

Abstract
While β-adrenergic receptor (β-AR) stimulation ensures adequate cardiac output during stress, it can also trigger life-threatening cardiac arrhythmias. We have previously shown that proarrhythmic Ca2+ waves during β-AR stimulation temporally coincide with augmentation of reactive oxygen species (ROS) production. In this study, we tested the hypothesis that increased energy demand during β-AR stimulation plays an important role in mitochondrial ROS production and Ca2+-wave generation in rabbit ventricular myocytes. We found that β-AR stimulation with isoproterenol (0.1 μM) decreased the mitochondrial redox potential and the ratio of reduced to oxidated glutathione. As a result, β-AR stimulation increased mitochondrial ROS production. These metabolic changes induced by isoproterenol were associated with increased sarcoplasmic reticulum (SR) Ca2+ leak and frequent diastolic Ca2+ waves. Inhibition of cell contraction with the myosin ATPase inhibitor blebbistatin attenuated oxidative stress as well as spontaneous SR Ca2+ release events during β-AR stimulation. Furthermore, we found that oxidative stress induced by β-AR stimulation caused the formation of disulfide bonds between two ryanodine receptor (RyR) subunits, referred to as intersubunit cross-linking. Preventing RyR cross-linking with N-ethylmaleimide decreased the propensity of Ca2+ waves induced by β-AR stimulation. These data suggest that increased energy demand during sustained β-AR stimulation weakens mitochondrial antioxidant defense, causing ROS release into the cytosol. By inducing RyR intersubunit cross-linking, ROS can increase SR Ca2+ leak to the critical level that can trigger proarrhythmic Ca2+ waves.
Introduction
The working myocardium highly depends on mitochondrial respiration to maintain a sufficient energy supply. At an increased workload, a higher rate of ATP hydrolysis stimulates oxidative phosphorylation, thus accelerating mitochondrial NADH oxidation and electron flux via the electron transport chain (ETC; Fig. 1). As a result, there is a higher rate of single electron leakage from the ETC and formation of superoxide anions (O2−). To prevent accumulation of mitochondrial reactive oxygen species (ROS), cardiomyocytes possess an elaborate antioxidant defense. This defense is composed of molecules with different complexity, ranging from small ROS scavengers to complex antioxidant enzymes (1). Reduced glutathione (GSH) plays a particularly important role in the mitochondrial antioxidant defense. GSH can prevent oxidative stress directly, by scavenging ROS, and indirectly, by acting as a substrate for glutathione peroxidase (GP). The high ratio of GSH to oxidized glutathione (GSSG) (GSH/GSSG) is maintained by the mitochondrial reducing equivalent of NADH, generated in the Krebs cycle. Antioxidant activity of another mitochondrial enzyme, peroxiredoxin (PR), relies on the same NADH pool. Thus, increased cardiac demand would not only elevate mitochondrial ROS production by the ETC, but could also reduce the ROS defense at the GP and PR levels due to the increased NADH consumption (Fig. 1). However, work-induced NADH depletion can be partially restored by the stimulation of Ca2+-dependent dehydrogenases of the Krebs cycle (2, 3).
Figure 1.

The mechanism of oxidative stress induced by increased workloads. Increase in workload is always associated with a higher ATP hydrolysis rate. Products of ATP hydrolysis ([ADP] and [Pi]) would stimulate mitochondrial oxidative phosphorylation and thus NADH consumption. This would increase electron flux via the ETC, leading to a higher rate of ROS production. NADH consumption by the ETC would also weaken the ROS defense at GP and PR levels. These changes in mitochondrial metabolism can lead to oxidative stress. Thus, inhibition of a myosin ATPase with blebbistatin should limit ROS production during increased workloads.
Under certain pathological conditions, increased mitochondrial ROS production can overwhelm the antioxidant defense, resulting in mitochondrial ROS spillover into the cytosol (1, 3, 4). In the cytosol, unopposed mitochondrial ROS can modify the function of different proteins by oxidizing their redox-sensitive cysteine residues. Among many ROS targets, the ryanodine receptor (RyR) plays a particularly important role in an initial cellular response to oxidative stress (5, 6, 7, 8). Foremost, alteration of RyR activity has a significant impact on Ca2+ homeostasis and myocardial contraction. Second, the RyR contains several highly redox-sensitive cysteine residues that can link oxidative stress and Ca2+ signaling (9). Although redox status of these cysteines affects RyR function (5, 6, 7, 8, 9), specific mechanisms of RyR dysfunction during oxidative stress remain largely unknown. There is strong structural evidence that RyR gating is associated with a large-scale intersubunit dynamic (10, 11, 12, 13). Previous work from our laboratory described, to our knowledge, a novel mechanism by which oxidative stress can affect RyR function in the heart. We found that oxidants increased RyR activity by promoting disulfide bond formation between two neighboring subunits, referred to as intersubunit cross-linking (14). Although the cross-linking has a significant impact on RyR activity, it remains less clear whether this novel posttranslation modification contributes to abnormal sarcoplasmic reticulum (SR) Ca2+ handling observed during pathological conditions associated with oxidative stress.
