

Abstract
Gemcitabine has long been the standard of care for treating pancreatic ductal adenocarcinoma (PDAC), despite its poor pharmacokinetics/dynamics and rapid development of drug resistance. In this study, we have developed a novel nanoparticle platform based on nanoscale coordination polymer-1 (NCP-1) for simultaneous delivery of two chemotherapeutics, oxaliplatin and gemcitabine monophosphate (GMP), at 30 wt.% and 12 wt.% drug loadings, respectively. A strong synergistic therapeutic effect of oxaliplatin and GMP was observed in vitro against AsPc-1 and BxPc-3 pancreatic cancer cells. NCP-1 particles effectively avoid uptake by the mononuclear phagocyte system (MPS) in vivo with a long blood circulation half-life of 10.1±3.3 h, and potently inhibit tumor growth when compared to NCP particles carrying oxaliplatin or GMP alone. Our findings demonstrate NCP-1 as a novel nanocarrier for the co-delivery of two chemotherapeutics that have distinctive mechanisms of action to simultaneously disrupt multiple anticancer pathways with maximal therapeutic efficacy and minimal side effects.
Keywords: Nanoscale coordination polymers, Oxaliplatin, Gemcitabine, Synergistic effect, Pancreatic ductal adenocarcinoma
Graphical abstract
1. Introduction
Pancreatic cancer has one of the poorest prognoses of all cancer types, with a five-year survival rate of less than 6% [1, 2]. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, and accounts for 95% of all cases of these tumors. Upon diagnosis, 80% of pancreatic cancer cases are deemed inoperable due to the high risk of surgically resecting tumors connected to surrounding blood vessels and digestive ducts [3, 4]. Developing effective chemotherapy is thus of great importance in treating this deadly cancer.
Gemcitabine (gem) alone has long been the standard care for PDAC in the clinic [5, 6]. As a nucleotide analog [7], gem enters the cells through nucleotide transporters [8] and is then phosphorylated to gemcitabine monophosphate (GMP) by deoxycytidine kinase [9, 10]. GMP is further phosphorylated by uridine/cytidine monophosphate (UMP/CMP) kinase and nucleoside diphosphate kinase (NDK) to generate pharmacologically active gemcitabine diphosphate (GDP) and gemcitabine triphosphate (GTP) [11]. Although gem is the standard of care for PDAC, the gem treatment has many drawbacks. First, free gem lacks tumor specificity, and enters cancerous and healthy cells indiscriminately, leading to high general toxicity and narrow therapeutic windows [12]. Second, about 90% of gem is rapidly deactivated with a short half-life of 32 minutes in blood circulation due to deamination to the inactive 2′,2′-difluorodeoxyuridine (dFdU). Third, many pancreatic cancer cells develop resistance to gem, making repeat treatments with gem ineffective.
Combination therapy with multiple chemotherapeutics is a successful strategy for treating many cancers [13-16]. In particular, several different multidrug combination regiments have emerged for the treatment of pancreatic cancer, such as FOLFIRNOX and the combination of gemcitabine and nab-paclitaxel [17, 18]. Compared with conventional single-agent treatment, multi-agent therapy can promote synergism of different drugs, increase therapeutic target selectivity, and overcome drug resistance through distinct mechanisms of action [19-21]. However, combination therapy has its own drawbacks as the drugs typically have different pharmacokinetic properties, which often makes it difficult to obtain the optimal dose and increases the chances of adverse side effects [22, 23]. As a result, there is a great need in developing a combination drug delivery system that would specifically and selectively deliver multiple chemotherapeutics to tumor sites.
Nanoparticle drug delivery has been shown to promote therapeutic effectiveness and reduce side effects by improving pharmacokinetics [24-27]. It is even more advantageous to develop nanocarriers that are able to deliver multiple chemotherapeutics with controlled release characteristics and optimal pharmacokinetic profiles. Oxaliplatin has been used in combination with gem to treat metastatic pancreatic cancer patients with significantly enhanced response rates and tumor growth inhibition than their monotherapy counterparts [28-31]. However, this combination is not safe in patients with advanced solid tumors due to serious adverse side effects [32]. In view of this clinical need, we sought to develop a novel nanoparticle system for simultaneous delivery of oxaliplatin and gem for synergistic combination therapy of PDAC.
We recently reported the development of nanoscale coordination polymers (NCPs) as versatile nanocarriers for both cisplatin and oxaliplatin prodrugs [33]. NCP nanoparticles are constructed from polydentate bridging ligands and metal ions or clusters through self-assembly processes [34, 35]. NCPs possess many beneficial characteristics as drug delivery vehicles, including chemical diversity, high loading capacity, and intrinsic biodegradability [36-38]. NCPs showed long blood circulation half-lives and significantly enhanced tumor growth inhibition over their free drug counterparts [33]. We hypothesized that NCPs could incorporate multiple chemotherapeutics, in particular platinum drugs and gem, for their selective delivery to PDAC cells to elicit synergistic therapeutic effects. In this work, NCP-1 particles carrying both oxaliplatin (30 wt.%) and GMP (12 wt.%) were synthesized in reverse microemulsions, and extensively characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM), and in vitro release profiles. Synergistic effects of oxaliplatin and gem of NCP-1 on pancreatic cancer cells were demonstrated in vitro by cytotoxicity assays, flow cytometry analysis, and confocal scanning laser microscopic (CLSM) imaging. Pharmacokinetics and biodistributions of intravenously injected particles of NCP-1 were evaluated in subcutaneous xenograft mouse model of colon cancer CT26, whereas in vivo efficacy studies were carried out on subcutaneous xenograft mouse models of human PDACs including BxPc-3 and AsPc-1. The low general toxicity of NCP-1 was indicated by histology analysis and lack of immunogenic responses. Our results indicate that the co-delivery of oxaliplatin and GMP in NCP-1 leads to synergistic therapeutic effects and much enhanced antitumor efficacy as compared to their single drug counterparts in human pancreatic cancer xenograft mouse models.
2. Materials and methods
2.1. Materials, cell lines, and animals
Please see supplementary materials for details.
