

Abstract
Inhibition of nucleoside metabolism is an important principle in cancer therapy as evidenced by the role of fluoropyrimidines, such as 5-fluorouracil (5-FU), and antifolates in the treatment of many cancers. TAS-102 is an oral combination therapy consisting of trifluridine (FTD), a thymidine-based nucleoside analog, plus tipiracil hydrochloride (TPI), a novel thymidine phosphorylase inhibitor that improves the bioavailability of FTD. TAS-102 has demonstrated efficacy in 5-FU-refractory patients based on a different mechanism of action and has been approved for the treatment of metastatic colorectal cancer in Japan. This review describes the mechanism of action of TAS-102, highlighting key differences between TAS-102 and 5-FU-based therapies. While both FTD and 5-FU inhibit thymidylate synthase (TS), a central enzyme in DNA synthesis, sufficient TS inhibition by FTD requires continuous infusion; therefore, it is not considered a clinically relevant mechanism with oral dosing. Instead, the primary cytotoxic mechanism with twice-daily oral dosing, the schedule used in TAS-102 clinical development, is DNA incorporation. FTD incorporation into DNA induces DNA dysfunction, including DNA strand breaks. Uracil-based analogs such as 5-FU may also be incorporated into DNA; however, they are immediately cleaved off by uracil-DNA glycosylases, reducing their ability to damage DNA. Moreover, the TPI component may enhance the durability of response to FTD. With its distinct mechanism of action and metabolism, TAS-102 is a promising treatment option for patients resistant to or intolerant of 5-FU-based fluoropyrimidines.
Keywords: 5-fluorouracil, fluoropyrimidines, mechanism of action, metastatic colorectal cancer, TAS-102, thymidylate synthase
Introduction
Inhibition of nucleoside metabolism is an important concept in cancer therapy. Fluoropyrimidines, such as 5-fluorouracil (5-FU) and its derivatives, are uracil-based nucleic acid analogs that inhibit thymidylate synthase (TS), which is a key enzyme in DNA synthesis, and are also incorporated into nucleic acids, causing RNA damage [1,2]. Antifolates, such as raltitrexed and pemetrexed, are another class of antimetabolites that act by inhibiting the TS pathway [2-4]. Agents that target nucleoside metabolism have been pivotal to the treatment of cancer for decades and are still the basis of chemotherapeutic treatment in multiple neoplasms, such as 5-FU for colon and breast cancer and pemetrexed for lung cancer, and a number of new antimetabolites are currently in development for clinical use [2,5].
5-FU and its derivatives are commonly used in the treatment of metastatic colorectal cancer (mCRC) as well as other cancers, including breast cancer [2,5]. However, additional agents are needed due to the development of secondary resistance [5]. TAS-102 is an oral combination drug consisting of trifluridine (FTD), which is a thymidine-based nucleoside analog, and tipiracil hydrochloride (TPI), which improves the bioavailability of FTD by inhibiting its catabolism by thymidine phosphorylase (TP) [6]. TAS-102 has been approved for the treatment of mCRC in Japan, and recently demonstrated positive results in overall and progression-free survival with a favorable safety profile in the global phase 3 RECOURSE trial, which was conducted in patients with mCRC refractory or intolerant to standard therapies [7]. This review describes and discusses the mechanism of action of TAS-102, particularly noting how this new drug demonstrates efficacy in patients with 5-FU-refractory cancers.
Targeting nucleoside metabolism
Thymidylate synthase pathway and DNA synthesis
TS plays a central role in the synthesis of DNA. The importance of the TS pathway in cancer is underscored by the overexpression of TS in many different human malignancies, including breast and colorectal cancers, and the association between TS overexpression and poor prognosis [8]. The TS enzyme catalyzes the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) [2,9]. The conversion of dUMP to dTMP depends on 5,10-methylenetetrahydrofolate (5,10-CH2-THF), which acts as a methyl-group donor for the reaction [2,9]. dTMP is subsequently phosphorylated to form deoxythymidine diphosphate and, ultimately, deoxythymidine triphosphate (dTTP) for incorporation into DNA [2]. The reaction catalyzed by TS is essential to the synthesis of DNA, as it is the only source for the production of dTMP in the cell [2,8,9].
Mechanism of action of 5-FU and 5-FU derivatives
The anticancer activity of 5-FU requires intracellular conversion of 5-FU to the active metabolites fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP) [2,5] (Figure 1; Table 1). FdUMP is a tight-binding inhibitor of TS, and TS inhibition by FdUMP requires the formation of an irreversible ternary complex with TS and the methyl-group donor 5,10-CH2-THF [2,5,10,11]. The downstream effects of TS inhibition include the depletion of dTTP and thymine-less cell death [2,5,12]. This may be accompanied by the accumulation of uracil nucleotides, including dUTP [13]. Under normal conditions, the enzyme deoxyuridine pyrophosphatase (dUTPase) catalyzes the hydrolysis of dUTP, forming dUMP and preventing the incorporation of dUTP into DNA [14,15]. The level of dUTPase expression is inversely related to dUTP accumulation [16]. The increased intracellular levels of dUTP as well as FdUTP following 5-FU treatment can exceed the ability of dUTPase to hydrolyze these nucleotides, leading to their misincorporation into DNA [14]. Misincorporated FdUTP or dUTP is rapidly excised from DNA by uracil-DNA-glycosylase enzymes [2,5]. However, without dTTP available for incorporation into DNA for DNA repair, the excision of uracil nucleotides is futile [2,5]. As a result, DNA strand breaks occur, leading to cell death [5,13].
Figure 1.
