

Abstract
Many tumors over express tumor-associated antigens relative to normal tissue, such as epidermal growth factor receptor (EGFR). This limits targeting by human T cells modified to express chimeric antigen receptors (CARs) due to potential for deleterious recognition of normal cells. We sought to generate CAR+ T cells capable of distinguishing malignant from normal cells based on the disparate density of EGFR expression by generating two CARs from monoclonal antibodies which differ in affinity. T cells with low affinity Nimo-CAR selectively targeted cells over-expressing EGFR, but exhibited diminished effector function as the density of EGFR decreased. In contrast, the activation of T cells bearing high affinity Cetux-CAR was not impacted by the density of EGFR. In summary, we describe the generation of CARs able to tune T-cell activity to the level of EGFR expression in which a CAR with reduced affinity enabled T cells to distinguish malignant from non-malignant cells.
Keywords: EGFR, glioblastoma, immunotherapy, CAR, affinity
INTRODUCTION
T cells genetically modified to express a chimeric antigen receptor (CAR) to redirect specificity for desired tumor-associated antigen (TAA) can be infused into patients as immunotherapy. Because many TAAs over-expressed on tumors also have low density expression on normal tissue, use of CAR-modified T cells to specifically target such antigens has been limited by the potential for on-target, normal tissue toxicity. Indeed, prolonged B-cell aplasia in patients with B-lineage leukemias and lymphomas after infusion of CD19-specifc CAR+ T cells highlights inability to distinguish between CD19 on malignant and normal B cells (1–3).
Glioblastoma (GBM) is the most common and aggressive malignancy of the brain and central nervous system with a median survival of ~1 year from diagnosis (4,5). Epidermal growth factor receptor (EGFR) is aberrantly overexpressed in more than 60% of adult GBM and contributes to tumor progression by promoting cell division, invasion, angiogenesis, and inhibiting apoptosis (6). Clinical efforts to target EGFR on GBM with tyrosine kinase inhibitors (TKIs) and monoclonal antibodies (mAbs) have had limited success, apparently due to signaling through compensatory pathways and poor blood-brain barrier penetration (7,8). Furthermore, targeting EGFR via TKIs or mAb can cause toxicity, primarily observed in skin, gastrointestinal system, and kidney, due distribution of EGFR on these normal tissues (9,10).
EGFR is over-expressed on GBM relative to normal cells, which raises the possibility that CAR+ T cells may be generated to distinguish malignant from normal cells based on EGFR density. This is based upon published observations that T-cell activation initiated through the endogenous αβ T-cell receptor (TCR) may result from achieving an activation threshold, which can be triggered by binding by a few high-affinity TCRs to or greater numbers of low-affinity TCR (11,12). We hypothesized that the affinity of a single-chain variable fragment (scFv), which imparts CAR specificity, may be modified to impact the effector response to the density of EGFR, such that T cells would be activated when docked with over-expressed EGFR on tumor cells and not when low, basal EGFR expression on normal cells.
To accomplish this, we derived two CAR species from cetuximab and nimotuzumab, which bind to highly overlapping epitopes on EGFR, yet exhibit different affinities (13). Nimotuzumab has a 10-fold lower Kd than cetuximab resulting from a 59-fold reduced on-rate of binding, which imparts a requirement for (at least) bivalent binding to EGFR and restricts the binding to cells expressing high density EGFR (13–15). Therefore, we generated two 2nd EGFR-specific CARs, which derive their specificity from the scFv regions of nimotuzumab (Nimo-CAR) or cetuximab (Cetux-CAR), and are activated by chimeric CD28 and CD3-ζ cytosolic domains. We observed that T cells could be fully activated by both Nimo-CAR and Cetux-CAR when EGFR was expressed at high density on glioma cells. However, at reduced levels of EGFR expression, the lower affinity Nimo-CAR+ T cells exhibited reduced T-cell activation while Cetux-CAR+ T cells maintained efficient activation independent of EGFR density. In summary, we describe a strategy to engineer T cells to distinguish tumor from normal cells based on tuning the functional affinity of CAR to selectively target cells over-expressing EGFR.
MATERIALS AND METHODS
DNA plasmids
The derivation of Cetux-CAR and Nimo-CAR is described in Supplementary Materials and Methods. Cetux-CAR and Nimo-CAR were cloned as Sleeping Beauty (SB) transposons under control of hEF1-α promoter, as previously described (16). Codon-optimized truncated human EGFR (amino acids 1–668, NP_005219.2; GeneArt, Regensburg, Germany) was cloned under expression of hEF1-α promoter followed by F2A cleavable peptide and neomycin phosphotransferase. CARL was derived from hybridoma clone 2D3 (17).
Cell lines
Cell lines obtained from ATCC include: EL4 (2009), NALM-6 (2011), U87 (2012), T98G (2012), LN18 (2012) and A431 (2012). K562 clone 9 were generated by stable expression of 41BB-L, CD86, CD64 and tCD19 (18) and were a kind gift from Dr. Carl June (University of Pennsylvania), obtained in 2007. U87-172b cells, designated U87high in this study, overexpressing wild-type EGFR were a kind gift from Dr. Oliver Bolger (MDACC) and obtained in 2012. Human renal cortical epithelial (HRCE) cells were obtained from Lonza in 2012. Details of propagation and genetic modification are described in Supplementary Materials and Methods. All cell line identities were validated by STR DNA fingerprinting in 2012, at the time of the study, using the AmpF_STR Identifier kit according to manufacturer’s instructions (Applied Biosystems, cat #4322288). The STR profiles were compared to known ATCC fingerprints (ATCC.org), and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (19). The STR profiles matched known DNA fingerprints.
T-cell modification and culture
Peripheral blood mononuclear cells (PBMC) from healthy donors were purchased from Gulf Coast Regional Blood Bank and isolated by Ficoll-Paque (GE Healthcare) and cryopreserved. T cells were cultured in RPMI-1640 (HyClone) supplemented with 10% FBS (HyClone) and 2mM Glutamax (Gibco). Electro-transfer of SB plasmids coding for CAR is detailed in Supplementary Materials and Methods and as previously described (16).
Surface immunostaining and flow cytometry
Data were collected on FACS Calibur (BD Biosciences) using CellQuest software (version 3.3, BD Biosciences) and analyzed using FlowJo software (version x.0.6, TreeStar). Up to 106 cells were stained with mAbs (Supplementary Table S1) in FACS buffer (PBS, 2% FBS, 0.5% sodium azide) for 30 minutes in the dark at 4°C. Quantitative flow cytometry was performed using Quantum Simply Cellular polystyrene beads, according to manufacturer’s instructions (cat #814, Bangs Laboratories) using mAb specific for EGFR (cat #555997, BD Biosciences). A standard curve relating MFI to EGFR molecules/cell was made using QuickCal Data Analysis Program (version 2.3, Bangs Laboratories). EGFR molecules/cell was calculated by subtracting MFI of isotype antibody from MFI of EGFR and extrapolating from standard curve.
