

Abstract
Metabolic engineering of polyketide synthase (PKS) pathways represents a promising approach to natural products discovery. The dehydratase (DH) domains of PKSs, which generate an α,β-unsaturated bond through a dehydration reaction, have been poorly studied compared to other domains, likely due to the simple nature of the chemical reaction they catalyze and lack of a convenient assay to measure substrate turnover. Herein we report the first steady-state kinetic analysis of a PKS DH domain employing LC-MS/MS analysis for product quantitation. PikDH2 was selected as a model DH domain. Its substrate specificity and mechanism was interrogated with a systematic series of synthetic triketide substrates containing a nonhydrolyzable thioether linkage as well as by site-directed mutagenesis, evaluation of the pH dependence of catalytic efficiency (Vmax/KM), and through kinetic characterization of a mechanism-based inhibitor. These studies revealed PikDH2 converts d-alcohol substrates to trans-olefin products. The reaction was reversible with equilibrium constants ranging from 1.2–2. Moreover, the enzyme activity was robust and PikDH2 was used on a preparative scale for the chemoenzymatic synthesis of unsaturated triketide products. PikDH2 was shown to possess remarkably strict substrate specificity and was unable to turnover substrates epimeric at the β, γ or δ-positions. We also demonstrated PikDH2 has a key ionizable group with a pKa of 7.0 and can be irreversibly inactivated through covalent modification by a mechanism-based inhibitor, which provides a foundation for future structural studies to elucidate substrate–protein interactions.
Polyketides derived from modular type I polyketide synthases (PKSs) have drawn enormous attention and interest from chemists for decades due to their intricate structures, stereochemical complexity and diverse pharmacological activities. Sharing the same evolutionary history with fatty acid synthases (FASs),1 modular type I PKSs employ similar assembly-line molecular machinery wherein the chain intermediates remain covalently attached to acyl carrier protein (ACP) domains during biosynthesis. Unlike FASs, the presence of the three processing domains in PKSs: ketoreductase (KR), dehydratase (DH) and enoylreductase (ER), are varied in each module, leading to a fully reduced, partially reduced or unreduced segment on the polyketide chain. Owing to its assembly-line attribute and varying combination of processing domains, PKSs have been exploited through combinatorial biosynthesis and metabolic engineering to provide large libraries of polyketide analogs.2
Many polyketides such as the archetypical macrolide antibiotic pikromycin, the antifungal polyene amphotericin B, the linear polyketide discodermolide, and the mixed nonribosomal peptide-polyketide curacin, contain one or more double bonds that serve as conformational constraints and are essential for biological activity (Figure 1).3 The double bonds are formed by DH domains through abstraction of the α-proton and concomitant protonation of the β-hydroxyl group of the nascent β-hydroxyacyl-ACP polyketide intermediate, resulting in loss of one water molecule. As observed in FAS DHs, the characteristic double hotdog fold is also found in PKS DHs to form the active site with two catalytic residues, aspartic acid and histidine.4 The olefin geometry of DH products cannot be predicted through a signature fingerprint as in the KRs5 since the active site residues appear quite similar in both cis- and trans-olefin generating DHs.4d,6 Initial evidence suggested the geometry of a double bond was exclusively dependent on the stereochemistry of the DH substrate, provided by the upstream KR module wherein A-type KR products (l-alcohols) lead to cis-double bonds while trans-olefins arise from B-type KR products (d-alcohols);7 although exceptions have recently been observed.4e,8 Post-PKS tailoring enzymes such as enoyl reductases or isomerases can further obscure the original olefin geometry.4e,8,9
Figure 1.
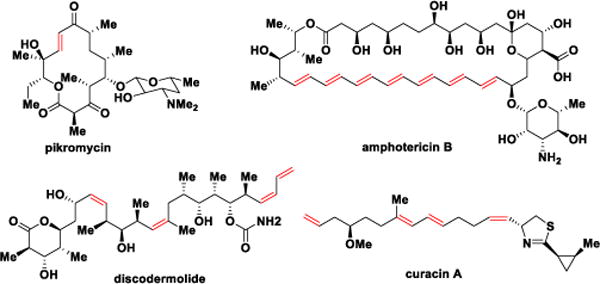
Structures of pikromycin, amphotericin B, discodermolide and curacin A.