2.2. Preparation of NCP particles
A 5 mL mixture of 0.3 M Triton X-100/1.5 M 1-hexanol in cyclohexane consisting of 0.2 mL of an aqueous 25 mg/mL (dach)Pt(BP) sodium salt solution (7.6 μmol) and 0.030 mL of an aqueous 15 mg/mL GMP sodium salt solution (1.3 μmol) was stirred vigorously at room temperature. Another 5 mL of 0.3 M Triton X-100/1.5 M 1-hexanol in cyclohexane containing 0.2 mL of an aqueous 100 mg/mL Zn(NO3)2 solution (67 μmol) was stirred in a similar manner. Twenty μL of dioleoyl-sn-glycero-3-phosphate sodium salt (DOPA, 11 μmol in CHCl3) was added to the solution containing (dach)Pt(BP) and GMP. The two microemulsions were stirred continuously for 15 min until clear solutions were formed. The resulting W=7.4 microemulsions were combined and stirred for an additional 30 minutes. NCP-1 particles were obtained by the addition of 20 mL ethanol, and washed once with ethanol, once with 50% (v/v) ethanol/cyclohexane and once with 50% (v/v) ethanol/tetrahydrofuran (THF), and redispersed in THF. The nanoparticles were purified by filtration through 200 nm syringe filter.
NCP-1 was synthesized at a 20× scale, which shows similar prodrug loading, morphology, and size as those obtained at 1× scale. NCP-2, the nanoparticle carrying oxaliplatin, and Zn control nanoparticles were synthesized according to our previous report [33]. NCP-3 particles carrying GMP monotherapy were prepared similarly as NCP-1.
The lipid-coated and PEGylated particles were obtained by adding a THF solution of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), cholesterol (1:1 molar ratio), and 1,2-distearoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2k, 20 mol%) to the DOPA-capped NCP nanoparticles. The resulting mixture was added to 500 μL of 30% (v/v) ethanol/H2O at 50°C. THF was evaporated, and the dispersion was allowed to cool to room temperature before use.
2.3. Characterization of NCP particles
Please see supplementary materials for details.
2.4. In vitro stability studies
Please see supplementary materials for details.
2.5. In vitro cytotoxicity assays and synergistic effects of drug combinations
In vitro cytotoxicity assays were performed on AsPc-1 and BxPc-3 cancer cell lines. Confluent AsPc-1 and BxPc-3 cells were trypsinized and counted with a hemocytometer. Cells were plated in 96-well plates at a cell density of 2000 cells/well and a total of 100 μL fresh culture media, followed by further incubating at 37 °C and 5 % CO2 for 24 h. The culture medium was replaced by 100 μL of fresh RPMI 1640 containing 10% FBS, and different concentrations of oxaliplatin, GMP, free oxaliplatin/GMP mixture (at the same NCP-1 drug dose), Zn Control, NCP-1, NCP-2, and NCP-3 were added. The cells were incubated at 37 °C and 5 % CO2 for 48 h, and cell viability was determined by MTS assay (Promega, USA) according to manufacturer's instructions. The concentrations of oxaliplatin or GMP required to inhibit cell growth by 50% (IC50 values) were calculated.
The combination index (CI) was calculated using the following equation[39, 40]
where D1 and D2 are concentrations of drug 1 and drug 2, respectively, in combination at a specific drug effect level (e.g. 50% inhibition concentration), while Dm1 and DmB are the concentrations of the drugs dosed individually to achieve that same drug effect level. CI values were plotted against drug effect level (ICx values), with CI values lower than, equal to, and greater than 1 indicating synergism, additivity, and antagonism, respectively.
2.6. Cell apoptosis by Annexin V staining
Please see supplementary materials for details.
2.7. Flow cytometry
Please see supplementary materials for details.
2.8. Pharmacokinetics of NCP-1
Nude mice bearing C26 tumors were intravenously injected with NCP-1 at an oxaliplatin dose of 3 mg/kg. The mice were randomly distributed into different time period groups (n = 3 for each time point). At 5 min, 1 h, 3 h, 8 h, 24 h, and 48 h post injection time point, the mice were sacrificed, and the liver, lung, spleen, kidney, bladder, tumor, and blood were collected. Organs were digested in concentrated nitric acid overnight and then diluted with water and filtered for ICP-MS measurements of the Pt. Blood circulation half-life was fitted by an one-compartment model with nonlinear elimination using PK solver.[41]
Pharmacokinetics of GMP was analyzed using high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS, Agilent 6460 QQQ MS-MS) following a literature procedure.[42] The initial sample was prepared on ice to minimize enzyme-mediated degradation. To 50 μL of blood, 200 μL of ice-cold acetonitrile was added, vortex mixed, and centrifuged. The resulting supernatant was evaporated and the residue was reconstituted in 100 μL of water. Calibration standard was prepared in mouse blood at GMP concentrations of 10, 25, 50, 100, 250, 500, 1,000, and 2,500 ng/mL. An injection volume of 20 μL was used. The separation was achieved using a PGC Hypercarb column (100 × 2.1 ID, 5 μm, Thermo Fisher Scientific) fitted with a guard column (Hypercarb 10 × 2.1, 5 μm, Thermo Fisher Scientific) and a gradient mobile phase consisted of (A) 10 mM ammonium acetate at pH 10 and (B) acetonitrile. The initial mobile phase consisted of 95% solvent A and 5% solvent B at a flow rate of 0.3 mL/min for 2 min. Solvent A was decreased to 80% in 0.2 min and held at this composition for 5.6 min. The gradient was returned to 95% solvent A over 0.2 min and held at this composition for 7 min to give a total run-time of 15 min. The autosampler and column temperatures were kept at 4°C and 30°C, respectively. The mass to charge transition was monitored from 342 to 231. A dwell time of 50 ms was used.
2.9. Tumor Growth Inhibition
Tumor growth inhibition studies were conducted on AsPc-1 or BxPc-3 subcutaneous xenograft mouse models. AsPc-1 or BxPc-3 cells (5×106 cells in 200 μL of RPMI-1640) were subcutaneously injected in the right flank regions of mice. When the tumor volumes reached around 100 mm3, the mice were randomly distributed into 6 groups (n=6), and intravenously injected with different formulations. For the mice bearing BxPc-3 tumors, the formulations included PBS, free oxaliplatin/gem (5 mg oxaliplatin/kg, 50 mg gem/kg), Zn Control, NCP-1 (2 mg oxaliplatin/kg, 0.8 mg GMP/kg), NCP-2 (2 mg oxaliplatin/kg), and NCP-3 (0.8 mg GMP/kg). For the mice bearing AsPc-1 tumors, the formulations included PBS, free oxaliplatin/gem (5 mg oxaliplatin/kg, 50 mg gem/kg), and NCP-1 (2 mg oxaliplatin/kg, 0.8 mg GMP/kg). The drugs were administered every four days. Tumor sizes were calculated by measuring two perpendicular diameters with a caliper with the formula of V =0.5 × (a × b2), where V = tumor volume, a = the larger perpendicular diameter and b = the smaller perpendicular diameter. The tumor volumes were measured every other day. Body weight of each mouse was recorded every other day. All mice were sacrificed when tumor volume reached the maximum allowed size.