Intracellular cytokine production
T cells were co-cultured with target cells at a ratio of 1:1 for 4–6 hours in the presence of GolgiStop diluted 1:4,000 (cat #554724, BD Biosciences). T cells treated with Leukocyte Activation Cocktail (cat #550583, BD Biosciences) diluted 1:1,000 served as positive control. An EGFR-specific mAb (clone LA1, Millipore) was used to block binding of CAR+ T cells with EGFR on target cells at a concentration of 5 μg/mL. Following incubation, surface staining and intracellular cytokine staining were performed using Cytofix/Cytoperm Fixation and Permeabilization kit (cat #554714, BD Biosciences) according to manufacturer’s instructions.
Phospho-flow cytometry
T cells were co-cultured with target cells at a ratio of 1:1 for 45 minutes, then lysed and fixed using PhosFlow Lyse/Fix buffer (cat #558049, BD Biosciences), detailed in Supplementary Materials and Methods, and analyzed via flow cytometry.
Measurement of down regulation of CAR expression
T cells and targets were mixed at a 1:1 ratio in a 12-well plate and CAR surface expression was measured by flow cytometry at each time point. T cells plated without targets served as negative controls. Percent down regulation of CAR was calculated as: (stimulated CAR expression)/(unstimulated CAR expression) × 100.
Chromium release assay
Specific cytotoxicity was assessed via standard 4-hour chromium release assay, as previously described (16).
Evaluation of T-cell efficacy in intracranial glioma xenograft
Animal experiments were carried out under regulation from the Institutional Animal Care and Use Committee (IACUC) at MDACC (ACUF 11-11-13131). Female NOD.Cg-PrkdcscidIL2Rγtm1Wjl/Sz (NSG, Jackson Laboratory) mice aged 6–8 weeks were anesthetized by intraperitoneal injection of cocktail of 10 mg/mL ketamine and 0.5 mg/mL xylazine dosed at 0.1 mL/10 grams. Implantation of guide-screw was performed is detailed in Supplementary Materials and Methods. Two weeks following guide-screw implantation, 250,000 U87-ffLuc-mKate or U87med-ffLuc-mKate cells were implanted through guide screw in 5 μL PBS. Burden of intracranial glioma was serially imaged using Xenogen IVIS Spectrum (Caliper Life Sciences,) 10 minutes after sub-cutaneous injection of 215μg D-luciferin potassium salt (cat #122796, Caliper Life Sciences). Tumor flux (photons/s/cm2/steradian) was measured using Living Image software (version 2.50, Caliper Life Sciences) in a delineated region encompassing entire cranial region. Mice were stratified into three groups to evenly distribute tumor burden based on BLI measurements 4 days after tumor injection and treated with 4×106 T cells administered once a week for three weeks through the guide-screw in 5 μL of PBS. Mice were sacrificed when they displayed progressive weight loss (>25% loss), rapid weight loss (>10% loss within 48 hours), hind limb paralysis, or any two of the following clinical symptoms of illness: ataxia, hunched posture, irregular respiration rate, ulceration of exposed tumor, or palpable tumor diameter exceeding 1.5 cm.
Statistical Analyses
Statistical analyses were performed in GraphPad Prism, version 6.03. Statistical analyses of in vitro assays were undertaken by two-way ANOVA with donor-matching and Sidak’s or Tukey’s post-test for multiple comparisons or two-tailed student’s t-test, as indicated in figure legends. Correlation of T-cell effector functions with EGFR density was performed by one-way ANOVA with post-test for linear trend. Analyses of in vivo bioluminescent imaging of tumor were performed using two-way ANOVA with repeated measures and Sidak’s post-test for multiple comparisons. Survival of mice was performed by log-rank (Mantel-Cox) test. Significance of findings defined as follows: ns = not significant, p>0.05, * p<0.05, ** p<0.01, *** p<0.001, **** p <0.0001.
RESULTS
Cetux-CAR+ and Nimo-CAR+ T cells are phenotypically similar
Cetux-CAR and Nimo-CAR were generated in Sleeping Beauty (SB) vectors (Supplementary Fig. S1A) expressed in T cells via electroporation, and propagated by weekly stimulations with EGFR+ activating and propagating cells (AaPC) (Supplementary Fig. S1B). Cetux- and Nimo-CAR+ T cells expanded ~1,000-fold over 28 days (Fig. 1A). There was no difference in the kinetics or number of resultant T cells (p = 0.92), and similar CAR expression (Cetux-CAR = 91 ± 6%, Nimo-CAR = 91 ± 6%; mean ± SD, n=7) (Fig. 1B). The density of CAR expression, represented by median fluorescence intensity, was statistically similar on Cetux-CAR+ and Nimo-CAR+ T-cell populations (Cetux-CAR = 119 ± 25 arbitrary units (A.U.), Nimo-CAR = 113 ± 21 A.U.; mean ± SD, n=7, p = 0.74) (Fig. 1C). While individual donors yielded variable proportions of CD4+ and CD8+ T cells (Supplementary Table S2), there was no statistical difference in the CD4/CD8 ratio between Cetux-CAR+ and Nimo-CAR+ T cells (p = 0.44) (Fig. 1D). Expression of cell-surface markers associated with T-cell differentiation (CD45RO, CD45RA, CD28, CD27, CCR7, CD62L, CD57, KLRG1, and PD-1) were not significantly different (p > 0.05) (Fig. 1E, F).
Figure 1.

Cetux-CAR+ and Nimo-CAR+ T cells are phenotypically similar. (A) Numeric expansion of Cetux-CAR+ and Nimo-CAR+ T cells was determined by calculating fold expansion of CD3+CAR+ T cells, determined by flow cytometry, during each stimulation cycle. Data represented as mean ± SD, n=5. Statistical analysis by two-tailed student’s t-test. (B) Expression of CAR in CD3+ T cells was determined 24 hours after electroporation of CAR and after 28 days of expansion by flow cytometry for the IgG portion of CAR. Data represented as mean, n=7. Statistical analysis performed by two-tailed student’s t-test. (C) Median fluorescence intensity (MFI of CAR expression was determined by flow cytometry for the IgG portion of CAR after 28 days of expansion. Data represented as mean ± SD, n=7. Statistical analysis performed by two-tailed student’s t-test. (D) Proportion of CD4 and CD8 T cells in total T-cell population after 28 days of expansion measured by flow cytometry on gated CD3+CAR+ cells. Data represented as mean ± SD, n=7. (E, F) Expression of T-cell memory and differentiation markers after 28 days of T-cell expansion measured by flow cytometry in gated CD4+ and CD8+ T-cell populations. Data represented as mean ± SD, n=4. Statistical analysis performed by two-way ANOVA with Sidak’s post-test correction.