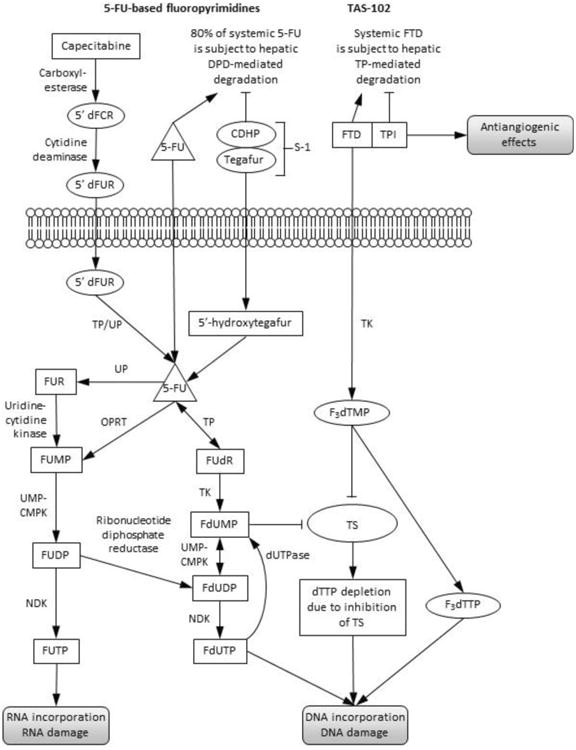
Mechanism of action of TAS-102: comparison with 5-FU-based fluoropyrimidines. Figure adapted from Wilson PM et al. Nat Rev Clin Oncol. 2014;11(5):282-298. Permission pending.
5′dFCR: 5′-deoxy-5-fluorocytidine; 5′dFUR: 5′-deoxy-5-fluorouridine; 5-FU: 5-fluorouracil; CDHP: 5-chloro-2,4-dihydroxypyridine; DPD: dihydropyrimidine dehydrogenase; dTTP: thymidine triphosphate; F3dTMP: trifluoromethyl deoxyuridine 5′-monophosphate; F3dTTP: trifluoromethyl deoxyuridine 5′-triphosphate; FdUDP: fluorodeoxyuridine diphosphate; FdUMP: fluorodeoxyuridine monophosphate; FdUTP: fluorodeoxyuridine triphosphate; dUTPase: deoxyuridine pyrophosphatase; FTD: α,α,α-trifluorothymidine (trifluridine); FUDP: fluorouridine diphosphate; FUMP: fluorouridine monophosphate; FUR: fluorouridine; FUTP: fluorouridine phosphate; NDK: nucleoside diphosphate kinase; OPRT: orotate phosphoribosyltransferase; TK: thymidine kinase; TP: thymidine phosphorylase; TPI: tipiracil hydrochloride; TS: thymidylate synthase; UMP-CMPK: uridine monophosphate-cytidine monophosphate kinase; UP: uridine phosphorylase.
Table 1.
Overview of TAS-102 and 5-FU-based chemotherapy agents. [2]
| Agent | Route of Administration | Active Metabolites and Their Functions |
|---|---|---|
| 5-FU-based agents | ||
|
| ||
| 5-FU | IV | FdUMP: Irreversible inhibitor of TS FUTP: Incorporated into RNA FdUTP: Incorporated into DNA |
| Capecitabine | Oral | |
| Tegafur-uracil | Oral | |
| S-1 | Oral | |
|
| ||
| FTD-based agents | ||
|
| ||
| TAS-102 (FTD + TPI) | Oral | F3dTMP: Reversible inhibitor of TS F3dTTP: Incorporated into DNA TPI: TP inhibition |
5-FU: 5-fluorouracil; F3dTMP: trifluoromethyl deoxyuridine 5′-monophosphate; F3dTTP: trifluoromethyl deoxyuridine 5′-triphosphate; FdUMP: fluorodeoxyuridine monophosphate; FdUTP: fluorodeoxyuridine triphosphate; FTD: α,α,α-trifluorothymidine (trifluridine); FUTP: fluorouridine triphosphate; IV: intravenous; TP: thymidine phosphorylase; TPI: tipiracil hydrochloride; TS: thymidylate synthase.
Incorporation into RNA is another cytotoxic mechanism of 5-FU. The 5-FU metabolite FUTP can be incorporated into RNA, leading to RNA damage [2,5,17,18]. RNA incorporation has been correlated with 5-FU cytotoxicity in breast cancer and CRC cell lines [17,18]. It appears that continuous infusion (CI) of 5-FU is associated with greater TS inhibition, while bolus administration is associated with greater RNA incorporation [19]. Based on data showing a more favorable efficacy and toxicity profile with CI 5-FU vs bolus 5-FU, CI 5-FU is the preferred regimen today [2,20]. In a meta-analysis of randomized trials of CI 5-FU and bolus 5-FU, overall survival was significantly longer with CI 5-FU (hazard ratio [HR]=0.88, P=0.04), and the rate of grade 3/4 hematologic toxicity was significantly higher with bolus 5-FU (31% vs 4%, P<0.0001) [20].
As 5-FU is an intravenous drug, a number of oral fluoropyrimidine prodrugs have also been developed, including capecitabine, tegafur-uracil, and S-1; all of these are converted to 5-FU in vivo [2]. Capecitabine is converted to 5-FU by three enzymes preferentially localized in tumor tissue: Carboxylesterase, cytidine deaminase, and TP [21].
5-FU and capecitabine are effective components of anticancer treatment regimens, particularly for mCRC [2,5]. However, the safety and tolerability of these agents are important considerations. It has been observed that there are ethnic and regional differences in the tolerability of 5-FU and capecitabine. In particular, US patients in phase 3 mCRC trials had a significantly higher relative risk of grade 3/4 adverse events (AEs), grade 3/4 gastrointestinal (GI) toxicity, and treatment discontinuations with 5-FU or capecitabine compared with patients from other countries; similar results were seen in an adjuvant colon cancer trial, with East Asian patients having the lowest risk [22]. The probability of grade 3/4 GI toxicity (diarrhea, nausea, vomiting, and stomatitis) for US patients was more than 3.5 times than that of East Asian patients in the adjuvant trial [22]. Another factor related to tolerability of 5-FU and/or capecitabine is the enzyme dihydropyrimidine dehydrogenase (DPD), which catalyzes the initial, rate-limiting step in the catabolism of 5-FU: The conversion of 5-FU to dihydrofluorouracil [5,23,24]. Patients with a DPD deficiency experience severe 5-FU-related AEs, including cytopenia and GI toxicity [25].