2.10. In vivo immunogenic response, hypersensitivity, and general toxicity evaluation of NCP-1
Please see supplementary materials for details.
2.11. In vivo tumor cell apoptosis
Please see supplementary materials for details.
2.12. Statistical analysis
Results were expressed as means ± standard deviation (S.D.). Student t-tests were used to determine statistical significance. A P value < 0.05 was considered statistically significant.
3. Results and Discussion
3.1. Synthesis and Characterization of NCP Particles
DOPA-capped NCP-1 particles were synthesized in reverse microemulsions by crosslinking (dach)Pt(BP), a Pt4+ prodrug, and gemcitabine monophosphate (GMP) with Zn2+ ions (Scheme 1); the Zn2+ ions formed coordination bonds with the phophonate groups of (dach)Pt(BP) and phosphate groups of GMP. DOPA-capped NCP-1 has hydrophobic surface, and is further coated with a DSPE/DSPE-PEG layer via hydrophobic/hydrophobic interactions to form NCP-1. The particles could then enter the cells through endocytosis and subsequently release oxaliplatin and GMP to disrupt DNA replication (Scheme 1). As shown in Fig. 1A-B, TEM images of DOPANCP-1 and NCP-1 showed well-dispersed, spherical nanoparticles of 26.4 ± 3.4 and 29.9 ± 1.7 nm in diameter, respectively. The average sizes of DOPA-NCP-1 and NCP-1 are 39.7 ± 0.8 and 49.5 ± 0.6 nm in diameter, respectively, as determined by DLS (Table 1 and Table S1). The polydispersity indexes (PDI) for the two particles were 0.032 and 0.062, respectively. NCP-1 has a near neutral zeta potential, indicating that PEG chains shield the nanoparticle surface charge and the possibility for NCP-1 to resist opsonization and to evade the mononuclear phagocytic system (MPS). The synthesis of NCPs has been scale up to hundreds of milligrams and each batch shows consistent prodrug loading, morphology, size, zeta potentials, and pharmacokinetic properties.
Scheme 1.
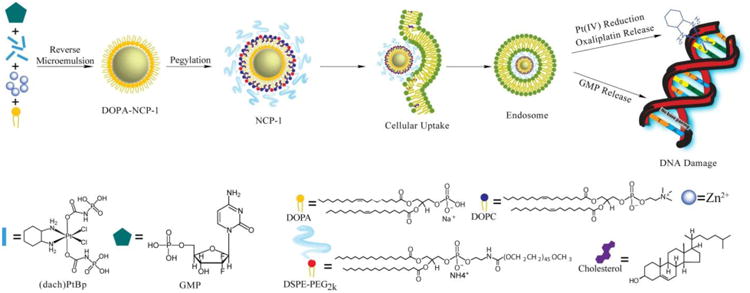
Schematic representation of the synthesis, composition, and mechanism of NCP-1 showing the endocytosis of NCP-1 to release oxaliplatin and GMP and the mechanisms by which oxaliplatin and GMP disrupt DNA replication.
Fig. 1.
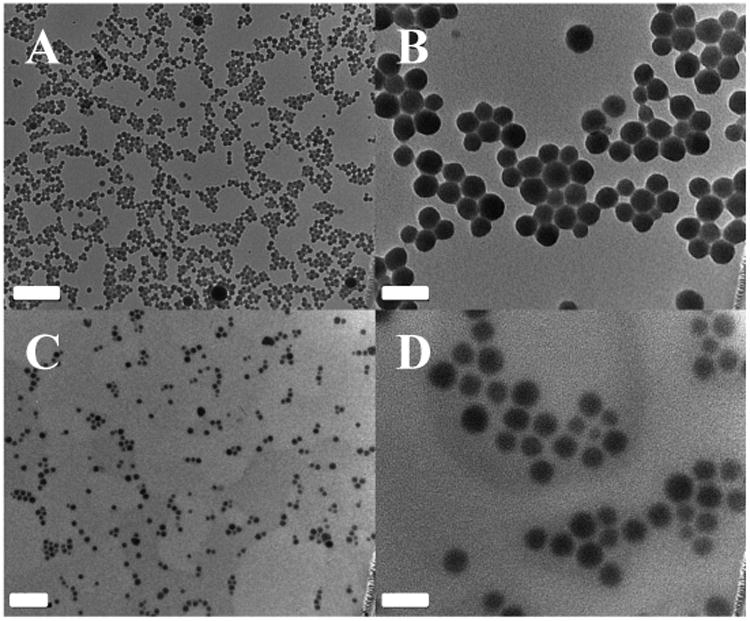
TEM micrographs of DOPA-NCP-1 (A, B) and NCP-1 (C, D). Scale bars represent 200 nm for A and C and 50 nm for B and D.
Table 1.
Sizes, polydispersities, and zeta potentials of NCP and Zn control particles.
| NCPs | Drug | Number-Ave diameter (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|
| NCP-1 | Oxaliplatin & gem | 49.5±0.6 | 0.062 | -1.3±0.2 |
| NCP-2 | Oxaliplatin | 56.7±6.1 | 0.130 | -1.0±0.6 |
| NCP-3 | gem | 97.3±8.2 | 0.161 | -6.6±0.7 |
| Zn Control | none | 51.6±12.2 | 0.179 | -1.2±0.7 |
Measured in PBS buffer. Data are expressed as means±SD.
NCP-2, NCP-3, and Zn control particles were similarly formulated for comparison purposes. The particle diameters, PDIs, and zeta potentials of these particles are shown in Table 1. NCP-2 containing only (dach)Pt(BP) and Zn control nanoparticles exhibited similar particle sizes of ∼50 nm in diameter and near neutral zeta potential. NCP-3 containing only GMP was also formulated and exhibited slightly larger particle size of 97 nm.
ICP-MS measurements of DOPA-NCP-1 and DOPA-NCP-2 gave oxaliplatin loadings of 30 ± 3 wt.% (corresponding to 50 ± 6 wt.% prodrug loading) and 25 ± 2 wt.% (corresponding to 42 ± 5 wt.% prodrug loading), respectively. UV-Vis and TGA analysis of NCP-1 and NCP-3 showed GMP loadings of 12 ± 2 wt.% and 57 ± 2 wt.%, respectively (Fig. S3). These levels of drug loadings are exceptionally high among all of the nanotherapeutics. Further, NCP-1 was shown to be stable in PBS buffer in the presence of bovine serum albumin (BSA) at 37°C (Fig. S4).
