzheimer’s Disease Research Center. This study made use of the NIH/NIGMS Biomedical Mass Spectrometry Resource at Washington University in St. Louis, MO, which is supported by National Institutes of Health\ National Institute of General Medical Sciences Grant # 8P41GM103422. We would like to thank personnel of the Washington University Cyclotron Facility for [18F]fluoride production and the Department of Chemistry staff and the Washington University High Resolution NMR Facility for assistance with NMR spectra. Purchase of the 400 MHz NMR instrument was partially supported by S10 RR027207 from the NIH Shared Instrument Grant program.
ABBREVIATIONS USED
- α-syn
alpha synuclein
- BCA assay
bicinchoninic acid assay
- DEAE ion exchange
diethylaminoethyl ion exchange
- DLB
dementia with Lewy bodies
- LB
Lewy body
- LN
Lewy neurite
- MSA
multiple system atrophy
- NFT
neurofibrillary tangles
- QC
quality control
- Thio-T
thioflavin-T
Footnotes
ASSOCIATED CONTENT
Methods for the 2D NMR study.
Figure S1. NOE in E,E configuration of 41b and 3D structure of 41a and 41b.
Figure S2. 1H NMR spectrum of 41b.
Figure S3. 2D NMR spectrum of 41b
Figure S4. Stability of 41a and 41 b in dichloromethane/methanol solution.
Figure S5. Saturation binding studies to determine binding affinity of Thio- T for α-syn, Aβ and tau fibrils
Figure S6. Analytical HPLC of [18F]46a
1H NMR of compounds: 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 18, 19, 20, 21, 22, 23, 24, 25, 27, 28, 29, 30, 31, 32, 33, 35, 36, 38a, 38b, 39a, 39b, 41a, 41b, 42a, 42b, 43a, 43b, 44a, 44b, 45a, 45b, 46a, 46b, 47, 48, 49
References
- 1.Mach RH. New targets for the development of PET tracers for imaging neurodegeneration in Alzheimer Disease. J Nucl Med. 2014;55:1221–1224. doi: 10.2967/jnumed.114.127811. [DOI] [PubMed] [Google Scholar]
- 2.Skovronsky DM, Lee VM, Trojanowski JQ. Neurodegenerative diseases: New concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol. 2006;1:151–170. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- 3.Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT, Holt DP, Wang Y, Huang GF, Debnath ML, Klunk WE. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett. 2002;12:295–298. doi: 10.1016/s0960-894x(01)00734-x. [DOI] [PubMed] [Google Scholar]
- 4.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 5.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 6.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC Dominantly Inherited Alzheimer N. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vlassenko AG, Benzinger TL, Morris JC. PET amyloid-beta imaging in preclinical Alzheimer’s disease. Biochim Biophys Acta. 2012;1822:370–379. doi: 10.1016/j.bbadis.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, Krautkramer MJ, Kung HF, Skovronsky DM, Hefti F, Clark CM. Correlation of amyloid PET ligand florbetapir F 18 (18F-AV-45) binding with β-amyloid aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord. 2012;26:8–16. doi: 10.1097/WAD.0b013e31821300bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, Krautkramer MJ, Kung HF, Coleman RE, Doraiswamy PM, Fleisher AS, Sabbagh MN, Sadowsky CH, Reiman EP, Zehntner SP, Skovronsky DM, Group AAS. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Becker GA, Ichise M, Barthel H, Luthardt J, Patt M, Seese A, Schultze-Mosgau M, Rohde B, Gertz HJ, Reininger C, Sabri O. PET quantification of 18F-florbetaben binding to β-amyloid deposits in human brains. J Nucl Med. 2013;54:723–731. doi: 10.2967/jnumed.112.107185. [DOI] [PubMed] [Google Scholar]
- 11.Serdons K, Verduyckt T, Vanderghinste D, Cleynhens J, Borghgraef P, Vermaelen P, Terwinghe C, Van Leuven F, Van Laere K, Kung H, Bormans G, Verbruggen A. Synthesis of 18F-labelled 2-(4′-fluorophenyl)-1,3-benzothiazole and evaluation as amyloid imaging agent in comparison with [11C]PIB. Bioorg Med Chem Lett. 2009;19:602–605. doi: 10.1016/j.bmcl.2008.12.069. [DOI] [PubMed] [Google Scholar]
- 12.Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, Shankle WR, Lerner AJ, Su MY, Elizarov A, Kolb HC. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimer’s Dis. 2014;38:171–184. doi: 10.3233/JAD-130098. [DOI] [PubMed] [Google Scholar]
- 13.Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M, Mulligan RS, Villemagne VL, Akatsu H, Yamamoto T, Arai H, Iwata R, Yanai K, Kudo Y. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med. 2013;54:1420–1427. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 14.Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Su H, Szardenings AK, Walsh JC, Wang E, Yu C, Zhang W, Zhao T, Kolb HC. [18F]T807 a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013;9:666–676. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological α-synuclein: New targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 16.Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res Ther. 2014;6:73. doi: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marques O, Outeiro TF. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. CSF α-synuclein improves diagnostic and prognostic performance of CSF tau and Aβ in Alzheimer’s disease. Acta Neuropathol. 2013;126:683–697. doi: 10.1007/s00401-013-1148-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen JR, Cisek K, Honson NS, Kuret J. Ligand polarizability contributes to tau fibril binding affinity. Bioorg Med Chem. 2011;19:5147–5154. doi: 10.1016/j.bmc.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol Dis. 2007;28:251–260. doi: 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neal KL, Shakerdge NB, Hou SS, Klunk WE, Mathis CA, Nesterov EE, Swager TM, McLean PJ, Bacskai BJ. Development and screening of contrast agents for in vivo imaging of Parkinson’s disease. Mol Imaging Biol. 2013;15:585–595. doi: 10.1007/s11307-013-0634-y. [DOI] [PubMed] [Google Scholar]
- 22.Yu L, Cui J, Padakanti PK, Engel L, Bagchi DP, Kotzbauer PT, Tu Z. Synthesis and in vitro evaluation of α-synuclein ligands. Bioorg Med Chem. 2012;20:4625–4634. doi: 10.1016/j.bmc.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bagchi DP, Yu L, Perlmutter JS, Xu J, Mach RH, Tu Z, Kotzbauer PT. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8:e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Jin H, Padakanti P, Li J, Yang H, Fan J, Mach R, Kotzbauer P, Tu Z. Radiosynthesis and in vivo evaluation of two PET radioligands for imaging α-synuclein. Appl Sci (Basel) 2014;4:66–78. doi: 10.3390/app4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: A novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- 26.Ariza M, Kolb HC, Moechars D, Rombouts F, Andres JI. Tau positron emission tomography (PET) imaging: Past, present, and futures. J Med Chem. 2015;58:4365–4382. doi: 10.1021/jm5017544. [DOI] [PubMed] [Google Scholar]
- 27.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–1484. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- 28.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, A fluorescent derivative of Congo red: A novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48:1223–1232. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 30.Kung HF, Choi SR, Qu W, Zhang W, Skovronsky D. 18F Stilbenes and styrylpyridines for PET imaging of Aβ plaques in Alzheimer’s disease: A miniperspective. J Med Chem. 2010;53:933–941. doi: 10.1021/jm901039z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W, Lee VM. Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry. 2006;45:15692–15701. doi: 10.1021/bi061422+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.