To verify Cetux-CAR and Nimo-CAR were functional in response to EGFR stimulation, we measured production of the inflammatory cytokine IFN-γ in response to A431, an epidermoid carcinoma cell line expressing ~1×106 molecules of EGFR/cell (20). Both Cetux-CAR+ and Nimo-CAR+ T cells produced IFN-γ in response to A431 and production was titrated in response to an EGFR-blocking antibody (clone LA1, Millipore) with an overlapping epitope (Fig. 2B, C). Reduction in IFN-γ production by Nimo-CAR+ T cells was observed at lower concentrations of antibody than Cetux-CAR+ T cells, supporting differences in scFv binding affinity are preserved in CAR structure. Cetux-CAR+ and Nimo-CAR+ T cells both lysed A431 cells, however, neither population significantly lysed EGFRneg NALM-6 cells (Fig. 2D). Cetux-CAR+ and Nimo-CAR+ T cells are phenotypically similar, including percentage and density of CAR expression, and are activated upon encounter with EGFR.
Figure 2.
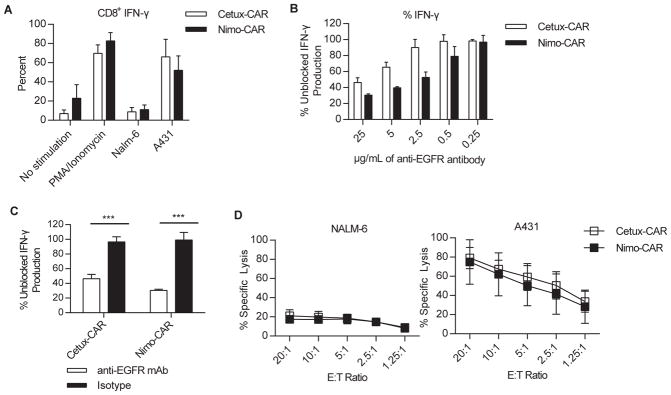
Cetux-CAR+ and Nimo-CAR+ T cells are activated in response to EGFR. (A) Production of IFN-γ by CD8+CAR+ T cells in response to co-culture with A431 and NALM-6 cell lines measured by intracellular flow cytometry gated on CD8+ cells. Data represented as mean ± SD, n=4,*** p < 0.001, two-way ANOVA (Sidak’s post-test). (B) Production of IFN-γ in response to EGFR+ A431 in the presence of titrating concentrations of EGFR-blocking mAb (clone LA1, Millipore). CAR+ T cells were co-cultured with A431 with EGFR-specific mAb and IFN-γ production was measured by intracellular flow cytometry. Percent of production was calculated as MFI of IFN-γ in gated CD8+ T cells relative to unblocked CD8+ T-cell production. Data represented as mean ± SD, n=3. (C) Production of IFN-γ as measured by intracellular flow cytometry in response to EGFR+ A431 in the presence 25 μg/mL of EGFR-blocking mAb (clone LA1, Millipore). Percent of production was calculated as MFI of IFN-γ in gated CD8+ T cells relative to unblocked CD8+ T-cell production. Data represented as mean ± SD, n=3, *** p<0.001, two-way ANOVA (Sidak’s post-test). (D) Specific lysis of A431 and NALM-6 by CAR+ T cells measured by standard 4 hour chromium release assay. Data represented as mean ± SD, n=4.
Nimo-CAR+ T cells have impaired response to low density EGFR
We next determined if affinity of scFv within the CARs could impact effector functions of genetically modified T cells as a function of density of EGFR expression. The activation of Cetux-CAR+ and Nimo-CAR+ T cells in response to EL4 cell line genetically modified to express tEGFR (Supplementary Fig. S1C) at a low density, similar to normal human fibroblasts (21), (Fig. 3A), and compared to T-cell activation by EL4 cells capable of cross-linking CARs independently of scFv through expression of CAR-L, an activating scFv of mAb specific for the IgG4-derived domain of CAR (17). A similar proportion of Cetux-CAR+ and Nimo-CAR+ T cells produced IFN-γ in response to CAR-L+ EL4, (p > 0.05) (Fig. 3B). While Cetux-CAR+ T cells increased IFN-γ production in response to tEGFR+ EL4 relative to tCD19+ EL4 (%IFN-γ, p<0.0001; MFI IFN-γ, p<0.01), Nimo-CAR+ T cells did not (%IFN-γ, p>0.05, MFI IFN-γ, p>0.05). There was no statistical difference in activation as assessed by phosphorylation of Erk1/2 (p > 0.05) or p38 (p > 0.05) between Cetux-CAR+ and Nimo-CAR+ T cells in response to CAR-L+ EL4 (Fig. 3C). In contrast, Cetux-CAR+ T cells exhibited phosphorylation of Erk1/2 and p38 in response to tEGFR+ EL4, while Nimo-CAR+ T cells did not. Cetux-CAR+ and Nimo-CAR+ T cells demonstrated equivalent specific lysis against CAR-L+ EL4 (10:1 E:T ratio, Cetux-CAR = 65 ± 7%, Nimo-CAR = 58 ± 13%, mean ± SD, n=4, p>0.05) (Fig. 3D). However, differences were observed in capacity to lyse tEGFR+ EL4, where Cetux-CAR+ T cells demonstrated significant lysis of tEGFR+ EL4 compared to irrelevant antigen (tEGFR+EL4 = 58 ± 9%, tCD19+EL4 = 17 ± 13%, mean ± SD, n=4; p < 0.0001) and Nimo-CAR+ T cells did not (tEGFR+EL4 = 21 ± 17%, CD19+EL4 = 12 ± 13%, mean ± SD, n=4, p > 0.05). These data demonstrate that both CARs are capable of activating T cells when triggered outside of scFv, yet differ in ability to activate T cells to low density EGFR.
Figure 3.

