

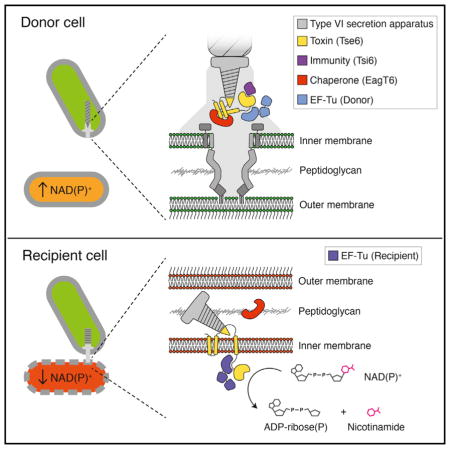
SUMMARY
Type VI secretion (T6S) influences the composition of microbial communities by catalyzing the delivery of toxins between adjacent bacterial cells. Here, we demonstrate that a T6S integral membrane toxin from Pseudomonas aeruginosa, Tse6, acts on target cells by degrading the universally essential dinucleotides NAD+ and NADP+. Structural analyses of Tse6 show that it resembles mono-ADP-ribosyltransferase proteins, such as diphtheria toxin, with the exception of a unique loop that both excludes proteinaceous ADP-ribose acceptors and contributes to hydrolysis. We find that entry of Tse6 into target cells requires its binding to an essential housekeeping protein, translation elongation factor Tu (EF-Tu). These proteins participate in a larger assembly that additionally directs toxin export and provides chaperone activity. Visualization of this complex by electron microscopy defines the architecture of a toxin-loaded T6S apparatus and provides mechanistic insight into intercellular membrane protein delivery between bacteria.
Graphical Abstract
INTRODUCTION
Bacteria utilize a diverse group of secreted toxins to establish and defend their niche. Among these are the effectors exported by the type VI secretion system (T6SS), which are delivered to target cells in a contact-dependent manner (Hood et al., 2010; LeRoux et al., 2012; Russell et al., 2011). Despite the tremendous number and predicted diversity of T6 effectors, few activities have been ascribed to this important group of proteins.
The majority of characterized T6 effectors act in the periplasm of target Gram-negative cells. Within this compartment, the proteins disrupt essential structures, such as cell-wall peptidoglycan (via amidase and glycoside hydrolase activity), and cellular membranes (via phospholipase and poreforming activity) (Russell et al., 2014). Although a large number of cytotoxic T6 effectors have been identified, the mechanisms by which they influence recipient cells are not well understood (Fritsch et al., 2013; Hood et al., 2010; Whitney et al., 2014). Indeed, a group of related effectors that exhibit DNase activity are the only such proteins yet characterized (Ma et al., 2014).
Unlike other proteinaceous toxins, such as the colicins, T6S effectors do not possess cell-entry mechanisms. Rather, they transit the T6SS, which breaches the outer membrane of recipient cells and thereby grants its substrates access to the cell interior (Russell et al., 2011). Many components of the T6SS bear structural and functional relatedness to tail proteins of contractile bacteriophage (Silverman et al., 2012). The delivery of T6 effectors into recipient cells has not been directly visualized; however, it is likely that they are propelled into recipient cells during phage-like contraction events of the apparatus (Basler et al., 2012). How T6 effectors are recruited to the secretory apparatus is not completely understood. Evidence suggests at least two genetically distinct mechanisms operate. One subset of T6 effectors requires direct interaction with the interior of ring-shaped phage tail tube-like haemolysin co-regulated proteins (Hcp) for export (Silverman et al., 2013). Hcp proteins themselves are abundantly secreted in a T6-dependent manner, leading to the proposal that these toxins are delivered to recipient cells in complex with Hcp. The relatively low molecular weight of Hcp-associated effectors suggests that interaction with the pore of Hcp places constraints on the size of toxins that can be delivered via this pathway.
A second subset of effectors, including many that are high-molecular-weight, multi-domain proteins, require specific valineglycine repeat protein G (VgrG) type proteins for export (Hachani et al., 2014; Whitney et al., 2014). VgrG proteins form homotrimeric assemblies that have extensive structural homology with phage tail spike proteins, and, like Hcp, are secreted in a T6-dependent manner. Also analogous to Hcp, the requirement for VgrG proteins in effector export is thought to reflect a physical association of these proteins with cognate effectors. The biochemical basis for VgrG-effector interaction is not well studied; however, modular adaptor domains—present either as domains within the effector protein or as independent polypeptides—appear to mediate binding. One such domain harbors PAAR repeat sequences, which fold into a pyramidal structure that interacts with the tip of the VgrG spike (Shneider et al., 2013). Despite recent advances in our understanding of the mechanisms underlying T6S-dependent interbacterial interactions, the structure of a T6 effector in complex with a VgrG family protein has remained elusive.
The genome of Pseudomonas aeruginosa encodes three T6SSs; each mediate antagonistic interactions with contacting Gram-negative bacterial cells (Hood et al., 2010; Jiang et al., 2014; Russell et al., 2013). The most extensively studied of these is the Hcp secretion island I-encoded T6SS (H1-T6SS), which delivers at least six effectors to recipients. Prior work established that one of these, type VI secretion exported 6 (Tse6), is a predicted transmembrane protein that contains a PAAR repeat domain, is exported in a VgrG-dependent manner, and is active in the cytoplasm of target cells (Whitney et al., 2014). Here, we demonstrate that Tse6 intoxicates by depleting cells of the related co-factors β-nicotinamide adenine dinucleotide (NAD+) and NAD+ phosphate (NADP+), thereby simultaneously inhibiting anabolic and catabolic processes required for homeostasis and growth. We make the surprising discovery that Tse6 requires interaction with translation elongation factor Tu for delivery into recipient cells and define the structural and biochemical basis for interbacterial transfer of this membrane-associated toxin.
RESULTS
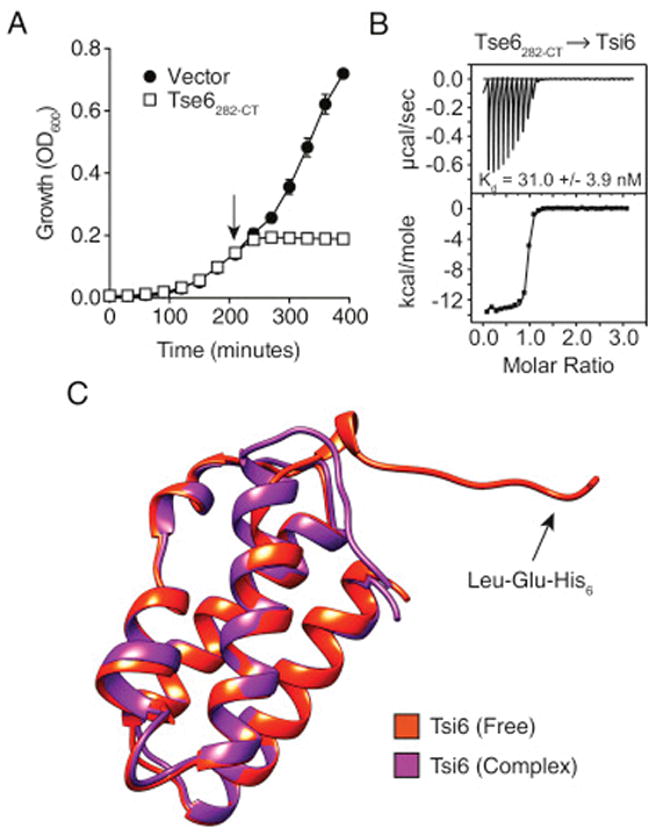
Tse6 Is a Bacteriostatic Toxin
We previously found that Tse6 is an H1-T6SS-dependent antibacterial effector that requires vgrG1 for intercellular delivery (Whitney et al., 2014). We further demonstrated that the toxic activity of Tse6 resides in its C terminus and can be neutralized by expression of a cognate immunity protein, Tsi6 (Figures 1A and 1B). Additionally, sequence and structural prediction algorithms identify a PAAR domain (Tse6PAAR) flanked by transmembrane segments in the N terminus of the protein.