Steady-state kinetic analysis using simple diffusible N-acetylcysteamine (NAC) thioester precursors, successfully employed to interrogate the substrate specificity of other PKS domains and modules,10 has thus far been ineffective for studying isolated DH domains. This is likely due to the extremely low activity of the excised DH domains, which necessitates overnight incubations to generate sufficient product for detection. Moreover, the simple nature and ready reversibility of the dehydration reaction cannot be easily monitored by conventional radio-TLC or spectrophotometric assays. Cane and co-workers have also shown, at least in one example, that a NAC thioester substrate was not properly delivered to a DH active site resulting in reversal of diastere-ospecificity.4e This result highlights the potential importance of the ACP for chaperoning the polyketide intermediate for proper recognition and processing by DHs. Despite these aforementioned challenges, significant progress has been made in PKS functional characterization using NAC and ACP-bound substrates, which has provided useful insight into the substrate specificity and compelling evidence that (E)-unsaturated polyketide intermediates involve a stereospecific syn-elimination.4e,8,11 To further advance our understanding of DH domains, we report herein the first detailed steady-state kinetic characterization of an individual DH domain through monitoring reaction progress by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The substrate specificity was studied with a systematic series of synthetic triketide analogs to probe the impact of both vicinal and distal stereochemistry and through mutagenesis of active-site residues. The ability to monitor reaction progress also permitted the study of pH dependence on catalytic activity and kinetic characterization of a mechanism-based irreversible inhibitor designed to facilitate co-crystallization and elucidation of substrate-protein interactions.
The pikromycin biosynthetic pathway is one of the most well-explored type I PKSs and has served as a model system for fundamental investigations of PKS catalysis.10d–f,12 We selected the DH domain from pikromycin PKS module 2 (PikDH2) for our studies since it is responsible for the trans-olefin in pikromycin (Figure 1) and Cane et al. revealed that the cryptic β-hydroxy stereochemistry of the triketide substrate possesses the d-configuration.10f,13 We predicted a larger, multi-domain portion of the PKS would create a more native context for in vitro analysis.13 The region of pikAI encoding the PikKR2-DH2 didomain comprising residues 3579–4365 was cloned into a pMCSG7 expression vector and the resulting protein was overexpressed and purified using standard methods (Supporting Information). Substrate mimics 1a and 2a for PikDH2 are based on its natural substrate (Figure 2).13 While maintaining the same stereochemistry on the triketide moiety, they possess two major modifications compared to the natural substrate including replacement of the ACP-phosphopantetheinyl arm with N-acetylcysteamine (NAC) and insertion of one or two methylene spacers between the carbonyl and sulfur atom to avoid undesired, intramolecular lactonization while not significantly perturbing the pKa of the α-protons.14 These two modifications were tolerated by PikKR2, instilling confidence the downstream DH would accept these substrates.13 Overnight incubation of substrates 1a and 2a with PikKR2-DH2 didomain in relatively large scale generated the corresponding enzymatic products 3 and 4, respectively, in slightly more than 50 isolated yield (Scheme S4). The structures of 3 and 4 were unambiguously confirmed by NMR spectroscopy and exhibited diagnostic 13C chemical shifts at 150 and 130 ppm and a 3JHH coupling of 16 Hz for the vinyl protons. These results are consistent with the empirical rule that d-alcohols provide trans-olefins.
Figure 2.
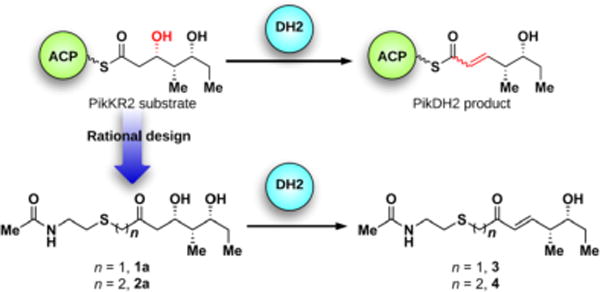
Rational design of PikDH2 substrate mimics 1a, 2a and their enzymatic products 3, 4.