β-adrenergic receptor (β-AR) stimulation produces the most important inotropic effect in the heart. It is also known that excessive adrenergic stimulation can trigger life-threatening arrhythmias (15, 16, 17, 18). The transition from positive inotropy to arrhythmias during β-AR stimulation is mediated by several mechanisms, including the augmentation of RyR activity. It has been shown that abnormally increased RyR-mediated Ca2+ leak (19, 20, 21), particularly in the form of Ca2+ waves (22, 23), can generate arrhythmogenic delayed afterdepolarizations (24). β-AR stimulation also increases cellular metabolism and mitochondrial respiration. This can lead to a higher rate of mitochondrial ROS production (3, 4, 25). We have shown previously that the generation of proarrhythmic Ca2+ waves during β-AR stimulation is associated with oxidative stress and augmentation of SR Ca2+ leak (4). However, the specific molecular mechanisms that cause oxidative stress and SR Ca2+ leak during β-AR stimulation have yet to be characterized. In this study, we tested the hypothesis that increased energy demand during β-AR stimulation plays an important role in the mitochondria-mediated oxidative stress. We also tested whether this oxidative stress increases SR Ca2+ leak by promoting RyR cross-linking. Thus, we expected that reducing ATP consumption during β-AR stimulation would decrease mitochondrial ROS production, preventing subsequent RyR cross-linking and SR Ca2+ leak.
Materials and Methods
Myocyte isolation
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Loyola University and comply with U.S. regulations on animal experimentation. Ventricular myocytes were isolated from hearts of New Zealand White rabbits according to the procedure described previously (26). Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. All experiments were performed at room temperature (20–24°C).
Confocal microscopy
Changes in flavin adenine dinucleotide (FAD), ROS, and cytosolic and luminal free Ca2+ concentration ([Ca2+]i) were measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, Hertfordshire, United Kingdom, and LSM 410, Zeiss, Oberkochen, Germany) equipped with a 40× oil-immersion objective lens (NA 1.3). In all intact cell experiments, myocytes were electrically stimulated (field stimulation) using a pair of platinum electrodes connected to a Grass stimulator (Astro-Med, Warwick, RI). Action potentials (APs) and cell contraction were triggered by a voltage ∼50% above the threshold. The threshold was defined as the point at which half of the myocytes in the suspension contracted in response to an electrical pulse.
Measurements of endogenous FAD autofluorescence
The method for measuring FAD autofluorescence was described in our earlier publications (27, 28). FAD autofluorescence was excited at 488 nm and fluorescence was measured at wavelengths >515 nm. Two-dimensional (2-D) images were acquired at 15 s intervals. Recordings were made only during the diastolic phase to avoid motion artifacts caused by cell contraction. Changes in FAD fluorescence were presented as F/F0, where F0 is the basal FAD signal recorded under steady-state conditions at the beginning of an experiment.
Measurements of intracellular ROS level
ROS production was measured with an ROS-sensitive fluorescent dye, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Molecular Probes/Invitrogen, Carlsbad, CA) according to a previously described protocol (4). Myocytes were loaded with 20 μM H2DCFDA for 30 min at room temperature. H2DCFDA was excited by the 488 nm line of an argon laser and emitted fluorescence was collected at wavelengths >600 nm. 2-D images were acquired at 15 s intervals. Recordings were made only during the diastolic phase to avoid motion artifacts. Fluorescence intensity, F, was integrated over the entire volume of the cell. Because H2DCFDA irreversibly reacts with free oxygen radicals, ROS production was presented as the first derivative of the H2DCFDA signal (ΔF/Δt).
Measurements of [Ca2+]i
[Ca2+]i was measured with the high-affinity Ca2+ indicator Rhod-2 (Molecular Probes/Invitrogen). To load the cytosol with Ca2+ indicator, cells were incubated at room temperature with 10 μM Rhod-2/AM for 15 min in Tyrode solution (in mM, NaCl 140; KCl 4,CaCl2 2, MgCl2 1, glucose 10, and HEPES 10, pH 7.4), followed by a 20 min wash. Rhod-2 was excited with the 543 nm line of an argon laser and fluorescence was measured at >610 nm. Rhod-2 images were acquired in line-scan mode (3 ms per scan; pixel size 0.12 μm). Ca2+ transients were presented as background-subtracted normalized fluorescence (F/F0), where F0 is the fluorescence intensity during diastole. Ca2+ wave propensity was analyzed as the percentage of waves per cardiac cycle.
Ca2+ sparks were studied after membrane permeabilization with saponin (29). The saponin-free internal solution was composed of K aspartate 100 mM; KCl 15 mM, KH2PO4 5, MgATP 5 mM, EGTA 0.35 mM, CaCl2 0.12 mM, MgCl2 0.75 mM, phosphocreatine 10 mM, HEPES 10 mM, Rhod-2 pentapotassium salt 0.04 mM, creatine phosphokinase 5 U/mL, and dextran (40,000 mol wt) 8%, pH 7.2 (KOH). Free [Ca2+] and [Mg2+] of this solution were 150 nM and 1 mM, respectively. Ca2+ sparks were detected and analyzed using SparkMaster (30).