Figure 1. Tse6 Causes Stasis from the Cytoplasm of P. aeruginosa.
(A) Genomic context of vgrG1, tsi6, tse6, and eagT6 in P. aeruginosa PAO1. Locus tag numbers are provided below each gene. The color of each gene corresponds to the color of its encoded protein shown in subsequent figures.
(B) Domain organization of P. aeruginosa Tse6. The boundaries for the PAAR (residues 64–175) and toxin (residues 282–430) domains are indicated. Predicted transmembrane domains are shown as dark gray rectangles.
(C) Intoxication of P. aeruginosa by Tse6 severely reduces growth. Data were derived from single-cell analysis of a parental strain (ΔretS ΔsspB pPSV38::sspB) and a derivative depleted of Tsi6 (ΔretS ΔsspB tsi6-D4 pPSV38::sspB). Bin size is 20 min and is normalized to total cells (parental, n = 15,042; tsi6–D4, n = 5,568).
(D) Tsi6 depletion strains undergo Tse6-based toxicity independent of inter-cellular toxin delivery by a functional H1-T6SS. Patches of the indicated P. aeruginosa strains grown for 24 hr at 37°C under Tsi6 depletion-inducing (+IPTG) or non-inducing (−IPTG) conditions are shown. The parental strain is the same as in (C).
See also Movies S1 and S2 and Tables S2 and S3.
The toxin domain of Tse6 does not bear homology to characterized proteins. The majority of studied antibacterial T6S effectors act on structures that are important for cellular integrity (Russell et al., 2014). Accordingly, intoxication by these effectors promotes morphological changes and cell lysis (LeRoux et al., 2012). We examined P. aeruginosa cells undergoing Tse6-based intoxication via depletion of Tsi6. The H1-T6SS is quiescent in monoculture, thus we performed this and subsequent experiments in a background with activated expression of the system (ΔretS) (LeRoux et al., 2015). Single-cell analyses showed Tse6-intoxicated cells displayed a dramatic increase in division time, but generally maintained their structural integrity (Figure 1C; Movies S1 and S2). The markedly slower growth of these cells was also apparent macroscopically; strains depleted of Tsi6 failed to form visible colonies after 24 hr of incubation (Figure 1D). Depletion of Tsi6 from P. aeruginosa cells lacking H1-T6SS function (ΔtssM1), and thus the capacity to transport effectors intercellularly, yielded indistinguishable effects, indicating that the toxin domain of Tse6 accesses the cytoplasm of donor cells prior to export.
Tse6 Resembles Mono-ADP-Ribosyltransferase Toxins
To gain further insight into Tse6 function, we determined the 1.4 Å resolution crystal structure of its C-terminal toxin domain (residues 282–430, Tse6282-CT) in complex with Tsi6 (Figure 2A; Table S1). Importantly, expression of Tse6282-CT alone induced stasis in Escherichia coli, recapitulating the phenotype of the full-length toxin in P. aeruginosa (Figure S1A). Tse6282-CT adopts a mixed α/β fold comprised of two N-terminal α helices and a central core that is formed by two perpendicularly oriented β sheets (Figure 2A). A search of the PDB using DALI indicated that the closest structural homologs of Tse6282-CT are the catalytic domains of bacterial mono-ADP-ribosyltransferase (mART) toxins (Holm and Rosenström, 2010; Simon et al., 2014). Identified members of this family include diphtheria toxin (DT) from Corynebacterium diphtheriae (Z score, 4.7; Cα root-mean-square deviation [RMSD] of 4.3 Å over 87 equivalent positions) and Exotoxin A (ExoA) from P. aeruginosa (Z score, 3.0; Cα RMSD of 2.8 Å over 73 equivalent positions). These and other characterized bacterial mART enzymes are secreted virulence factors that transfer the ADP-ribose moiety of NAD+ onto eukaryotic proteins, typically leading to target protein inactivation, dramatic changes in cellular physiology, and, often, cell death (Simon et al., 2014).
Figure 2. The Toxin Domain of Tse6 Adopts a mART Fold and Harbors a Putative NAD+ Binding Site.
(A) Overall structure of the Tse6282-CT–Tsi6 complex. Tse6282-CT is shown in ribbon (left) and space-filling (right) representations. Secondary structure elements are labeled. Dots denote a disordered segment (amino acids 400–408) of Tse6282-CT that was not modeled.
(B) Tse6282-CT resembles mART toxins. Structural alignment of Tse6282-CT with the catalytic domain of diphtheria toxin (PDB: 4AE1). Inset shows a structural alignment of the three conserved NAD+ binding residues (circled numbers) of diphtheria toxin and Tse6. The numbers correspond to amino acid positions within Tse6.
(C) Tsi6 interacts with the putative NAD+ binding pocket of Tse6. Structural alignment of free Tsi6 and Tsi6 bound to Tse6282-CT. The structure of Tsi6 does not change significantly upon complex formation (e.g., Glu63), except for Lys62, which rotates ~120° and interacts with Gln413 of Tse6.
See also Figure S1 and Tables S1–S3.
Despite a high degree of sequence divergence within mART proteins, they possess a structurally conserved β sheet core that harbors the molecular determinants for NAD+ binding (Fieldhouse et al., 2010; Zhang et al., 2014). Structural alignment of Tse6282-CT with DT shows that peripheral secondary structure elements differ significantly, while the two β sheets that comprise the core overlay well (Figure 2B). Characterized mART enzymes are subdivided into two main groups based on the identities of amino acid residues at three positions involved in NAD+ binding (Fieldhouse and Merrill, 2008). Members of the DT group use His, Tyr, and Glu, whereas Cholera toxin-type (CT) proteins retain Glu at position 3, but use Arg and Ser at positions 1 and 2, respectively. Strict conservation of the glutamate between the two groups may reflect its role in stabilizing the oxocarbenium intermediate that forms upon nicotinamide dissociation from ADP-ribose during the catalytic cycle (Yates et al., 2006). Our structure indicates that Tse6 residues differ from those of both mART groups at each position involved in NAD+ binding, including the placement of a non-acidic residue at position 3 (Gln413) (Figure 2B). Nonetheless, the pocket lined by these residues is the principal site of Tsi6 binding, suggesting its importance for the toxic activity of Tse6. In total, our structuralanalyses suggest that Tse6282-CT is a mART fold enzyme with unique substrate binding and catalytic motifs.
Tsi6 assumes an all α-helical fold that arranges into a four-helix bundle (Figure 2A). A search of the PDB indicates that Tsi6 shares structural similarity with several proteins of unknown function including Nmul_A1745 from Nitrosospira multiformis (Z score, 10.4; Cα RMSD of 2.2 Å over 86 equivalent positions) and PA2107 from P. aeruginosa (Z score, 9.5; Cα RMSD of 2.1 Å over 82 equivalent positions). The Tse6282-CT–Tsi6 interaction involves extensive contacts between α3 of Tsi6 and the putative NAD+ binding pocket of Tse6282-CT. Interface analysis indicates that complex formation between Tse6282-CT and Tsi6 buries 1,348 Å2 of solvent-accessible surface area. Isothermal titration calorimetry (ITC) measurements yielded a dissociation constant of 31 nM for the complex (Figure S1B).
To identify the conformational changes within Tsi6 required for inhibition of Tse6 activity, we determined the 1.9 Å crystal structure of Tsi6 in isolation (Table S1). Overall, the structure of free Tsi6 does not differ significantly from that of Tsi6 in complex with Tse6282-CT (Cα RMSD of 0.4 Å) (Figure S1C). This includes amino acid side chains of Tsi6 involved in the interaction with Tse6282-CT, with the notable exception of Lys62, which rotates approximately 120° around Cβ to form a hydrogen bond with the putative NAD+ binding residue at position 3, Gln413 (Figure 2C). Taken together, our structural data suggest that Tsi6 inhibits the activity of Tse6 through direct occlusion of its putative NAD+ binding site.