Additionally, we investigated the reason for incomplete reaction. Since most FAS DHs conduct the reverse hydration reactions, enones 3 and 4 were incubated with PikKR2-DH2 to test the reversibility of the enzymatic reaction. We observed PikKR2-DH2 stereoselectively converted 3 and 4 exclusively to their hydrated products 1a and 2a, respectively, whose identities were confirmed by LC-MS/MS with authentic synthetic standards (Figure S2–S3). When equilibrium was reached, the ratio of 1a to 3 was 1:1.2 and the ratio of 2a to 4 was 1:2, slightly favoring the dehydration products in both cases. In the biosynthetic pathway, this equilibrium is pushed towards dehydration, driven by downstream module activities.
To interrogate the stereospecificity of PikDH2, we altered the β-stereocenter of substrates 1a and 2a as well as every stereogenic center in 2a through chemical synthesis (Scheme S1–S3) resulting in substrate stereoisomers 1b and 2b–e (Table 1).13 As LC-MS/MS showed high sensitivity and selectivity for detection of KR products,13 we continued using this technique to examine the formation of DH products by their unique fragmentation patterns and/or retention times. Initial velocity was linear up to 40 min with 10 μM PikKR2-DH2 didomain (Figure S4), thus we chose an end-point quench after 15 min incubation for our kinetic studies. The kinetic parameters of all substrates were obtained by fitting the saturation curves to the Michaelis-Menten equation (Table 1). Substrates 1a and 2a, maintaining the native stereochemistry, were accepted and processed rapidly by the enzyme. The KM (6.9 ± 1.7 mM and 5.7 ± 1.4 mM) and kcat values (0.67 ± 0.11 min−1 and 1.28 ± 0.19 min−1) were comparable, suggesting the methylene spacer does not adversly impact active site binding and catalysis. Substrate 1b and 2b, epimeric at the β-position were not processed. This strict substrate specificity has been similarly observed with other DHs.4e,8a,11b To inspect the sensitivity of PikDH2 to changes in distal stereochemistry, we next investigated substrates 2c and 2d, epimeric at the γ- and δ-position as well as 2e wherein both the γ- and δ-positions are inverted. Enzymatic products of 2c–e were not detected by LC-MS/MS, indicating an unprecedented degree of discrimination of these distal stereocenters. Moreover, racemic diketide 5, similar to widely used diketide substrates to study other DHs,4e,8a was unexpectedly not converted to the corresponding dehydration product.
Table 1.
Structures of substrate analogs 1a–b, 2a–e, 5 and their steady state kinetic parameters.
| No. | Substrate Structure | KM (mM) | kcat (min−1) | kcat/KM (min−1M−1) |
|---|---|---|---|---|
| 1a |
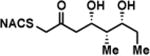
|
6.9 ± 1.7 | 0.67 ± 0.11 | 98 ± 39 |
| 1b |
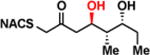
|
> 8 | <0.01 | < 1 |
| 2a |
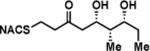
|
5.7 ± 1.4 | 1.28 ± 0.19 | 225 ± 88 |
| 2b |
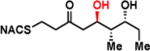
|
> 8 | <0.01 | < 1 |
| 2c |
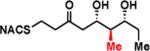
|
> 8 | <0.01 | < 1 |
| 2d |
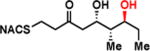
|
> 8 | <0.01 | < 1 |
| 2e |
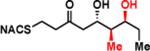
|
> 8 | <0.01 | < 1 |
| (±)-5 |
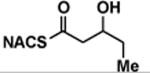
|
> 8 | <0.01 | < 1 |
To study the chemical mechanism of PikDH2, the pH dependence of catalytic efficiency (Vmax/KM) with substrate 2a was obtained from pH 6.6 to 9.0. The hyperbolic curve implicates at least one key ionizable group responsible for binding and catalysis (from either the free enzyme or the free substrate)15 with a pKa value of 7.0 ± 0.1 (Figure 3A). The protonation of this ionizable group at low pH values abolished the enzyme activity. The pKa values for the two hydroxyl groups of substrate 2a are expected to be ~14–16. Thus we expect the observed ionizable group is a general base in the enzyme, presumably the conserved histidine residue (His3611), which abstracts the α-proton. An aspartic acid residue protonates the β-hydroxyl group to facilitate the loss of a water molecule (Figure 3B).4 The slightly perturbed pKa of 7.0 for the catalytic histidine, compared to pKa of 6.0 for the free amino acid, indicates the influence of adjacent residues to form a slightly negatively charged cavity.16
Figure 3.