Measurements of SR Ca2+ leak
SR Ca2+ leak was measured according to the previously described protocol (31, 32). The SR was loaded with the low-affinity Ca2+ indicator Fluo-5N (Molecular Probes/Invitrogen), as described before (26, 33). Fluo-5N was excited with the 488 nm line of an argon laser and fluorescence was collected at >515 nm. The Fluo-5N fluorescence was collected with an open pinhole and averaged over the entire cellular width of 2-D images. [Ca2+]SR was calculated using Eq. 7: [Ca2+]SR = Kd × R/(Kd/[Ca2+]SR diast − R + 1), where R is the normalized Fluo-5N fluorescence (R = (F − Fmin)/(F0 − Fmin)); F0 and Fmin are the fluorescence level during diastole and after depletion of the SR with caffeine, respectively. Kd (the Fluo-5N Ca2+ dissociation constant) was 390 μM based on in situ calibrations (32) and diastolic [Ca2+]SR at 1 Hz was 1000 μM (26, 31, 34). SR Ca2+ leak was measured as the rate of total [Ca2+]SR ([Ca2+]SRT) decline during SR calcium transport ATPase (SERCA) inhibition with thapsigargin (10 μM). [Ca2+]SRT was calculated as [Ca2+]SRT = Bmax/(1 + Kd/[Ca2+]SR) + [Ca2+]SR; where Bmax and Kd were 2700 μM and 630 μM, respectively (35).
Western blotting and RyR cross-linking
Alteration in the electrophoretic mobility of the 560 kDa subunit of RyR was used to detect the RyR cross-linking level (14). After different experimental protocols, myocytes were lysed in nonreducing Laemmli buffer containing N-ethylmaleimide (NEM, 5 mM) to block free sulfhydryl groups. The addition of 200 mM dithiothreitol was used to confirm reversibility of RyR cross-linking and to be used as a reference for total RyR. After incubating at 70°C for 10 min, samples were run on 4–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membrane using the Turbo-transfer system (Bio-Rad). Immunoblot against RyR was carried out using the monoclonal 34C primary antibody (1:1000; Developmental Studies Hybridoma Bank, Iowa City, IA) and anti-mouse HRP-conjugated secondary antibody (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA). Western blots were quantified using the UVP EpiChemi imaging system and ImageJ software. Relative RyR cross-linking was analyzed as (Total RyR − Monomeric RyR)/(Total RyR).
Measurements of the GSH/GSSG level
The GSH/GSSG ratio was measured in control conditions and after β-AR stimulation. After different experimental protocols, cells were quickly settled down and homogenized using a bead-beater homogenizer (Biospec Products, Bartlesville, OK). The addition of 5-sulfo-salicylic acid dihydrate (5% w/v) was used to precipitate protein from homogenate samples. Samples were centrifuged at 14,000 rpm at 4°C for 10 min. The resulting supernatant was used for analysis of GSH/GSSG content. The concentration of GSH was defined using the fluorometric glutathione detection assay DetectX (Arbor Assays, Ann Arbor, MI), utilizing a fluorescent label that covalently binds to GSH. The sample fluorescence was measured using a fluorometer (OLIS DM 45, On-Line Instrument Systems, Athens, GA) at an excitation/emission of 390/510 nm. A standard plot of known GSH concentrations was developed before each set of experiments. After GSH fluorescence was determined for each sample, the subsequent reduction of GSSG by glutathione reductase would yield the total GSH fluorescence. Absolute concentrations were determined based on the linear fit of the standard fluorescence. Absolute GSSG concentration was quantified as (Total GSH − Free GSH)/2. The values presented are percentage changes in GSH/GSSG.
Statistical analysis
Data were presented as the mean ± SE of n measurements. In the case of single-cell experiments (such as FAD, ROS, and Ca measurements), n represents the number of cells isolated from at least three different animals. In the case of RyR cross-linking and GSH/GSSG measurements, n represents the number of animals used in these experiments. When only two groups were compared, the statistical significance was determined by Student’s t-test. Significance between multiple groups was determined by one-way analysis of variance followed by a Newman-Keuls post hoc test. p < 0.05 was considered statistically significant.
Results
β-AR stimulation decreases mitochondrial redox potential and increases ROS formation
Within the mitochondrial matrix, there is an equilibrium between the redox potential of NADH/NAD+ and FADH2/FAD pools (36). Thus, NADH or FAD autofluorescence can be used as an index of the redox state of the mitochondria. In contrast to an NADH signal, which originates from both the cytosol and mitochondria, FAD autofluorescence originates primarily from mitochondria (37). Thus, we measured changes of the FAD signal as the index of mitochondrial redox state. Application of isoproterenol (ISO; 0.1 μM) initially decreased the FAD signal in electrically stimulated (at 0.75 Hz) myocytes (Fig. 2 A, black circles), indicating an increase of the mitochondrial reducing power. This can be explained by increased activity of Ca2+-dependent dehydrogenases of the Krebs cycle, which enhance NADH synthesis (2, 3). However, after ∼6 min of ISO application, the FAD signal gradually increased, suggesting that the mitochondrial redox potential became more oxidized. Cyanide (CN−) and FCCP were applied in series at the end of each experiment to define maximum reduction and oxidation, respectively, of FADH2. Inhibition of cell contraction with a myosin ATPase inhibitor, blebbistatin (5 μM), abolished the late rising phase of FAD signal during β-AR stimulation (Fig. 2 A, gray squares). The relative changes of the FAD signal induced by ISO in control conditions and in the presence of blebbistatin are summarized in Fig. 2 B. In parallel experiments, the GSH/GSSG ratio was measured in control conditions and after 12 min β-AR stimulation. In these experiments, myocytes were continuously stimulated at 0.75 Hz before homogenization. We found that a prolonged β-AR stimulation decreased the GSH/GSSH ratio by 22% (n = 5; Fig. 2 C).