3.2. In Vitro Drug Release
In vitro drug release of DOPA-NCP-1 and NCP-1 was studied in phosphate buffered saline (PBS) at 37°C at pH 7.4. Only about 7% of the platinum was released from NCP-1 after 72 h, while DOPA-NCP-1 showed a rapid burst release, with 70% of the total platinum released before 12 h (Fig. S5A & Fig. S5C). Similarly, GMP release experiments revealed only 21% of the GMP released from NCP-1 after 12 h, as compared to DOPA-NCP-1 which has a GMP release of 86% after 12 h (Fig. S5B & Fig. S5D). No initial burst release was observed for nanoparticles after pegylation, suggesting that the lipid coating strategy adopted herein would prevent premature drug release prior to reaching the tumor sites, while stabilizing the nanoparticles in systemic circulation for a prolonged circulation lifetime.
To verify the stability of NCP-1 under reducing environment, the drug release was also simulated in presence of 5 mM cysteine. The results revealed that the addition of 5 mM cysteine triggered a faster drug release of DOPA-NCP-1 with 95% of platinum and 96% of GMP release after 12 h, indicating that the NCPs would rapidly undergo reductive degradation to release the drugs. However, NCP-1 exhibited similar drug release pattern in 5 mM PBS and 5 mM PBS supplemented with 5 mM cysteine. PEGylation of the particles made it difficult for cysteine to penetrate the lipid layer, improving the stability of the particle in blood circulation. On the other hand, once the NCP-1 particles enter cancer cells via endocytosis, some of the lipid coatings might be incorporated into the cell and plasma membranes, making the particle coatings more permeable to cysteine or other endogenous reducing agents to lead to rapid drug release via reductive degradation of the particles.
3.3. In vitro cytotoxicity and combination index
MTS assays of drug-loaded NCPs, Zn Control, and free drugs were carried out against AsPc-1 and BxPc-3 cells. After 48 h of incubation, NCP-1 exhibited significantly enhanced anticancer efficacy against AsPc-1 (Fig. 2A and B) and BxPc-3 (Fig. 2E and F) cells, with IC50 values that are about 5-fold, 2-fold, and 4-fold lower than free oxaliplatin, NCP-2, free GMP, and NCP-3, respectively. NCP-1 and free combination drugs showed comparable cytotoxicity in AsPc-1 (Oxaliplatin IC50=3.5 ± 0.5 μM vs. 3.6 ± 0.6 μM and GMP IC50=1.4 ± 0.1 μM vs.1.4 ± 0.3 μM, respectively) and BxPc-3 (Oxaliplatin IC50=4.8 ± 2.2 μM vs. 3.0 ± 0.2 μM and GMP IC50=1.9 ± 0.9 μM vs. 1.2 ± 0.1 μM, respectively). The results demonstrated that the NCP-1 carrying two chemotherapeutics significantly outperformed their platinum and GMP drug counterparts and their NCP monotherapy counterparts in terms of cytotoxicity, which could be ascribed to the synergistic effect exerted by oxaliplatin and GMP (Fig. 2C-D,G-H).
Fig. 2.

In vitro cytotoxicity plots and combination index (CI) of oxaliplatin/GMP combinations on AsPc-1 (A-D) and BxPc-3 (E-H) cells. The cell viabilities on AsPc-1 and BxPc-3, cells were measured after 48 h exposure to Zn Control, NCP-1, NCP-2, or free drugs (oxaliplatin, GMP, or oxaliplatin/GMP). Data are mean ± S.D. (n=6).
When comparing the NCP-1 and oxaliplatin/GMP mixture with their free drug and NCP counterparts, most of the CI values were below 1, indicating synergy between oxaliplatin and GMP. Due to this synergistic effect, oxaliplatin and GMP inside a single nanocarrier exhibited better efficacy than the single free drug alone and single drug nanoparticle formulation over the same concentrations. The synergistic effect between oxaliplatin and GMP in NCP-1 can be explained by their different mechanisms of action, leading to much enhanced anticancer efficacy against pancreatic tumor models than monotherapy NCPs alone. We have shown here and previously [33] that NCP-2 has tumor inhibition effect on PDAC. Oxaliplatin exerts its effect by forming DNA adduct that interferes with DNA replication [43, 44]. However, only 5-10% of platin complex are covalently bounded to DNA, while 75-85% of drug is bounded to proteins, such as cysteine and methionine [45, 46]. Furthermore, tumor cells develop acquired platinum resistance, primarily from high expression levels of resistance genes [47]. Likewise, gem resistance can disrupt signaling pathways during cell apoptosis and the conversion of gem to the triphosphate active form [48]. GTP works differently from oxaliplatin in that it induces apoptosis by replacing cytidine during DNA replication. Combining oxaliplatin and GMP in a single nanocarrier thus brings together the benefits of the combination therapy, while overcoming the hypersensitivity of platinum or gem alone due to their reduced doses.
3.4. Intracellular Uptake and Cell apoptosis in vitro
Dye-doped particles of NCPs (Ce6-NCP-x, where x = 1, 2, and 3) were synthesized by incorporating chlorin-6 into the particles for confocal imaging studies. The bare and lipid-coated Ce6-NCP-x showed similar morphologies as well as particle sizes and distributions (Table S2 and Fig. S6). The Ce6 loading was determined to be 0.42 wt.% by fluorimetry. Cellular uptake and intracellular drug release behaviors were observed by CLSM. AsPc-1 (Fig. S7) or BxPc-3 (Fig. S8) cells were incubated with free oxaliplatin, GMP, oxaliplatin/GMP, Zn Control, NCP-1, NCP-2, or NCP-3 for 48 h. Internalization of the nanoparticles was observed, as shown by strong Ce6 fluorescence found in the cells. Significant FITC-Annexin V signals were found for both AsPc-1 and BxPc-3 cells incubated with oxaliplatin/GMP and Ce6-NCP-1, indicating the combination of oxaliplatin and GMP drugs induced substantial cell apoptosis. In comparison, Zn Control showed no cytotoxicity which was supported by the absence of apoptosis marker in CLSM imaging.