Tse6 Exhibits NAD(P)+ Glycohydrolase Activity
The structure of Tse6282-CT implies that the toxin may exert its effects within recipient cells via mono-ADP-ribosylation of an unknown bacterial protein. An important feature of characterized mART enzymes that facilitates their transferase activity is an open active site that allows docking of the acceptor protein. This concept is exemplified by the co-crystal structure of P. aeruginosa ExoA in complex with eukaryotic elongation factor 2 (Figure 3A) (Jørgensen et al., 2005). However, structural super-position of Tse6282-CT with ExoA predicts a steric clash between Tse6282-CT and a proteinaceous ADP-ribose acceptor (Figure 3B). Interestingly, the structural element of Tse6282-CT that prohibits accommodation of a high-molecular-weight acceptor is comprised of a motif conserved among Tse6 orthologs ([K/R]STxxPxxDxx[S/T]), implying that this region is important for Tse6 function (Zhang et al., 2012). Consistent with these data, incubation of purified Tse6 with P. aeruginosa or E. coli cell lysates containing 32P-NAD+ did not lead to observable transfer of 32P-ADP-ribose to a protein target (data not shown).
Figure 3. Tse6 Is an NAD(P)+ Glycohydrolase Toxin.

(A) mART toxins possess open active sites that allow for docking of their protein targets. Co-crystal structure of P. aeruginosa ExoA and eukaryotic elongation factor 2 (eEF2) from Jørgensen et al. (2005). The diphthamide moiety of eEF2 that is ADP-ribosylated by ExoA is shown in pink as a stick representation.
(B) Structural superposition of Tse6282-CT with ExoA predicts a steric clash with eEF2. The clash occurs through the conserved [K/R]STxxPxxDxx [S/T] motif of Tse6 (red).
(C and D) Tse6282-CT exhibits NAD(P)+ glycohydrolase activity. Rate of NAD+ (C) and NADP+ (D) consumption by purified Tse6282-CT in the presence and absence of Tsi6. Each enzyme concentration was assayed in triplicate, and error bars represent ± SD.
(E) Mass spectra of the products generated by Tse6-catalyzed breakdown of NAD+. Peaks corresponding to nicotinamide ([M+H]+, m/z = 123.1) and ADP-ribose ([M−H]−, m/z = 558.3) were identified in the reaction containing Tse6282-CT, whereas NAD+ ([M+H]+, m/z = 664.4) was identified in the reaction containing the Tse6282-CT–Tsi6 complex.
(F) NAD(P)+ levels in E. coli cells expressing Tse6282-CT (−Tsi6) or co-expressing Tse6282-CT and Tsi6 (+Tsi6) relative to empty vector. Cellular NAD(P)+ levels were assayed 60 min after induction of Tse6282-CT expression.
(G) Relative NAD(P)+ levels in the indicated P. aeruginosa strains 45 min after induction of Tsi6 degradation. Strains correspond to those used in Figure 1C. Error bars represent ± SD (n = 3).
See also Figure S2 and Tables S2 and S3.
Given the limited accessibility of the Tse6 active site, we hypothesized that the protein might instead function as an NAD+ glycohydrolase. Although it is less common within the mART superfamily of enzymes, NAD+ glycohydrolase activity has been observed for the SPN toxin of Streptococcus pyogenes (Ghosh et al., 2010). To test our hypothesis that Tse6 is an NAD+ glycohydrolase enzyme, we performed kinetic analyses of NAD+ consumption by Tse6282-CT. Whereas mART enzymes exhibit only low levels of NAD+ hydrolysis (<10 min−1), we found that purified Tse6282-CT catalyzes NAD+ breakdown at a rate of approximately 1.2 × 105 min−1 (Figure 3C) (Ghosh et al., 2010). This activity was reduced to background by the addition of 1.5 molar equivalents of Tsi6 to the reaction mixture, suggesting NAD+ degradation is a physiologically relevant activity of the toxin. Given the structural similarity between NAD+ and its phosphorylated derivative NADP+, we also tested the ability of Tse6282-CT to consume NADP+. Breakdown of this dinucleotide occurred at a comparable rate (6.0 × 104 min−1), suggesting that Tse6 degrades both NAD+ and NADP+ (NAD(P)+) (Figure 3D).
Rather than hydrolytically cleaving their substrates, some NAD+-degrading enzymes generate a cyclic product that is a characterized signaling molecule in eukaryotes (Guse, 2000). The fluorescence assay we employed does not distinguish between cyclized and non-cyclized forms of ADP-ribose, thus we used mass spectrometry (MS) to analyze the reaction products of Tse6 and NAD+. Nicotinamide and ADP-ribose were the only detectable products, defining Tse6 as an NAD(P)+-glycohydrolase enzyme (Figure 3E).
Tse6 Induces Bacteriostasis by Depleting Cellular NAD(P)+ Levels
Our biochemical data show that Tse6 rapidly hydrolyzes NAD(P)+ in vitro; however, it is possible that we observe this activity due to the absence of an appropriate ADP-ribose acceptor molecule. To address this possibility, we expressed Tse6282-CT in E. coli and measured endogenous NAD(P)+ levels. Upon induction of Tse6282-CT expression, we found that E. coli cells contained vastly reduced cellular concentrations of NAD+ and NADP+ relative to the vector control (Figure 3F). Co-expression with Tsi6 restored NAD(P)+, indicating that the loss of the dinucleotides is a direct consequence of Tse6282-CT activity.
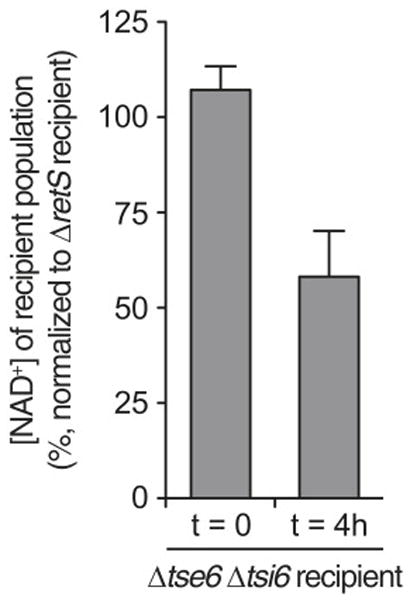
Next, we sought to measure the influence of endogenous Tse6 on NAD(P)+ levels in intoxicated P. aeruginosa cells. In agreement with our findings in E. coli, intracellular intoxication caused by depletion of Tsi6 led to a profound decrease in NAD(P)+ (Figure 3G). The precise measurement of Tse6-catalyzed NAD(P)+ depletion during intercellular intoxication is complicated by high background levels of the dinucleotides derived from donor cells, which as intoxication of recipient cells proceeds, constitute an increasingly large proportion of the total cellular population. To partially overcome this, we examined a time point at which recipient cells have begun to experience intoxication, but are not yet depleted from the population. Comparing total NAD+ levels in conjunction with donor and recipient colony-forming units (CFU) to a reference experiment, we confirmed a significant reduction in NAD+ within recipient cells (Figure S2). Based on these findings, we propose that the toxicity elicited by Tse6 is due to depletion of cellular NAD(P)+ pools.