Figure 2.

Effects of β-AR stimulation on FAD autofluorescence and the GSH/GSSG ratio. (A) Changes of FAD signal during ISO (0.1 μM) application in control conditions (black circles) and in the presence of 5 μM blebbistatin (Bleb; gray squares). Myocytes were electrically stimulated at 0.75 Hz. At the end of each experiment, CN− (4 mM) and FCCP (1 μM) were applied to estimate maximum reduction and oxidation, respectively, of the FADH2/FAD couple. The FAD signal was normalized to CN− and FCCP levels. (B) Changes in the FAD signal during ISO application in control conditions (n = 11 cells from 4 animals) and in the presence of blebbistatin (n = 10 cells from 4 animals). (C) Changes of the GSH/GSSG ratio during ISO application (n = 5 animals). ∗p < 0.05 versus control.
In the next experiments, we studied whether an increase in energy demand during β-AR stimulation contributes to mitochondrial ROS production. The intracellular ROS level was measured using an ROS-sensitive fluorescent dye, H2DCFDA. We found that ISO (0.1 μM) application increased the rate of ROS production in electrically stimulated myocytes (Fig. 3 A, black circles). We have previously shown that the mitochondria-targeted ROS scavenger Mito-Tempo or the ETC inhibitor rotenone can prevent oxidative stress induced by ISO (4). Here, we found that β-AR-mediated ROS production can be also prevented by blebbistatin (Fig. 3 A, gray squares). The average data for the blebbistatin effect on ROS production are illustrated in Fig. 3 B. Oxidative stress induced by ISO positively correlated with the time needed for β-AR stimulation to increase the FAD signal (Fig. 2 A). On the contrary, inhibition of nitric oxide synthase (NOS) with L-NAME (100 μM) did not prevent an increase in H2DCFDA fluorescence in the presence of ISO (data not shown). These results suggest that increased energy demand during prolonged β-AR activation decreases the mitochondrial redox potential and thus the ROS defense. As a result, β-AR activation can lead to mitochondrial ROS release into the cytosol.
Figure 3.

Effects of β-AR stimulation on ROS production in the presence of blebbistatin. (A) Changes in the H2DCFDA signal during ISO (0.1 μM) application in control conditions (black circles) and in the presence of 5 μM blebbistatin (Bleb; gray squares). Myocytes were electrically stimulated at 0.75 Hz. At the end of each experiment, H2O2 (1 mM) was applied to estimate the maximal rate of ROS production. (B) Relative changes in the ROS production rate during ISO application in control conditions (n = 12 cells from 5 animals) and in the presence of blebbistatin (n = 10 cells from 5 animals). ∗p < 0.05 versus control.
Limiting energy demand decreases the propensity of Ca2+ waves during β-AR stimulation
β-AR stimulation increased the systolic Ca2+ transient amplitude, but it also increased the propensity of diastolic Ca2+ waves. Although the positive inotropic effect reached a maximum within 3 min, Ca2+ waves did not appear until 6 min after ISO application (Fig. 4 A). We have shown previously that the generation of Ca2+ waves during β-AR stimulation temporally coincides with ROS formation (4). Since a decrease in energy demand with blebbistatin reduces mitochondrial ROS production (Fig. 3), we tested whether blebbistatin can prevent the occurrence of Ca2+ waves during ISO application. Fig. 4 A shows the effect of ISO (0.1 μM) on Ca2+ transients in control conditions (upper) and after inhibition of cell contraction with blebbistatin (lower). We found that pretreatment of myocytes with blebbistatin (5 μM) significantly reduced the occurrence of Ca2+ waves in the presence of ISO (Fig. 4 B). This protective effect of blebbistatin was associated with inhibition of SR Ca2+ leak (Fig. 4 C). SR Ca2+ leak was measured as the rate of [Ca2+]SR decline (with Fluo-5N) during SERCA inhibition (14, 31, 32). At the same time, blebbistatin did not affect the Ca2+ transient amplitude: ΔF/F0 was 2.3 ± 0.2 (n = 18) in control conditions and 2.2 ± 0.2 (n = 12) after cell incubation with blebbistatin. Also, blebbistatin did not limit the stimulatory effect of ISO on Ca2+ transient amplitude (Fig. 4 A) and SR Ca2+ load (Fig. 4 D). We also studied the effect of blebbistatin on spontaneous Ca2+ sparks in saponin-permeabilized myocytes. This protocol allowed us to study SR Ca2+ release under conditions of very limited energy demand, since cytosolic [ATP] was kept constant in these experiments. We found that application of blebbistatin (5 μM) did not affect Ca2+ spark properties in control conditions (Fig. 5; n = 8 cells from 3 animals) and after stimulation of Ca2+ sparks with 5 μM cAMP (n = 7 cells from 3 animals; data not shown), indicating that blebbistatin by itself does not modulate SR Ca2+ handling. There was some tendency for Ca2+ sparks to decrease over time. This decline is presumably caused by washout of some important cytosolic molecules, not by the blebbistatin effect. Thus, these results suggest that blebbistatin suppresses Ca2+ waves during β-AR stimulation mainly by reducing energy demand and thus limiting mitochondria-mediated oxidative stress.