Flow cytometry analysis showed increased percentages of early and late apoptosis for oxaliplatin/GMP and NCP-1 for AsPc-1 and BxPc-3 cells (Fig. S9). Oxaliplatin/GMP induced 63.7% and 50.6% apoptosis for AsPc-1 and BxPc-3 cells, respectively, whereas NCP-1 induced 74.6% and 78.6% apoptosis for BxPc-3 cells, respectively. In comparison, oxaliplatin, GMP, and their corresponding monotherapy NCPs showed inferior cytotoxicity as evidenced in confocal microscopy imaging and flow cytometry analysis (Fig. 3, Fig. S7-9). Apoptotic cell percentages for oxaliplatin, GMP, NCP-2, and NCP-3 were determined to be 52.9%, 55.9%, 60.8%, and 54.2%, respectively, for AsPc-1 cells. Oxaliplatin, GMP, NCP-2, and NCP-3 induced 1.5%, 32.3%, 30.2%, and 35.1% apoptosis for BxPc-3 cells, respectively. These results showed an efficient intracellular uptake of NCPs and triggered release of both oxaliplatin and/or GMP from the nanoparticles to lead to high anticancer efficacy.
Fig. 3.
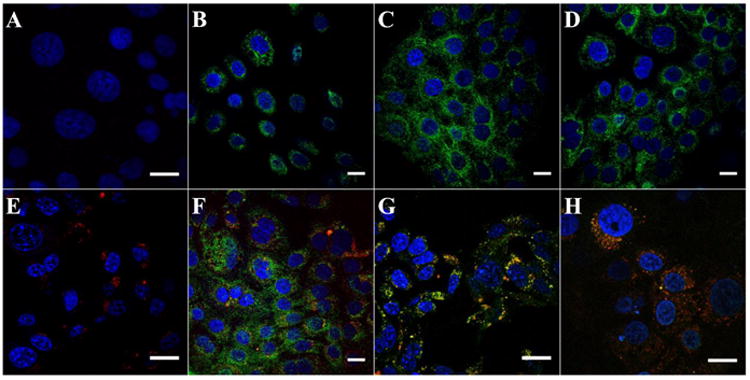
CLSM images showing the apoptosis induced by saline (A) oxaliplatin (B), GMP (C), oxaliplatin/GMP (D), Ce6-Zn Control (E), Ce6-NCP-1 (F), Ce6-NCP-2 (G), and Ce6-NCP-3 (H) in BxPc-3 pancreatic cancer cells. The nuclei were stained with DAPI (blue) and the cells were stained with Annexin V FIFC Conjugate (green). The nanoparticles were doped with chlorin e6 (red). Bar = 20 μm.
3.5. Pharmacokinetic Studies
We examined the biodistribution of NCP-1 on CT-26-tumor-bearting mice in order to assess its ability to evade the MPS and to accumulate in tumor tissues (Fig. 4). NCP-1 showed a prolonged blood circulation with Pt and GMP blood circulation half-lives of 10.1±3.3 h and 8.0±2.3 h, respectively, after intravenous injection (Table S3 & Fig. S10). This blood circulation half-life is more than 1000-fold longer than reported value of the oxaliplatin prodrug, which is rapidly cleared from blood circulation with an α-half-life of 0.01±0.004 h [49]. NCP-1 showed a comparable blood circulation half-life as our previously reported NCP-2 (t1/2=12.0±3.9 h) [33]. The tumor uptake of NCP-1 reached 8.8 ± 2.0% ID/g (percentage injected dose per gram) after 24 h (Fig. 4A), which is six times higher than that of oxaliplatin reported in the literature [50]. Very low Pt concentrations were observed in other organs such as the liver, spleen, and kidney (Fig. S10A). All of these results indicate that NCP-1 has the ability to evade the MPS system to lead to a prolonged circulation time and enhanced tumor uptake over small molecule therapeutics, presumably due to the enhanced permeability and retention (EPR) effect [51, 52]. Since the NCP-1 particles are smaller than 50 nm, they can readily accumulate in tumors through misaligned and defected vasculatures and by taking advantage of the poor lymphatic drainage of tumors. The dense PEG layer on NCP-1 is believed to allow the particles to evade the MPS system, which is indicated by the low Pt concentrations observed in the organs with high MPS activity such as the liver, spleen, and kidney (Fig. 4A).
Fig. 4.
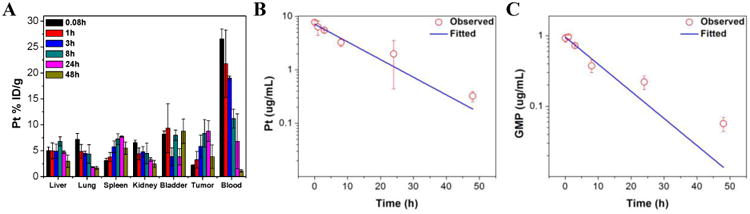
(A) Percentage injected dose per gram (% ID/g) of Pt in tissues and blood after intravenous administration of NCP-1 in CT26 tumor-bearing mice at time points 5 min, 1 h, 3 h, 8 h, 24 h, and 48 h. Data are mean ± S.D. (n=3). (B) Average observed and predicted time-dependent Pt distributions in blood after administration of NCP-1 (n=3). (C) Average observed and predicted time-dependent GMP distributions in blood after administration of NCP-1 (n=3). One-compartment model was used for fitting the Pt and GMP distributions in blood.
3.6. Tumor growth inhibition studies
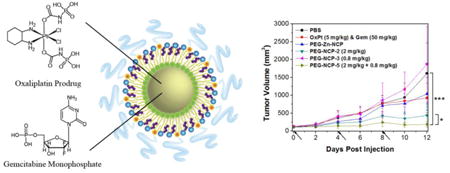
To determine whether NCP-1 possesses synergistic efficacy for pancreatic cancers in vivo, tumor growth inhibition was evaluated in BxPc-3 and AsPc-1 subcutaneous xenograft murine models. For mice bearing BxPc-3 tumors, free oxaliplatin/gem (5 mg oxaliplatin/kg, 50 mg gem/kg dose) and NCP-3 (0.8 mg/kg dose) did not show statistically significant anti-tumor efficacy. Though inhibition of tumor growth was achieved by NCP-2 (2 mg/kg dose) as compared to the control (p=0.0085), NCP-1, dosed at a dose of 2 mg/kg for oxaliplatin and 0.8 mg/kg GMP, showed the most potent anticancer efficacy in BxPc-3 models (Fig. 5A). The tumor inhibition was dramatically enhanced in the NCP-1 group in comparison to the monotherapy NCP groups (p=0.0319 vs. NCP-2 and p=0.0030 vs. NCP-3). The tumor growth in NCP-1 was effectively suppressed with the average tumor size increasing 1.7-fold, in comparison to the increase of 3.6∼14.3-folds in other groups. Tumor weight of NCP-1 was also significantly smaller compared with those in the other treatment groups; the average tumor weight in the NCP-1 group is more than 11-fold smaller than that of the PBS control group (p=0.0002) (Fig. 5B). Moreover, the tumor weight of the NCP-1 group was 2.7 times smaller than the NCP-2 group (p=0.0342) and 16.6 times smaller than the NCP-3 group (p=0.0031).