Tegafur-uracil and S-1 are oral combination drugs that contain N1-(2′-tetrahydrofuryl)-5-fluorouracil (tegafur), which is an oral 5-FU prodrug, plus an inhibitor of DPD, which increases the bioavailability of 5-FU; S-1 also contains a third agent to decrease GI toxicity. Tegafur-uracil contains tegafur plus uracil, which is an inhibitor of DPD, in a 1:4 molar ratio [26]. Coadministration of uracil slows the breakdown of 5-FU and results in increased tumor levels of 5-FU [26]. S-1 contains tegafur plus 5-chloro-2,4-dihydroxypyridine (CDHP; gimeracil) and oteracil potassium (potassium oxonate) in a molar ratio of 1:0.4:1 [27]. CDHP inhibits the degradation of 5-FU by inhibiting DPD, and oteracil potassium inhibits the enzyme orotate phosphoribosyltransferase (OPRT), which is responsible for the conversion of 5-FU to 5-fluorouridine 5′-monophosphate [27]. Because oteracil potassium is preferentially distributed to the GI tract, it can reduce the GI toxicity associated with 5-FU [27]. Still, ethnic differences in GI toxicity have been observed with S-1, with higher rates observed in Caucasian compared with East Asian patients [28], and the maximal tolerated dose established for Japanese patients is higher than that for Caucasian patients: Up to 80 mg/m2/day vs 60 mg/m2/day [29]. Tegafururacil is not approved by the US Food and Drug Administration but it is approved for CRC and other solid tumors in more than 50 countries. S-1 is an effective agent for the treatment of gastric cancer in both the adjuvant and metastatic settings. It is approved in both of these settings in Japan and in the metastatic setting in Europe.
5-FU and FTD: Pyrimidine-based nucleoside analogs
5-FU and FTD were first synthesized by Heidelberger et al in 1957 and 1964, respectively [30,31]. 5-FU is a uracil-based nucleic acid analog with fluorine replacing hydrogen at the C-5 position [1,5,30]. FTD is a thymidine-based nucleoside analog with a trifluoromethyl group (CF3) replacing the methyl group at the C-5 position [1,31]. Although both agents have the potential to inhibit TS and be incorporated into nucleic acids, the main clinically relevant cytotoxic mechanism of action of FTD (as a component of TAS-102) is distinct from that of 5-FU, as discussed below.
Mechanism of action of TAS-102
TAS-102 is an oral combination of an antineoplastic thymidine-based nucleoside analog (FTD, trifluridine) and a TP inhibitor (TPI, tipiracil hydrochloride) at a 1:0.5 molar ratio [6,32,33]. The elimination half-life of FTD after intravenous administration to humans is very rapid (18 minutes), due to the rapid degradation of FTD to its major metabolite, 5-trifluoromethyl-2,4(1H,3H)-pyrimidinedione [34]. In monkeys, the plasma FTD level after oral administration alone was very low, suggesting extensive first-pass metabolism by the liver and intestine TPase [32,35]. However, the addition of TPI was found to enable oral administration [35]. By inhibiting TP, TPI inhibits the degradation of FTD in the liver and intestines following oral administration and thereby improves its bioavailability [35]. The TP enzyme catalyzes the phosphorolysis of pyrimidine 2′-deoxynucleosides such as FTD [35]. TP is also involved in angiogenesis and is a resistance marker for 5-FU [36]. TPI was shown to selectively inhibit TP in human liver, small intestine, and tumor extracts and potentiated the antitumor activity of FTD in human stomach cancer xenografts [35]. TPI does not inhibit other pyrimidine-metabolizing enzymes such as uridine phosphorylase, thymidine kinase, OPRT, or DPD [35]. The optimum ratio of FTD to TPI for oral administration was determined using pharmacokinetic/pharmacodynamic studies in animal models. Studies using human CRC tumor xenografts in mice determined that the maximum antitumor activity was achieved with a 1:0.5 molar ratio, and studies in mice and monkeys showed that the maximum plasma concentration of FTD was almost achieved with the same ratio [32]. Moreover, this ratio produced a favorable balance between antitumor activity and toxicity. Lower toxicity in mice was observed with TPI coadministration than with FTD alone [32].
Phase 1 and phase 2 studies of FTD administered intravenously were initiated in the 1960s. Because the major antitumor activity was observed only with a dosing schedule (divided doses every 3 hours for 8 to 13 days) that was not feasible for long-term treatments, further development of intravenous FTD alone was discontinued [37]. This is due to the rapid elimination of FTD. TAS-102 allowed the clinically feasible administration via the oral route to maximize the plasma concentration and antitumor activity of FTD.
The mechanism of action of TAS-102 is dependent on its dosing schedule. DNA incorporation appears to be the primary anticancer mechanism associated with twice-daily oral dosing of TAS-102, which is the dosing schedule used in the clinical development of TAS-102 [38]. In tumor xenograft mouse models, the level of incorporation into DNA was higher with twice-daily oral dosing of FTD than with CI administration of FTD [38]. In contrast, TS inhibitory activity in tumor xenografts was greater with CI FTD [38]. An earlier study in FM3 A breast cancer cells also showed that inhibition of TS was lost without continuous exposure to FTD [39], which is comparable with what is observed with oral dosing.
The FTD monophosphate metabolite (trifluoromethyl deoxyuridine 5′-monophosphate [F3dTMP]) is a reversible tight-binding inhibitor that binds to the active site of TS and inhibits TS activity with a Ki of 0.38 nM [6,39-42] (Figure 1; Table 1). F3dTMP, in contrast to the 5-FU metabolite FdUMP, does not form a ternary complex with the TS enzyme and 5,10-CH2-THF [6,40,41] and inhibition of TS is rapidly reversed following the removal of FTD [38,39].
Further phosphorylation of F3dTMP results in the formation of the triphosphate metabolite, trifluoromethyl deoxyuridine 5′-triphosphate (F3dTTP), which can be incorporated into DNA [6,43,44] (Figure 1; Table 1). F3dTTP insertion into DNA is catalyzed by DNA polymerase α and occurs primarily at sites opposite adenine (A) [45]. Compared with 5-FU or FdUrd, FTD is incorporated into DNA to a considerably greater extent, with an approximately 300-fold higher concentration of FTD than 5-FU and a few hundred-fold higher concentration of FTD than FdUrd detected in DNA at 4 hours according to studies in HeLa or HCT 116 cells [46, 47]. In a study in a CRC cell line, the level of FTD retention in DNA at 72 hours following washout was comparable with that of thymidine [45]. This is consistent with earlier data from an in vitro study, in which more than 80% of the FTD incorporated into DNA was retained 24 hours after washout, indicating that FTD is efficiently incorporated into DNA [48]. Moreover, the incorporation of FTD into DNA in tumor tissues is significantly higher than its incorporation into DNA in normal tissues [45]. This may be related to the upregulation of the pyrimidine metabolic pathway observed in cancer [45]. Unlike FdUTP, F3dTTP is not a substrate for dUTPase, the enzyme that prevents the incorporation of uracil and uracil analogs into DNA through the conversion of the triphosphates to the monophosphate forms [45].