Nimo-CAR+ T cells have impaired response to low density of EGFR. (A) Representative histograms of expression of tEGFR on EL4 cells relative to cell lines negative for EGFR. Density of EGFR expression was determined by quantitative flow cytometry. (B) Production of IFN-γ by gated CD8+ CAR+ T cells after co-culture with CD19+, tEGFR+, or CAR-L+ EL4 cells measured by intracellular staining and flow cytometry. Data represented as mean percent ± SD, n=4 and mean fluorescence intensity (MFI) ± SD, ** p<0.01 two-way ANOVA (Sidak’s post-test) and mean fluorescence intensity ± SD, n=2. (C) Phosphorylation of p38 and Erk1/2 by phosflow cytometry in gated CD8+ CAR+ T cells 30 minutes after co-culture with CD19+, tEGFR+, or CAR-L+ EL4 cells. Data represented as mean ± SD, n=2, * p<0.05, two-way ANOVA (Sidak’s post-test). (D) Specific lysis of CD19+, tEGFR+ and CAR-L+ EL4 cells measured by standard 4-hour chromium release assay. Data represented as mean ± SD, n=4, **** p<0.0001, two-way ANOVA (Tukey’s post-test).
Nimo-CAR+ T cell activation is positively correlated with EGFR density
We sought to further evaluate the impact of TAA density on activation of CAR T cells in a single cellular background by modifying U87 cells to express varied levels of EGFR, reflective of densities found in glioblastoma cell lines (22) (Fig. 4A): unmodified parental U87, U87low, U87med, and U87high. Phosphorylation Erk1/2 and p38 in Cetux-CAR+ T cells exhibited no correlation with density of EGFR expression (one-way ANOVA with post-test for linear trend; Erk1/2, p = 0.88; p38, p = 0.09) (Fig. 4B). In contrast, phosphorylation of Erk1/2 and p38 by Nimo-CAR+ T cells positively correlated with EGFR expression (one-way ANOVA with post-test for linear trend; Erk1/2, p = 0.003 and p38, p = 0.004). We also noted that Nimo-CAR+ T cells demonstrated significantly less phosphorylation of Erk1/2 and p38 compared with Cetux-CAR+ T cells, even in response to high EGFR density on U87high (Erk1/2, p < 0.0001; p38, p < 0.01). Percentage and magnitude IFN-γ production by Cetux-CAR+ T cells did not correlate with EGFR density on target cells (one-way ANOVA with post-test for linear trend; %IFN-γ, p = 0.5703; MFI, p = 0.9508) (Fig. 4C). In contrast, both proportion of Nimo-CAR+ T cells producing IFN-γ and magnitude of production directly correlated with increasing EGFR density (one-way ANOVA with post-test for linear trend; %IFN-γ, p = 0.0124; MFI, p = 0.0175). Production of TNF-α by Cetux-CAR+ T cells and Nimo-CAR+ T cells showed a similar trend (Supplementary Fig. S2A). Lastly, Cetux-CAR+ T cells demonstrated significantly increased specific lysis of both parental U87 (10:1 E:T ratio, p < 0.0001) and U87low (10:1 E:T ratio, p < 0.05) targets compared with reduced killing by Nimo-CAR+ T cells. However, there was statistically similar lysis by T cells expressing both CAR designs targeting U87med (10:1 E:T ratio, p>0.05) and U87high (10:1 E:T ratio, p > 0.05) (Fig. 4D). These data were verified using cell lines with naturally varied EGFR expression (Supplementary Fig. S3). We investigated the ability of CAR T cells to be activated by low density EGFR on normal human renal cortical epithelial cells (HRCE), which express ~15,000 molecules of EGFR/cell (Fig. 4E). Cetux-CAR+ T cells produced IFN-γ in response to HRCE (%IFN-γ, p < 0.05; MFI, p < 0.01) whereas Nimo-CAR+ T cells did not (%IFN-γ, p > 0.05; MFI, p > 0.05) (Fig. 4F). Similarly, Cetux-CAR+ T cells, but not Nimo-CAR+ T cells, produced TNF-α in response to HRCE (Supplementary Fig. S2B). In summary, Cetux-CAR+ and Nimo-CAR+ T cells were both activated by target cells expressing high EGFR density, but only Nimo-CAR+ T cells showed diminished activity in response to low EGFR density. These observations support the premise that activation of Nimo-CAR+ T cells, in contrast to Cetux-CAR+ T cells, is dependent on density of EGFR expression.
Figure 4.

Nimo-CAR+ T-cell activation is positively correlated with EGFR density. (A) Representative histogram of EGFR expression on series of four U87-derived tumor cell lines (U87, U87low, U87med, and U87high) measured and quantified by flow cytometry. Data is representative from triplicates. (B) Phosphorylation of Erk1/2 and p38 in gated CD8+ T cells after 45 minutes of co-culture with U87 cell lines by phosflow cytometry. Data represented as MFI ± SD, n=4, **** p<0.0001, *** p<0.001, ** p<0.01, two-way ANOVA (Sidak’s post-test). (C) Production of IFN-γ by gated CD8+ CAR+ T cells in response to co-culture with U87 cell lines measured by intracellular staining and flow cytometry. Data represented as mean percent ± SD and mean fluorescence intensity ± SD, n=4, **** p<0.0001, *** p<0.001, ** p<0.01, two-way ANOVA (Sidak’s post-test). (D) Specific lysis of U87 cell lines by CAR+ T cells measured by standard 4-hour chromium release assay. Data represented as mean ± SD, n=5, **** p<0.0001, ** p<0.01, * p<0.05, two-way ANOVA (Tukey’s post-test). (E) Representative histogram of expression of EGFR on HRCE measured by flow cytometry. Number of EGFR molecules per cell determined by quantitative flow cytometry. Data are representative of triplicates. (F) Production of IFN-γ by CD8+ CAR+ T cells after co-culture with HRCE measured by intracellular staining and flow cytometry gated on CD8+ cells. Data represented as mean percent ± SD and mean fluorescence intensity ± SD, n=4, ** p<0.01, * p<0.05, ns p>0.05, two-way ANOVA (Sidak’s post-test).
Increasing duration of interaction does not restore Nimo-CAR+ T-cell response to low EGFR density
Because low-affinity TCR-mediated responses may be activated by prolonged interaction with antigen in context of MHC (23,24), we extended the duration of interaction of Nimo-CAR+ T cells with targets to 24 hours, based on demonstrations of saturated maximal killing at this time point in non-radioactive lysis assays (25,26). Over 24 hours of interaction, Nimo-CAR+ T cells failed to significantly produce IFN-γ in response to U87, however, maintained similar IFN-γ production to Cetux-CAR+ T cells in response to U87high (Fig. 5A). Furthermore, Cetux-CAR+ and Nimo-CAR+ T cells demonstrated a statistically similar ability to control the growth of U87high tumor cells in long-term co-culture, however Nimo-CAR+ T cells were unable to control parental U87 growth (Fig. 5B). While Cetux-CAR+ T cells demonstrated moderate, similar proliferation in response to U87 and U87high, Nimo-CAR+ T cells demonstrated significantly higher proliferation in response to U87high cells than parental U87 (Fig. 5C). In aggregate, these data show that reduced activation of Nimo-CAR+ T cells to low density EGFR is not restored by increasing duration of interaction.