Fig. 5.
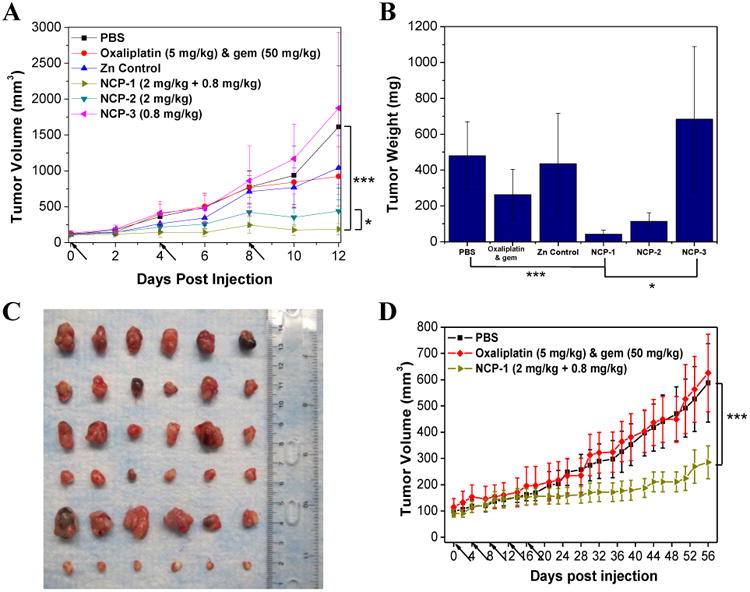
(A) In vivo tumor growth inhibition curves for PBS(■), oxaliplatin & gem (◆), Zn Control (▲), NCP-1(►), NCP-2 (▼), and NCP-3 (◄) on subcutaneous BxPc-3 xenografts. Oxaliplatin (dose, 5 mg/kg) and gem (dose, 50 mg/kg), NCP-1 (doses, 2 mg/kg + 0.8 mg/kg), NCP-2 (dose, 2 mg/kg), and NCP-3 (dose, 0.8 mg/kg) were administered on day 0, 4, and 8. Data are expressed as means±S.D. (n=6), *p<0.05, **p<0.01, ***p<0.001. (B) End-point tumor weights. Data are expressed as means±S.D. (n=6), *p<0.05, **p<0.01, ***p<0.001. (C) Photos of the resected BxPc-3 tumors from top to bottom: PBS, oxaliplatin & gem, Zn Control, NCP-2, NCP-3, and NCP-1. (D) In vivo tumor growth inhibition curves for PBS(■),oxaliplatin & gem (◆), and NCP-1(►) on subcutaneous AsPc-1 xenografts. Oxaliplatin (dose, 5 mg/kg) and gem (dose, 50 mg/kg) and NCP-1 (doses, 2 mg/kg + 0.8 mg/kg) were administered on day 0, 4, 8, 12, 16, and 20. Data are expressed as means±S.D. (n=6), *p<0.05, **p<0.01, ***p<0.001.
The mice were sacrificed 12 days post injection because the tumors in the PBS control and NCP-3 groups had exceeded the 2000 mm3 limit. No significant change in body weight was observed for the NCP-treated groups, demonstrating the safety and tolerance of the NCP vehicles (Fig. S12A). The general toxicity was further investigated by immunogenic responses (Fig. S13) and histological assessments (Fig. S14). No statistically significant difference was observed between the control and NCP-1 groups in terms of the pro-inflammatory cytokine production and Pt hypersensitivity. From the H&E-stained sectioned tissues of liver, lung, spleen, and kidney, no difference in general toxicity was observed between the control and NCP-1 groups.
Mice bearing AsPc-1 tumors were also intravenously administrated with free oxaliplatin & gem (5 mg/kg oxaliplatin and 50 mg/kg gem) and NCP-1 (2 mg/kg for oxaliplatin and 0.8 mg/kg for GMP) every four days for a total of five injected doses to compare their anticancer efficacy. NCP-1 showed superior tumor growth inhibition at lower drug doses compared with the combination of free drugs (Fig. 5D). For the NCP-1 treatment, the tumor size 56 days post injection showed only a 3-fold increase, whereas the PBS control and free drug combination groups show nearly a 6-fold increase in tumor size (p=0.00076). Mice treated with NCP-1 showed a slight decrease in weight during the treatment but quickly regained their weight after the last injection. No adverse effects from the NCP-1 were observed as indicated by the relatively stable body weights maintained during the experiment (Fig. S12B).
The oxaliplatin and GMP combination in the NCP-1 particle has thus not only shown synergistic effects in vitro, but also exhibited outstanding antitumor efficacy on pancreatic cancer cell subcutaneous xenografts in vivo. Even with much lower doses, NCP-1 thus achieved much higher potency than free drugs, which is still rare among all the existing nanotherapeutics. Higher antitumor efficacy of NCP-1 over monotherapy NCPs confirmed the synergistic effects of oxaliplatin and gem in NCP-1 in vivo.
3.7. In vivo tumor cell apoptosis
BxPc-3 tumors from the tumor inhibition efficacy study were sectioned for TUNEL assays to investigate cell apoptosis caused by different treatments (Fig. 6A). NCP-1 induced 77.8±7.5% apoptotic tumor cells, which was superior to 46.5±5.7% and 6.5±4.8% apoptotic tumor cells induced by NCP-2 and NCP-3, respectively. No apoptosis was triggered by the control, Zn Control, and free oxaliplatin & gem (Fig. 6B). The TUNEL assay results are consistent with the tumor growth inhibition results as shown in Fig. 5. The superior anticancer potency of NCP-1 is a result of the enhanced drug delivery to tumors and the synergistic effect of oxaliplatin and GMP. The lack of apoptosis shown in the free oxaliplatin & gem treatment group indicated that the free drugs are rapidly cleared away from the body, leading to sub-therapeutic accumulation of the drugs in the tumors.
Fig. 6.