The role of the base excision repair pathway and DNA glycosylases in response to FTD-induced DNA damage is different from the response to 5-FU-induced DNA damage. The main mammalian DNA glycosylases involved in the excision of uracil and 5-FU from DNA include UNG, SMUG1, thymine DNA glycosylase (TDG), and methyl-CpG binding domain 4 (MBD4; also known as MED1) [49-55]. FTD inserted at T-sites (paired to A) is not excised by UNG, SMUG1, TDG, or MBD4 [46]. FTD paired to guanine (G) is excised by TDG and MBD4, but most of the FTD incorporated into DNA is paired with A, not G [46]. Although a deficiency in TDG or MBD4 has been linked to 5-FU resistance [53,56], knockdown of these enzymes using siRNA did not generate resistance to FTD [46].
Along with FTD, TPI may also contribute to the anticancer activity of TAS-102. By inhibiting the enzyme TP, TPI has the potential to induce antiangiogenic effects. TP is identical to platelet-derived endothelial cell growth factor and has the ability to promote tumor growth in vivo by mechanisms including endothelial cell migration [57,58]. In mouse models, TPI was shown to inhibit TP-induced angiogenesis and the development of metastases [32,59,60]. The clinical significance of the antiangiogenic effects of TPI has not been determined; additionally, at the intended dosage of TAS-102, this effect is not likely to be apparent.
TAS-102: Overcoming resistance to 5-FU-based agents
FTD and 5-FU appear to have different mechanisms for resistance. While a number of different molecular mechanisms have been shown to mediate 5-FU resistance, we will focus on the resistance mechanisms that have been investigated for TAS-102 in order to draw comparisons between the two drugs. The enzymes involved in nucleoside metabolism are an important determinant of resistance to fluoropyrimidines.
The main mechanism of 5-FU resistance in DLD-1/5-FU (CRC cells) was a significant decrease in OPRT activity, leading to a decrease in the cellular uptake of 5-FU in the RNA fraction [61]. The main mechanism of 5-FU resistance in NUGC3/5-FU (gastric cancer cells) was due to reduced OPRT and uridine phosphorylase activities, but not to a change in TS levels or a mutation in TS [62]. FTD was able to overcome resistance to 5-FU in MKN-74/5-FU and KATOIII/5-FU [63]. In these 5-FU-resistant gastric cell lines, TS expression was higher than parent line [64]. Collectively, these results indicate that TAS-102 (FTD) can overcome acquired resistance to 5-FU because the main mechanism of TAS-102 is not associated with main metabolic enzymes of 5-FU, such as TS and OPRT.
Another mechanism for resistance to 5-FU and other chemotherapeutic drugs is autophagy [65,66]. Autophagy is a cellular process that occurs in response to stress and involves the degradation of proteins and organelles in lysosomes [66]. Although autophagy can enhance the cytotoxic effects of some anticancer drugs, for 5-FU and other anticancer drugs it serves as a survival mechanism that contributes to resistance [66]. In a study in which FTD was more potent than 5-FU at inducing cytotoxicity in human CRC cell lines, autophagy was upregulated by 5-FU and inhibition of autophagy led to increased cytotoxicity induced by 5-FU but not FTD [67]. These results from in vitro studies suggest that autophagy may play a role for resistance to 5-FU but not FTD [67].
Preclinical studies of TAS-102 in combination with other anticancer agents
A number of preclinical studies have been performed to investigate the combination of TAS-102 with other drugs commonly used for the treatment of mCRC [68-70]. In studies using human CRC cell lines, FTD combined with oxaliplatin or SN-38, the active metabolite of irinotecan, demonstrated synergistic effects on growth inhibition [68,69,71]. The tumor growth-inhibitory activity of TAS-102 in combination with oxaliplatin was significantly greater than that of either agent as monotherapy for mice with HCT 116, SW-48, SC-2, MKN74 and MKN74/5FU GI cancer xenografts [72]. The tumor growth-inhibitory activity of TAS-102 in combination with irinotecan was significantly greater than that of either agent as monotherapy for mice with KM12C, KM12C/5-FU, DLD-1/5-FU, and SC-2 GI cancer xenografts [73]. Increased antitumor activity was also observed when TAS-102 was combined with the monoclonal antibodies cetuximab, panitumumab, or bevacizumab in CRC xenograft mouse models [70]. In a KRAS wild-type CRC xenograft, oral administration of TAS-102 in combination with the anti– epidermal growth factor receptor (EGFR) antibodies cetuximab or panitumumab was associated with increased antitumor effects compared with monotherapy [70]. TAS-102 in combination with the anti–vascular endothelial growth factor (VEGF) antibody bevacizumab also showed increased antitumor effects compared with monotherapy in CRC xenografts; this was not affected by either KRAS mutation status or BRAF mutation status [70]. Moreover, coadministration of TAS-102 with bevacizumab resulted in the detection of higher levels of FTD and its phosphorylated metabolites in the tumor compared with TAS-102 administered alone [70]. Together, the results of these studies suggest that TAS-102 combinations with other agents approved for mCRC treatment deserve further development and clinical investigation.