Tse6 Participates in a Five-Protein Complex Containing Elongation Factor Tu
Bioinformatic analyses predict that Tse6 is a PAAR domain-containing integral membrane protein (Figure 1B). To test whether Tse6 resides in membranes, we generated a P. aeruginosa strain producing a functional fusion of Tse6 to the vesicular stomatitis virus glycoprotein epitope from the native tse6 locus (tse6-V) (Figure S3A). Western blot analysis of the soluble and membrane fractions of this strain revealed that despite high-confidence prediction of transmembrane domains within Tse6, the majority of the protein is soluble (Figure 4A). Based on structural studies of PAAR domains in complex with VgrG-like chimeras, it has been speculated that effectors containing this domain interact with VgrG proteins (Shneider et al., 2013). We hypothesized that Tse6 could be solubilized within donor cells by virtue of association with VgrG1 via its PAAR domain. Indeed, in the absence of VgrG1, we observed significant repartitioning of Tse6 to the membrane fraction of cells. Tse6 remained soluble in a strain lacking tssM1, indicating that the localization of the toxin is not generally sensitive to T6 function.
Figure 4. Tse6 Participates in a Multi-protein Complex and Binds Helix D of EF-Tu through Residues N-Terminal to Its Toxin Domain.

(A) Tse6 is a membrane protein that is solubilized by VgrG1. Western blot analysis of the soluble (S) and membrane (M) fractions of the indicated P. aeruginosa strains. Tse1 and OprF serve as soluble and membrane controls, respectively.
(B) Silver-stained SDS-PAGE analysis of proteins enriched by anti-VSV-G immunoprecipitation from P. aeruginosa strains encoding Tse6 (parental) and Tse6-V. The labels indicate the identities of proteins that specifically co-precipitate with Tse6-V as determined by MS. In addition to their monomeric forms, VgrG1 and Tse6 form a high-molecular-weight complex that is resistant to heat and SDS denaturation.
(C) A 17-amino-acid segment of Tse6 mediates interaction with EF-Tu. Coomassie-stained SDS-PAGE analysis of purified Tse6 truncations. All truncations were expressed with Tsi6 and assessed for co-purification with endogenous EF-TuEC.
(D) Overall structure of the Tse6265-CT–EF-TuPA complex. Secondary structure elements involved in the interaction are labeled.
(E) The Tse6 activation loop harbors Asp396 and rotates toward the active site of Tse6 in the Tse6265-CT –EF-TuPA structure relative to its position in the Tse6282-CT–Tsi6 structure. Dots denote a disordered segment (amino acids 400–408) of Tse6282-CT that was not modeled.
(F) Tse6282-CT D396A exhibits significantly reduced NAD(P)+ glycohydrolase activity. Rate of NAD+ (left) and NADP+ (right) consumption by purified Tse6282-CT D396A.
(G) Asp396 is critical for Tse6-based intercellular toxicity. Growth competition experiments between the indicated P. aeruginosa donor and recipient strains. Donor and recipient strains were mixed 1:1, grown for 24 hr on solid media, and differentiated using blue/white screening.
(H) Helix D of EF-Tu is the site of interaction for both Tse6265-CT (left) and the guanine exchange factor EF-Ts (right). In all panels, error bars represent ± SD (n = 3).
See also Figures S3 and S4 and Tables S1–S3.
Motivated by the finding that Tse6 is a soluble protein in the presence of VgrG1, we used co-immunoprecipitation to probe for a physical interaction between the proteins. Surprisingly, this led to the identification of a putative complex containing Tse6, Tsi6, VgrG1, PA0094, and translation elongation factor Tu (EF-Tu) (Figure 4B). PA0094 is a member of a recently described group of effector-specific accessory factors that facilitate delivery of their cognate effectors (Alcoforado and Coulthurst, 2015). Henceforth, we refer to PA0094 as effector-associated gene with tse6 (EagT6).
The identification of EF-Tu in a complex containing Tse6 was unexpected. This conserved bacterial protein is a GTPase that delivers newly charged aminoacyl-tRNA molecules to the ribosome during translation elongation (Voorhees and Ramakrishnan, 2013). Interactions between T6 effectors and essential bacterial proteins have not been described; therefore, we decided to probe the functional significance of this observation. To test whether Tse6 and EF-Tu interact directly, we initiated experiments to evaluate P. aeruginosa EF-Tu (EF-TuPA) binding to the soluble region of Tse6 (Tse6222-CT) in vitro. Interestingly, during the course of this work, we noted Tse6222-CT associates with E. coli EF-Tu (EF-TuEC), which is 88% identical to EF-TuPA. Purification of N-terminal Tse6 truncations narrowed the region responsible for EF-Tu binding to the last 165 amino acids of the toxin (Figure 4C). Since EF-Tu does not bind the glycohydrolase domain of Tse6 (residues 282-CT), we reasoned that the interaction with EF-Tu requires amino acids 265–282 of the toxin. Despite considerable sequence divergence from P. aeruginosa Tse6, orthologs of the toxin from P. putida and P. syringae also co-purified with EF-TuEC (Figures S3B and S3C).
Next, we measured the binding affinity of Tse6265-CT to EF-TuEC and EF-TuPA using ITC. Guanosine triphosphate (GTP) hydrolysis by EF-Tu is coupled to significant structural changes in the protein; therefore, we investigated both GTP- and GDP- (EF-Tu•GDP) bound conformations of the molecule (Clark and Nyborg, 1997). In line with our purification results, we found that Tse6265-CT interacts tightly with both EF-TuEC•GDP (Kd =81 nM) and EF-TuPA•GDP (Kd = 23 nM) (Figures S3D and S3E). Application of the antibiotic Aurodox, which locks EF-Tu into its GTP-bound conformation, reduced the affinity for Tse6 by approximately 10-fold (Figure S3F) (Vogeley et al., 2001). In summary, these data indicate that Tse6 binds directly to the GDP form of EF-Tu within a larger, multiprotein complex.
Structure of the Tse6-EF-Tu Complex
Though all cells require NAD(P)+, the process of translation does not rely on these co-factors. Thus, the significance of Tse6 interaction with EF-Tu was not apparent. As a first step toward defining the relevance of the Tse6-EF-Tu complex, we determined the 3.5 Å crystal structure of Tse6265-CT bound to EF-TuPA•GDP (Figure 4D; Table S1).
Overall, the structure of Tse6265-CT is highly similar to Tse6282-CT (Cα RMSD of 0.7 Å). The most striking divergence between the two structures is the ordering and 60° hinge-like movement of the [K/R]STxxPxxDxx[S/T] motif-containing loop, henceforth referred to as the Tse6 activation loop. This results in a ~15- Å displacement of Asp396 that directs its side chain into the putative NAD(P)+ binding site (Figure 4E). Asp396 is the sole invariant acidic residue among Tse6 orthologous proteins, leading us to postulate that it serves a role analogous to the conserved glutamic acid at position 3 within the DT and CT mART families (Figure S4) (Zhang et al., 2012). Consistent with this hypothesis, purified Tse6282-CTD396A displayed approximately 225-fold reduced NAD(P)+ glycohydrolase activity relative to the wild-type protein, and a P. aeruginosa strain producing Tse6D396A from the native tse6 locus did not exhibit Tse6-based intercellular intoxication (Figures 4F and 4G).
In accordance with our Tse6 truncation studies, interaction with EF-TuPA is mediated by residues immediately N-terminal to the toxin domain (residues 265–291). This basic segment forms two α helices (α0 and α1) that engage in numerous salt bridges with acidic side chains on the GTPase domain (G domain) of EF-TuPA. Interestingly, the EF-TuPA residues involved in this interaction are found on helix D, which functions as the key interaction site for both the guanine exchange factor EF-Ts and the ribosome (Figure 4H) (Kawashima et al., 1996).
Interaction with EF-Tu Is Required for the Delivery of Tse6 into Recipient Cells
Our structure of Tse6265-CT–EF-TuPA shows that a spatially confined cluster of electrostatic interactions facilitates binding of the proteins (Figures 5A and 5B). We reasoned that this interaction mechanism might afford an opportunity to dissect the functional significance of the interaction via site-directed mutagenesis. A non-conserved leucine residue (Leu270) was identified within the patch of basic amino acids on α0 that mediate EF-Tu binding (Figure 5C). We postulated that an acidic residue substituted at this position would disrupt charge complementarity between the proteins. As predicted, Tse6222-CTL270E did not co-purify with EF-TuEC, whereas variants containing a more conservative substitution at this site (L270A) or an analogous substitution on the opposite face of α0 (A268E) retained EF-TuEC binding (Figure 5D).