Figure 4.

Effects of β-AR stimulation on SR Ca2+ handling in the presence of blebbistatin. (A) Action-potential (AP)-induced Ca2+ transients (F/F0) recorded with Rhod-2 in electrically stimulated (at 0.75 Hz) myocytes. In two separate experiments, the effect of ISO on Ca2+ transients (after 3 and 12 minutes) was studied in control conditions (upper) and in the presence of 5 mM blebbistatin (lower). In the case of Ca2+ waves, the asterisks were used to indicate AP-induced Ca2+ transients. (B–D) Average effects of ISO on Ca2+ wave frequency (B), SR Ca2+ leak (C), and SR Ca2+ load (D) in control conditions and in the presence of blebbistatin. Ca2+ wave propensity was presented as numbers of waves per cardiac cycle. In Ca2+ wave experiments (B), 18 cells were analyzed in control conditions and 12 cells in the presence of blebbistatin (6 animals were used). SR Ca2+ load was measured from the peak of the Ca2+ transient induced by the application of 10 mM caffeine. SR Ca2+ leak was measured with the low-affinity Ca2+ dye Fluo-5N within the SR (see Materials and Methods). In Ca2+ leak/load experiments (C and D), 14 cells were analyzed in control conditions, 12 in the presence of ISO and 9 in the presence of ISO + blebbistatin (5 animals were used). ∗p < 0.05 versus control; #p < 0.05 ISO versus ISO + Bleb.
Figure 5.

Effect of blebbistatin on Ca2+ sparks in permeabilized myocytes. Confocal line-scan images of Rhod-2 fluorescence recorded in control conditions and in the presence of 5 μM blebbistatin (upper). Effects of blebbistatin on Ca2+ spark frequency, amplitude, and width (lower).
β-AR stimulation promotes RyR cross-linking
We have recently shown that oxidative stress increases RyR-mediated Ca2+ leak by promoting RyR intersubunit cross-linking (14). In this study, we performed Western blot experiments with nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis to measure relative changes in RyR cross-linking level before and after β-AR stimulation. Before homogenization, myocytes were electrically stimulated at 0.75 Hz. In control conditions, RyR was always detected as a single band with a molecular mass of ∼560 kDa (Fig. 6 A, first lane). After treating cells for 15 min with ISO (0.1 μM), RyR can be detected as two separate bands: as a monomer and as a dimer with a molecular mass of ∼1 MDa (Fig. 6 A, second lane). On average, ISO increased the fraction of cross-linked RyR by 42.4 ± 10.5% (n = 4 animals). ISO did not produce a significant RyR cross-linking in quiescent myocytes or myocytes treated with blebbistatin (Fig. 6 A, third lane). Inhibition of mitochondrial ROS production by rotenone (Rot; 5 μM) partially prevented RyR cross-linking during β-AR stimulation (Fig. 6 A, fourth lane). The RyR cross-linking was reversed with the reducing agent dithiothreitol or was prevented by blocking free thiols with 100 μM NEM (Fig. 6 B). Because intersubunit dynamics within the RyR complex determines the channel gating (10, 11, 12), we suggested that RyR cross-linking should play an important role in SR Ca2+ mishandling during β-AR stimulation.
Figure 6.
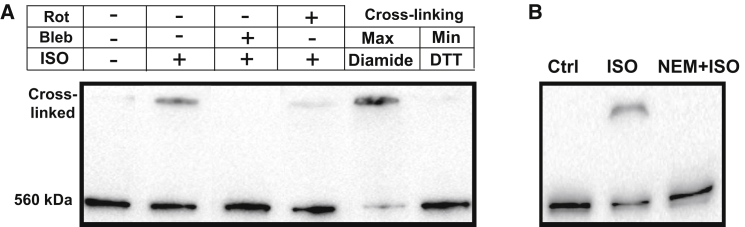
Effects of β-AR stimulation on RyR cross-linking. (A) Representative Western immunoblots (nonreducing) against RyR from control myocytes, myocytes treated with 0.1 μM ISO, myocytes treated with ISO in the presence of 5 μM blebbistatin (Bleb), and myocytes treated with ISO in the presence of 4 μM rotenone (Rot). Myocytes were electrically stimulated (at 0.75 Hz) before homogenization. Maximal and minimal RyR cross-linking were obtained after treatment of cells with diamide (100 μM) and dithiothreitol (200 mM), respectively. (B) Western immunoblots (nonreducing) against RyR from control myocytes, myocytes treated with ISO, and myocytes treated with ISO in the presence of 100 μM NEM.