Pharmacokinetic data with TAS-102
Based on the preclinical findings above, initial dose-finding phase 1 studies employed daily dosing of TAS-102 in order to facilitate FTD incorporation into tumor cells. The results of these initial clinical studies indicated that TAS-102 was better tolerated when administered for 5 consecutive days rather than 14 consecutive days, and the dose regimen of 5 days a week with 2 days rest for 2 weeks, repeated every 4 weeks, was determined to be the optimal dose regimen [74-77]. A phase 1 dose-finding study conducted in Japanese patients with advanced solid tumors showed that TAS-102 was well tolerated at doses up to 70 mg/m2/day (35 mg/m2 twice daily) with the same schedule [78]. In the study, FTD in plasma reached Cmax at 1.2 to 1.9 hours after dose, and declined with a short half-life of 1.39 to 2.44 hours [78]. The urinary excretion of FTD and TPI were 1% to 8% and approximately 20%, respectively, of the administered dose [78]. The urinary excretion of unchanged FTD was limited; thus, the major elimination pathway of FTD is metabolism by TP. Clinical studies of TAS-102 in patients with hepatic or renal impairment are currently being conducted (ClinicalTrials.gov Identifier: NCT02301117, NCT02301104).
Efficacy and safety data with TAS-102
TAS-102 had been investigated in a number of phase 1 studies in patients with solid tumors [33,74-77]. In a phase 1 dose escalation study conducted in Japanese patients with solid tumors (N=21; n=18 CRC), TAS-102 at 30, 40, 50, 60, or 70 mg/m2 per day was given twice daily on days 1 to 5 and 8 to 12 in a 28-day cycle; median treatment duration was 68 days [76]. All patients with mCRC were refractory to 5-FU, irinotecan, and oxaliplatin; three patients with mCRC were refractory to anti-EGFR or anti-VEGF monoclonal antibodies [78]. TAS-102 demonstrated efficacy in patients with mCRC, with a 50% disease control rate, median progression-free survival of 2.4 months, and median overall survival of 9.8 months [78]. Dose-limiting toxicities included grade 4 leukopenia, neutropenia, and thrombocytopenia. A statistically significant relation was observed between the decrease in neutrophil count and FTD PK parameters, Cmax and AUC [78]. The recommended phase 2 dose, 70 mg/m2 per day, was also found to be acceptable in a phase 1 study in Western patients with refractory mCRC [78,79].
In a multicenter, double-blind, phase 2 trial of TAS-102 conducted in Japan, patients with mCRC who were resistant to or intolerant of a fluoropyrimidine, irinotecan, and oxaliplatin were randomized 2:1 to best supportive care plus either TAS-102 (35 mg/m2 twice daily on days 1 to 5 and 8 to 12 of a 28-day cycle) or placebo [33]. Among the 169 patients included in the efficacy analysis (n=112 TAS-102; n=57 placebo), the majority were refractory to fluoropyrimidine, oxaliplatin, irinotecan, bevacizumab, and cetuximab [33]. Median overall survival was 9.0 months in the TAS-102 group vs 6.6 months in the placebo group (HR=0.56, P=0.0011), and median progression-free survival was 2.0 months in the TAS-102 group vs 1.0 month in the placebo group (HR=0.41, P<0.0001) [33]. The rates of grade 3/4 neutropenia, leukopenia, anemia, and lymphopenia were ≥10% in the TAS-102 group compared with ≤5% in the placebo group; the most common grade 3/4 nonhematologic AEs in the TAS-102 group were fatigue and diarrhea (6% for each) [33]. Results from the phase 3 RECOURSE trial, a global, multicenter trial conducted in patients with mCRC with at least 2 prior lines of treatment showed improvement in the primary endpoint of overall survival compared with placebo [7].
TAS-102 is also being investigated for the treatment of gastric cancer. In a multicenter, single-arm phase 2 trial, Japanese patients with pretreated advanced gastric cancer (N=29) received TAS-102 at the same dose as in the phase 2 mCRC trial described above (35 mg/m2 twice daily on days 1 to 5 and 8 to 12 of a 28-day cycle) [80]. All patients had previously received a fluoropyrimidine and a platinum agent; the patients' treatment history also included either irinotecan or a taxane [80]. Disease control rate at 8 weeks was 67.9% and median progression-free survival was 2.9 months [80]. The most common grade 3/4 AEs were neutropenia, leukopenia, anemia, and anorexia [80]. The results of this trial indicate that TAS-102 may also be a potential treatment option for patients with advanced gastric cancer, but additional confirmative studies in this patient population are needed.
Toxicity considerations with TAS-102 compared with other 5-FU-based agents
The toxicity profile of TAS-102 differs from the known toxicity profiles of 5-FU and its derivatives. The incidence of 5-FU-associated AEs such as stomatitis, hand-foot syndrome, or cardiac toxicity is rather low with TAS-102 [7]. In addition, TAS-102, unlike 5-FU, can be administered in patients with a DPD deficiency. In contrast to 5-FU, FTD does not appear to be metabolized by DPD in humans; instead, TP is the primary enzyme involved in the catabolism of FTD [5,23,24,34]. Therefore, it may be an alternative for DPD-deficient patients with mCRC.
Conclusions
TAS-102 is a promising oral fluorothymidine agent for the treatment of mCRC. It has a unique mechanism of action compared with the 5-FU-based fluoropyrimidines currently used in anticancer treatments. The intracellular metabolic pathway for FTD, the trifluorothymidine component of TAS-102, is distinct from that of 5-FU and the oral 5-FU prodrugs S-1 and capecitabine. The second component, TPI, enhances the bioavailability of FTD by inhibiting its degradation by TP and may be beneficial in producing a more durable and sustainable response; it may also have antiangiogenic effects. TAS-102 has demonstrated preclinical and clinical activity against cancers resistant to 5-FU and its derivatives.
TAS-102 is an oral fluorothymidine agent for the treatment of cancer
TAS-102 is a combination of the thymidine analog trifluridine (FTD) and tipiracil
Tipiracil, a thymidine phosphorylase inhibitor, improves FTD bioavailability
Compared with 5-fluorouracil (5-FU), TAS-102 has a distinct mechanism of action and metabolism
TAS-102 has demonstrated efficacy in 5-FU-refractory cancers
Acknowledgments
The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this publication. All authors contributed to the research, writing, and reviewing of all drafts of this manuscript. All authors approved the final draft. Editorial support in the preparation of this publication was provided by Phase Five Communications, supported by Taiho Oncology Inc.
Heinz-Josef Lenz has reported clinical trial support from Taiho.