Figure 5. Interaction with EF-Tu Is Required for Tse6-Based Intercellular Toxicity.
(A and B) An electrostatic patch mediates interaction between EF-Tu and Tse6265-CT. Electrostatic surface representation of EF-TuPA (A) and Tse6265-CT (B). Residues participating in the interaction are labeled and outlined in black.
(C) Close-up view of the Tse6265-CT –EF-TuPA interaction. Secondary structure elements referred to in the text are labeled.
(D and E) An L270E variant of Tse6 does not interact with EF-TuEC or EF-TuPA. (D) Coomassie-stained SDS-PAGE analysis of purified Tse6265-CT variants. All variants were expressed with Tsi6 and assessed for their ability to co-purify with endogenous EF-TuEC. (E) Silver-stained SDS-PAGE analysis of proteins enriched by anti-VSV-G immunoprecipitation from P. aeruginosa strains encoding Tse6-V (parental) and Tse6-VL270E. Enriched low-molecular-weight proteins (EagT6 and Tsi6) not shown.
(F) Tse6 requires interaction with EF-Tu to intoxicate recipient cells. Outcome of growth competition experiments between the indicated P. aeruginosa donor strains and a parental (ΔretS) or Tse6-susceptible (Δtse6 Δtsi6) recipient. The competitive index is calculated as the change (final/initial) in ratio of donor to recipient CFU.
(G) Interaction with EF-TuPA does not enhance NAD+ glycohydrolase activity of Tse6222-CT. Reactions were performed using 500 pM Tse6222-CT in the presence or absence of 1 μM EF-TuPA.
(H) Interaction with EF-Tu is not required for Tse6-based intracellular intoxication. NAD+ levels in the indicated P. aeruginosa strains 45 min after induction of Tsi6 degradation (top). Patches of the indicated P. aeruginosa strains grown for 24 hr at 37°C under Tsi6 depletion-inducing (+IPTG) conditions (bottom). The parental strain is the same as in Figure 1C. Error bars represent ± SD (n = 3).
See also Figure S5 and Tables S2 and S3.
Encouraged by our in vitro data, we next generated a P. aeruginosa strain expressing Tse6L270E-V from the native tse6 locus. An immunoprecipitation and growth competition experiment utilizing this strain showed that Tse6L270E-V displays a specific defect in EF-Tu interaction and is unable to intoxicate recipient cells (Figures 5E and 5F). We conclude that association with EF-Tu is essential for Tse6-based toxicity.
Tse6-based intercellular intoxication can be viewed as a number of discrete processes. We considered the involvement in and requirement for EF-Tu in (1) the stability of Tse6, (2) the enzymatic activity of Tse6, (3) Tse6 export from donor cells, and (4) entry of Tse6 into recipient cells. Since Tse6L270E is present at equal concentrations as the wild-type protein, we ruled out a requirement for EF-Tu in Tse6 stability. Our biochemical data show that the catalytic domain of Tse6 degrades NAD+ rapidly, at a rate consistent with a known cytotoxic NAD+ glycohydrolase enzyme (Ghosh et al., 2010). Therefore, one possibility is that residues N-terminal to the toxin domain are auto-inhibitory and that EF-Tu binding to this region relieves this inhibition. Indeed, the activation loop of Tse6 differs significantly in position between the Tse6282-CT–Tsi6 and Tse6265-CT–EF-TuPA structures, suggesting that either EF-Tu induces a conformational change in the toxin or immunity protein binding excludes this loop from the active site (Figure 4E). We found that a purified Tse6 variant that includes the EF-Tu binding region (Tse6222-CT) catalyzes NAD+ hydrolysis at a rate indistinguishable to the toxin domain alone (Figure 5G). Furthermore, the activity of this protein was unaffected by the addition of excess EF-TuPA.
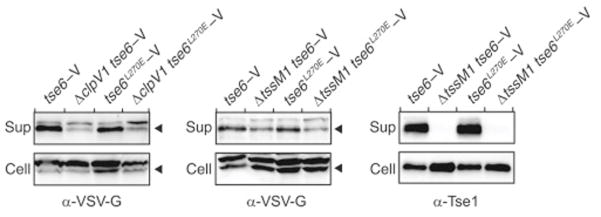
Next, we considered the possibility that association of Tse6 with EF-Tu is required for export of the toxin from donor cells. However, we found that both cellular and extracellular levels of Tse6L270E-V are similar to the wild-type protein (Figure S5). These experiments further showed that unlike the Hcp-associated effector Tse1, Tse6 accumulation in the exo-proteome of P. aeruginosa is only partially dependent on H1-T6SS function. The significance of this is not yet understood; however, strains lacking the T6S ATPase ClpV1 or the core integral membrane protein TssM1 yielded similar results.
Since interaction with EF-Tu is dispensable for Tse6 catalytic activity and export, we deduced that interaction with the translation factor must be required for Tse6 to reach the cytoplasm of recipient cells. In further support of this contention, we found Tse6L270E-V is as active in intracellular intoxication triggered by Tsi6 depletion as the wild-type protein (Figure 5H). Together with our findings that Tse6L270E-V is incapable of Tse6-based intercellular intoxication despite its unencumbered transit of the T6SS, these data indicate that interaction with EF-Tu grants Tse6 access to the cytoplasm of recipient cells.
Ultrastructure of a T6 Effector-VgrG Complex
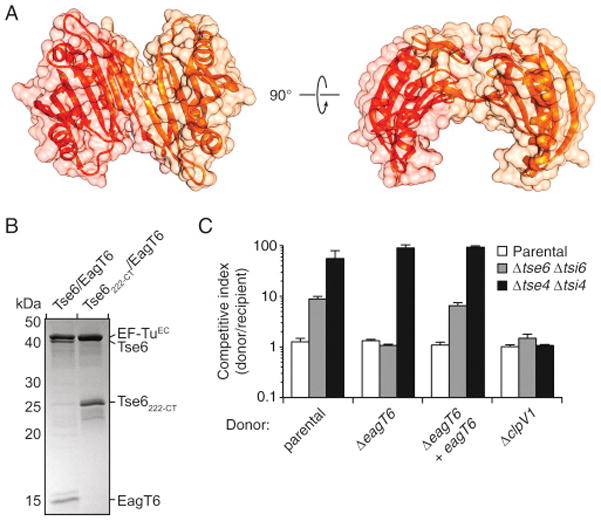
VgrG is thought to serve as the T6S protein that pierces the outer membrane of recipient cells, granting its bound cognate effector(s) access to the periplasm of target cells (Silverman et al., 2012). The PAAR domain of effectors associates with the tip of VgrG; however, the placement of additional effector domains, as well as accessory proteins, in this particle is not known (Shneider et al., 2013). To gain insight into the topology of an effector-loaded VgrG complex, we examined purified C-terminally octa-histidine-tagged Tse6 in complex with Tsi6, VgrG1, EagT6, and EF-TuEC using negative-stain electron microscopy (EM) (Figures 6A and 6B, left).
Figure 6. Two Conformations of the Tse6 Secretory Particle Revealed by Electron Microscopy.

(A and B) Addition of detergent dissociates EagT6 from the Tse6 secretion particle and causes a conformational change. (A) Coomassie-stained SDS-PAGE analysis and (B) representative class averages of purified Tse6-containing complex in the presence and absence of 0.03% β-D-dodecylmaltopyranoside.
(C) 3D density map and molecular fitting of the Tse6-Tsi6-VgrG1-EagT6-EF-TuPA complex. The identity of each subunit is indicated. The model for Tse6PAAR was generated using Phyre (Kelley and Sternberg, 2009).