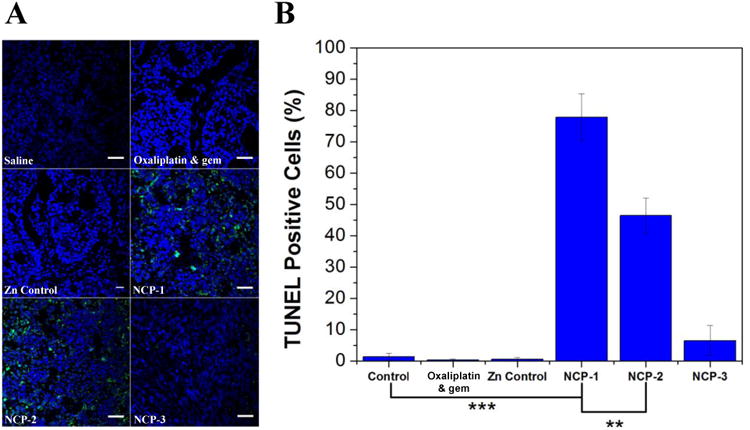
(A) Representative CLSM images of TUNEL assays of tumor tissues. DNA fragment in apoptotic cells was stained with fluorescein-conjugated deoxynucleotides (green) and the nuclei were stained with DAPI (blue). Scale = 40 μm. (B) The percentages of TUNEL-positive cells in tumor tissues.
4. Conclusions
We have developed a NCP-based formulation for the co-delivery of oxaliplatin and GMP as a combination therapy for the treatment of pancreatic cancers. We have shown the synergistic effect of oxaliplatin and GMP against pancreatic cancer cell lines in in vitro studies. The combination NCP particle, NCP-1, showed a long blood circulation half-life with high drug accumulation in tumors. NCP-1 effectively inhibits tumor growth in vivo when compared to monotherapy NCPs and free combination drugs. As the combination of oxaliplatin and gem is FDA approved for the treatment of pancreatic cancer, this work highlights the potential of combination NCPs as highly effective delivery vehicles for PDAC treatment in the clinic. The versatility of NCP to incorporate other combinations of drugs opens many possibilities in the treatment of a variety of difficult-to-treat cancers.
Supplementary Material
Table 2.
Oxaliplatin IC50 values of oxaliplatin, GMP, oxaliplatin/GMP, NCP-1, NCP-2, and NCP-3 against AsPc-1 and BxPc-3 cells (the numbers in parenthesis refer to GMP concentrations).
| Oxaliplatin | GMP | Oxaliplatin/GMP (μM) | Zn Control* (μM) | NCP-1 (μM) | NCP-2 (μM) | NCP-3 (μM) | |
|---|---|---|---|---|---|---|---|
| AsPc-1 | 18.1±2.1 | (2.6±0.4) | 3.6±0.6 (1.4±0.3) | >50 (>20) | 3.5±0.5 (1.4±0.1) | 22.4±1.4 | (6.1±4.1) |
| BxPc-3 | 29.0±7.1 | (2.7±2.1) | 3.0±0.2 (1.2±0.1) | >50 (>20) | 4.8±2.2 (1.9±0.9) | 7.8±2.5 | (25.8±6.3) |
Zn Control does not contain oxaliplatin or GMP as they are used to study the toxicity of NCP excipients. The amount of Zn Control particle was the same as NCP-1 under the studied concentrations. Data are expressed as means±SD.
Acknowledgments
This work was supported by National Cancer Institute (U01–CA151455) and startup funds from the University of Chicago. We thank Mr. Alexander Chen for experimental help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bunt S, Mohr A, Bailey J, Grandgenett P, Hollingsworth M. Rosiglitazone and Gemcitabine in combination reduces immune suppression and modulates T cell populations in pancreatic cancer. Cancer Immunology Immunotherapy. 2013;62:225–236. doi: 10.1007/s00262-012-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.A.C. Society. Cancer Facts & Figures 2013. Atlanta: American Cancer Society; 2013. [Google Scholar]
- 3.El Kamar FG, Grossbard ML, Kozuch PS. Metastatic pancreatic cancer: emerging strategies in chemotherapy and palliative care. Oncologist. 2003;8:18–34. doi: 10.1634/theoncologist.8-1-18. [DOI] [PubMed] [Google Scholar]
- 4.Geer RJ, Brennan MF. Prognostic indicators for survival after resection of pancreatic adenocarcinoma. Am J Surg. 1993;165:68–72. doi: 10.1016/s0002-9610(05)80406-4. discussion 72-63. [DOI] [PubMed] [Google Scholar]
- 5.King RS. Gemcitabine, New first-line therapy for pancreatic cancer. Cancer Pract. 1996;4:353–354. [PubMed] [Google Scholar]
- 6.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von HDD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 7.Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, Grindey GB. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) Cancer Res. 1990;50:4417–4422. [PubMed] [Google Scholar]
- 8.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–4357. [PubMed] [Google Scholar]
- 9.Oguri T, Achiwa H, Sato S, Bessho Y, Takano Y, Miyazaki M, Muramatsu H, Maeda H, Niimi T, Ueda R. The determinants of sensitivity and acquired resistance to gemcitabine differ in non-small cell lung cancer: a role of ABCC5 in gemcitabine sensitivity. Mol Cancer Ther. 2006;5:1800–1806. doi: 10.1158/1535-7163.MCT-06-0025. [DOI] [PubMed] [Google Scholar]
- 10.Plunkett W, Huang P, Gandhi V. Preclinical characteristics of gemcitabine. Anticancer Drugs. 1995;6(Suppl 6):7–13. doi: 10.1097/00001813-199512006-00002. [DOI] [PubMed] [Google Scholar]
- 11.Mackey JR, Yao SY, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst. 1999;91:1876–1881. doi: 10.1093/jnci/91.21.1876. [DOI] [PubMed] [Google Scholar]
- 12.Zhou BBS, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nature Reviews Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- 13.Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, III, Staunton JE, Jin X, Lee MS, Zimmermann GR, Borisy AA. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–2019. doi: 10.1200/JCO.2007.14.9930. [DOI] [PubMed] [Google Scholar]
- 15.Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A, Johnson DH. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial - INTACT 2. J Clin Oncol. 2004;22:785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 16.Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A, Johnson DH. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial - INTACT 1. J Clin Oncol. 2004;22:777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M. FOLFIRINOX versus gemcitabine or metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 18.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeVita VT, Jr, Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 1975;35:98–110. doi: 10.1002/1097-0142(197501)35:1<98::aid-cncr2820350115>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 20.Shah MA, Schwartz GK. The relevance of drug sequence in combination chemotherapy. Drug Resistance Updates. 2000;3:335–356. doi: 10.1054/drup.2000.0165. [DOI] [PubMed] [Google Scholar]
- 21.Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, 3rd, Staunton JE, Jin X, Lee MS, Zimmermann GR, Borisy AA. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer LD, Harasym TO, Tardi PG, Harasym NL, Shew CR, Johnstone SA, Ramsay EC, Bally MB, Janoff AS. Ratiometric dosing of anticancer drug combinations: Controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5:1854–1863. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 23.Ismael GFV, Rosa DD, Mano MS, Awada A. Novel cytotoxic drugs: Old challenges, new solutions. Cancer Treat Rev. 2008;34:81–91. doi: 10.1016/j.ctrv.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 25.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 26.Devalapally H, Chakilam A, Amiji MM. Role of nanotechnology in pharmaceutical product development. J Pharm Sci. 2007;96:2547–2565. doi: 10.1002/jps.20875. [DOI] [PubMed] [Google Scholar]