Footnotes
Sebastian Stintzing and Fotios Loupakis have reported no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sebastian Stintzing, Email: sebastian.stintzing@med.uni-muenchen.de.
Fotios Loupakis, Email: fotiosloupakis@gmail.com.
References
- 1.Heidelberger C. On the rational development of a new drug: the example of the fluorinated pyrimidines. Cancer Treat Rep. 1981;65(Suppl 3):3–9. [PubMed] [Google Scholar]
- 2.Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11:282–98. doi: 10.1038/nrclinonc.2014.51. [DOI] [PubMed] [Google Scholar]
- 3.Jackman AL, Taylor GA, Gibson W, Kimbell R, Brown M, Calvert AH, et al. ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of L1210 tumor cell growth in vitro and in vivo: a new agent for clinical study. Cancer Res. 1991;51:5579–86. [PubMed] [Google Scholar]
- 4.Shih C, Chen VJ, Gossett LS, Gates SB, MacKellar WC, Habeck LL, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997;57:1116–23. [PubMed] [Google Scholar]
- 5.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 6.Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007;98:779–89. doi: 10.1111/j.1349-7006.2007.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–19. doi: 10.1056/NEJMoa1414325. [DOI] [PubMed] [Google Scholar]
- 8.Rahman L, Voeller D, Rahman M, Lipkowitz S, Allegra C, Barrett JC, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell. 2004;5:341–51. doi: 10.1016/s1535-6108(04)00080-7. [DOI] [PubMed] [Google Scholar]
- 9.Hardy LW, Finer-Moore JS, Montfort WR, Jones MO, Santi DV, Stroud RM. Atomic structure of thymidylate synthase: target for rational drug design. Science. 1987;235:448–55. doi: 10.1126/science.3099389. [DOI] [PubMed] [Google Scholar]
- 10.Santi DV, McHenry CS. 5-Fluoro-2′-deoxyuridylate: covalent complex with thymidylate synthetase. Proc Natl Acad Sci U S A. 1972;69:1855–7. doi: 10.1073/pnas.69.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santi DV, McHenry CS, Sommer H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry. 1974;13:471–81. doi: 10.1021/bi00700a012. [DOI] [PubMed] [Google Scholar]
- 12.Goulian M, Bleile BM, Dickey LM, Grafstrom RH, Ingraham HA, Neynaber SA, et al. Mechanism of thymineless death. Adv Exp Med Biol. 1986;195(Pt B):89–95. doi: 10.1007/978-1-4684-1248-2_15. [DOI] [PubMed] [Google Scholar]
- 13.Curtin NJ, Harris AL, Aherne GW. Mechanism of cell death following thymidylate synthase inhibition: 2′-deoxyuridine-5′-triphosphate accumulation, DNA damage, and growth inhibition following exposure to CB3717 and dipyridamole. Cancer Res. 1991;51:2346–52. [PubMed] [Google Scholar]
- 14.Wyatt MD, Wilson DM., 3rd Participation of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci. 2009;66:788–99. doi: 10.1007/s00018-008-8557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grafstrom RH, Tseng BY, Goulian M. The incorporation of uracil into animal cell DNA in vitro. Cell. 1978;15:131–40. doi: 10.1016/0092-8674(78)90089-2. [DOI] [PubMed] [Google Scholar]
- 16.Webley SD, Welsh SJ, Jackman AL, Aherne GW. The ability to accumulate deoxyuridine triphosphate and cellular response to thymidylate synthase (TS) inhibition. Br J Cancer. 2001;85:446–52. doi: 10.1054/bjoc.2001.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kufe DW, Major PP. 5-Fluorouracil incorporation into human breast carcinoma RNA correlates with cytotoxicity. J Biol Chem. 1981;256:9802–5. [PubMed] [Google Scholar]
- 18.Glazer RI, Lloyd LS. Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol Pharmacol. 1982;21:468–73. [PubMed] [Google Scholar]
- 19.Aschele C, Sobrero A, Faderan MA, Bertino JR. Novel mechanism(s) of resistance to 5-fluorouracil in human colon cancer (HCT-8) sublines following exposure to two different clinically relevant dose schedules. Cancer Res. 1992;52:1855–64. [PubMed] [Google Scholar]
- 20.Meta-analysis Group In Cancer. Piedbois P, Rougier P, Buyse M, Pignon J, Ryan L, et al. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol. 1998;16:301–8. doi: 10.1200/JCO.1998.16.1.301. [DOI] [PubMed] [Google Scholar]
- 21.Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34:1274–81. doi: 10.1016/s0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 22.Haller DG, Cassidy J, Clarke SJ, Cunningham D, Van Cutsem E, Hoff PM, et al. Potential regional differences for the tolerability profiles of fluoropyrimidines. J Clin Oncol. 2008;26:2118–23. doi: 10.1200/JCO.2007.15.2090. [DOI] [PubMed] [Google Scholar]
- 23.Heggie GD, Sommadossi JP, Cross DS, Huster WJ, Diasio RB. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987;47:2203–6. [PubMed] [Google Scholar]
- 24.Lu ZH, Zhang R, Diasio RB. Purification and characterization of dihydropyrimidine dehydrogenase from human liver. J Biol Chem. 1992;267:17102–9. [PubMed] [Google Scholar]
- 25.Saif MW, Syrigos K, Mehra R, Mattison LK, Diasio RB. Dihydropyrimidine dehydrogenase deficiency (DPD) in GI malignancies: experience of 4-years. Pak J Med Sci. 2007;23:832–9. [PMC free article] [PubMed] [Google Scholar]
- 26.Ho DH, Pazdur R, Covington W, Brown N, Huo YY, Lassere Y, et al. Comparison of 5- fluorouracil pharmacokinetics in patients receiving continuous 5-fluorouracil infusion and oral uracil plus N1-(2′-tetrahydrofuryl)-5-fluorouracil. Clin Cancer Res. 1998;4:2085–8. [PubMed] [Google Scholar]
- 27.Shirasaka T, Nakano K, Takechi T, Satake H, Uchida J, Fujioka A, et al. Antitumor activity of 1 M tegafur-0.4 M 5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res. 1996;56:2602–6. [PubMed] [Google Scholar]