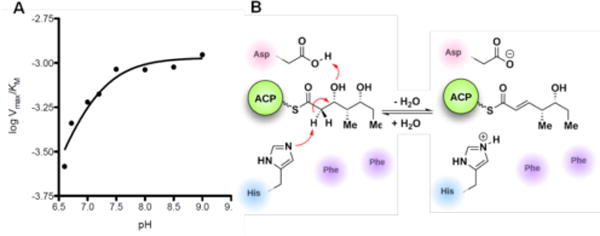
A) pH dependence of logVmax/KM. B) Mechanism and key residues involved in the dehydration reaction.
In order to understand the basis for the remarkable selectivity of PikDH2 we next aligned the amino acid sequence of PikDH2 with other DHs of known 3D structure.4c–e Two catalytic residues, the histidine (His3611) in an HXXGXXXP motif and the aspartic acid (Asp3800) in an HPALLD motif, are also found in PikDH2. However, the tyrosine in a YGP motif (observed in most DHs), whose role was proposed to assist the protonation of the β-hydroxyl group,6 is absent in this DH. Instead, a phenylalanine (Phe3746, FGP motif) is located only 3.8 Å from the catalytic histidine in a PikDH2 homology model (Figure S7), suggesting that this residue could impact substrate binding at the active site. The F3746L point mutant was generated and tested with substrate surrogate 2a to verify this possibility. The specificity constant dropped to 1/40 of the wild type, indicating the important role this phenylalanine plays for substrate binding (Table 2). Another phenylalanine, residue 3750, conserved in most DHs, is also near the catalytic residues (3.2 Å from the aspartic acid) in the homology model. Mutants F3750Y and F3750L had similar KM values to the wild type, but 15–30-fold decreased kcat values, demonstrating that change of Phe3750 hampers the catalytic activity of PikDH2. We hypothesize that the two phenylalanine residues (F3746 and F3750) may shape a hydrophobic pocket at the active site to position the substrate as well as maintain the catalytically active state.
Table 2.
Kinetic parameters of PikDH2 mutants.
| Mutants | KM (mM) | kcat (min−1) | kcat/KM (min−1M−1) |
|---|---|---|---|
| Wild | 5.7 ± 1.4 | 1.28 ± 0.19 | 225 ± 88 |
| F3746L | > 40 | N/A | 5.2 ± 0.3 |
| F3750Y | 4.1 ± 1.4 | 0.086 ± 0.013 | 21 ± 9 |
| F3750L | 7.1 ± 2.6 | 0.046 ± 0.011 | 6.5 ± 3.9 |
Co-crystallography of PKSs is particularly challenging since substrate affinity is often low due to the intramolecular nature of substrate presentation. With the hope of aiding structural studies and potentially allowing visualization of protein interfaces between ACP and DH domains,17 we set out to screen for a suitable PikDH2 suicide substrate that would covalently react within the enzyme active site. 3-Decynoyl-N-acetylcysteamine (6) developed by Bloch was the first described mechanism-based inhibitor for any enzyme, which coincidentally inactivates FAS DHs.18 Burkart and co-workers recently developed second-generation DH probes with improved chemical stability and demonstrated these could inactivate functionally and structurally related PKS DHs.17 Therefore, we evaluated the ability of 6 to irreversibly inhibit PikDH2 using our discontinuous assay monitoring the dehydration of substrate 2a by LC-MS/MS. Compound 6 was shown to exhibit time-dependent inhibition and the kinetic constants for inactivation were determined using the Kitz–Wilson method.19 The kinetic parameters KI and kinact were 156 ± 34 μM and 0.36 ± 0.06 min−1 (Figure 4B and 4C). Enzyme activity could not be recovered by dialysis and substrate protected PikDH2 from inactivation. Consequently, we expect 6 covalently labels PikDH2 in analogy to FAS DHs through abstraction of an α-proton by the catalytic histidine residue of the enzyme to form a reactive allene via propargylic rearrangement, which subsequently undergoes nucleophilic attack by the catalytic histidine (Figure 4A).20
Figure 4.