Figure 5.

Increasing duration of interaction does not restore Nimo-CAR+ T-cell response to low EGFR density. (A) Production of IFN-γ was measured by intracellular staining and flow cytometry following stimulation with U87 or U87high over time in CD8+ gated cells. Data represented as mean percent producing ± SD, n=4 and mean fluorescence intensity of IFN-γ ± SD, n=4, **** p<0.0001, two-way ANOVA (Sidak’s post-test). (B) Fraction of U87 and U87high cells remaining after co-culture with Cetux-CAR+ or Nimo-CAR+ T cells. U87 cell lines were co-cultured with CAR+ T cells at an E:T ratio of 1:5 in triplicate. Percent surviving was calculated as [cell number harvested after co-culture]/[cell number without T cells]*100. Data represented as mean ± SD, n=3, *** p<0.001, two-way ANOVA (Sidak’s post-test). (C) Proliferation of CAR+ T cells measured by intracellular flow cytometry for Ki-67 in CD8+ gated T cells following 36 hours of co-culture. Data represented as mean fluorescence intensity of Ki-67 ± SD, n=4, *** p<0.001, two-way ANOVA (Tukey’s post-test). (D) Surface expression of CAR during co-culture (E:T 1:5) with parental U87 or U87high measured by flow cytometry for IgG portion of CAR. Data represented as mean ± SD, n=3, *** p<0.001, * p<0.05, two-way ANOVA (Sidak’s post-test) (E) Representative histograms of Intracellular and surface expression of CAR determined by flow cytometry after 24 hours of co-culture with U87 or U87high in CD8+ gated T cells. Data representative of three independent donors. (F) After 24-hours of incubation with U87 or U87high, CAR+ T cells were rechallenged with U87 or U87high and production of IFN-γ CAR+ T cells measured by intracellular staining and flow cytometry gated on CD8+ cells. Data represented as mean ± SD, n=3, *** p<0.001, ** p<0.01, * p<0.05, two-way ANOVA (Sidak’s post-test).
Cetux-CAR exhibits enhanced down regulation upon EGFR docking
The cell surface expression of endogenous TCR and CAR may be decreased following interaction with antigen, and for endogenous TCR, the degree of down regulation is correlated with the strength of TCR binding (27). We investigated whether Cetux-CAR+ and Nimo-CAR+ T cells have a differing propensities for TAA-induced down regulation. When co-cultured with parental U87, Cetux-CAR was reduced from T-cell surface after 12 hours and returned by 48 hours, but expression of Nimo-CAR did not appreciably reduce from the cell surface (Fig. 5D). In response to high EGFR density on U87high, Cetux-CAR was significantly reduced after 12 hours of co-culture and remained reduced over 48 hours. Again, Nimo-CAR did not appreciably downregulate (12 hours; Cetux-CAR = 37 ± 12%, Nimo-CAR= 124 ± 15%, mean ± SD, n=3). Cetux-CAR was detectable within T cells when reduced from the T-cell surface, demonstrating that reduced CAR expression was due to internalization of CAR and not outgrowth of genetically unmodified T cells (Fig. 5E). Similarly, when stimulated with tEGFR+ EL4, Cetux-CAR is reduced from T-cell surface, but Nimo-CAR is not, and both CARs are down-regulated to a similar degree when stimulated with CAR-L+ EL4 (Supplementary Fig. S4). To determine if down-regulation of CAR impacts subsequent activation to antigen, we re-challenged CAR+ T cells after initial encounter with EGFR. While Cetux-CAR+ T cells had reduced production of IFN-γ, Nimo-CAR+ T cells retained IFN-γ production in response to re-challenge with the glioma cells expressing low and high levels of EGFR (Fig. 5F). Thus, downregulation of CAR is function of affinity and antigen density, enabling Nimo-CAR+ T cells to maintain function to repeated challenge with elevated levels of EGFR, while Cetux-CAR+ T cells have reduced capacity to respond to under conditions of serial stimulation.
Nimo- and Cetux-CAR+ T cells inhibit high-density EGFR+ glioma growth in vivo
Because glioma cell lines express a median density of 240,000 molecules of EGFR/cell (22), U87med expressing ~340,000 molecules of EGFR/cell modified to co-express ffLuc-mKate were established as intracranial tumors in NSG mice to evaluate anti-tumor efficacy. Mice were stratified to receive no treatment, Cetux-CAR+ T cells, or Nimo-CAR+ T cells to evenly distribute relative tumor burden (Supplementary Fig. S5A). CAR+ T cells were infused once a week for 3 weeks and relative tumor growth was monitored by BLI. CAR expression was similar Cetux-CAR+ T cells and Nimo-CAR+ T cells prior to infusion (Supplementary Fig. S5B). Cetux-CAR+ T cells and Nimo-CAR+ T cells both significantly inhibited tumor growth (day 18; Cetux-CAR, p < 0.01 and Nimo-CAR, p < 0.05) (Fig. 6A, B) and were not significantly different (p > 0.05). Mice receiving Cetux-CAR+ T cells exhibited toxicity manifested as rapid weight loss and lethargy, resulting in significant death of mice within 7 days of T-cell infusion. (6/14 mice, p = 0.0006; Fig. 6C). Overall, adoptive transfer of Cetux-CAR+ T cells did not statistically improve survival compared to untreated mice (untreated median survival = 88 days, Cetux-CAR median survival = 105 days, p = 0.19) (Fig. 6D). When only considering mice surviving 7 days after Cetux-CAR+ T cell infusion, we noted improvement in survival of 75% of mice (p = 0.0065 relative to untreated mice). In contrast, Nimo-CAR+ T cells mediated effective tumor regression and extended survival in 4/7 of mice without apparent toxicity (untreated median survival = 88 days, Nimo-CAR median survival = 158 days, p = 0.0269). Following 225 days, 2/7 mice in each treatment group were still living. These results demonstrate that Cetux-CAR+ T cells and Nimo-CAR+ T cells are both capable of controlling glioma with an intermediate density of EGFR.
Figure 6.