- 28.Chuah B, Goh BC, Lee SC, Soong R, Lau F, Mulay M, et al. Comparison of the pharmacokinetics and pharmacodynamics of S-1 between Caucasian and East Asian patients. Cancer Sci. 2011;102:478–83. doi: 10.1111/j.1349-7006.2010.01793.x. [DOI] [PubMed] [Google Scholar]
- 29.Hoff PM, Saad ED, Ajani JA, Lassere Y, Wenske C, Medgyesy D, et al. Phase I study with pharmacokinetics of S-1 on an oral daily schedule for 28 days in patients with solid tumors. Clin Cancer Res. 2003;9:134–42. [PubMed] [Google Scholar]
- 30.Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179:663–6. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 31.Heidelberger C, Parsons DG, Remy DC. Syntheses of 5-trifluoromethyluracil and 5- trifluoromethyl-2′-deoxyuridine. J Med Chem. 1964;7:1–5. doi: 10.1021/jm00331a001. [DOI] [PubMed] [Google Scholar]
- 32.Emura T, Suzuki N, Fujioka A, Ohshimo H, Fukushima M. Potentiation of the antitumor activity of alpha, alpha, alpha-trifluorothymidine by the co-administration of an inhibitor of thymidine phosphorylase at a suitable molar ratio in vivo. Int J Oncol. 2005;27:449–55. [PubMed] [Google Scholar]
- 33.Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13:993–1001. doi: 10.1016/S1470-2045(12)70345-5. [DOI] [PubMed] [Google Scholar]
- 34.Dexter DL, Wolberg WH, Ansfield FJ, Helson L, Heidelberger C. The clinical pharmacology of 5-trifluoromethyl-2′-deoxyuridine. Cancer Res. 1972;32:247–53. [PubMed] [Google Scholar]
- 35.Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′-deoxyribonucleosides. Biochem Pharmacol. 2000;59:1227–36. doi: 10.1016/s0006-2952(00)00253-7. [DOI] [PubMed] [Google Scholar]
- 36.Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–7. [PubMed] [Google Scholar]
- 37.Ansfield FJ, Ramirez G. Phase I and II studies of 2′-deoxy-5-(trifluoromethyl)-uridine (NSC-75520) Cancer Chemother Rep. 1971;55:205–8. [PubMed] [Google Scholar]
- 38.Tanaka N, Sakamoto K, Okabe H, Fujioka A, Yamamura K, Nakagawa F, et al. Repeated oral dosing of TAS-102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32:2319–26. doi: 10.3892/or.2014.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Temmink OH, Comijn EM, Fukushima M, Peters GJ. Intracellular thymidylate synthase inhibition by trifluorothymidine in FM3A cells. Nucleosides Nucleotides Nucleic Acids. 2004;23:1491–4. doi: 10.1081/NCN-200027707. [DOI] [PubMed] [Google Scholar]
- 40.Santi DV, Sakai TT. Thymidylate synthetase. Model studies of inhibition by 5-trifluoromethyl-2′-deoxyuridylic acid. Biochemistry. 1971;10:3598–607. doi: 10.1021/bi00795a018. [DOI] [PubMed] [Google Scholar]
- 41.Eckstein JW, Foster PG, Finer-Moore J, Wataya Y, Santi DV. Mechanism-based inhibition of thymidylate synthase by 5-(trifluoromethyl)-2′-deoxyuridine 5′-monophosphate. Biochemistry. 1994;33:15086–94. doi: 10.1021/bi00254a018. [DOI] [PubMed] [Google Scholar]
- 42.Reyes P, Heidelberger C. Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Mol Pharmacol. 1965;1:14–30. [PubMed] [Google Scholar]
- 43.Heidelberger C, Boohar J, Kampschroer B. Fluorinated pyrimidines. XXIV. In vivo metabolism of 5-trifluoromethyluracil-2-C-14 and 5-trifluoromethyl-2′-deoxyuridine-2-C-14. Cancer Res. 1965;25:377–81. [PubMed] [Google Scholar]
- 44.Fujiwara Y, Oki T, Heidelberger C. Fluorinated pyrimidines. XXXVII. Effects of 5-trifluoromethyl-2′-deoxyuridine on the synthesis of deoxyribonucleic acid of mammalian cells in culture. Mol Pharmacol. 1970;6:273–80. [PubMed] [Google Scholar]
- 45.Oguchi K, Sakamoto K, Kazuno H, Ueno H, Ishida K, Yokogawa T, et al. TAS-102 treatment results in high trifluridine incorporation into DNA with pyrimidine metabolic pathway markedly up-regulated in cancer. Eur J Cancer. 2014;50(suppl 6) Abstract 27. [Google Scholar]
- 46.Suzuki N, Emura T, Fukushima M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int J Oncol. 2011;39:263–70. doi: 10.3892/ijo.2011.1003. [DOI] [PubMed] [Google Scholar]
- 47.Matsuoka K, Iimori M, Niimi S, Tsukihara H, Watanabe S, Kiyonari S, et al. Trifluridine induces p53-dependent sustained G2 phase arrest with its massive misincorporation into DNA and few DNA strand breaks. Mol Cancer Ther. 2015;14:1004–13. doi: 10.1158/1535-7163.MCT-14-0236. [DOI] [PubMed] [Google Scholar]
- 48.Emura T, Nakagawa F, Fujioka A, Ohshimo H, Yokogawa T, Okabe H, et al. An optimal dosing schedule for a novel combination antimetabolite, TAS-102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med. 2004;13:249–55. [PubMed] [Google Scholar]
- 49.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, et al. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–36. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 50.Grogan BC, Parker JB, Guminski AF, Stivers JT. Effect of the thymidylate synthase inhibitors on dUTP and TTP pool levels and the activities of DNA repair glycosylases on uracil and 5-fluorouracil in DNA. Biochemistry. 2011;50:618–27. doi: 10.1021/bi102046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettersen HS, Visnes T, Vågbø CB, Svaasand EK, Doseth B, Slupphaug G, et al. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011;39:8430–44. doi: 10.1093/nar/gkr563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–5. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 53.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, et al. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7:e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turner DP, Cortellino S, Schupp JE, Caretti E, Loh T, Kinsella TJ, et al. The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 2006;66:7686–93. doi: 10.1158/0008-5472.CAN-05-4488. [DOI] [PubMed] [Google Scholar]
- 55.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, et al. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275:32422–9. doi: 10.1074/jbc.M004535200. [DOI] [PubMed] [Google Scholar]