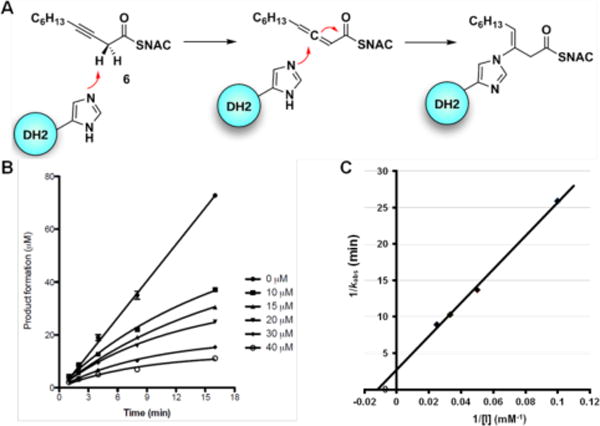
A) Mechanism of DH domain inactivation by 3-decynoyl-N-acetylcysteamine (6). B) Time course of the inactivation of PikDH2 with 10 to 40 μM 6. Symbols represent the mean ± S.D. from duplicate experiments. C) Kitz and Wilson plot19 of the inhibition data (1/kobs vs. 1/[I]).
In summary, we have performed the first steady-state kinetic analysis of a PKS DH domain by using LC-MS/MS analysis for product quantitation. PikDH2 was found to process only d-alcohols to furnish trans-olefin products. The enzyme showed an unforeseen extremely high stereospecificity at the β-, γ- and δ-positions of the substrate, potentially revealing a challenge for future metabolic engineering efforts. The pH dependence of kcat/KM identified a general base with a pKa of 7.0 critical for substrate binding and catalysis, which we assign to histidine (His3611). Mutagenesis and kinetic studies uncovered a critical hydrophobic region shaped by two phenylalanine residues that is essential for substrate binding as well as catalytic activity and suggest the tyrosine in a YGP motif (observed in most DHs) implicated in DH catalysis is not required for activity. In the search for a co-crystallography candidate for PikDH2, we demonstrated that an alkyne-based probe irreversibly modified this non-iterative type I PKS DH. These studies provide a foundation for future efforts aimed to uncover the protein-substrate interactions that govern binding and catalysis. Our findings further reinforce the utility of diffusible NAC substrate mimics containing a ketone isostere in place of the labile thioester for studying intrinsically unstable PKS chain intermediates.
Supplementary Material
Acknowledgments
The authors thank B. Witthuhn (Center for Mass Spectrometry and Proteomics, University of Minnesota) for assistance with LC-MS/MS. Financial support from NIH GM081544 (to JLS), and the Department of Medicinal Chemistry (University of Minnesota) (to YL and WDF) is gratefully acknowledged.
Footnotes
Supporting Information
Experimental details and additional data. This material is available free of charge via the Internet at http:/pubs.acs.org.
The authors declare no competing financial interests.
References
- 1.(a) Smith S, Tsai S-C. Nat Prod Rep. 2007;24:1041. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smith JL, Sherman DH. Science. 2008;321:1304. doi: 10.1126/science.1163785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Proc Natl Acad Sci USA. 1999;96:1846. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tang L, Shah S, Chung L, Camey J, Katz L, Khosla C, Julien B. Science. 2000;287:640. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]; (c) Werneburg M, Busch B, He J, Richter ME, Xiang L, Moore BS, Roth M, Dahse HM, Hertweck C. J Am Chem Soc. 2010;132:10407. doi: 10.1021/ja102751h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol Pharmacol. 1998;53:62. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]; (b) Martello LA, LaMarche MJ, He L, Beauchamp TJ, Smith AB, III, Horwitz SB. Chem biol. 2001;8:843. doi: 10.1016/s1074-5521(01)00055-2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure. 1996;4:253. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]; (b) Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, Fiebig KM, Schmid M, Rock CO. J Biol Chem. 2004;279:52593. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]; (c) Keatinge-Clay A. J Mol Biol. 2008;384:941. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Akey DL, Razelun JR, Tehranisa J, Sherman DH, Gerwick WH, Smith JL. Structure. 2010;18:94. doi: 10.1016/j.str.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gay D, You YO, Keatinge-Clay A, Cane DE. Biochemistry. 2013;52:8916. doi: 10.1021/bi400988t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caffrey P. ChemBioChem. 2003;4:654. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 6.Keatinge-Clay AT. Nat Prod Rep. 2012;29:1050. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 7.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 8.(a) Vergnolle O, Hahn F, Baerga-Ortiz A, Leadlay PF, Andexer JN. ChemBioChem. 2011;12:1011. doi: 10.1002/cbic.201100011. [DOI] [PubMed] [Google Scholar]; (b) Kandziora N, Andexer JN, Moss SJ, Wilkinson B, Leadlay PF, Hahn F. Chem Sci. 2014;5:3563. [Google Scholar]