Nimo- and Cetux-CAR+ T cells inhibit high-density EGFR+ glioma growth in vivo. (A) Serial BLI assessed relative size of tumor (introduced on day 0). (B) Relative tumor growth as assessed by serial BLI of tumor. Background luminescence (gray shading) was defined by BLI of mice with no tumors. Significant difference in BLI between mice with no treatment vs. treatment (n=7) with Cetux-CAR+ T cells (n=7, p < 0.01) and no treatment (n=7) vs. treatment with Nimo-CAR+ T cells (n=7, p < 0.05) at day 18, two-way ANOVA (Sidak’s post-test). (C) Survival of mice with U87med-ffLuc-mKate intracranial xenografts from two independent experiments within 7 days of T-cell administration. Significant reduction in survival in Cetux-CAR+ T cell treated mice 8/14 surviving at day 26) relative to untreated mice (14/14 surviving at day 26) determined by Mantel-Cox log-rank test, p = 0.0006. (D) Survival of mice with U87med-ffLuc-mKate intracranial xenografts receiving no treatment, Cetux-CAR+ T cells or Nimo-CAR+ T cells. Significant extension in survival in Nimo-CAR+ T cell treatment group determined by Mantel-Cox log-rank test, p = 0.027.
Nimo-CAR+ T cells exhibit impaired targeting of low-density EGFR+ cells in vivo
We next sought to determine whether CAR+ T cells might exhibit a potential for undesired targeting of low EGFR density, by using parental U87 cells with low EGFR expression as a mimic for normal cells (Supplementary Fig. S5C). Treatment of mice with Cetux-CAR+ T cells resulted in significant reduction of U87 cells compared to untreated mice (day 25, p < 0.01) (Fig. 7A, B), and significantly extended survival in 3/6 mice compared to mice receiving no treatment (untreated median survival = 38.5 days, Cetux-CAR median survival = 53 days, p = 0.015; Fig. 7C). In contrast, administration of Nimo-CAR+ T cells did not significantly impact the growth of U87 compared to untreated mice (Nimo-CAR, p > 0.05) and thus did not significantly improve survival (untreated median survival 38.5 days, Nimo-CAR median survival 46 days, p = 0.0969). These data demonstrate that Nimo-CAR+ T cells, in contrast to Cetux-CAR+ T cells, do not mediate significant activity against parental U87 in vivo, which serve as a surrogate for normal tissue, demonstrating an effector profile consistent with reduced ability to target EGFR on normal cells.
Figure 7.

Nimo-CAR+ T cells exhibit impaired targeting of low-density EGFR cells in vivo. (A) Serial BLI assessed relative size of tumor. (B) Relative tumor growth as assessed by serial BLI of tumor. Significant difference in BLI between mice with no treatment vs. treatment (n=6) with Cetux-CAR+ T cells (n=6, p < 0.01) reached at day 25, two-way ANOVA (Sidak’s post-test). (C) Survival of mice with U87-ffLuc-mKate intracranial xenografts receiving no treatment, Cetux-CAR+ T cells or Nimo-CAR+ T cells. Significant extension in survival in Cetux-CAR+ T-cell treatment group determined by Mantel-Cox log-rank test, p=0.0150.
DISCUSSION
Aberrantly-expressed antigens on tumors, such as EGFR on glioblastoma, can be overexpressed relative to lower, basal levels on normal tissues. Taking advantage of this observation, we tuned CAR affinity to activate T cells based on the density of EGFR expression and demonstrated that a CAR with reduced affinity rendered T cells preferentially activated by high, but not low, density of EGFR. Our approach was based on the clinical toxicity exhibited by cetuximab and nimotuzumab, which recognize overlapping epitopes and exhibit different kinetics of binding to EGFR (13,14). The lower affinity of nimotuzumab has been credited with absence of adverse events relative to cetuximab (15). Indeed, we revealed that a CAR species recognizing EGFR via the scFv derived from nimotuzumab exhibited a gradient of T-cell activation that directly correlated with density of expression of EGFR. In fact, as predicted by studies with nimotuzumab, Nimo-CAR+ T cells exhibited no apparent T-cell activation (absence of phosphorylation of MAP kinase molecules and cytokine production) in response to densities of EGFR found on normal tissues.
There are few studies addressing the role of scFv affinity in the therapeutic aptitude of CAR designs. Most recently, and in support of our findings, an EGFRvIII-specific CAR with reduced affinity has been reported with reduced binding to wild type EGFR, and therefore reduced potential for normal tissue toxicity (28). A ROR1-specific CAR with increased affinity resulted in augmented T-cell function in response to TAA, which led to superior anti-tumor activity in vivo (29). Our study supports these findings in the situation of low antigen density on target cells, however, when antigen density is high on target cells, we found no benefit of increased scFv affinity. A series of CARs derived from a panel of HER2-specific mAbs with a range of Kd values demonstrated that the affinity of CAR above a minimum threshold required for activation did not improve T-cell response to targets with a range of HER2 densities (30).
While the affinity constant (Kd) is one measure used to describe the functional affinity of a CAR species, the relative contributions of on- and off-rates may account for disparate findings between these studies, as is likely the case with αβ TCR affinity in governing endogenous T-cell responses (31,32). The reduced affinity constant of nimotuzumab is most impacted by a reduced on-rate of binding, requiring at least bivalent interaction to bind to target cells, which is more likely to occur in the presence of high antigen density. Thus, the capacity of the CAR to bind antigen at differing densities likely accounts for the superior ability of Cetux-CAR compared with Nimo-CAR to recognize low density EGFR. Nimotuzumab and cetuximab bind highly overlapping epitopes, making it unlikely that magnitude of differences in CAR function are due solely to differences in epitope location; however, the contribution of this factor cannot be excluded.
Two parameters of TCR binding have been described that contribute to T-cell functional avidity: quality (binding affinity) and quantity (number of receptors engaged) (12,33). Increasing quantity of lower affinity TCRs can restore some T-cell functions, but not all, highlighting that some functions are dependent on quality versus quantity of activation signal. We observed a distinction between the quality and quantity of signaling mediated by Cetux-CAR versus Nimo-CAR. Nimo-CAR+ T cells docking with glioma cells expressing high EGFR density demonstrated reduced phosphorylation of Erk1/2 and p38 compared to Cetux-CAR+ T cells, in spite of equivalent phosphorylation of Erk1/2 and p38 in response to CAR-L+ EL4, yet maintained comparable functional responses. Because low affinity TCRs can signal through transient, undetectable phosphorylation intermediates to culminate in functional T-cell response, it is possible a similar mechanism is responsible for this observation in Nimo-CAR+ T cells (23).
High affinity TCRs and CARs can elicit impaired functional T cell responses, due to prolonged interactions prohibiting serial triggering, which is necessary to generate a fully functional T cell response (34–37). In CAR-modified T cells, however, the requirement for serial triggering has been overcome by expressing CAR at higher densities, reducing the need for serial triggering to activate T cells. Thus, any theoretical impairment of Cetux-CAR function due to its high affinity may be masked by expression at high density on T cells. We observed that Cetux-CAR was less capable of mounting a T-cell response upon re-exposure to EGFR, which was predicted by the down-regulation of Cetux-CAR after initial docking with EGFR. Similarly, Cetux-CAR+ T cells exhibited less proliferative capacity than Nimo-CAR+ T cells in response to high EGFR density. Signaling through engineered and endogenous TCRs of high affinity has been reported to cause functional anergy in T cells (38–40), so whether a reduced secondary response of Cetux-CAR+ T cells is due to loss of CAR and/or functional anergy remains to be elucidated.