To define the contribution of RyR cross-linking to the generation of Ca2+ waves during β-AR stimulation, we studied the effects of ISO on SR Ca2+ release in the presence of an NEM concentration that effectively prevents RyR cross-linking (Fig. 6 B). Fig. 7 A shows representative examples of AP-induced Ca2+ transients recorded 6 min after treatment with 100 μM NEM and after subsequent ISO application. The propensity of Ca2+ waves induced by ISO decreased from 54 ± 6% (n = 18 cells) in control cells to 10 ± 8% (n = 10 cells from 4 animals) after treatment of cells with NEM (Fig. 7 C). At the same time, NEM did not limit the positive inotropic effect during ISO application. These results indicate that the blocking of redox-mediated RyR cross-linking can normalize Ca2+ dynamics during β-AR activation. Contrarily, inhibition of ROS with L-NAME (100 μM) did not prevent Ca2+ waves during ISO application, but rather increased the frequency of these spontaneous SR Ca2+-release events (Fig. 7, B and C; n = 7 cells from 3 animals).
Figure 7.

Effects of β-AR stimulation on Ca2+ waves in the presence of NEM and L-NAME. (A) Effect of ISO on AP-induced Ca2+ transients (F/F0) in the presence of 100 μM NEM. (B) Effect of ISO on AP-induced Ca2+ transients (F/F0) in the presence of 100 μM L-NAME. In the case of Ca2+ waves, asterisks were used to indicate AP-induced Ca2+ transients. (C) Average effect of ISO on Ca2+ wave frequency in control conditions and in the presence of NEM and L-NAME. ∗p < 0.05 versus control.
Discussion
In the working heart, mitochondria are constantly producing free oxygen radicals as a result of single electron leakage from the ETC. It has been suggested that up to 2% of electrons flowing through the ETC do not reach their terminal point at complex IV, but instead react with oxygen to produce O2− (38). There are several lines of antioxidant defense in the mitochondria, however, that maintain ROS at low levels. This keeps ROS highly restricted to mitochondria. The antioxidant defenses include reduced coenzyme Q, cytochrome c, type 2 superoxide dismutase, catalase, GP, and PR (1, 38). Activity of the latter two enzymes is highly dependent on the mitochondrial redox potential, which is determined by the ratio of NADH and NADPH to their oxidized equivalents. In the unstressed heart, a balance between ROS production and scavenging maintains an optimal intracellular redox status. Thus, by working in the optimal redox environment, ion channels and pumps can maintain regular Ca2+ cycling and cardiac contraction.
Enhanced mitochondrial respiration during increased workloads, however, can shift the intracellular redox balance toward its oxidation (2, 39). It is widely accepted that a high rate of ATP hydrolysis during increased workloads stimulates electron flux via the ETC. This will not only increase mitochondrial ROS formation due to electron leakage but will also reduce the mitochondrial ROS defense by consuming more NADH and NADPH for ATP production (Fig. 1). Our measurements of FAD and ROS dynamics, however, revealed that the positive inotropic effect of β-AR stimulation caused an initial increase of the mitochondrial redox potential (Fig. 2). This effect can be explained by increased mitochondrial Ca2+ turnover that stimulates Ca2+-dependent dehydrogenases of the Krebs cycle and thus NADH production (2, 3). As a result, we did not detect any significant increase in ROS formation in the beginning of the β-AR action. These results suggest that the mitochondrial antioxidant defense is sufficient to prevent ROS accumulation during a short period of increased workload. Prolonged β-AR stimulation, however, increased ATP hydrolysis and NADH consumption, which subsequently weakened the mitochondrial antioxidant defense. Indeed, we found that after 12 min of β-AR stimulation at constant electrical pacing, the mitochondrial redox potential starts to decline. This time point was associated with a decrease in the GSH/GSSG ratio (Fig. 2). Although mitochondrial NADH can be partially recovered due to Ca2+-dependent mechanisms (2), it seems that this recovery cannot match the rate of NADH oxidation by the ETC. Consequently, this compromises the antioxidant activity of GP and PR, leading to mitochondrial ROS release into the cytosol (Fig. 3). To determine if energy demand is a major cause in initiating oxidative stress during β-AR stimulation, we used the myosin ATPase inhibitor blebbistatin as an experimental tool to decrease energy consumption by the contractile apparatus. In accordance with previous studies (40), blebbistatin effectively inhibited cell contraction without significant effect on Ca2+ handling. We found that inhibition of cell contraction prevents the decline of mitochondrial redox potential (Fig. 2) as well as the increase in ROS formation (Fig. 3) during β-AR stimulation. These findings suggest that β-AR-mediated oxidative stress is a result of increased ATP consumption, but not Ca2+ turnover. These findings are in good agreement with previously published work showing that an increase in energy demand at high pacing rates (>2 Hz) can deplete NADH and increase ROS production (39). On the contrary, another study showed that β-AR stimulation neither decreased NADH nor promoted oxidative stress (3). The authors of the latter study suggested that Ca2+-dependent mechanisms can effectively prevent the mitochondrial NADH depletion and ROS production. The discrepancy between these studies can be explained by the fact that in the latter publication, NADH and ROS measurements were conducted in patch-clamped myocytes perfused with an ATP-containing solution. These conditions would significantly minimize energy demand and NADH consumption during β-AR stimulation.