(D) Tse6 requires EagT6 for intracellular accumulation. Western blot analysis of Tse6 levels in the indicated P. aeruginosa strains. RNA polymerase (RNAP) is used as a loading control.
(E) 3D density map and molecular fitting of the detergent-bound Tse6–Tsi6–VgrG1–EF-TuPA complex. Scale bars, 20 nm.
See also Figures S6 and S7 and Tables S2 and S3.
Analysis of 12,000 single particles permitted the calculation of a 3D map of the complex resolved to 22 Å (Figures 6C and S6A–S6D). VgrG proteins have a characteristic structure that was readily apparent within the map (Shneider et al., 2013). Fortuitously, the unpublished X-ray crystal structure of P. aeruginosa VgrG1 is available in the PDB (PDB: 4MTK); the location of this trimeric assembly in our structure was unambiguous. For estimating the placement of Tse6, Tsi6, and EF-Tu, we were able to utilize our assorted high-resolution structures of these proteins to produce a ternary complex that largely conformed to regions of density found near the end of the complex predicted to initiate contact with recipient cells. An additional constraint on the location of Tse6 is its PAAR domain, which could be modeled with high confidence bound to the tip of VgrG1 (Figure 6C). The 91 residues connecting Tse6PAAR to the first residue included in our modeled ternary complex (residue 265) are predicted to form a transmembrane helix, followed by a disordered glycine-rich span (not modeled). Ni-NTA-nanogold labeling of the C-terminal His8-tag of Tse6 provided support for our placement of this linchpin protein within the complex (Figure S6E).
The positions of VgrG, Tse6, Tsi6, and EF-TuPA left a protruding region of density surrounding the PAAR domain of Tse6 unoccupied. We postulated that this region of the map corresponds to EagT6. The structure of EagT6 was determined in a high-throughput X-ray crystallography project and made publicly available. The domain-swapped homodimeric protein bears a distinctive horseshoe configuration that we placed in this unoccupied density (Figures 6C and S7A). In this configuration, EagT6 would be predicted to bind Tse6PAAR and its buttressing hydrophobic segments. To test this prediction, we performed co-purification experiments of the proteins in E. coli. In contrast to the complex isolated with full-length toxin, Tse6222-CT did not co-purify with EagT6 (Figure S7B). Although the X-ray crystal structure of EagT6 does not fully agree with the calculated map in this region, these data provide biochemical support for our placement of EagT6 in proximity to the N-terminal domains of Tse6.
To garner additional insight into EagT6 function, we probed the capacity of P. aeruginosa ΔeagT6 to intoxicate Tse6-sensitive recipient cells. This strain failed to elicit Tse6-based toxicity, but retained the capacity to intoxicate recipients using another H1-T6S effector (Figure S7C). EagT6 associates with a region of Tse6 rich in transmembrane domains, suggesting that the accessory factor could act as a chaperone for the toxin. As predicted for substrate-chaperone systems, we found that accumulation of Tse6 is markedly diminished by the absence of EagT6 (Figure 6D). Genetic complementation of this phenotype was achieved with ectopic expression of eagT6. Taken together with the findings of Alcoforado and Coulthurst (2015) pertaining to a EagT6-related protein in Serratia, we propose that EagT6 functions as a Tse6-specific chaperone.
Tse6 is a transmembrane protein; thus, its transport between cells likely requires shielding of its hydrophobic domains. Based on its orientation and position relative to Tse6 in our model, we posited that EagT6 might chaperone Tse6 by shielding its hydrophobic segments from aqueous mediums during intercellular transport. In support of this hypothesis, EagT6 dissociates from the effector complex in the presence of detergent (Figure 6A, right). To gain structural insight into the consequence of EagT6 release, we examined the Tse6 particle depleted of this protein by negative-stain EM and single-particle reconstruction (Figure 6B, right). Analysis of 11,000 particles permitted the calculation of a 3D map of the complex resolved to 19 Å (Figures 6E and S6F–S6J). Remarkably, we observed a ~40 Å movement of the EF-Tu–Tse6265-CT–Tsi6 sub-complex from VgrG-Tse6PAAR. The localization of Tse6265-CT within the displaced density was verified by Ni-NTA-nanogold labeling (Figure S6K). Accompanying this reorganization, we observed a region of unoccupied density in proximity to the predicted site of the hydrophobic segments of Tse6. Given the capacity of detergent to compete for EagT6 binding to the complex, we hypothesize that ordered detergent molecules bound to the hydrophobic domains of Tse6 occupy this density.
The significance of Tsi6 and EF-Tu within the effector complex is not understood. The toxicity conferred by depletion of Tsi6 from donor cells shows that the catalytic domain of Tse6 is present in the cytoplasm and is in complex with Tsi6 prior to export by the H1-T6SS. Therefore, EF-Tu also likely interacts with the toxin prior to export. If these proteins do not dissociate from the toxin during transit, the structures we obtained would represent the secreted complex. Alternatively, Tsi6 and EF-Tu could be removed during secretion and re-engage the complex in the cytoplasm of recipient cells. In this scenario, the complex we isolated would represent that found in recipient cells with immunity. In total, our ultrastructural analyses of the Tse6 secretory particle define the architecture of an effector-loaded VgrG and suggest a mechanism for deployment of a membrane-associated toxin (Figure 7).
Figure 7. Proposed Model for Tse6 Transport by the T6S Apparatus.
The configuration of the Tse6 particle subunits in donor and recipient cells represent those determined in this study in the absence and presence of detergent, respectively. A representative EM class average for each is provided for reference. In the donor cell, Tse6 and associated proteins are bound to the T6S sheath complex (gray; PDB: 3J9O) (Clemens et al., 2015). The T6 trans-envelope complex (composed of TssL, M, and J) is schematized to approximate its recently determined EM structure (Durand et al., 2015), whereas the T6 baseplate-like assembly is depicted in filled gray. In the model, donor cell EF-Tu (light blue) and Tsi6 disengage from the Tse6 secretion particle upon export from the donor cell. Upon crossing the outer membrane (OM) of the recipient cell, EagT6 dissociation frees the hydrophobic domains of Tse6 (yellow rectangles) for incorporation into the recipient inner membrane (IM). Recipient cell EF-Tu (dark blue) facilitates transfer of the NAD(P)+ glycohydrolase domain into the recipient cell cytoplasm by an unknown mechanism. Several other possibilities consistent with all available data are not presented. Most notably, donor-cell-derived EF-Tu may be exported as part of the secretion particle and facilitate Tse6 delivery into recipient cells, or donor-cell-derived EF-Tu may be excluded from the secretion complex.
See also Tables S2 and S3
DISCUSSION
We have discovered that Tse6 intoxicates recipient cells by catalyzing the hydrolytic removal of the nicotinamide moiety from NAD+ and NADP+. This mechanism has not been described for an interbacterial toxin, but it is consistent with the general observation that T6 effectors act on target molecules that are both essential and highly conserved among bacteria (Russell et al., 2014). The consequence of NAD(P)+ degradation by Tse6 is stasis in most cells, rather than death. The relative benefit(s) of inhibiting the growth of target cells is not yet understood; however, the H1-T6SS Tse2 toxin also induces stasis in recipients (Li et al., 2012). In instances of self-intoxication, the exchange of bacteriostatic toxins could promote the formation of persister cells. A non-mutually exclusive possibility is that when delivered within an effector cocktail, bacteriostatic and bacteriocidal toxins act synergistically.