Because cetuximab and nimotuzumab do not cross-react with murine EGFR, we measured deleterious binding of CAR+ T cells to low density EGFR based upon implantation of parental U87 cells, which express low levels of EGFR and thus serve as a surrogate for normal human cells. We observed that adoptive transfer of Cetux-CAR+ T cells, but not Nimo-CAR+ T cells, could control growth of parental U87 cells. Therefore, T cells expressing a CAR with increased affinity for EGFR are likely to result in recognition of low levels of EGFR expression in vitro and in vivo leading to on-target, off-tissue toxicity. It is possible that Nimo-CAR+ T cells may result in outgrowth of low density EGFR tumor escape variants, as described in studies with other CARs targeting GBM-associated antigens (41). Computational modeling based on extent and intensity of multiple antigens expressed on resected patient GBMs can inform design of a combination therapy utilizing CARs with multiple specificities to overcome this limitation.
Alternative approaches exist for reducing or avoiding the potential for CAR-mediated toxicity to TAA expressed on normal tissues. Expression of CAR from introduced mRNA species or activation of a suicide switch to result in death of infused T cells temporally limit CAR+ T cell presence to reduce toxicity, but both permanently negate CAR T cell function and may undermine therapeutic efficacy (42–45). Limiting T-cell activation to CAR binding malignant cells can be achieved by co-expression of CARs with dissociated signaling domains targeting TAA only mutually expressed on malignant cells, such that binding of both CARs is needed to achieve a fully-competent T-cell activation signal (46–48). However, requirement for expression of two antigens for efficient T-cell activation reduces the fraction of tumor capable of triggering CAR T cells, and a partial signal received from interaction of one CAR with TAA on normal tissue may be sufficient to mediate toxicity. Co-expression of a CAR activated in response to TAA and a CAR inhibitory in response to normal tissue antigen could minimize normal tissue toxicity (49), however, careful consideration of stoichiometric ratios of antigens on normal tissue is required. Our approach has merit in simplicity as only one TAA is targeted.
In conclusion, the weakness of the Nimo-CAR affinity is its apparent strength. Our data support a new approach to designing genetically modified T cells to help them separate friend from foe by tuning scFv affinity to preferentially target the increased density of TAA. The relative inability of Nimo-CAR+ T cells to control U87 xenografts with low EGFR density in vivo combined with in vitro evidence that Nimo-CAR+ T cells are not activated in the presence of low density EGFR suggest that Nimo-CAR+ T cells are favorably impaired in their ability to damage normal cells, especially relative to Cetux-CAR+ T cells. We cannot eliminate the possibility of Nimo-CAR+ T cells mediating some degree of on-target, off-tissue toxicity, therefore, initial clinical evaluation of Nimo-CAR+ T cells should be undertaken with suicide genes in patients with high-grade glioma via \ intracranial delivery of T cells as the central nervous system lacks notable EGFR expression (9), and may be extended to other cancers overexpressing EGFR, including bladder, cervical, esophageal, head and neck squamous cell, ovarian, breast, colorectal, gastric, endometrial, and non-small lung cell carcinomas (50).
Supplementary Material
Acknowledgments
Funding: Cancer Center Core Grant (CA16672); RO1 (CA124782, CA120956, CA141303; CA141303); P01 (CA148600); Center for Clinical and Translational Sciences, which is funded by National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number TL1TR000369 (The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health); SPORE (P50 CA136411); Albert J Ward Foundation; Alex Lemonade Stand Foundation; American Legion Auxiliary, Burroughs Wellcome Fund; Cancer Answers; Cancer Prevention and Research Institute of Texas; Charles B. Goddard Foundation of Texas; CLL Global Research Foundation; DARPA (Defense Sciences Office); Department of Defense; Estate of Noelan L. Bibler; Gillson Longenbaugh Foundation; Harry T. Mangurian, Jr., Fund for Leukemia Immunotherapy; Khalifa Bin Zayed Al Nahyan Foundation; Leukemia and Lymphoma Society; Lymphoma Research Foundation; Miller Foundation; Moon Shot program at MDACC, Mr. Herb Simons; Mr. and Mrs. Joe H. Scales; Mr. Thomas Scott; National Foundation for Cancer Research; Pediatric Cancer Research Foundation; Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy; R.W. Butcher Foundation, MDACC Sister Institution Network Fund and Moon Shot Fund; William Lawrence and Blanche Hughes Children’s Foundation.
We thank flow cytometry and cellular imaging core facilities at MDACC (supported by CA016672). We thank P. Hackett (University of Minnesota) for his assistance with SB system, O. Bogler (MDACC) for his gift of U87 cell line overexpressing EGFR (U87high), F. Lang, V. Henry and L. Holmes for training and implementation of guide-screw glioma xenograft model, and J. Moyes and G. McNamara for editing. In memory of Patricia Ann Cooper.
Footnotes
Competing Interests: Some of the technology described in this presentation was advanced through research conducted at the MD Anderson Cancer Center under the direction of Laurence J.N. Cooper, M.D., Ph.D. In January 2015, the technology was licensed for commercial application to ZIOPHARM Oncology., and Intrexon Corporation in exchange for equity interests in each of these companies for which the following authors. MD Anderson Cancer Center, and the following authors have a financial interest in ZIOPHARM Oncology, Inc., and Intrexon Corporation: HG Caruso, LV Hurton, A Najjar, D Rushworth, S Ang, S Olivares, T Mi, K Switzer, H Singh, H Huls, DA Lee, RE Champlin and LJN Cooper. On May 7, 2015, Dr. Cooper was appointed as the Chief Executive Officer at ZIOPHARM. Dr. Cooper is now a Visiting Scientist at MD Anderson where he will continue to supervise the development of this technology. Patent application based on this manuscript filed April 23, 2014.
Author Contributions: HGC conceived project concept, planned and performed experiments, analyzed and interpreted data, and wrote the manuscript. LVH provided intellectual input, assisted with analyzing data, and assisted with animal experiments. AN assisted with design of animal model and assisted with animal experiments. SA and SO designed Cetux-CAR and Nimo-CAR structures in Sleeping Beauty vectors. TM and KS assisted with animal experiments. HS, HH, and DAL provided guidance in designing and interpreting experiments and writing the manuscript. AH and REC provided advice and support. LJNC conceived and directed the study, interpreted data, as well as wrote the manuscript.