In the cytosol, mitochondrial ROS can affect the function of a variety of proteins, including those that are involved in SR Ca2+ handling. Typically, sulfhydryl groups of cysteine residues are the main targets for redox modification of ion channels and pumps (8). Because the RyR contains a large number of cysteines (9), it is no surprise that the RyR play a key role in the cellular response to oxidative stress. Here, we focused on a novel redox modification of the RyR that is particularly relevant to SR Ca2+ mishandling during oxidative stress. In our recent publication, we found that activation of RyRs during oxidative stress is mediated by the formation of disulfide bonds between two neighboring subunits, i.e., intersubunit cross-linking (14). Because prolonged β-AR stimulation is associated with increased ROS production and RyR thiol oxidation (4), we suggested that this novel posttranslation modification should play an important role in redox-mediated RyR dysfunction during β-AR stimulation. Indeed, we found that β-AR stimulation induces RyR cross-linking (Fig. 6), similar to what was measured after treatment of cells with the thiol oxidant diamide (14). Inhibiting mitochondrial ROS production with rotenone or reducing ATP consumption with blebbistatin effectively prevented β-AR-mediated RyR cross-linking (Fig. 6 A). Consequently, pretreating myocytes with blebbistatin partially normalized SR Ca2+ leak and decreased Ca2+-wave frequency during β-AR stimulation (Fig. 4). This protective effect of blebbistatin was not due to reduced SR Ca2+ load, since Ca2+ load became even more elevated in the presence of blebbistatin (Fig. 4 D). Similar to the blebbistatin effect, blocking cysteine sulfhydryl groups on the RyR with NEM prevented both RyR cross-linking and Ca2+ waves during β-AR stimulation (Fig. 7). These findings suggest that cross-linking acts as an important posttranslation modification of RyR that transforms SR Ca2+ release during β-AR activation from inotropic to arrhythmogenic. However, this transition can only occur at elevated SR Ca2+ loads, since RyR cross-linking alone was not sufficient to produce Ca2+ waves in ventricular myocytes (14).
It was reported previously that augmentation of SR Ca2+ leak during β-AR stimulation is the result of increased nitric oxide (NO) production, CaMKII activation by NO, and consecutive RyR phosphorylation by CaMKII (41). In our previous publication, we found a significant level of RyR phosphorylation at the CaMKII site after a short period of electrical stimulation in control conditions (4). We also detected a small increase in CaMKII-RyR phosphorylation after prolonged β-AR stimulation. Because H2DCFDA can react with nitrogen free radicals (42), the observed increase in H2DCFDA signal during β-AR activation (Fig. 3) may be a result of stimulation of NO production. However, we found that the NOS inhibitor L-NEMA was ineffective in preventing the increase in H2DCFDA signal, whereas the ROS scavengers MnTBPA and Mito-Tempo abolished it (4). Moreover, the NOS inhibitor did not decrease the frequency of Ca2+ waves during β-AR activation, but instead made them more frequent (Fig. 7). This finding is in agreement with the idea that basal NO production and protein S-nitrosylation play a protective role against further oxidation by ROS (43). It has been reported that a decrease in the RyR S-nitrosylation level makes the channel more susceptible to activation during oxidative stress (44, 45). Thus, the observed increase in RyR phosphorylation at the CaMKII site can be explained by Ca2+-independent activation of CaMKII by ROS (46).
Conclusion
In conclusion, this study describes, to our knowledge, a novel mechanism that plays an important role in the generation of proarrhythmic Ca2+ waves during β-AR stimulation. Namely, an increase in energy demand during prolonged β-AR stimulation weakens the mitochondrial antioxidant defense that leads to a release of mitochondrial ROS into the cytosol. This mitochondria-related oxidative stress in turn can increase SR Ca2+ leak by promoting intersubunit cross-linking within the RyR tetramer. The increased SR Ca2+ leak would further enhance energy demand, because more ATP will be used by SERCA to maintain the SR Ca2+ load. This self-amplification process (i.e., energy demand → ROS → Ca2+ leak → energy demand) will ultimately increase RyR activity to a critical level that can produce Ca2+ waves. This mechanism can be particularly important in developing oxidative stress and SR Ca2+ mishandling in the failing heart, because this clinical syndrome is commonly associated with an increased adrenergic tone.
Author Contributions
All authors (E.B., S.R.M., P.d.T., and A.V.Z.) contributed to the conception and design of the study, interpretation of the data, and writing of the manuscript. E.B. and S.R.M. performed the experimental work and analyzed the results. All authors have approved the version to be published.
Acknowledgment
This work was supported by National Institutes of Health grants HL62426 and HL75494 to P.d.T., a Research Career Development Award from the Schweppe Foundation and an RFC grant from Loyola University Chicago to A.V.Z., and an American Heart Association fellowship grant to E.B.
Editor: Mark Cannell.