- 27.Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol: Semin Orig Invest. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 28.Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, Zaniboni A, Ducreux M, Aitini E, Taieb J, Faroux R, Lepere C, de Gramont A. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:3509–3516. doi: 10.1200/JCO.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Poplin E, Feng Y, Berlin J, Rothenberg ML, Hochster H, Mitchell E, Alberts S, O'Dwyer P, Haller D, Catalano P, Cella D, Benson AB., III Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the eastern cooperative oncology group. J Clin Oncol. 2009;27:3778–3785. doi: 10.1200/JCO.2008.20.9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demols A, Peeters M, Polus M, Marechal R, Gay F, Monsaert E, Hendlisz A, Van Laethem JL. Gemcitabine and oxaliplatin (GEMOX) in gemcitabine refractory advanced pancreatic adenocarcinoma: a phase II study. Br J Cancer. 2006;94:481–485. doi: 10.1038/sj.bjc.6602966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louvet C, Andre T, Lledo G, Hammel P, Bleiberg H, Bouleuc C, Gamelin E, Flesch M, Cvitkovic E, De Gramont A. Gemcitabine combined with oxaliplatin in advanced pancreatic adenocarcinoma: final results of a GERCOR multicenter phase II study. J Clin Oncol. 2002;20:1512–1518. doi: 10.1200/JCO.2002.20.6.1512. [DOI] [PubMed] [Google Scholar]
- 32.Wong E, Giandomenico CM. Current Status of Platinum-Based Antitumor Drugs. Chem Rev (Washington, D C) 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Poon C, Lu K, He C, Lin W. Self-assembled nanoscale coordination polymers with trigger release properties for effective anticancer therapy. Nat Commun. 2014;5 doi: 10.1038/ncomms5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rieter WJ, Pott KM, Taylor KML, Lin W. Nanoscale Coordination Polymers for Platinum-Based Anticancer Drug Delivery. Journal of the American Chemical Society. 2008;130:11584–11585. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]
- 35.Taylor-Pashow KML, Della Rocca J, Xie Z, Tran S, Lin W. Postsynthetic Modifications of Iron-Carboxylate Nanoscale Metal-Organic Frameworks for Imaging and Drug Delivery. Journal of the American Chemical Society. 2009;131:14261–14263. doi: 10.1021/ja906198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yavuz MS, Cheng Y, Chen J, Cobley CM, Zhang Q, Rycenga M, Xie J, Kim C, Song KH, Schwartz AG, Wang LV, Xia Y. Gold nanocages covered by smart polymers for controlled release with near-infrared light. Nature Materials. 2009;8:935–939. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. Uniform Mesoporous Dye-Doped Silica Nanoparticles Decorated with Multiple Magnetite Nanocrystals for Simultaneous Enhanced Magnetic Resonance Imaging, Fluorescence Imaging, and Drug Delivery. Journal of the American Chemical Society. 2010;132:552–557. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]
- 38.Liu R, Zhang Y, Zhao X, Agarwal A, Mueller LJ, Feng P. pH-Responsive Nanogated Ensemble Based on Gold-Capped Mesoporous Silica through an Acid-Labile Acetal Linker. Journal of the American Chemical Society. 2010;132:1500–1501. doi: 10.1021/ja907838s. [DOI] [PubMed] [Google Scholar]
- 39.Chou TC. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Research. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 40.Xiao H, Li W, Qi R, Yan L, Wang R, Liu S, Zheng Y, Xie Z, Huang Y, Jing X. Co-delivery of daunomycin and oxaliplatin by biodegradable polymers for safer and more efficacious combination therapy. J Controlled Release. 2012;163:304–314. doi: 10.1016/j.jconrel.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99:306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Bapiro TE, Richards FM, Goldgraben MA, Olive KP, Madhu B, Frese KK, Cook N, Jacobetz MA, Smith DM, Tuveson DA, Griffiths JR, Jodrell DI. A novel method for quantification of gemcitabine and its metabolites 2′,2′-difluorodeoxyuridine and gemcitabine triphosphate in tumour tissue by LC-MS/MS: comparison with 19F NMR spectroscopy. Cancer Chemother Pharmacol. 2011;68:1243–1253. doi: 10.1007/s00280-011-1613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol. 1998;25:4–12. [PubMed] [Google Scholar]
- 44.Woynarowski JM, Faivre S, Herzig MC, Arnett B, Chapman WG, Trevino AV, Raymond E, Chaney SG, Vaisman A, Varchenko M, Juniewicz PE. Oxaliplatin-induced damage of cellular DNA. Mol Pharmacol. 2000;58:920–927. doi: 10.1124/mol.58.5.920. [DOI] [PubMed] [Google Scholar]
- 45.Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- 46.Alian OM, Azmi AS, Mohammad RM. Network insights on oxaliplatin anti-cancer mechanisms. Clin Transl Med. 2012;1:26. doi: 10.1186/2001-1326-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooke SL, Brenton JD. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011;12:1169–1174. doi: 10.1016/S1470-2045(11)70123-1. [DOI] [PubMed] [Google Scholar]
- 48.Fryer RA, Barlett B, Galustian C, Dalgleish AG. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD® lenalidomide. Anticancer Res. 2011;31:3747–3756. [PubMed] [Google Scholar]
- 49.Levi F, Metzger G, Massari C, Miano G. Oxaliplatin: Pharmacokinetics and Chronopharmacological Aspects. Clin Pharmacokinet. 2000;38:1–21. doi: 10.2165/00003088-200038010-00001. [DOI] [PubMed] [Google Scholar]
- 50.Rafi M, Cabral H, Kano MR, Mi P, Iwata C, Yashiro M, Hirakawa K, Miyazono K, Nishiyama N, Kataoka K. Polymeric micelles incorporating (1,2-diaminocyclohexane)platinum (II) suppress the growth of orthotopic scirrhous gastric tumors and their lymph node metastasis. J Controlled Release. 2012;159:189–196. doi: 10.1016/j.jconrel.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 51.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 52.Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.