The requirement for NAD(P)+ extends to all forms of life, raising the possibility that Tse6, and related proteins exported by the T6SS, could intoxicate archaeal and eukaryotic cells. The SPN toxin of S. pyogenes provides precedent for the action of a bacterial NAD+ glycohydrolase against a eukaryotictarget, although this is a structurally distinct toxin that utilizes pores introduced by Streptolysin O to gain entry into host cells (Madden et al., 2001; Smith et al., 2011). The H3-T6SS of P. aeruginosa has been shown to deliver a phospholipase D toxin to both bacterial and eukaryotic cells, implying that there is not a fundamental barrier to inter-domain targeting of effectors by this bacterium (Jiang et al., 2014).
To our knowledge, the requirement for a housekeeping protein in the function of a T6S effector has not previously been observed. Likely owing to the central role of EF-Tu in translation, tufA, which encodes EF-Tu, is a slowly evolving bacterial gene (Lathe and Bork, 2001). Thus, if the role of the Tse6 interaction with a cellular housekeeping protein is to grant the toxin access to recipient cells as our data suggest, EF-Tu would allow the toxin to target phylogenetically diverse bacteria. The high concentration of EF-Tu within cells could also contribute to a wide target range for Tse6, as there would be more tolerance for weakened association between the two proteins driven by EF-Tu sequence divergence.
Although the site of Tse6 binding to EF-Tu would preclude binding of the translation factor to EF-Ts, it is unlikely that Tse6 affects translation (Kawashima et al., 1996). We find Tse6 present at low levels in P. aeruginosa; therefore, the yet lower levels within recipient cells would not sequester a functionally significant portion of EF-Tu. Indeed, there is growing evidence that the large pool of EF-Tu is exploited for multiple purposes within bacteria, including P. aeruginosa (Balasubramanian et al., 2008; Barel et al., 2008; Defeu Soufo et al., 2010; Kunert et al., 2007; Mohan et al., 2014). Barbier et al. (2013) have found that EF-TuPA is posttranslationally modified by trimethylation at Lys5. These authors also found that this form of the protein localizes to the cell surface, where it mediates interactions with airway epithelial cells. Whether EF-Tu is actively secreted to the cell surface or if its presence there is a consequence of cell lysis was not determined. It is worth noting that the high concentration of EF-Tu present in culture supernatants through T6-independent mechanisms precluded measurement of the contribution of T6 to EF-Tu export in our study.
Given the changing chemical and physical environments that necessarily accompany translocation across multiple membranes, it is without doubt that effectors delivered intercellularly assume multiple states en route. We have captured just two of these for a T6S effector. Tse6 is the hub of a multi-protein complex in our structures; however, our biochemical data show that it need not interact with any of these proteins in order to catalyze NAD(P)+ degradation. This leaves many open questions, including how does EF-Tu facilitate Tse6 translocation into recipient cells? From our current data, we cannot determine whether EF-Tu derived from donor cells, recipient cells, or both is critical for Tse6 activity. One appealing model consistent with our data holds that Tse6 is delivered to the target cell periplasm, whereupon EagT6 is released and the exposed transmembrane segments of the toxin spontaneously insert into the inner membrane. At this point, translocation of residues N-terminal to the toxin domain and ensuing EF-Tu-binding could serve as a molecular ratchet that favors passage of the remaining toxin domain into the cytoplasm. Interestingly, the EF-Tu binding domain of Tse6 is rich in basic residues, a property of many known cell-penetrating peptides (Bechara and Sagan, 2013).
While this study provides two snapshots of interbacterial protein transport, it also highlights the challenges in understanding this intricate, multi-step process. The Tse6 particle we describe may provide a tractable system for the characterization of additional secretory intermediates. A complete understanding of toxin entry into recipient cells could define novel routes for the delivery of antimicrobials.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
All P. aeruginosa strains generated were derived from the sequenced strain PAO1 (Stover et al., 2000). P. aeruginosa mutants and chromosomal fusions were generated by allelic exchange as described previously (Hood et al., 2010). E. coli strains DH5α, BL21(DE3) pLysS, and SM10 were used for plasmid maintenance, gene expression, andconjugative transfer, respectively. A detailed list of strains and plasmids used in this study can be found in Tables S2 and S3.
Crystallization and Structure Determination
Details for the crystallization of Tse6282-CT–Tsi6, Tsi6, and Tse6265-CT–EF-Tu are described in the Supplemental Experimental Procedures. The structures of Tse6282-CT–Tsi6 and Tsi6 were solved by Se-SAD. The Tse6265-CT-EF-Tu structure was solved by molecular replacement using EF-TuEC•GDP (PDB: 1EFC) as a search model. Details for structure determination and model refinement are described in the Supplemental Experimental Procedures (Table S1).
Biochemical Assays
Hydrolysis rates of NAD(P)+ by Tse6 were measured using a fluorescence endpoint assay as described previously (Johnson and Morrison, 1970). Determination of relative NAD+ and NADP+ levels from cell lysates was performed using the NAD/NADH-Glo and NADP/NADPH-Glo bioluminescence assays, respectively, as per the instructions of the manufacturer (Promega). Details can be found in the Supplemental Experimental Procedures.
Bacterial Competition Assays
Intraspecific competition assays between P. aeruginosa strains were performed as previously described (Whitney et al., 2014). Briefly, overnight cultures of P. aeruginosa strains were mixed in a 1:1 ratio and spotted onto 0.2-μm nitrocellulose membranes overlaid on a 3% agar Luria broth no-salt plate. Competitive indices were calculated by enumerating donor/recipient CFU after 24 hr of growth at 37°C. All competitive indices were adjusted by the donor/recipient ratio in the initial inoculum. Recipient strains express the lacZ gene from a neutral phage attachment site to enable their differentiation from unlabeled donor via blue/white screening.
Electron Microscopy and Image Analysis
Protein samples were negatively stained with uranyl formate (SPI Supplies/Structure Probe) and imaged using a JEOL1400 microscope equipped with a LaB6 cathode operated at 120 kV. Images were recorded at a magnification of 50,000× on a 4k × 4k CMOS camera F416 (TVIPS). Data analysis and further processing was done in SPARX (Hohn et al., 2007). Details can be found in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Type VI secretion effector Tse6 acts by depleting bacteria of NAD+ and NADP+
Entry of Tse6 into target cells requires its binding to elongation factor Tu
Tse6 is a membrane protein that requires a chaperone for intercellular transport
EM structures reveal the mechanism for Tse6 deployment to recipient cells
Acknowledgments
The authors would like to thank H. Kulasekara for assistance with membrane fractionation, C. Outten for providing pRSFDuet-1, W. Catterall for use of the ITC instrument, J. Woodward for assistance with radioactivity experiments and for providing Aurodox, C. Ralston for assistance with X-ray data collection, I. Attree for providing α-OprF antibody, C. Gatsogiannis for electron microscopy expertise, and S. Dove, C. Goulding, C. Hayes, D. Low, A. Merz, D. Veesler, and members of the S.R. and J.D.M. laboratories for helpful discussions. This work was supported by grants from the NIH (AI080609) (to J.D.M.) and by the University of Maryland Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014) (to D.R.G.). J.C.W. was supported by a postdoctoral research fellowship from the Canadian Institutes of Health Research, D.Q. was supported by a Chemiefonds fellowship from the Fonds der Chemischen Industrie, S.S. was supported by a Mary Gates Research Scholarship, and J.D.M. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
ACCESSION NUMBERS
The accession numbers for the atomic coordinates of Tse6282-CT—Tsi6, Tsi6, and Tse6265-CT—EF-TuPA are PDB: 4ZV0, 4ZUY, and 4ZV4, respectively. The accession numbers for the negative-stain EM maps of the detergent-bound and detergent-free Tse6 secretion particle are EMDB: EMD-3112 and EMD-3113, respectively.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, three tables, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.09.027.