- 9.Kusebauch B, Busch B, Scherlach K, Roth M, Hertweck C. Angew Chem Int Ed. 2010;49:1460. doi: 10.1002/anie.200905467. [DOI] [PubMed] [Google Scholar]
- 10.(a) Holzbaur IE, Harris RC, Bycroft M, Cortes J, Bisang C, Staunton J, Rudd BA, Leadlay PF. Chem Biol. 1999;6:189. doi: 10.1016/S1074-5521(99)80035-0. [DOI] [PubMed] [Google Scholar]; (b) Wu N, Kudo F, Cane DE, Khosla C. J Am Chem Soc. 2000;122:4847. [Google Scholar]; (c) Cane DE, Kudo F, Kinoshita K, Khosla C. Chem Biol. 2002;9:131. doi: 10.1016/s1074-5521(02)00089-3. [DOI] [PubMed] [Google Scholar]; (d) Lu H, Tsai S-C, Khosla C, Cane DE. Biochemistry. 2002;41:12590. doi: 10.1021/bi026006d. [DOI] [PubMed] [Google Scholar]; (e) Yin Y, Lu H, Khosla C, Cane DE. J Am Chem Soc. 2003;125:5671. doi: 10.1021/ja034574q. [DOI] [PubMed] [Google Scholar]; (f) Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Guo X, Liu T, Valenzano CR, Deng Z, Cane DE. J Am Chem Soc. 2010;132:14694. doi: 10.1021/ja1073432. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Valenzano CR, You YO, Garg A, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2010;132:14697. doi: 10.1021/ja107344h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Berkhan G, Hahn F. Angew Chem Int Ed. 2014;53:14240. doi: 10.1002/anie.201407979. [DOI] [PubMed] [Google Scholar]
- 12.(a) Xue Y, Zhao Li, Liu H-W, Sherman DH. Proc Natl Acad Sci USA. 1998;95:12111. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beck BJ, Aldrich CC, Fecik RA, Reynolds KA, Sherman DA. J Am Chem Soc. 2003;125:12551. doi: 10.1021/ja034841s. [DOI] [PubMed] [Google Scholar]; (c) Kittendorf JD, Sherman DH. Bioorg Med Chem. 2009;17:2137. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Håkansson K, Sherman DH, Smith JL, Skiniotis G. Nature. 2014;510:512. doi: 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Fiers WD, Bernard SM, Smith JL, Aldrich CC, Fecik RA. ACS Chem Biol. 2014;9:2914. doi: 10.1021/cb5006883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The pKa of the α-protons of thioesters and ketones are approximately the same due to the poor orbital overlap between the sulfur 3d lone-pairs and the carbonyl π* orbital. For example, ethyl thioacetate has a pKa of 21, whereas ethyl acetate is 26.5 and acetone is 20, see:; Aymes TL, Richard JP. J Am Chem Soc. 1992;114:10297. [Google Scholar]
- 15.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W. H. Freeman; 1999. p. 175. [Google Scholar]
- 16.Purich DL, Allison RD. Handbook of Biochemical Kinetics: A Guide to Dynamic Processes in the Molecular Life Sciences. Elsevier Science; 1999. p. 405. [Google Scholar]
- 17.(a) Ishikawa F, Haushalter RW, Burkart MD. J Am Chem Soc. 2012;134:769. doi: 10.1021/ja2082334. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ishikawa F, Haushalter RW, Lee DJ, Finzel K, Burkart MD. J Am Chem Soc. 2013;135:8846. doi: 10.1021/ja4042059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Brock DJ, Kass LR, Bloch K. J Biol Chem. 1967;242:4432. [PubMed] [Google Scholar]; (b) Kass LR, Bloch K. Proc Natl Acad Sci USA. 1967;58:1168. doi: 10.1073/pnas.58.3.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245. [PubMed] [Google Scholar]
- 20.(a) Endo K, Helmkamp GM, Jr, Bloch K. J Biol Chem. 1970;245:4293. [PubMed] [Google Scholar]; (b) Schwab JM, Li WB, Ho CK, Townsend CA, Salituro GM. J Am Chem Soc. 1984;106:7293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.