References
- 1.Turrens J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandes R., Bers D.M. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ. Res. 1997;80:82–87. doi: 10.1161/01.res.80.1.82. [DOI] [PubMed] [Google Scholar]
- 3.Kohlhaas M., Liu T., Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bovo E., Lipsius S.L., Zima A.V. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hool L.C., Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2007;9:409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- 6.Kourie J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998;275:C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki Y.J., Ford G.D. Redox regulation of signal transduction in cardiac and smooth muscle. J. Mol. Cell. Cardiol. 1999;31:345–353. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 8.Zima A.V., Blatter L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 9.Xu L., Eu J.P., Stamler J.S. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 10.Orlova E.V., Serysheva I.I., Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat. Struct. Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 11.Serysheva I.I., Ludtke S.J., Chiu W. Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc. Natl. Acad. Sci. USA. 2008;105:9610–9615. doi: 10.1073/pnas.0803189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tung C.C., Lobo P.A., Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 13.Zalk R., Clarke O.B., Marks A.R. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazurek S.R., Bovo E., Zima A.V. Regulation of sarcoplasmic reticulum Ca2+ release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic. Biol. Med. 2014;68:159–167. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Katra R.P., Oya T., Laurita K.R. Ryanodine receptor dysfunction and triggered activity in the heart. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H2144–H2151. doi: 10.1152/ajpheart.00924.2006. [DOI] [PubMed] [Google Scholar]
- 16.Myles R.C., Wang L., Ripplinger C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam G.B., Burashnikov A., Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venetucci L.A., Trafford A.W., Eisner D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 19.Curran J., Hinton M.J., Shannon T.R. β-Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 20.Ellison G.M., Torella D., Nadal-Ginard B. Acute β-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J. Biol. Chem. 2007;282:11397–11409. doi: 10.1074/jbc.M607391200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogrodnik J., Niggli E. Increased Ca2+ leak and spatiotemporal coherence of Ca2+ release in cardiomyocytes during β-adrenergic stimulation. J. Physiol. 2010;588:225–242. doi: 10.1113/jphysiol.2009.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curran J., Brown K.H., Shannon T.R. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venetucci L.A., Trafford A.W., Eisner D.A. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ. Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 24.Schlotthauer K., Bers D.M. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ. Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 25.Andersson D.C., Fauconnier J., Westerblad H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J. Physiol. 2011;589:1791–1801. doi: 10.1113/jphysiol.2010.202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domeier T.L., Blatter L.A., Zima A.V. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J. Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zima A.V., Copello J.A., Blatter L.A. Effects of cytosolic NADH/NAD+ levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J. Physiol. 2004;555:727–741. doi: 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zima A.V., Kockskämper J., Blatter L.A. Pyruvate modulates cardiac sarcoplasmic reticulum Ca2+ release in rats via mitochondria-dependent and -independent mechanisms. J. Physiol. 2003;550:765–783. doi: 10.1113/jphysiol.2003.040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zima A.V., Picht E., Blatter L.A. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of Ca release. Biophys. J. 2008;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picht E., Zima A.V., Bers D.M. SparkMaster: automated calcium spark analysis with ImageJ. Am. J. Physiol. Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 31.Bovo E., de Tombe P.P., Zima A.V. The role of dyadic organization in regulation of sarcoplasmic reticulum Ca2+ handling during rest in rabbit ventricular myocytes. Biophys. J. 2014;106:1902–1909. doi: 10.1016/j.bpj.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zima A.V., Bovo E., Blatter L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zima A.V., Picht E., Blatter L.A. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon T.R., Guo T., Bers D.M. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 35.Shannon T.R., Ginsburg K.S., Bers D.M. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys. J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vuorinen K.H., Ala-Rämi A., Hassinen I.E. Respiratory control in heart muscle during fatty acid oxidation. Energy state or substrate-level regulation by Ca2+? J. Mol. Cell. Cardiol. 1995;27:1581–1591. doi: 10.1016/s0022-2828(95)90458-1. [DOI] [PubMed] [Google Scholar]
- 37.Hassinen I., Chance B. Oxidation-reduction properties of the mitochondrial flavoprotein chain. Biochem. Biophys. Res. Commun. 1968;31:895–900. doi: 10.1016/0006-291x(68)90536-6. [DOI] [PubMed] [Google Scholar]
- 38.Balaban R.S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Heinzel F.R., Luo Y., Heusch G. Formation of reactive oxygen species at increased contraction frequency in rat cardiomyocytes. Cardiovasc. Res. 2006;71:374–382. doi: 10.1016/j.cardiores.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Farman G.P., Tachampa K., de Tombe P.P. Blebbistatin: use as inhibitor of muscle contraction. Pflugers Arch. 2008;455:995–1005. doi: 10.1007/s00424-007-0375-3. [DOI] [PubMed] [Google Scholar]
- 41.Curran J., Tang L., Shannon T.R. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One. 2014;9:e87495. doi: 10.1371/journal.pone.0087495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Possel H., Noack H., Wolf G. 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Lett. 1997;416:175–178. doi: 10.1016/s0014-5793(97)01197-6. [DOI] [PubMed] [Google Scholar]
- 43.Sun J., Murphy E. Protein S-nitrosylation and cardioprotection. Circ. Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cutler M.J., Plummer B.N., Laurita K.R. Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc. Natl. Acad. Sci. USA. 2012;109:18186–18191. doi: 10.1073/pnas.1210565109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez D.R., Treuer A.V., Hare J.M. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erickson J.R., Joiner M.L., Anderson M.E. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]