References
- Alcoforado DJ, Coulthurst SJ. Intra-species competition in Serratia marcescens is mediated by type VI secretion Rhs effectors and a conserved effector-associated accessory protein. J Bacteriol. 2015;197:2350–2350. doi: 10.1128/JB.00199-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Kannan TR, Baseman JB. The surface-exposed carboxyl region of Mycoplasma pneumoniae elongation factor Tu interacts with fibronectin. Infect Immun. 2008;76:3116–3123. doi: 10.1128/IAI.00173-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier M, Owings JP, Martínez-Ramos I, Damron FH, Gomila R, Blázquez J, Goldberg JB, Albertí S. Lysine trimethylation of EF-Tu mimics platelet-activating factor to initiate Pseudomonas aeruginosa pneumonia. MBio. 2013;4:e00207–e00213. doi: 10.1128/mBio.00207-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barel M, Hovanessian AG, Meibom K, Briand JP, Dupuis M, Charbit A. A novel receptor - ligand pathway for entry of Francisella tularensis in monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 2008;8:145. doi: 10.1186/1471-2180-8-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature. 2012;483:182–186. doi: 10.1038/nature10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- Clark BF, Nyborg J. The ternary complex of EF-Tu and its role in protein biosynthesis. Curr Opin Struct Biol. 1997;7:110–116. doi: 10.1016/s0959-440x(97)80014-0. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Ge P, Lee BY, Horwitz MA, Zhou ZH. Atomic structure of T6SS reveals interlaced array essential to function. Cell. 2015;160:940–951. doi: 10.1016/j.cell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defeu Soufo HJ, Reimold C, Linne U, Knust T, Gescher J, Graumann PL. Bacterial translation elongation factor EF-Tu interacts and colocalizes with actin-like MreB protein. Proc Natl Acad Sci USA. 2010;107:3163–3168. doi: 10.1073/pnas.0911979107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand E, Nguyen VS, Zoued A, Logger L, Péhau-Arnaudet G, Aschtgen MS, Spinelli S, Desmyter A, Bardiaux B, Dujeancourt A, et al. Biogenesis and structure of a type VI secretion membrane core complex. Nature. 2015;523:555–560. doi: 10.1038/nature14667. [DOI] [PubMed] [Google Scholar]
- Fieldhouse RJ, Merrill AR. Needle in the haystack: structure-based toxin discovery. Trends Biochem Sci. 2008;33:546–556. doi: 10.1016/j.tibs.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Fieldhouse RJ, Turgeon Z, White D, Merrill AR. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput Biol. 2010;6:e1001029. doi: 10.1371/journal.pcbi.1001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch MJ, Trunk K, Diniz JA, Guo M, Trost M, Coulthurst SJ. Proteomic identification of novel secreted antibacterial toxins of the Serratia marcescens type VI secretion system. Mol Cell Proteomics. 2013;12:2735–2749. doi: 10.1074/mcp.M113.030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh J, Anderson PJ, Chandrasekaran S, Caparon MG. Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: reevaluation of enzymatic properties associated with pathogenesis. J Biol Chem. 2010;285:5683–5694. doi: 10.1074/jbc.M109.070300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse AH. Cyclic ADP-ribose. J Mol Med. 2000;78:26–35. doi: 10.1007/s001090000076. [DOI] [PubMed] [Google Scholar]
- Hachani A, Allsopp LP, Oduko Y, Filloux A. The VgrG proteins are “à la carte” delivery systems for bacterial type VI effectors. J Biol Chem. 2014;289:17872–17884. doi: 10.1074/jbc.M114.563429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Singh P, Hsu F, Güvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010;7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Waterfield NR, Yang J, Yang G, Jin Q. A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe. 2014;15:600–610. doi: 10.1016/j.chom.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Johnson SL, Morrison DL. The alkaline reaction of nicotinamide adenine dinucleotide, a new transient intermediate. J Biol Chem. 1970;245:4519–4524. [PubMed] [Google Scholar]
- Jørgensen R, Merrill AR, Yates SP, Marquez VE, Schwan AL, Boesen T, Andersen GR. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature. 2005;436:979–984. doi: 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- Kawashima T, Berthet-Colominas C, Wulff M, Cusack S, Leberman R. The structure of the Escherichia coli EF-Tu. EF-Ts complex at 2.5 A resolution. Nature. 1996;379:511–518. doi: 10.1038/379511a0. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kunert A, Losse J, Gruszin C, Hühn M, Kaendler K, Mikkat S, Volke D, Hoffmann R, Jokiranta TS, Seeberger H, et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol. 2007;179:2979–2988. doi: 10.4049/jimmunol.179.5.2979. [DOI] [PubMed] [Google Scholar]
- Lathe WC, 3rd, Bork P. Evolution of tuf genes: ancient duplication, differential loss and gene conversion. FEBS Lett. 2001;502:113–116. doi: 10.1016/s0014-5793(01)02639-4. [DOI] [PubMed] [Google Scholar]
- LeRoux M, De Leon JA, Kuwada NJ, Russell AB, Pinto-Santini D, Hood RD, Agnello DM, Robertson SM, Wiggins PA, Mougous JD. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci USA. 2012;109:19804–19809. doi: 10.1073/pnas.1213963109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoux M, Kirkpatrick RL, Montauti EI, Tran BQ, Peterson SB, Harding BN, Whitney JC, Russell AB, Traxler B, Goo YA, et al. Kin cell lysis is a danger signal that activates antibacterial pathways of Pseudomonas aeruginosa. eLife. 2015;4:4. doi: 10.7554/eLife.05701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Le Trong I, Carl MA, Larson ET, Chou S, De Leon JA, Dove SL, Stenkamp RE, Mougous JD. Structural basis for type VI secretion effector recognition by a cognate immunity protein. PLoS Pathog. 2012;8:e1002613. doi: 10.1371/journal.ppat.1002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LS, Hachani A, Lin JS, Filloux A, Lai EM. Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microbe. 2014;16:94–104. doi: 10.1016/j.chom.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden JC, Ruiz N, Caparon M. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell. 2001;104:143–152. doi: 10.1016/s0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- Mohan S, Hertweck C, Dudda A, Hammerschmidt S, Skerka C, Hallström T, Zipfel PF. Tuf of Streptococcus pneumoniae is a surface displayed human complement regulator binding protein. Mol Immunol. 2014;62:249–264. doi: 10.1016/j.molimm.2014.06.029. [DOI] [PubMed] [Google Scholar]
- Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475:343–347. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature. 2013;496:508–512. doi: 10.1038/nature12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Peterson SB, Mougous JD. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature. 2013;500:350–353. doi: 10.1038/nature12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012;66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JM, Agnello DM, Zheng H, Andrews BT, Li M, Catalano CE, Gonen T, Mougous JD. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell. 2013;51:584–593. doi: 10.1016/j.molcel.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon NC, Aktories K, Barbieri JT. Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol. 2014;12:599–611. doi: 10.1038/nrmicro3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Ghosh J, Elam JS, Pinkner JS, Hultgren SJ, Caparon MG, Ellenberger T. Structural basis of Streptococcus pyogenes immunity to its NAD+ glycohydrolase toxin. Structure. 2011;19:192–202. doi: 10.1016/j.str.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Vogeley L, Palm GJ, Mesters JR, Hilgenfeld R. Conformational change of elongation factor Tu (EF-Tu) induced by antibiotic binding. Crystal structure of the complex between EF-Tu GDP and aurodox. J Biol Chem. 2001;276:17149–17155. doi: 10.1074/jbc.M100017200. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- Whitney JC, Beck CM, Goo YA, Russell AB, Harding BN, De Leon JA, Cunningham DA, Tran BQ, Low DA, Goodlett DR, et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 2014;92:529–542. doi: 10.1111/mmi.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SP, Jørgensen R, Andersen GR, Merrill AR. Stealth and mimicry by deadly bacterial toxins. Trends Biochem Sci. 2006;31:123–133. doi: 10.1016/j.tibs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, Burroughs AM, Aravind L. Resilience of biochemical activity in protein domains in the face of structural divergence. Curr Opin Struct Biol. 2014;26:92–103. doi: 10.1016/j.sbi.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.