- 56.Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071–6. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moghaddam A, Zhang HT, Fan TP, Hu DE, Lees VC, Turley H, et al. Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Natl Acad Sci U S A. 1995;92:998–1002. doi: 10.1073/pnas.92.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hotchkiss KA, Ashton AW, Schwartz EL. Thymidine phosphorylase and 2-deoxyribose stimulate human endothelial cell migration by specific activation of the integrins alpha 5 beta 1 and alpha V beta 3. J Biol Chem. 2003;278:19272–9. doi: 10.1074/jbc.M212670200. [DOI] [PubMed] [Google Scholar]
- 59.Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, et al. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res. 1999;59:1911–6. [PubMed] [Google Scholar]
- 60.Takao S, Akiyama SI, Nakajo A, Yoh H, Kitazono M, Natsugoe S, et al. Suppression of metastasis by thymidine phosphorylase inhibitor. Cancer Res. 2000;60:5345–8. [PubMed] [Google Scholar]
- 61.Murakami Y, Kazuno H, Emura T, Tsujimoto H, Suzuki N, Fukushima M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int J Oncol. 2000;17:277–83. doi: 10.3892/ijo.17.2.277. [DOI] [PubMed] [Google Scholar]
- 62.Inaba M, Mitsuhashi J, Sawada H, Miike N, Naoe Y, Daimon A, et al. Reduced activity of anabolizing enzymes in 5-fluorouracil-resistant human stomach cancer cells. Jpn J Cancer Res. 1996;87:212–20. doi: 10.1111/j.1349-7006.1996.tb03161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuoka K, Kobunai T, Takechi T. Efficacy of trifluridine for 5- fluorouracil-resistant human gastric cancer cell lines and their mechanisms. Cancer Res. 2014;74(19 Suppl) Abstract 781. [Google Scholar]
- 64.Nakamura A, Nakajima G, Okuyama R, Kuramochi H, Kondoh Y, Kanemura T, et al. Enhancement of 5-fluorouracil-induced cytotoxicity by leucovorin in 5-fluorouracil-resistant gastric cancer cells with upregulated expression of thymidylate synthase. Gastric Cancer. 2014;17:188–95. doi: 10.1007/s10120-013-0249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370. doi: 10.1186/1471-2407-10-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010;126:2457–68. doi: 10.1002/ijc.24943. [DOI] [PubMed] [Google Scholar]
- 68.Temmink OH, Hoebe EK, Fukushima M, Peters GJ. Irinotecan-induced cytotoxicity to colon cancer cells in vitro is stimulated by pre-incubation with trifluorothymidine. Eur J Cancer. 2007;43:175–83. doi: 10.1016/j.ejca.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 69.Temmink OH, Hoebe EK, van der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin-induced cytotoxicity to colorectal cancer cells. Br J Cancer. 2007;96:231–40. doi: 10.1038/sj.bjc.6603549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishida K, Sakamoto K, Tanaka N, Oguchi K, Yamamura K, Fujioka A, et al. Novel combination therapy, TAS-102 combined with the anti-EGFR antibody or the anti-VEGF antibody showed therapeutic benefit toward colorectal cancer xenografts. Eur J Cancer. 2014;50(supplement 6) abstract 22. [Google Scholar]
- 71.Tsukihara H, Nakagawa F, Sakamoto K, Ishida K, Tanaka N, Okabe H, et al. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, together with bevacizumab, cetuximab, or panitumumab on human colorectal cancer xenografts. Oncol Rep. 2015;33:2135–42. doi: 10.3892/or.2015.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nukatsuka M, Nakagawa F, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with oxaliplatin on human colorectal and gastric cancer xenografts. Anticancer Res. in press. [PubMed] [Google Scholar]
- 73.Nukatsuka M, Nakagawa F, Saito H, Sakata M, Uchida J, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with irinotecan hydrochloride on human colorectal and gastric cancer xenografts. Anticancer Res. 2015;35:1437–45. [PubMed] [Google Scholar]
- 74.Hong DS, Abbruzzese JL, Bogaard K, Lassere Y, Fukushima M, Mita A, et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer. 2006;107:1383–90. doi: 10.1002/cncr.22125. [DOI] [PubMed] [Google Scholar]
- 75.Overman MJ, Kopetz S, Varadhachary G, Fukushima M, Kuwata K, Mita A, et al. Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest. 2008;26:794–9. doi: 10.1080/07357900802087242. [DOI] [PubMed] [Google Scholar]
- 76.Overman MJ, Varadhachary G, Kopetz S, Thomas MB, Fukushima M, Kuwata K, et al. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008;26:445–54. doi: 10.1007/s10637-008-9142-3. [DOI] [PubMed] [Google Scholar]
- 77.Green MC, Pusztai L, Theriault LR, Adinin RB, Hofweber M, Fukushima M, et al. Phase I study to determine the safety of oral administration of TAS-102 on a twice daily (BID) schedule for five days a week (wk) followed by two days rest for two wks, every (Q) four wks in patients (pts) with metastatic breast cancer (MBC) Proc Am Soc Clin Oncol. 2006;24 Abstract 10576. [Google Scholar]
- 78.Doi T, Ohtsu A, Yoshino T, Boku N, Onozawa Y, Fukutomi A, et al. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107:429–34. doi: 10.1038/bjc.2012.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Patel MR, Bendell JC, Mayer RJ, Benedetti FM, Rosen LS. A phase I dose-escalation study of TAS-102 in patients (pts) with refractory metastatic colorectal cancer (mCRC) J Clin Oncol. 2012;30(suppl) abstract 3631. [Google Scholar]
- 80.Muro K, Doi T, Bando H, Yasui H, Nishina T, Yamaguchi K, et al. Multicenter phase 2 study of TAS-102 monotherapy in patients with pretreated advanced gastric cancer. Eur J Cancer. 2013;49(suppl 2) doi: 10.1016/j.ejca.2016.04.009. Abstract 2621. [DOI] [PubMed] [Google Scholar]