References
- 1.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365(8):725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England journal of medicine. 2013;368(16):1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371(16):1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-oncology. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. Journal of neuro-oncology. 2012;107(2):359–64. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 6.Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal growth factor receptor - mediated signal transduction in the development and therapy of gliomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(24):7261–70. doi: 10.1158/1078-0432.CCR-06-0874. [DOI] [PubMed] [Google Scholar]
- 7.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 8.Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Current molecular pharmacology. 2010;3(1):37–52. doi: 10.2174/1874467211003010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yano S, Kondo K, Yamaguchi M, Richmond G, Hutchison M, Wakeling A, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer research. 2003;23(5A):3639–50. [PubMed] [Google Scholar]
- 10.Lenz HJ. Anti-EGFR mechanism of action: antitumor effect and underlying cause of adverse events. Oncology (Williston Park) 2006;20(5 Suppl 2):5–13. [PubMed] [Google Scholar]
- 11.Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol. 2011;186(9):5039–45. doi: 10.4049/jimmunol.1003650. [DOI] [PubMed] [Google Scholar]
- 12.Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, et al. TCR binding affinity governs the functional profile of cancer-specific CD8 T cells. Clinical and experimental immunology. 2014 doi: 10.1111/cei.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talavera A, Friemann R, Gomez-Puerta S, Martinez-Fleites C, Garrido G, Rabasa A, et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer research. 2009;69(14):5851–9. doi: 10.1158/0008-5472.CAN-08-4518. [DOI] [PubMed] [Google Scholar]
- 14.Garrido G, Tikhomirov IA, Rabasa A, Yang E, Gracia E, Iznaga N, et al. Bivalent binding by intermediate affinity of nimotuzumab: a contribution to explain antibody clinical profile. Cancer biology & therapy. 2011;11(4):373–82. doi: 10.4161/cbt.11.4.14097. [DOI] [PubMed] [Google Scholar]
- 15.Perez R, Moreno E, Garrido G, Crombet T. EGFR-Targeting as a Biological Therapy: Understanding Nimotuzumab’s Clinical Effects. Cancers. 2011;3(2):2014–31. doi: 10.3390/cancers3022014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer research. 2008;68(8):2961–71. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rushworth D, Jena B, Olivares S, Maiti S, Briggs N, Somanchi S, et al. Universal artificial antigen presenting cells to selectively propagate T cells expressing chimeric antigen receptor independent of specificity. J Immunother. 2014;37(4):204–13. doi: 10.1097/CJI.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Molecular therapy : the journal of the American Society of Gene Therapy. 2007;15(5):981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell Line Data Base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic acids research. 2009;37(Database issue):D925–32. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz Miqueli A, Blanco R, Garcia B, Badia T, Batista AE, Alonso R, et al. Biological activity in vitro of anti-epidermal growth factor receptor monoclonal antibodies with different affinities. Hybridoma (Larchmt) 2007;26(6):423–31. doi: 10.1089/hyb.2007.0516. [DOI] [PubMed] [Google Scholar]
- 21.Hollenberg MD, Cuatrecasas P. Insulin and epidermal growth factor. Human fibroblast receptors related to deoxyribonucleic acid synthesis and amino acid uptake. The Journal of biological chemistry. 1975;250(10):3845–53. [PubMed] [Google Scholar]
- 22.Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer research. 1990;50(24):8017–22. [PubMed] [Google Scholar]
- 23.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, et al. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15(1):59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 24.Yachi PP, Ampudia J, Zal T, Gascoigne NR. Altered peptide ligands induce delayed CD8-T cell receptor interaction--a role for CD8 in distinguishing antigen quality. Immunity. 2006;25(2):203–11. doi: 10.1016/j.immuni.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Karawajew L, Jung G, Wolf H, Micheel B, Ganzel K. A flow cytometric long-term cytotoxicity assay. Journal of immunological methods. 1994;177(1–2):119–30. doi: 10.1016/0022-1759(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 26.Stanke J, Hoffmann C, Erben U, von Keyserling H, Stevanovic S, Cichon G, et al. A flow cytometry-based assay to assess minute frequencies of CD8+ T cells by their cytolytic function. Journal of immunological methods. 2010;360(1–2):56–65. doi: 10.1016/j.jim.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Cai Z, Kishimoto H, Brunmark A, Jackson MR, Peterson PA, Sprent J. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. The Journal of experimental medicine. 1997;185(4):641–51. doi: 10.1084/jem.185.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science translational medicine. 2015;7(275):275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(12):3153–64. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647–53. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 31.Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009;126(2):165–76. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(19):8724–9. doi: 10.1073/pnas.1000966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottschalk RA, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan-Bonnet G, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(3):881–6. doi: 10.1073/pnas.1119763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas S, Xue SA, Bangham CR, Jakobsen BK, Morris EC, Stauss HJ. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood. 2011;118(2):319–29. doi: 10.1182/blood-2010-12-326736. [DOI] [PubMed] [Google Scholar]
- 35.Chervin AS, Stone JD, Soto CM, Engels B, Schreiber H, Roy EJ, et al. Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene therapy. 2013;20(6):634–44. doi: 10.1038/gt.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene therapy. 2000;7(1):35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- 37.Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. 2007;30(7):684–93. doi: 10.1097/CJI.0b013e3180de5d90. [DOI] [PubMed] [Google Scholar]
- 38.Corse E, Gottschalk RA, Krogsgaard M, Allison JP. Attenuated T cell responses to a high-potency ligand in vivo. PLoS biology. 2010;8(9) doi: 10.1371/journal.pbio.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer research. 2008;68(8):2993–3000. doi: 10.1158/0008-5472.CAN-07-5008. [DOI] [PubMed] [Google Scholar]
- 40.Lim DG, Hollsberg P, Hafler DA. Strength of prior stimuli determines the magnitude of secondary responsiveness in CD8+ T cells. Cellular immunology. 2002;217(1–2):36–46. doi: 10.1016/s0008-8749(02)00511-7. [DOI] [PubMed] [Google Scholar]
- 41.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21(11):2087–101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Human gene therapy. 2013;24(8):717–27. doi: 10.1089/hum.2013.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, et al. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer immunology research. 2014;2(11):1059–70. doi: 10.1158/2326-6066.CIR-14-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific Chimeric Antigen Receptor mRNA-Engineered T cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer immunology research. 2014;2(2):112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine. 2011;365(18):1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. Journal of clinical immunology. 2012;32(5):1059–70. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 47.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused anti-tumor activity with reduced potential for toxicity. Cancer immunology research. 2013;1(1) doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nature biotechnology. 2013;31(1):71–5. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Science translational medicine. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. European journal of cancer. 2001;37 (Suppl 4):S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.